

Abstract
Metastatic breast cancer is a leading cause of cancer-related deaths in women worldwide. Patients with triple negative breast cancer (TNBCs), a highly aggressive tumor subtype, have a particularly poor prognosis. Multiple reports demonstrate that altered content of the multicopy mitochondrial genome (mtDNA) in primary breast tumors correlates with poor prognosis. We earlier reported that mtDNA copy number reduction in breast cancer cell lines induces an epithelial-mesenchymal transition associated with metastasis. However, it is unknown whether the breast tumor subtypes (TNBC, Luminal and HER2+) differ in the nature and amount of mitochondrial defects and if mitochondrial defects can be used as a marker to identify tumors at risk for metastasis. By analyzing human primary tumors, cell lines and the TCGA dataset, we demonstrate a high degree of variability in mitochondrial defects among the tumor subtypes and TNBCs, in particular, exhibit higher frequency of mitochondrial defects, including reduced mtDNA content, mtDNA sequence imbalance (mtRNR1:ND4), impaired mitochondrial respiration and metabolic switch to glycolysis which is associated with tumorigenicity. We identified that genes involved in maintenance of mitochondrial structural and functional integrity are differentially expressed in TNBCs compared to non-TNBC tumors. Furthermore, we identified a subset of TNBC tumors that contain lower expression of epithelial splicing regulatory protein (ESRP)-1, typical of metastasizing cells. The overall impact of our findings reported here is that mitochondrial heterogeneity among TNBCs can be used to identify TNBC patients at risk of metastasis and the altered metabolism and metabolic genes can be targeted to improve chemotherapeutic response.
Keywords: Mitochondrial DNA copy number, Mitochondrial DNA sequence imbalance, Metabolic gene expression, Triple negative breast cancer, ESRP1
1. Introduction
Metastatic breast cancer accounts for the highest mortalities in women worldwide [1]. It is estimated that 20–30% of all newly diagnosed breast cancers progress to metastatic breast cancer in which case the median survival is < 3 years. Furthermore, tumor heterogeneity and the existence of several molecular subtypes of breast tumors [2–6] make it difficult to design individualized therapeutics, and to identify patients at risk to develop metastatic disease. While the response to targeted therapies for receptor positive tumors (ER+, PR+, HER2+) has improved significantly, 10–15% patients with the highly aggressive triple negative tumors (TNBC: ER−, PR−, HER2−) have not benefitted from current therapies which emphasize the need for identifying effective drug targets in TNBCs [7]. While transcriptional (oncogene activation/suppressor gene loss) and environmental (local hypoxia, oxidative stress) changes drive progression within the primary tumor, such environmental stressors also select for cells that have adapted to these changes by altering their metabolism to meet nutrient availability. Mitochondria, principle regulators of these metabolic changes, are now recognized as the cellular stress-sensors that promote adaptation during tumorigenic transition [8]. However, such stress also promotes the acquisition of mitochondrial defects that in turn exacerbate stress factors favoring metastasis. Mutations in mitochondrial genes and mtDNA as well as reduced mtDNA copy number often reported in cancer cells do not completely inactivate mitochondrial functions but they alter the mitochondrial bioenergetics, metabolic and biosynthetic state to adapt to the substrate requirements of the tumor cell [9–15]. It has been well documented that while reduced mtDNA copy number and impaired mitochondrial functions are associated with tumorigenicity, complete loss of mitochondrial functions, such as elimination of mitochondrial respiration, inhibits tumor cell growth [12,16–20].
The number of mitochondria and copies of the mitochondrial genome per cell varies according to the tissue type and energy demand [21]. Numerous cellular and environmental factors including reactive free radicals, the hypoxic environment within solid tumors and defects in mtDNA transcriptional machinery (such as mtPOLG and TFAM mutations/deletions) result in loss of mtDNA copy number. Ironically, many chemotherapeutic drugs targeting primary tumors, including doxorubicin and other anthracycline derivatives, damage mtDNA and impair mitochondrial functions among breast cancer patients [22]. Reports suggest over half of all breast cancer patients have markedly reduced mtDNA copy number [23–29]. A study observed that the 5 year-disease free survival post-surgery for patients without adjuvant chemotherapy was only 59% with low mtDNA content compared to 91.7% for patients with high mtDNA content [30]. However, whether mitochondrial genome defects are more prevalent in any particular subtype of breast tumors has not been investigated.
Over 90% of primary breast tumors undergo cellular reprogramming during metastasis involving an epithelial-to-mesenchymal transition (EMT) which gives rise to tumor initiating breast cancer stem cells (brCSCs) and disseminated tumor cells in the bone marrow (BM-DTCs) with self-renewal potential that drive metastasis [31–33]. We previously demonstrated that 70–80% reduction in mtDNA copy number impaired cellular respiration in breast cancer cell line MCF7 as well as normal human mammary epithelial cell line MCF10A and activates a mitochondria-to-nucleus retrograde signal leading to cellular and transcriptional reprogramming similar to an EMT [34]. These effects were reversed by restoring mtDNA content, indicating that changes in mtDNA content are a signal of metastatic transition [34]. We also reported that experimentally reducing mtDNA copy number diminished the expression of the epithelial splicing regulatory protein-1 (ESRP1) suggestive of changes in gene splicing favoring metastasis [34]. Notably, ectopic expression of ESRP1 in low mtDNA cells reversed the EMT suggesting that mtDNA loss induced EMT in cell lines is mediated at least in part via loss of ESRP1 [34].
Based on our previous observations that mtDNA loss induces EMT and a metastatic phenotype in mammary cells [34], combined with the reported molecular tumor heterogeneity and variability in treatment responses among patients, we hypothesized that the different breast tumor subtypes have varying patterns of mitochondrial genome and functional defects which possibly determines their metastatic potential and response to therapy. However, there is no report analyzing the exact nature of mitochondrial defects in primary tumors of TNBC and non-TNBC (Luminal and HER2+) human patients. To test this hypothesis, we first analyzed The Cancer Genome Atlas (TCGA) breast cancer dataset, and subsequently validated our findings using primary breast tumors from an ethnically diverse patient population as well as human breast cancer cell lines from the four tumor subtypes (TNBC and non-TNBC). Here, we report that in primary tumors, mitochondrial defects (mtDNA copy number reduction, low basal respiratory capacity and mtDNA sequence imbalance) are more pronounced in the highly aggressive triple negative tumors compared to other subtypes, and low mtDNA content in primary tumors reported in TCGA correlates with tumor metastasis. We observed that TNBCs showed marked variability in mitochondrial metabolism gene expression compared to non-TNBCs, indicating that mitochondrial defects and loss of ESRP1 expression are molecular markers associated with aggressive TNBCs. Interestingly, we observed that the glycolytic flux is higher in TNBC cells compared to LumA and HER2+ cells and the in vivo tumorigenicity in a xenograft model correlated with the mitochondrial dysfunction and higher glycolysis. Understanding the fundamental mitochondrial and metabolic differences among breast tumors, is critical to new advances in precision medicine. We report here: (1) unique mitochondrial aberrations in TNBCs which can potentially serve as a diagnostic marker of TNBC metastasis (2) a novel mtDNA-RNRl ratiometric sequence imbalance in TNBCs. In future, mtDNA defects and altered metabolic profile can serve as new molecular targets to design effective chemotherapeutics for TNBC patients.
2. Materials and methods
2.1. Human tissue samples
Malignant breast tumors from patients in three different populations were used in this study. We analyzed 20 tumors with matched non-tumor tissue (n = 20) from the same patient from a cohort of Japanese, Caucasian, Hispanic and African-American patients and 22 tumors from the TNBC (ER−, PR−, HER2−) and non-TNBC (ER+, PR+, HER2+) subtypes obtained from the Comparative Human Tissue Network (CHTN). Tumor biopsies used in this study were confirmed histopathologically to be > 70% tumor. Because mtDNA is polymorphic and the resulting haplogroups segregate with ethnicity, we included tumors from individuals of Asian, Caucasian, Hispanic and African American ancestry to enhance the generality of our findings. Sample numbers are indicated in the figures. We did not exclude samples based on age, race and treatment regimen. This study was carried out in compliance with the regulations of the Institutional Review Board (IRB), IACUC (Protocol # 805964) and Environmental Health and Radiation Safety (EHRS) at the University of Pennsylvania.
2.2. Cell lines
Receptor triple negative basal-like cells (TNBC: ER−, PR−, HER2−) Hs578T, SUM1315, MDA-MB-231 and HCC1806 and receptor positive cells (non-TNBC: ER+, PR+ and ER+, PR+, HER2+) T47D, MCF7 and SUM52PE cell lines were obtained either from ATCC or collaborators and cultured in their recommended growth medium. All cell lines used in this study have been authenticated by STR profiling using Promega’s GenePrint® 10 kit. None of the cell lines used in this study is included in the database of misidentified and cross-contaminated cell lines (http://iclac.org/databases/cross-contaminations).
2.3. TCGA dataset analyses
In this study we analyzed The Cancer Genome Atlas (TCGA) dataset for 825 breast cancer patients. For mtDNA copy number estimation of the TCGA dataset, we used a computational approach using the Sanders algorithm [35]. Data for mtDNA tumor levels were obtained from supplementary material of Reznik et al. [35]. Briefly, Sander’s algorithm for mtDNA copy number estimation in tumors was designed based on comparing the number of sequencing reads aligning to [1] the mitochondrial (MT) genome and [2] a nuclear genome of known ploidy (which for normal tissues is 2 and for tumor tissues varies based on a correction factor influenced by the copy number of the tumor genome and stromal contamination). TCGA breast cancer expression data for ESRP1 was obtained from the Genomics Data Commons Portal (https://gdc.nci.nih.gov/). TCGA breast cancer PAM50 molecular classification and BRCA1 mutation data was obtained from the cBioPortal (www.cbioportal.org). Data was processed, integrated, analyzed and visualized in R Bioconductor using the ‘readxl’, ‘biomaRt’, ‘jsonlite’ and ‘ggplot2’ packages.
2.4. mtDNA copy number estimation
At least three punch sections were taken from each tissue for replicates to rule out the effects of intratumor heterogeneity and each sample was analyzed in triplicate. MtDNA copy number was measured from total genomic DNA isolated from normal and tumor tissues using DNeasy Blood and Tissue kit™ (Qiagen Cat # 69504). Using SYBR Green assay and quantitative real time PCR (qPCR) we amplified mtDNA (50 ng template) using primers specific for mtDNA encoded gene Cytochrome Oxidase subunit I (MT-COI) and normalized using a nuclear encoded single copy gene Cytochrome c Oxidase subunit IVi1 (CcO IVi1). Data analysis was done using the ΔΔCT method. This method of analysis estimates mtDNA copy number relative to a single copy nuclear gene as an internal reference gene [34,36,37]. Taking into consideration that tumors often have nuclear genome polyploidy due to chromosomal instability, in preliminary experiments, we have used multiple nuclear encoded single copy genes coded on different chromosomes, 18S, 36B4, RPL13A and CcOIVi1 as reference genes for normalization. Although there are possible inter individual differences in the transcripts of nuclear coded genes in tumors, the overall patterns of mtDNA contents among our comparisons remained unchanged irrespective of the gene of reference.
2.5. Analysis of variations in mtDNA sequence amplifications
For estimating mtDNA sequence variations, multiplex qPCR assays were completed using TaqMan® Fast Advanced Master Mix (Thermo Fisher Scientific Inc.). 5 μM primer/probe were used in each 8 μl reactions, which were performed on a Quant Studio 5 Thermocycler (Thermo Fisher Scientific Inc.). Cycling parameters were optimized for each multiplex primer/probe assays: for ND1pl-B2M, CYTBpl-GUSB, ND1-ND4, ND1-RNR1, or ND4pl-RNR1 assays, 95 °C for 20 s followed by 40 cycles of 95 °C for 1 s, 63 °C for 20 s and 60 °C for 20 s; for RNR1pl-hPRT assays, 95 °C for 20 s followed by 40 cycles of 95 °C for 1 s, 62 °C for 20 s and 60 °C for 20 s; for ND4pl-hPPIA assays, 95 °C for 20 s followed by 40 cycles of 95 °C for 1 s, 59 °C for 20 s and 60 °C for 20 s. Standard curves were generated using 4–5 serial dilutions performed for every primer/probe set and both CT and ΔCT were analyzed to ensure linearity to input DNA. Relative mtDNA content was calculated using ΔΔCT normalized to the average ΔCT of the standard curve. This allows averaging across different probe sets, which was employed to limit the impact of genome sequence variation. The ratio of specific mtDNA sequences was calculated as previously described [38]. In the current study, we have included a TAQMAN probe for RNR1 sequence, and performed pairwise comparison of all three probe sets to identify regions of instability. Alterations in ND4 abundance was calculated by subtracting the Ct value of ND1 from ND4 in each replicate, while normalizing back to the average of non-TNBC samples. Similarly, alteration of RNR1 abundance was calculated where Ct values of ND1 or ND4 were subtracted from RNR1 Ct values, then normalizing as above.
2.6. Cellular respiration assays
Oxygen consumption rate (OCR) and extracellular rate of acidification rate (ECAR) was carried out in a XF24 Seahorse Analyzer (Seahorse Bioscience, Billerica, MA, USA) using 5 × 104 cells as described before [34]. Equal number of cells were counted and seeded on the XF24 culture plates 6 h prior to performing the assay to rule out the differences in proliferation rates between cell types. Thirty minutes prior to the OCR and ECAR measurements, the growth medium of the cells was replaced with unbuffered assay medium containing 10 mM glucose in the absence of CO2. Cellular protein was estimated after the assay and oxygen consumption rates are represented per 0.1 mg cellular protein.
2.7. Tumor xenotransplantation
Mammary fat pad injections for tumor xenotransplantation were performed following standard procedure [39]. Hs578T and SUM1315 cells (1 × 105) suspended in Matrigel:PBS (1:1) solution were injected in the mammary fat pad of 6 weeks old female immunocompromised NSG™ mice in an orthotopic tumor model. Tumor growth was monitored weekly for 8 weeks. In the initial two weeks, there were no detectable tumors. Cells were labeled only numerically during this study and researchers and technician who measured the tumors remained blind to the cell identity during the course of this study. For statistical significance of the observed differences, we used sample size of 4 animals per group as determined by power analysis calculation, based on one tailed alpha value of 0.05, a beta value of 0.8, and effect size of 0.9. All animal studies were performed in accordance with IACUC regulations.
2.8. Gene expression analysis
Total cellular RNA was prepared using the RNeasy mini kit™ (Qiagen Cat # 74104). Genomic DNA was eliminated from the RNA preparations using Turbo DNA Free kit™ (Thermo Fisher Scientific). 1 μg RNA was reverse transcribed into cDNA using High Capacity reverse transcription kit (Applied Biosystems). 10 ng cDNA was used in each Taqman Assay reaction for the ESRP1 (Life technologies ID # Hs00214472_m1) and 18S (Life Technologies ID # Hs03928985_g1) TAQMAN Assay reactions. 25 ng cDNA was used for each SYBR Green reaction for expression analysis of TFAM, SLC25A25, HK2, UQCR11. RPL13A was used as an endogenous control. Quantitative Real Time PCR assays were run on an ABI Quant Studio 6 real time thermocycler (Applied Biosystems). All real time PCR assays were run in triplicate. Data are presented as Relative Quantitation (RQ).
2.9. RT2 profiler array for mitochondrial metabolism pathway genes
The mitochondrial gene expression pattern was analyzed using the human mitochondrial metabolism Pathway RT2 pre-designed PCR profiler array. This array (catalog number PAHS-008Y, SA Biosciences, Qiagen, CA) is designed to quantify the mRNA transcript levels for 84 genes representative of cellular metabolism, mitochondrial biogenesis and mitochondrial structural and functional integrity pathways. Total RNA was extracted from the tumors and breast cancer cell lines as indicated in the figure. Data was analyzed using the SA Biosciences (Qiagen) web-based RT2 Profiler Array Data Analysis version 3.5 program. No template controls on the predesigned array plates ensured lack of contamination and set the threshold for the absent/present calls. Samples yielding Ct values above 35 (transcript abundance below detection threshold) were discarded. Genes were considered as altered if there was at least 2 fold change in expression. The resulting values are reported as fold change in gene expression in non-TNBC compared to TNBCs.
2.10. Primer sequences
mtDNA copy number analysis:
COX1 gene:
TGATCTGCTGCAGTGCTCTGA (forward)
TCAGGCCACCTACGGTGAA (reverse)
COX IV1i gene:
GAAAGTGTTGTGAAGAGCGAAGAC (forward)
GTGGTCACGCCGATCCAT (reverse)
mtDNA imbalance analysis:
hTRNP-RNR1 primer 1:
GGTTTGGTCCTAGCCTTTCTAT
hTRNP-RNR1 primer 2:
GTGCTTGATGCTTGTTCCTTT
hTRNP-RNR1:
HEX: /5HEX/CCCGTTCCA/ZEN/GTGAGTTCACCCTCTA/3IABkFQ/
ND1 assay:
Probe: 5′-HEX/CCATCACCC/ZEN/TCTACATCACCGCCC/3IABkFQ/-3′
Primer 1: 5′-GAGCGATGGTGAGAGCTAAGGT-3′
Primer 2: 5′-CCCTAAAACCCGCCACATCT-3′
ND4 assay:
Probe: 5′-FAM/CCGACATCA/ZEN/TTACCGGGTTTTCCTCTTG/3IABkFQ/-3′
Primer 1: 5′-ACAATCTGATGTTTTGGTTAAACTATATTT-3′
Primer 2: 5′-CCATTCTCCTCCTATCCCTCAAC-3′
mRNA quantitation by qReal Time PCR:
TFAM:
ACCGAGGTGGTTTTCATCTG (forward)
TTTGCATCTGGGTTCTGAGC (reverse)
RPL13A:
GCCATCGTGGCTAAACAGGTA (forward)
GTTGGTGTTCATCCGCTTGC (reverse)
SLC25A25:
TGACCATCGACTGGAACGAGT (forward)
ACATCAAAGATCGTGGAATGCTT (reverse)
UQCR11:
AACTGGGTCCCGACGGCCTA (forward)
ATCCTTCTTAAACTTGCCAT (reverse)
HK2:
TGCCACCAGACTAAACTAGACG (forward)
CCCGTGCCCACAATGAGAC (reverse)
ESRP1: Taqman probe Assay ID: Hs00214472_m1 (Thermo Fisher)
18S: Taqman Probe Assay ID: Hs03928985_g1 (Thermo Fisher)
2.11. Statistical analysis
We analyzed the statistical significance of differences using two-tailed t-test for normally distributed datasets. Welch’s t-test was used while comparing between two tumor types and human samples which have high variance within each type. We used ANOVA for analyses in experiments where we compared among multiple tumor types. Copy numbers/expression levels were not normally distributed and were log2 transformed for analyses to conform Gaussian distributions. Data are presented as the mean ± SEM (standard error of the mean) with individual data points (red cross) and the standard box-plot with median (red line), lower and upper quartile values (blue box), and 1.5 interquartile-ranges (black whiskers). Groups were compared by one-tailed Welch’s t-test (in the log2 scale) and statistical significance was set at 0.05. For mtDNA sequence imbalance in non-normally distributed tumor samples Mann-Whitney test was used to calculate the significance of the differences in the mtDNA sequence imbalance assays. * indicates p < .05, ** indicates p < .01, *** indicates p < .001.
3. Results
3.1. Mitochondrial DNA copy number variations among breast cancer patients analyzed from the TCGA dataset
Association between mtDNA content and prognosis has been previously reported [23,25–28,30] but these reports are contradictory as to whether high or low levels of mtDNA correlate with poor prognosis. Here we extracted the mtDNA copy number from 825 breast cancer patients in the TCGA dataset following the Sander’s algorithm [35]. We analyzed the TCGA dataset for associations between patient tumor mtDNA content and metastasis following the tumor staging TNM (Tumor size, lymph Nodes affected and Metastases) recommended by the American Joint Committee on Cancer (AJCC) (https://cancerstaging.org). Patients with most advanced Stage IV disease, who have at least one diagnosed metastatic site, had the lowest tumor mtDNA copy number (Fig. 1). Furthermore, of all tumors examined, those that progressed to metastasis had the lowest mtDNA copy number by two-tail Student’s t-test (p = .007) (Fig. 1). Although the TCGA dataset for metastatic tumors has a smaller sample size as there is no information on metastases for the majority of the TCGA breast tumors, based on the effect size, the difference in mtDNA content between non-metastatic and metastatic tumors were significant as noted above with p = .007. Although aging is associated with cancer incidences, we did not find any significant correlation in the mtDNA content of primary breast tumors in the TCGA with the age at initial diagnosis of the patients (Suppl. Fig. S1).
Fig. 1.
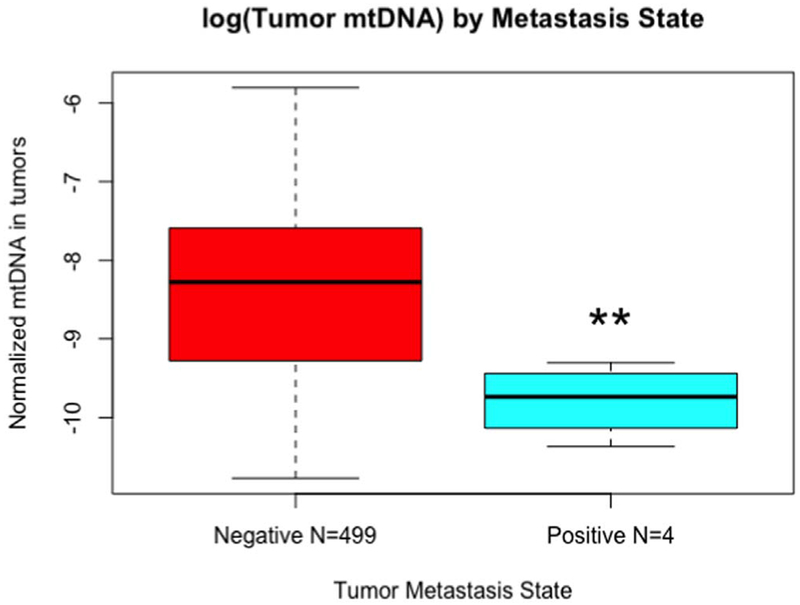
TCGA analysis of the mtDNA content among breast tumors.
mtDNA content of breast tumors in TCGA dataset correlating with the metastatic stage. Negative = tumors did not proceed to metastases; positive = tumors formed metastases. Significance analysis was done using two-tail Student’s t-test (p = .007) ** = p < .01.
3.2. Mitochondrial genome content is variable among the intrinsic subtypes of breast tumors
To analyze mtDNA copy numbers in tumors, we obtained tumor and matched normal (non-tumor) breast tissues from different patient cohorts. The tissues used in this study were obtained from diverse ethnic background of patients (Asian, Caucasian, Hispanics and African American) to circumvent potential affects due to mtDNA sequence differences (i.e., haplogroups) [40]. We observed the tumor mtDNA content was not statistically significant compared to the matched non-tumor tissue when we grouped all tumors and non-tumor tissues. The copy number distributions of non-tumor and tumor tissues have indistinguishable averages (mean ± S.E.M.: 10.7 ± 0.7, 10.2 ± 0.2, respectively; p = .23, n = 20, one-tail paired Student’s t-test). While tumors have lower mtDNA contents, we also observed that mtDNA content in tumors have less variability than non-tumor tissues which have a wider distribution (second momentum ± S.E.M.: 8.4 ± 4.4, 0.7 ± 0.3, respectively; p = .05, n = 20, one-tail paired Student’s t-test) (Fig. 2A).
Fig. 2.
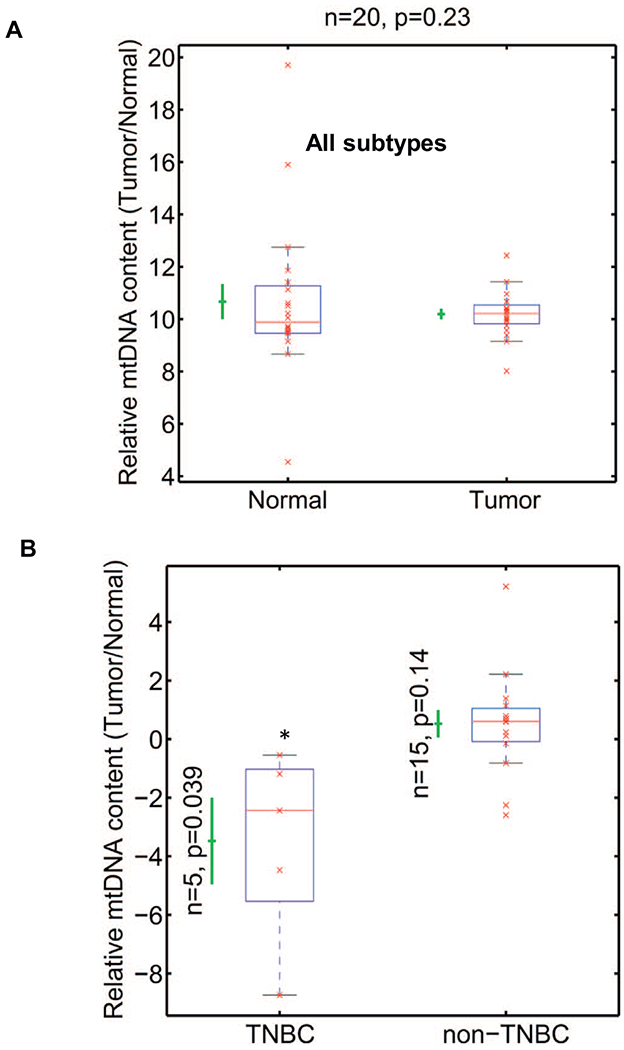
mtDNA content in human breast tumors.
(A) Real time PCR showing relative mtDNA levels in breast tumor tissues (n = 20) and matched non-tumor tissues (n = 20). The mtDNA content (y-axis) is quantified as the copy number of mtDNA gene CcO1, normalized by copy number of nuclear single copy gene CcOIVi1, plotted in log2 scale. P = .23 (B) Relative mtDNA content (y-axis) is the ratio of mtDNA contents of tumor and matched normal tissues in TNBC (n = 5; p < .039) and non-TNBC (n = 14, p = .14) plotted in Log2 scale. Statistical analysis was done using one-tail paired Student’s t-test. All p-values were calculated in the original −log2 scale, assuming two Gaussian distributions with different means, variances and different numbers of samples. P values are indicated in the figure. * indicates p < .05.
Our observation in Fig. 2A that there was no significant difference in mtDNA copy number in tumors when compared to matched normal (non-tumor) tissues in our cohort could account for the contradicting reports about tumor mtDNA levels because most prior studies included all breast tumor subtypes for their analyses. However, the molecular gene signatures are variable among the tumor subtypes: triple negative (TNBC: ER−, PR−, HER2−) and non-TNBC (Luminal A, B and HER2+: ER+, PR+, HER2+) [41] which could influence mitochondrial biogenesis, functions and mtDNA content. To evaluate the notion that mtDNA copy number loss is unique to specific tumor subtypes, we measured mtDNA copy number in tumors compared to their matched non-tumor tissues (Fig. 2B). We observed that mtDNA copy number was significantly (p = .039) lower in TNBC tumors compared to their matched non-tumor tissue. In contrast, mtDNA content in non-TNBC (Luminal A, B, and HER2+) tumors either tended to be higher or remained unchanged relative to the non-tumor matched tissue (Fig. 2B and Suppl. Fig. S2). We analyzed at least 3 punch biopsies from the tumor sample (as determined by a pathologist). The mtDNA copy numbers between the different punch biopsies from the same patient tumor remained similar with no statistical difference.
3.3. Triple negative basal-like breast cancer cells have impaired mitochondrial functions
We analyzed mtDNA content and mitochondrial functions in human TNBC and non-TNBC cell lines. We aimed to address if TNBC cell lines in general, similar to human tumors, had higher prevalence of mitochondrial defects. Even though we observed variability in mtDNA content among individual cell lines in both TNBC as well as non-TNBCs, which is reflective of inter-individual differences observed among human tumors, TNBC cells have lower mtDNA contents compared to non-TNBC cells (Fig. 3A). We next quantified the mRNA levels of the mtDNA regulatory nuclear-encoded transcription factor TFAM. TFAM levels have been reported to positively correlate with mtDNA levels [42]. Similar to breast cancer tissues, Tfam mRNA level was lower in TNBC basal-like cells when compared to the non-TNBC cells (Fig. 3B and Suppl. Fig. S3). It is likely that lower mtDNA contents in TNBC is related to lower TFAM mRNA levels.
Fig. 3.
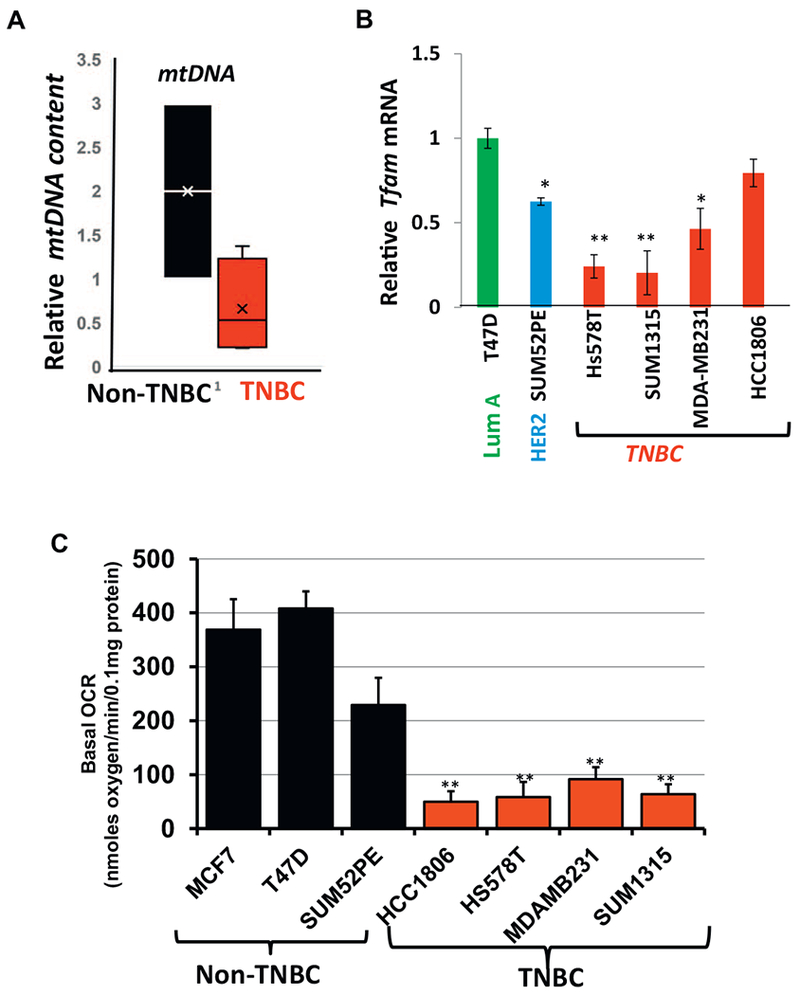
Mitochondrial functional defects in breast cancer cell lines.
(A) Relative mtDNA content in non-TNBC and TNBC cells. Box plot showing the distribution of the relative mtDNA content in TNBC and non-TNBC cells normalized to internal single gene CcOIVi1. The line inside each box indicates the mean. (B) Relative Tfam mRNA in TNBC (Hs578T, SUM1315, MDA-MB231, HCC1806) and non-TNBC (T47D, SUM52PE) cell lines. 20 ng of cDNA was used per reaction for real time PCR amplification using either Tfam or endogenous gene RPL13A. (C) Basal cellular oxygen consumption rate (OCR) in the cell lines (5 × 104 cells per well) as indicated in the figure measured using an XF24 analyzer. All experiments were done in triplicates and mean ± SEM are plotted. Statistical analysis for significance was done using Students t-test. Data is significant at p < .05.
We assessed cellular respiration as a measure of mitochondrial function in a panel of breast cancer cell lines (Fig. 3C). The basal oxygen consumption rate in the four TNBC basal-like cells were markedly lower than the non-TNBC cells (Fig. 3C). This is indicative of impaired mitochondrial functions in TNBC subtype compared to other subtypes.
3.4. MtDNA sequence imbalance in breast tumors is variable among the intrinsic subtypes
Mutations and deletions in mtDNA are widely reported in breast tumors and purported to contribute to tumor progression. However, there is no report examining the variations in mtDNA sequence contents among the four major molecular subtypes of breast tumors. This is important because variation among tumor subtypes in one mtDNA coding sequence region is evidence of abnormal mtDNA metabolism. To evaluate this point, we used quantitative PCR for multiplex reactions using multiple custom-designed primer-probe combinations for sequences spanning the mtDNA (Fig. 4A), some of which are imbalanced in other disease states [38].
Fig. 4.
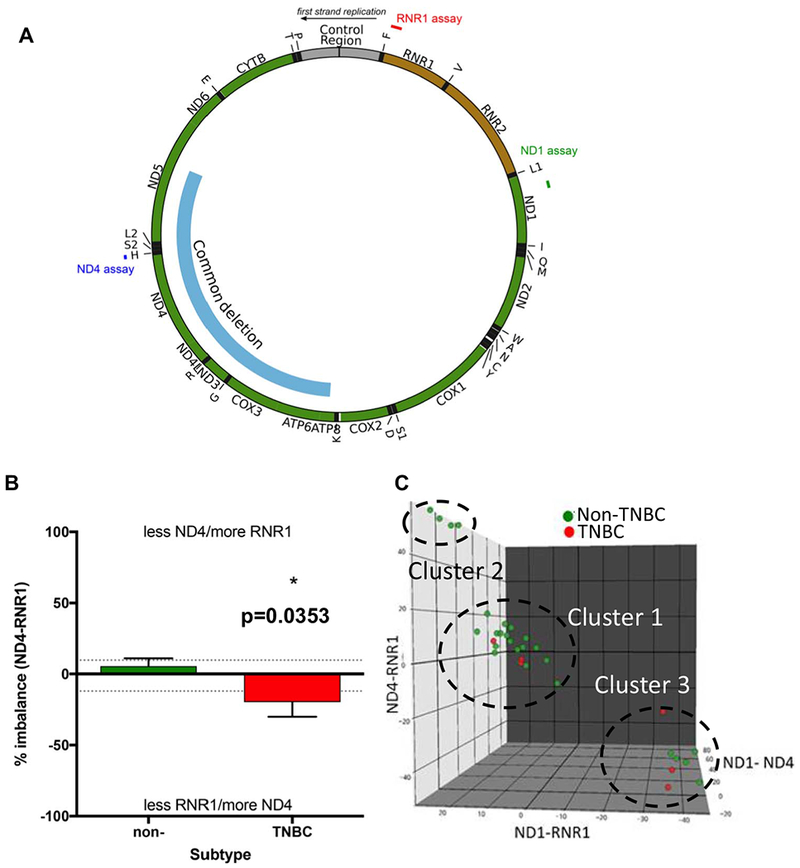
Relative abundance of mtDNA in primary human breast tumors.
(A) Circos plot of the mitochondrial DNA. The RNR1, ND1 and ND4 assay regions are indicated in the figure. (B) Isolated total DNA from TNBC (n = 6) and non-TNBC (Luminal and HER2+, n = 28) were analyzed by multiplex qPCR for all pairings of ND1, ND4, and RNR1 primer-probe sets using optimized conditions. Percent imbalance of ND4 and RNR1 probes. Values above upper dashed lines indicate a loss of ND4 and gain of RNR1 content. Values below the lower dashed lines indicate the inverse. (C) 3D scatter blot of ND1, ND4 and RNR1 for n = 33 samples. Visually determined clusters are indicated by ellipse and enumerated by text. Mann-Whitney of ND4-RNR1 shows significant difference between TNBC vs non-TNBC at p = .035.
We probed for relative abundance of RNR1 (ribosomal subunit 1), ND1 (NADH dehydrogenase subunit 1) and ND4 (NADH dehydrogenase subunit 4) sequences in all pairings (Fig. 4A–C and Suppl. Fig. S4). We normalized and compared the average values between TNBC and non-TNBC tumors. In Fig. 4 and Suppl. Fig. S4, plots show comparison of two probes, where a data point off of zero toward one marker (such as RNR1) indicates imbalance that could be decrease in RNR1 content, or increase in the other marker (such as ND4 or ND1). The so called “common” mtDNA deletion is evident when ND4 amplification is lower than ND1 [38] where ND1-ND4 would produce a negative stability value, but this was not evident in any of the tumor subtypes (Fig. 4). The lower amplification of RNR1 ratiometric to ND4 we observed in the TNBC tumors is statistically significant at p = .03 (Fig. 4B). This is interesting in light of the fact that previous studies with non-cancer post-mitotic tissues reported that the ND4 region is more vulnerable to deletions [43]. The bulk of samples for any one analysis did not show imbalance, but we observed some interesting trends centered around the RNR1 probe set in these pairwise comparisons (Fig. 4B,C). To combine these parameters, we plotted the data in a 3D scatter plot, which allows us to visualize three clusters (Fig. 4C). Cluster 1 contains the majority of cancer samples, all having normal sequence stability (within ~20% zero in X, Y, and Z axes). Cluster 2 is typified by ~25% ND4-RNR1 and ~50% ND4-RNR1 imbalance, with the common trend toward more RNR1 sequence amplification. Cluster 3 shows ~40% ND1-RNR1 and ~50% ND4-RNR1 imbalance, with the common aspect toward less RNR1. Although tumor samples from all groups appeared in Cluster 3, it was striking that 50% of the TNBCs were in this population. Tumor samples being limiting, for validating our assays we combined samples and performed dilution analysis to ensure linearity of each probe set, as well as test the consistency of clustering (data not shown). Considering the stringency of the assay, ratiometric imbalance of these mtDNA probe sets can potentially be used for stratifying primary breast tumors on the basis of mtDNA defects.
3.5. Differential OXPHOS modulation among the intrinsic subtypes of breast tumors
MtDNA defects can induce broad transcriptional changes in multiple metabolic pathways and modulate mitochondrial functions which is commonly observed in tumors [36,37,44–47]. However, it has not been investigated if there are metabolic differences among the intrinsic subtypes of breast tumors. We therefore tested the relationship between the mtDNA copy number variability among the intrinsic subtypes and the gene expression levels for 84 genes which regulate different pathways involved in mitochondrial energy metabolism using the Human Mitochondrial Energy Metabolism Pathway Plus RT2 (SA Biosciences, Qiagen) profiler array (Fig. 5 and Table 1). We explored the possibility that clinically challenging TNBCs, containing lower mtDNA content have metabolic differences that can be harnessed in future for therapeutic targeting. Scatter plot and Clustergram analyses of overall changes in gene signatures identified differential metabolic gene expression patterns and the pathways they regulate between TNBC when compared to non-TNBC subtypes (Fig. 5A–C). Because we compared among tumor tissues, we observed an overlap in metabolic gene expression patterns between TNBC and non-TNBCs (Fig. 5A). However, 52% genes (p = .05) involved in mitochondrial functions were down-regulated in TNBCs compared to non-TNBC subtypes (Fig. 5 and Table 1).
Fig. 5.
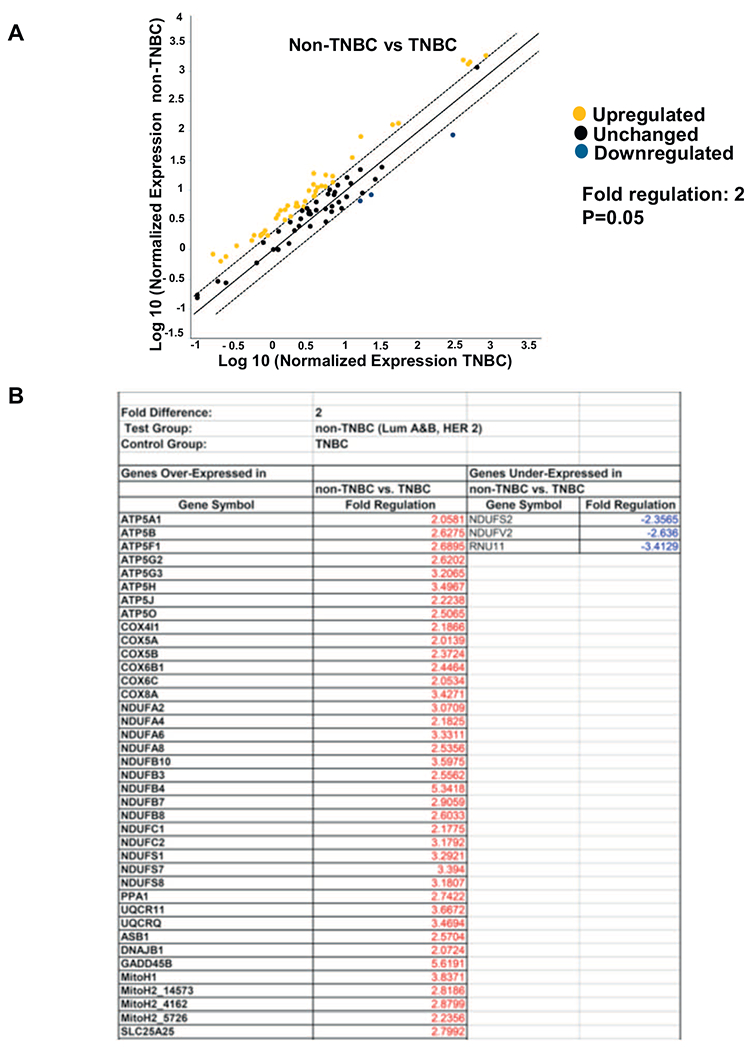
RT2 profiler array showing changes in mitochondrial metabolism genes in TNBC compared to non-TNBCs. Four non-TNBCs and two TNBCs were analyzed for the profiler array and run in duplicate. (A) Scatter plot showing distribution of the genes involved in the mitochondrial metabolism pathway compared between non-TNBC (Luminal A, Luminal B and HER2+) and TNBC (control group). Black dots indicate unchanged genes, yellow dots indicate genes activated non-TNBC. Fold-regulation represents fold-change results in a biologically relevant way. Fold-change values greater than one indicate a positive- or an up-regulation. The p value is calculated based on a Student’s t-test of the replicate 2^(−Delta Ct) values for each gene in the TNBC (control) group and non-TNBC groups. The fold change has significant difference at p value < .05.
Table 1.
List of genes at least 2-fold-change in non-TNBC (Lum A,B and HER2+) compared to TNBC. Fold change (2^(−Delta Delta Ct) is the normalized gene expression (2^(−Delta Ct)) in the test sample (Lum A, B and HER2+) divided the normalized gene expression (2^(−Delta Ct)) in the control sample (TNBC). The difference is significant at p = .05.
| Gene symbol | Fold difference | Fold up- or down-regulation |
|---|---|---|
| Non-TNBC/TNBC | Non-TNBC/TNBC | |
| ATP5A1 | 2.06 | 2.06 |
| ATP5B | 2.63 | 2.63 |
| ATP5C1 | 1.12 | 1.12 |
| ATP5F1 | 2.69 | 2.69 |
| ATP5G1 | 1.62 | 1.62 |
| ATP5G2 | 2.62 | 2.62 |
| ATP5G3 | 3.21 | 3.21 |
| ATP5H | 3.5 | 3.5 |
| ATP5I | 1.59 | 1.59 |
| ATP5J | 2.22 | 2.22 |
| ATP5J2 | 1.79 | 1.79 |
| ATP5L | 0.77 | −1.3 |
| ATP5O | 2.51 | 2.51 |
| COX4I1 | 2.19 | 2.19 |
| COX5A | 2.01 | 2.01 |
| COX5B | 2.37 | 2.37 |
| COX6A1 | 0.79 | −1.27 |
| COX6A2 | 1.7 | 1.7 |
| COX6B1 | 2.45 | 2.45 |
| COX6C | 2.05 | 2.05 |
| COX7A2 | 1.81 | 1.81 |
| COX7A2L | 1.7 | 1.7 |
| COX7B | 0.76 | −1.31 |
| COX8A | 3.43 | 3.43 |
| CYC1 | 0.85 | −1.17 |
| NDUFA1 | 1.89 | 1.89 |
| NDUFA10 | 1.33 | 1.33 |
| NDUFA11 | 1.52 | 1.52 |
| NDUFA2 | 3.07 | 3.07 |
| NDUFA3 | 1.41 | 1.41 |
| NDUFA4 | 2.18 | 2.18 |
| NDUFA5 | 1 | 1 |
| NDUFA6 | 3.33 | 3.33 |
| NDUFA8 | 2.54 | 2.54 |
| NDUFAB1 | 0.69 | −1.45 |
| NDUFB10 | 3.6 | 3.6 |
| NDUFB2 | 0.84 | −1.19 |
| NDUFB3 | 2.56 | 2.56 |
| NDUFB4 | 5.34 | 5.34 |
| NDUFB5 | 0.56 | −1.79 |
| NDUFB6 | 1.22 | 1.22 |
| NDUFB7 | 2.91 | 2.91 |
| NDUFB8 | 2.6 | 2.6 |
| NDUFB9 | 0.61 | −1.65 |
| NDUFC1 | 2.18 | 2.18 |
| NDUFC2 | 3.18 | 3.18 |
| NDUFS1 | 3.29 | 3.29 |
| NDUFS2 | 0.42 | −2.36 |
| NDUFS3 | 1.3 | 1.3 |
| NDUFS4 | 1.1 | 1.1 |
| NDUFS5 | 1.59 | 1.59 |
| NDUFS6 | 1.72 | 1.72 |
| NDUFS7 | 3.39 | 3.39 |
| NDUFS8 | 3.18 | 3.18 |
| NDUFV1 | 0.77 | −1.31 |
| NDUFV2 | 0.38 | −2.64 |
| NDUFV3 | 1.38 | 1.38 |
| PPA1 | 2.74 | 2.74 |
| SDHA | 1.22 | 1.22 |
| SDHB | 0.93 | −1.08 |
| SDHC | 0.57 | −1.75 |
| SDHD | 1.01 | 1.01 |
| UQCR11 | 3.67 | 3.67 |
| UQCRC1 | 1.69 | 1.69 |
| UQCRC2 | 1.32 | 1.32 |
| UQCRFS1 | 1.18 | 1.18 |
| UQCRH | 0.79 | −1.26 |
| UQCRQ | 3.47 | 3.47 |
| ARRDC3 | 1.22 | 1.22 |
| ASB1 | 2.57 | 2.57 |
| CYB561D1 | 1.68 | 1.68 |
| DNAJB1 | 2.07 | 2.07 |
| EDN1 | 1.42 | 1.42 |
| GADD45B | 5.62 | 5.62 |
| HSPA1A | 1.71 | 1.71 |
| HSPA1B | 0.54 | −1.86 |
| LRP5L | 0.88 | −1.14 |
| MitoH1 | 3.84 | 3.84 |
| MitoH2_12106 | 1.88 | 1.88 |
| MitoH2_14573 | 2.82 | 2.82 |
| MitoH2_4162 | 2.88 | 2.88 |
| MitoH2_5726 | 2.24 | 2.24 |
| RNU11 | 0.29 | −3.41 |
| SLC25A25 | 2.8 | 2.8 |
Genes downregulated in TNBCs compared to non-TNBC subtypes were mainly associated with mitochondrial ATP synthesis-coupled electron transport, including subunits of Complex I (NDUFA2, NDUFA4, NDUFA6, NDUFA8, NDUFB10, NDUFS1, NDUFS7,NDUFS8), Complex III (UQCR11, UQCRC1,UQCRC2, UQCRFS1, UQCRH, UQCRQ), ATP-Mg2+/Pi transporter (SLC25A25), ATP synthase, MitoH2_14573, MitoH2_4162. This is consistent with our observation that TNBCs have a reduction in mitochondrial respiratory capacity and decreased mtDNA copy number. A complete list of the genes expression changes in non-TNBC (Luminal A, Luminal B and HER2+) compared to TNBC tumors and cells is provided in Fig. 5B and Table 1.
We verified by quantitative real time PCR analysis the mRNA levels of an ETC component gene (UQCR11) and a solute carrier gene (SLC25A25), which we identified from the RT2 profiler array were downregulated in TNBCs. The mRNA levels of the ATP-Mg2+/Pi transporter SLC25A25 and the Ubiquinol-cytochrome c Reductase, Complex III, subunit 11 (UQCR11) are lower in TNBC cells compared to the non-TNBC cells (Fig. 6A, B and Suppl. Fig. S5A, B). The UQCR11 mRNA was lower in TNBC compared to the non-TNBC tumor tissues (Suppl. Fig. S5C).
Fig. 6.
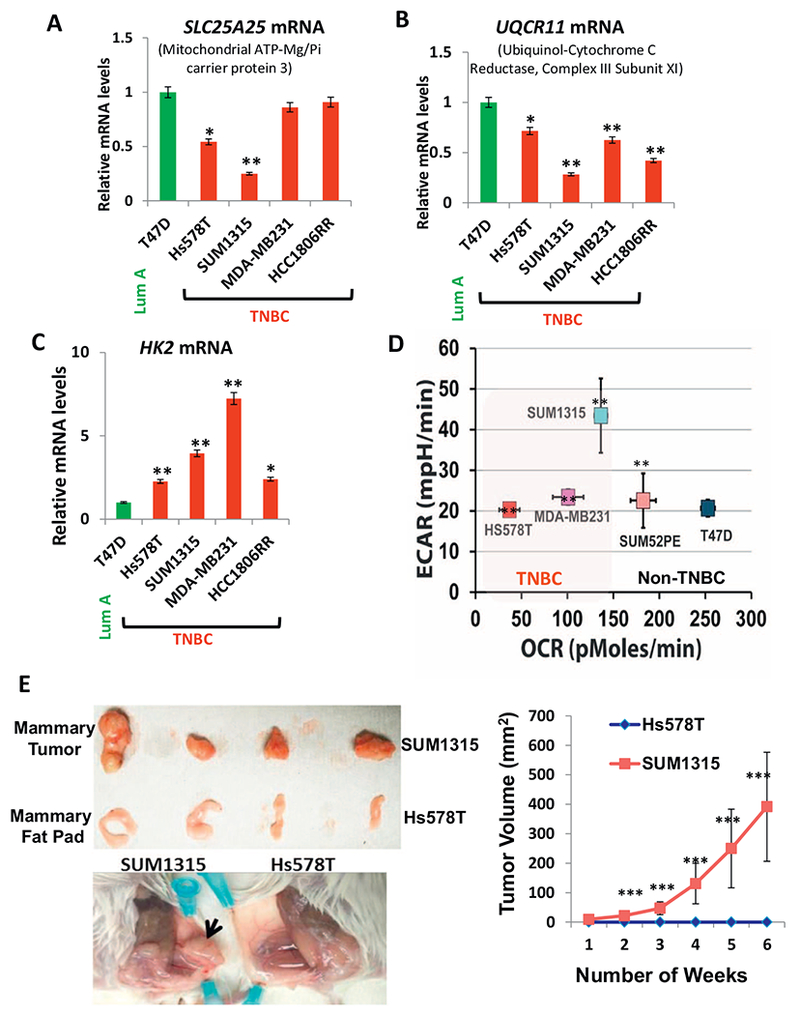
OXPHOS modulation among the breast tumor subtypes.
(A, B) Real time PCR showing alterations In genes encoding mitochondrial proteins, SLC25A25 and UQCR11 (as indicated in the figure) in TNBC cells compared to non-TNBC T47D cell line. (C) Real time PCR showing altered mRNA levels of glycolytic gene hexokinase 2 in TNBC cells compared to non-TNBC T47D cell line. (D) Extracellular acidification rate (ECAR) versus the OCR in the cell lines (5 × 104 cells per well) indicated in the figure. All experiments were done in triplicates and mean ± SEM are plotted. (E) Left panel: (Top) Representative xenograft tumors in 6 week old female NSG™ mice (n = 4 each category) derived from SUM1315 cells and mammary fat pad (MFP) of mice injected with Hs578T as indicated in the figure. (Below) Representative picture of the mouse mammary fat pad showing xenograft tumors formed from SUM1315 cells and no tumor formation by Hs578Tcells. Right panel: Graph showing tumor volume measured over six weeks. Statistical analysis for significance of differences was done using Students t-test. * indicates significance level at p < .05, ** = p < .01 and *** = p < .001.
Metabolic switch to higher glycolysis is widely accepted as a hallmark of many aggressive tumors [48–50]. This prompted us to investigate the expression of hexokinase 2 (HK2) gene, an irreversible enzyme at the start of glycolysis. We observed that TNBC cell lines and tumors expressed higher HK2 mRNA compared to the non-TNBCs (Fig. 6C and Suppl. Fig. S5D, E). Extracellular flux analysis measures the rate of acidification of the medium and is an indicator of glycolytic flux [51]. We observed that TNBC cells have higher extracellular flux (ECAR) indicative of higher glycolysis (Fig. 6D). To further test the hypothesis that higher glycolysis in TNBCs is an adaptive mechanism to meet their high proliferation, we selected two TNBC basal-like cell lines, Hs578T and SUM1315 with varying ECAR to test their tumorigenic potential in a xenograft model. SUM1315 which has higher extracellular acidification rate (ECAR) compared to Hs578T (Fig. 6D), formed higher tumor load when injected into immunocompromised NSG™ mice compared to Hs578T, which formed no tumors in the 6 weeks duration of the study (Fig. 6E). It is notable that although there are differences in the molecular signature between Hs578T and SUM1315, it is of potential clinical significance that tumorigenic potential can be additionally determined based on differences in their metabolic (glycolytic) status.
3.6. A subset of TNBCs has lower ESRP1 expression correlating with low mtDNA content
ESRP1 is involved in epithelial-mesenchymal transition and metastasis by regulating alternative splicing [52–54] and mediates the retrograde response toward epithelial to mesenchymal transition in low mtDNA containing mammary epithelial cells [34]. We therefore analyzed the breast tumor tissues with the goal to identify a subset of the tumors containing low mtDNA as well as low ESRP1 levels which based on our previous report would potentially be have higher metastatic risk. In agreement with our previous report [34] that ESRP1 levels positively correlate with low mtDNA content and inversely related to metastasis, we observed that ESRP1 mRNA levels were lower in the low mtDNA containing TNBC cells when compared to the non-TNBC cells (Fig. 7A and Suppl. Fig. S6A). By association analysis, we identified unique subset of tumors containing both low mtDNA content as well as low ESRP1 mRNA levels (Fig. 7B and Suppl. Fig. S6B). Additionally we identified a subset among TNBC tumors (marked in red box), which contained the lowest mtDNA content, as well as lower ESRP1 gene expression (Suppl. Fig. S6C).
Fig. 7.
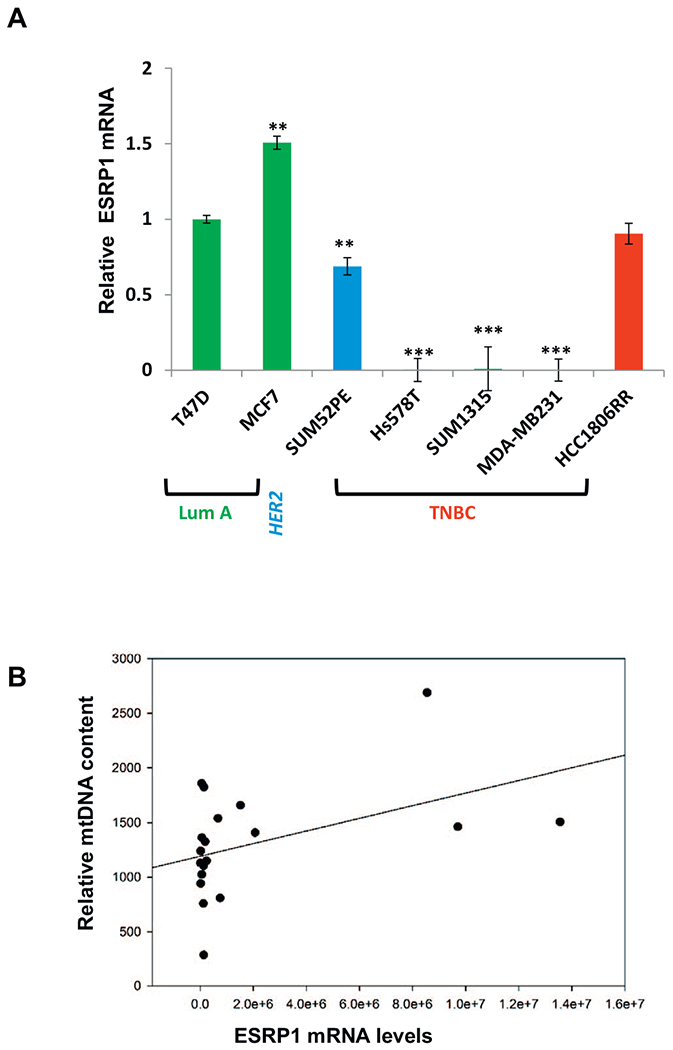
Relative levels of ESRP1 among the breast tumor subtypes.
(A) Relative levels of ESRP1 in TNBC cells (as indicated in the figure) compared to non-TNBC T47D cells analyzed by real time PCR using ESRP1 specific Taqman primer-probe pairs. 18S Taqman primer-probe pair was used as an internal control for normalization. Statistical analysis for significance of differences was done using Students t-test. * indicates p < .05, ** indicates p < .01. (B) Correlation plot for relative mtDNA (mtDNA encoded cytochrome oxidase subunit 1 Cco1) and ESRP1 levels in the tumors (n = 22). Pearson (linear) and Spearman (rank) methods were used to identify tumors containing low mtDNA content as well as low ESRP1.
4. Discussion
Mitochondrial genome defects have been associated with various aggressive cancers including breast cancer. Numerous reports show mtDNA copy number changes (high and low), mtDNA mutations and deletions in breast tumor tissues compared to normal tissues [25,55–60]. However, breast tumors are heterogeneous and have distinct molecular signatures depending on their subtypes, which dictate their progression and therapeutic outcome. In this study, we aimed to address if there are differences in mtDNA copy number and mitochondrial functional defects among the four major molecular subtypes of breast tumors [3,4] and identify the nature of mitochondrial defects that could be used for tumor stratification and metastasis-risk analysis. Our results suggest that reduced mtDNA copy number loss and lower cellular respiration occur preferentially in receptor triple negative tumors and basal-like cells is suggestive of the contribution of mitochondrial defects toward the aggressiveness frequently observed in TNBCs. Furthermore, lower amplification in the RNR1 region relative to ND4 is indicative of either higher mtDNA deletion, or replication defects specifically in the RNR1 loci and this needs to be investigated further. Tumor mtDNA content and specific mtDNA alterations should therefore be explored in larger retrospective and prospective studies to examine their potential as prognostic indicators of metastatic disease.
Triple negative tumors account for 5–10% of all breast cancers and affect mostly African Americans and younger women [7,61]. Moreover, a high percentage of individuals harboring defects (mutations or deletions) in the tumor suppressor gene BRCA1 have receptor triple negative tumors and are at an increased lifetime risk for developing high aggressive breast tumors [62,63]. Interestingly, it has been suggested that BRCA1, a nuclear DNA damage response protein, similar to ATM, is involved in mtDNA maintenance, mtDNA integrity and mtDNA damage repair and regulates cellular metabolism [64]. Our analysis of the TCGA dataset suggests that tumors containing low mtDNA content harbor a higher frequency of pathogenic BRCA1 mutations (Suppl. Fig. S7). Therefore, it is plausible that BRCA1 regulates mtDNA biogenesis in TNBCs and patients harboring mtDNA defects with associated BRCA1 loss/mutations may respond better with chemotherapeutics modulating tumor metabolism.
Recent reports suggest OXPHOS modulation in mammary tumors and the microenvironment is an adaptive mechanism by which tumor cells meet their energy demands [65–68]. However, it remained unclear if there were significant differences in metabolic gene expression among the four intrinsic breast tumor subtypes. We observed differences in the extracellular flux and OXPHOS gene expression among the tumor subtypes, which indicate that the metabolic reprogramming, is not a generalized phenomenon in all tumors but have unique differences among tumor subtypes. We identified genes involved in maintenance of mitochondrial structural and functional integrity that are differentially expressed in TNBCs compared to non-TNBCs which make them potential molecular candidates to therapeutically target in TNBCs. Reports show changes in one or more respiratory chain complexes in various pathologies including different cancers and because each of the respiratory chain complexes are comprised of many subunits, subtle differences in gene expression can potentially affect metabolic pattern of the tumor cell. Therefore, in our study we focused on identifying differences at the molecular level in mitochondrial OXPHOS gene expressions and identified differential expression of genes involved in mitochondrial biogenesis, ion transporters, solute carriers between TNBCs and non-TNBCs. A top candidate gene we identified is the reduced expression in TNBCs of the ATP-Mg/Pi carrier SLC25A25 which shuttles metabolites, nucleotides and cofactors through the mitochondrial membrane and modulates cytoplasm and matrix functions.
Changes in alternative splicing mediated by various RNA-binding proteins affect cancer progression. The regulation of splicing factors and alterations in splicing events mediated by these factors are dynamic and responsive to signals from tumor cells and their microenvironment [54]. In recent years, ESRP1 has emerged as a key regulator of cancer cell associated splicing network. Whole genome and transcriptome analysis show reduced ESRP1 expression in breast tumors compared to matched normal tissues [54]. We earlier reported that mitochondrial dysfunction modulates the expression of ESRP1 and associated splicing events of oncogenes and drives epithelial-mesenchymal transition in mammary cell lines [34]. Therefore it is clinically important that we identified a subset of TNBC tumors containing mtDNA defects as well as expressing low ESRP1 suggesting stratification of TNBCs further on the basis of the mitochondrial/metabolic defects and ESRP1 expression can be an effective diagnostic tool for metastasis risk assessment. Interestingly, in silico analysis indicates alternative splicing sites in the mitochondrial solute carrier SLC25A25 gene which is downregulated in TNBCs and the role of ESRP1 in its regulation remains to be investigated. Identification of additional splicing regulatory proteins that are uniquely modulated in TNBCs compared to the other subtypes will be beneficial in designing targeted therapeutics in TNBCs for metastasis ablation.
We postulate that mitochondrial genome defects provide the second line of assault in tumor cells, which dictate the pathogenesis and drug resistance in highly aggressive triple negative tumors. In the absence of targeted therapeutics for TNBCs, there is an unmet need to design new strategies to improve precision medicine. Our current study identified unique mitochondrial genome and functional defects and altered expression of metabolic genes specifically in the highly metastatic TNBCs. While it is essential to confirm these findings in larger cohorts and further investigate the mechanisms for the mtDNA changes our data suggest that the candidates identified in this study will potentially be useful to develop a combinatorial diagnostic platform for metastatic risk assessment and develop precision medicine.
Supplementary Material
Acknowledgements
We thank Dr. Douglas Wallace (Center for Mitochondrial and Epigenomic Medicine at Children’s Hospital of Philadelphia) for helpful discussions in data interpretation and critical comments. We thank Drs. Anna Kashina, Chris Lengner and Leslie King (University of Pennsylvania, School of Veterinary Medicine) for helpful edits on the manuscript. We thank Marie Fina for technical assistance. Tumor tissue samples were provided by the Comparative Human Tissue Network (CHTN), a National Cancer Institute supported resource. We thank Yoshiaki Shinden and Shoji Natsugoe from Kagoshima University for tissues from breast cancer patients.
Funding
This Research is supported by the Breast Cancer Alliance Young Investigator Grant (#568489), the University Research Foundation Grant (#000002-2199) and the PENN-CHOP Mitochondria Research Affinity Award (to MG); NIGMS grant R01GM110424 (to BAK); NCI grant CA22762 (to NGA).
Footnotes
Transparency document
The Transparency document associated with this document can be found, in online version.
Conflict of interest
The authors have no conflict of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbadis.2018.01.002.
References
- [1].O’Shaughnessy J, Extending survival with chemotherapy in metastatic breast cancer, Oncologist 10 (Suppl. 3) (2005) 20–29. [DOI] [PubMed] [Google Scholar]
- [2].Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. , A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes, Cancer Cell 10 (6) (December 2006) 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Comprehensive molecular portraits of human breast tumours, Nature 490 (7418) (2012) 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. , Comprehensive molecular portraits of invasive lobular breast cancer, Cell 163 (2) (2015) 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fournier MV, Martin KJ, Transcriptome profiling in clinical breast cancer: from 3D culture models to prognostic signatures, J. Cell. Physiol. 209 (3) (2006) 625–630. [DOI] [PubMed] [Google Scholar]
- [6].Wirapati P, Sotiriou C, Kunkel S, Farmer P, Pradervand S, Haibe-Kains B, et al. , Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures, Breast Cancer Res. 10 (4) (2008) R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hudis CA, Gianni L, Triple-negative breast cancer: an unmet medical need, Oncologist 16 (Suppl. 1) (2011) 1–11. [DOI] [PubMed] [Google Scholar]
- [8].Vyas S, Zaganjor E, Haigis MC, Mitochondria and cancer, Cell 166 (3) (July 28 2016) 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bardella C, Pollard PJ, Tomlinson I, SDH mutations in cancer, Biochim. Biophys. Acta 1807 (11) (2011) 1432–1443. [DOI] [PubMed] [Google Scholar]
- [10].Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. , Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma, Science 287 (5454) (2000) 848–851. [DOI] [PubMed] [Google Scholar]
- [11].Kurelac I, Romeo G, Gasparre G, Mitochondrial metabolism and cancer, Mitochondrion 11 (4) (2011) 635–637. [DOI] [PubMed] [Google Scholar]
- [12].Wallace DC, Mitochondria and cancer, Nat. Rev. Cancer 12 (10) (2012) 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Frezza C, The role of mitochondria in the oncogenic signal transduction, Int. J. Biochem. Cell Biol. 48 (2014) 11–17. [DOI] [PubMed] [Google Scholar]
- [14].Thompson CB, Metabolic enzymes as oncogenes or tumor suppressors, N. Engl. J. Med. 360 (8) (2009) 813–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. , The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate, Cancer Cell 17 (3) (2010) 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cavalli LR, Varella-Garcia M, Liang BC, Diminished tumorigenic phenotype after depletion of mitochondrial DNA, Cell Growth Differ. 8 (11) (1997) 1189–1198. [PubMed] [Google Scholar]
- [17].King MP, Attardi G, Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation, Science 246 (4929) (1989) 500–503. [DOI] [PubMed] [Google Scholar]
- [18].Magda D, Lecane P, Prescott J, Thiemann P, Ma X, Dranchak PK, et al. , mtDNA depletion confers specific gene expression profiles in human cells grown in culture and in xenograft, BMC Genomics 9 (2008) 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Morais R, Zinkewich-Peotti K, Parent M, Wang H, Babai F, Zollinger M, Tumor-forming ability in athymic nude mice of human cell lines devoid of mitochondrial DNA, Cancer Res. 54 (14) (1994) 3889–3896. [PubMed] [Google Scholar]
- [20].Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. , Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity, Proc. Natl. Acad. Sci. U. S. A. 107 (19) (2010) 8788–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, et al. , A mitochondrial protein compendium elucidates complex I disease biology, Cell 134 (1) (2008) 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ashley N, Poulton J, Mitochondrial DNA is a direct target of anti-cancer anthracycline drugs, Biochem. Biophys. Res. Commun. 378 (3) (2009) 450–455. [DOI] [PubMed] [Google Scholar]
- [23].Bai RK, Chang J, Yeh KT, Lou MA, Lu JF, Tan DJ, et al. , Mitochondrial DNA content varies with pathological characteristics of breast cancer, J. Oncol. 2011 (2011) 496189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Barekati Z, Radpour R, Kohler C, Zhang B, Toniolo P, Lenner P, et al. , Methylation profile of TP53 regulatory pathway and mtDNA alterations in breast cancer patients lacking TP53 mutations, Hum. Mol. Genet. 19 (15) (2010) 2936–2946. [DOI] [PubMed] [Google Scholar]
- [25].Fan AX, Radpour R, Haghighi MM, Kohler C, Xia P, Hahn S, et al. , Mitochondrial DNA content in paired normal and cancerous breast tissue samples from patients with breast cancer, J. Cancer Res. Clin. Oncol. 135 (8) (2009) 983–989. [DOI] [PubMed] [Google Scholar]
- [26].Tseng LM, Yin PH, Chi CW, Hsu CY, Wu CW, Lee LM, et al. , Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer, Genes Chromosom. Cancer 45 (7) (2006) 629–638. [DOI] [PubMed] [Google Scholar]
- [27].Verma M, Kumar D, Application of mitochondrial genome information in cancer epidemiology, Clin. Chim. Acta 383 (1–2) (2007) 41–50. [DOI] [PubMed] [Google Scholar]
- [28].Xia P, An HX, Dang CX, Radpour R, Kohler C, Fokas E, et al. , Decreased mitochondrial DNA content in blood samples of patients with stage I breast cancer, BMC Cancer 9 (2009) 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yu M, Zhou Y, Shi Y, Ning L, Yang Y, Wei X, et al. , Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients, IUBMB Life 59 (7) (2007) 450–457. [DOI] [PubMed] [Google Scholar]
- [30].McMahon S, LaFramboise T, Mutational patterns in the breast cancer mitochondrial genome, with clinical correlates, Carcinogenesis 35 (5) (2014) 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. , The epithelial-mesenchymal transition generates cells with properties of stem cells, Cell 133 (4) (2008) 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Polyak K, Weinberg RA, Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits, Nat. Rev. Cancer 9 (4) (2009) 265–273. [DOI] [PubMed] [Google Scholar]
- [33].Zhao Z, Zhu X, Cui K, Mancuso J, Federley R, Fischer K, et al. , In vivo visualization and characterization of epithelial-mesenchymal transition in breast tumors, Cancer Res. 76 (8) (2016) 2094–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Guha M, Srinivasan S, Ruthel G, Kashina AK, Carstens RP, Mendoza A, et al. , Mitochondrial retrograde signaling induces epithelial-mesenchymal transition and generates breast cancer stem cells, Oncogene 33 (45) (2013) 5238–5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Reznik E, Miller ML, Senbabaoglu Y, Riaz N, Sarungbam J, Tickoo SK, et al. , Mitochondrial DNA copy number variation across human cancers, elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Guha M, Tang W, Sondheimer N, Avadhani NG, Role of calcineurin, hnRNPA2 and Akt in mitochondrial respiratory stress-mediated transcription activation of nuclear gene targets, Biochim. Biophys. Acta 1797 (6–7) (2010) 1055–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Srinivasan S, Guha M, Dong DW, Whelan K, Ruthel G, Uchikado Y, et al. , Disruption of cytochrome c oxidase function induces Warburg effect and metabolic reprogramming, Oncogene 35 (12) (2015) 1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Belmonte FR, Martin JL, Frescura K, Damas J, Pereira F, Tarnopolsky MA, et al. , Digital PCR methods improve detection sensitivity and measurement precision of low abundance mtDNA deletions, Sci. Rep. 6 (2016) 25186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chakrabarti R, Kang Y, Transplantable mouse tumor models of breast cancer metastasis, Methods Mol. Biol. 1267 (2015) 367–380. [DOI] [PubMed] [Google Scholar]
- [40].Wallace DC, Mitochondrial DNA variation in human radiation and disease, Cell 163 (1) (2015) 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Eroles P, Bosch A, Perez-Fidalgo JA, Lluch A, Molecular biology in breast cancer: intrinsic subtypes and signaling pathways, Cancer Treat. Rev 38 (6) (2012) 698–707. [DOI] [PubMed] [Google Scholar]
- [42].Campbell CT, Kolesar JE, Kaufman BA, Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number, Biochim. Biophys. Acta 1819 (9–10) (2012) 921–929. [DOI] [PubMed] [Google Scholar]
- [43].Rygiel KA, Tuppen HA, Grady JP, Vincent A, Blakely EL, Reeve AK, et al. , Complex mitochondrial DNA rearrangements in individual cells from patients with sporadic inclusion body myositis, Nucleic Acids Res. 44 (11) (2016) 5313–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Guha M, Srinivasan S, Biswas G, Avadhani NG, Activation of a novel calcineurin-mediated insulin-like growth factor-1 receptor pathway, altered metabolism, and tumor cell invasion in cells subjected to mitochondrial respiratory stress, J. Biol. Chem. 282 (19) (2007) 14536–14546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Guha M, Pan H, Fang JK, Avadhani NG, Heterogeneous nuclear ribonucleo-protein A2 is a common transcriptional coactivator in the nuclear transcription response to mitochondrial respiratory stress, Mol. Biol. Cell 20 (18) (2009) 4107–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Guha M, Avadhani NG, Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics, Mitochondrion 13 (6) (2013) 577–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, et al. , Progressive increase in mtDNA 3243A > G heteroplasmy causes abrupt transcriptional reprogramming, Proc. Natl. Acad. Sci. U. S. A. 111 (38) (2014) E4033–E4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Brizel DM, Schroeder T, Scher RL, Walenta S, Clough RW, Dewhirst MW, et al. , Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer, Int. J. Radiat. Oncol. Biol. Phys. 51 (2) (2001) 349–353. [DOI] [PubMed] [Google Scholar]
- [49].Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfor K, Rofstad EK, et al. , High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers, Cancer Res. 60 (4) (2000) 916–921. [PubMed] [Google Scholar]
- [50].Dang CV, Links between metabolism and cancer, Genes Dev. 26 (9) (May 1 2012) 877–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].TeSlaa T, Teitell MA, Techniques to monitor glycolysis, Methods Enzymol. 542 (2014) 91–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, et al. , An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype, PLoS Genet. 7 (8) (2011) e1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, Shen S, et al. , An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition, EMBO J. 29 (19) (2010) 3286–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sebestyen E, Singh B, Minana B, Pages A, Mateo F, Pujana MA, et al. , Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks, Genome Res. 26 (6) (2016) 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Canter JA, Kallianpur AR, Parl FF, Millikan RC, Mitochondrial DNA G10398A polymorphism and invasive breast cancer in African-American women, Cancer Res. 65 (17) (2005) 8028–8033. [DOI] [PubMed] [Google Scholar]
- [56].Dimberg J, Hong TT, Nguyen LT, Skarstedt M, Lofgren S, Matussek A, Common 4977 bp deletion and novel alterations in mitochondrial DNA in Vietnamese patients with breast cancer, Spring 4 (2015) 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hsu CW, Yin PH, Lee HC, Chi CW, Tseng LM, Mitochondrial DNA content as a potential marker to predict response to anthracycline in breast cancer patients, Breast J 16 (3) (2010) 264–270. [DOI] [PubMed] [Google Scholar]
- [58].Lee HC, Yin PH, Lin JC, Wu CC, Chen CY, Wu CW, et al. , Mitochondrial genome instability and mtDNA depletion in human cancers, Ann. N. Y. Acad. Sci. 1042 (2005) 109–122. [DOI] [PubMed] [Google Scholar]
- [59].Ma Y, Bai RK, Trieu R, Wong LJ, Mitochondrial dysfunction in human breast cancer cells and their transmitochondrial cybrids, Biochim. Biophys. Acta 1797 (1) (2010) 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mambo E, Chatterjee A, Xing M, Tallini G, Haugen BR, Yeung SC, et al. , Tumor-specific changes in mtDNA content in human cancer, Int. J. Cancer 116 (6) (2005) 920–924. [DOI] [PubMed] [Google Scholar]
- [61].Chen L, Li CI, Tang MC, Porter P, Hill DA, Wiggins CL, et al. , Reproductive factors and risk of luminal, HER2-overexpressing and triple negative breast cancer among multiethnic women, Cancer Epidemiol. Biomark. Prev. 25 (9) (2016) 1297–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Peshkin BN, Alabek ML, Isaacs C, BRCA1/2 mutations and triple negative breast cancers, Breast Dis. 32 (1–2) (2010) 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Graeser MK, Engel C, Rhiem K, Gadzicki D, Bick U, Kast K, et al. , Contralateral breast cancer risk in BRCA1 and BRCA2 mutation carriers, J. Clin. Oncol. 27 (35) (2009) 5887–5892. [DOI] [PubMed] [Google Scholar]
- [64].Privat M, Radosevic-Robin N, Aubel C, Cayre A, Penault-Llorca F, Marceau G, et al. , BRCA1 induces major energetic metabolism reprogramming in breast cancer cells, PLoS One 9 (7) (2014) e102438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jiang HL, Sun HF, Gao SP, Li LD, Huang S, Hu X, et al. , SSBP1 suppresses TGFbeta-driven epithelial-to-mesenchymal transition and metastasis in triple-negative breast cancer by regulating mitochondrial retrograde signaling, Cancer Res. 76 (4) (2016) 952–964. [DOI] [PubMed] [Google Scholar]
- [66].Kannan A, Wells RB, Sivakumar S, Komatsu S, Singh KP, Samten B, et al. , Mitochondrial reprogramming regulates breast cancer progression, Clin. Cancer Res. 22 (13) (2016) 3348–3360. [DOI] [PubMed] [Google Scholar]
- [67].Park JH, Vithayathil S, Kumar S, Sung PL, Dobrolecki LE, Putluri V, et al. , Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer, Cell Rep. 14 (9) (2016) 2154–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Pelicano H, Zhang W, Liu J, Hammoudi N, Dai J, Xu RH, et al. , Mitochondrial dysfunction in some triple-negative breast cancer cell lines: role of mTOR pathway and therapeutic potential, Breast Cancer Res. 16 (5) (2014) 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.