

Abstract
Polycystic ovary syndrome (PCOS) is highly associated with cardiometabolic risk and the metabolic syndrome (MetS), predisposing women to increased risk of developing type 2 diabetes and cardiovascular disease. Metformin is commonly used to treat insulin resistance-glucose intolerance, and flutamide, an androgen receptor (AR) antagonist, is used to target hyperandrogenemia and dyslipidemia. Currently, the physiological mechanism of action of these treatments on androgen, lipidogenic, and insulin signaling pathways remains unclear in PCOS. The aim of this study was to investigate the effects and mechanisms of action of metformin and flutamide on plasma lipid-apolipoprotein (Apo)B-lipoprotein and insulin-glucose metabolism, and endocrine-reproductive indices in a PCOS-prone MetS rodent model. PCOS-prone rodents were treated with metformin (300 mg/kg body wt), flutamide (30 mg/kg body wt), or metformin + flutamide combination treatment for 6 wk. Metformin was shown to improve fasting insulin and HOMA-IR, whereas flutamide and combination treatment were shown to reduce plasma triglycerides, ApoB48, and ApoB100, and this was associated with decreased intestinal secretion of ApoB48/triglyceride. Flutamide and metformin were shown to reduce plasma androgen indices and to improve ovarian primary and preovulatory follicle frequency. Metformin treatment increased hepatic estrogen receptor (ER)α, and metformin-flutamide decreased intestinal AR and increased ERα mRNA expression. Metformin-flutamide treatment upregulated hepatic and intestinal insulin signaling, including insulin receptor, MAPK1, and AKT2. In conclusion, cardiometabolic risk factors, in particular ApoB-hypertriglyceridemia, are independently modulated via the AR, and understanding the contribution of AR and insulin-signaling pathways further may facilitate the development of targeted interventions in high-risk women with PCOS and MetS.
Keywords: flutamide, lipid and insulin metabolism, metformin, PCOS
INTRODUCTION
Polycystic ovary syndrome (PCOS) is the most common metabolic-endocrine disorder in women of reproductive age (4, 44). PCOS is highly associated with the metabolic syndrome (MetS) and increased cardiometabolic risk predisposing women to increased transition to the development of type 2 diabetes and premature cardiovascular disease (CVD) (9, 36, 93). Currently, the etiological causes of the disorder are not fully understood; however, hyperandrogenemia, obesity, and insulin resistance are implicated in the etiology of PCOS (9, 36, 51). It has been estimated that up to 85% of PCOS women exhibit clinical or biochemical hyperandrogenemia, and 70% of lean and 95% of overweight women diagnosed with PCOS have impaired glucose tolerance and elevated plasma insulin concentrations in the fasting and non-fasting state (114). Atherogenic dyslipidemia is common in PCOS, occurring in up to 70% of individuals, and is defined as elevated fasting plasma triglycerides (TG) and apolipoprotein (Apo)B-lipoproteins, lower high-density lipoprotein cholesterol (HDL-C), and elevated excursions in non-fasting TG and ApoB-lipoproteins (7, 54, 92, 100, 110, 111). The most frequent aberrations in lipid metabolism observed in PCOS include increased fasting plasma TG, total cholesterol, low-density lipoprotein cholesterol (LDL-C), and total ApoB concentrations (110, 111). Postprandial or non-fasting lipemia has also been reported in adolescents and women with PCOS (7, 100, 104). Atherogenic dyslipidemia predisposes these young women to the early development of atherosclerosis and CVD (83, 104, 111). This is evidenced by young women and adolescents with PCOS presenting with subclinical CVD, including increased carotid artery media thickness and endothelial and cardiac dysfunction (63, 75, 80, 93, 95, 108, 109, 112).
Insulin resistance and hyperandrogenemia have been proposed to act independently and synergistically in the development of atherogenic dyslipidemia and CVD risk in PCOS (3, 30). Testosterone and insulin-mediated effects appear to underpin the dysregulation of lipid metabolism in PCOS (30, 102). Testosterone induction of PCOS leads to unchanged or increased plasma TG and a reduction in HDL-C in animal models, including PCOS-like models in rodents, sheep, and rhesus monkeys (1, 76, 89). In our spontaneous PCOS-prone rodent model, plasma testosterone and insulin concentrations are both positively correlated with elevated fasting and non-fasting concentrations of plasma TG and ApoB48 lipoproteins (102). In PCOS patients, hyperandrogenemia is correlated with higher concentrations of plasma TG, lower HDL-C, and small, dense LDL particles, and this appears to be independent of body weight (13, 20, 26, 40, 90, 97). We have shown that obese adolescents with PCOS have early impairment in lipid metabolism, including elevated fasting and non-fasting plasma TG and ApoB-lipoproteins, and this is highly correlated with plasma free testosterone concentrations (104). Polymorphisms in the androgen receptor (AR) and single-nucleotide polymorphisms in the AR co-chaperone FK506-binding protein have been associated with the onset of PCOS (55, 62, 116). The presence of shorter and longer (CAG)n repeats in the AR gene sequence is a risk factor for PCOS development (62) and has been shown to positively correlate with higher plasma testosterone levels in PCOS patients (116). Indeed, flutamide, an AR antagonist, has been shown in clinical studies to lower fasting plasma lipids (TG, total cholesterol, and LDL-C) as well as increase HDL-C, and these observations are found independent of body weight (29, 41, 49, 67). The effects of flutamide on fasting and non-fasting plasma lipids and ApoB-lipoprotein metabolism and the mechanisms associated with decreased lipogenesis are currently unknown in PCOS. The AR has been shown in vitro to directly stimulate maturation of the sterol regulatory element-binding protein (SREBP) precursor to promote SREBP activation and to increase SREBP cleavage activating protein (SCAP) expression leading to an upregulation in lipogenesis (47).
It is well established that insulin resistance and hyperinsulinemia are associated with increased lipogenesis and overproduction of hepatic (ApoB100-VLDL) and intestinal (ApoB48-chylomicron) TG-rich lipoproteins and the remnants of these ApoB-lipoproteins (2, 32, 81, 103). Further evidence from the Copenhagen City Heart Study and ACCORD lipid study have shown that increased fasting and non-fasting concentrations of TG and ApoB-remnant lipoproteins are positively associated with ischemic cardiovascular events (21, 39, 95, 98, 99). We have shown that plasma insulin concentrations are positively correlated with fasting and non-fasting TG and ApoB-remnants in obese adolescents with PCOS (104). Non-fasting or postprandial hypertriglyceridemia has also been positively associated with insulin resistance in women with PCOS, independent of body weight (7, 104). Insulin sensitizing agents have been used to improve insulin resistance in PCOS, and this may include beneficial effects on lipid and ApoB-lipoprotein metabolism (10, 28). Metformin is a standard therapy to treat insulin resistance and impaired glucose tolerance in PCOS; however, alone or in combination with diet-lifestyle interventions, it has been shown to have no effect or inconsistent results in reducing fasting blood lipids, particularly in the obese-PCOS phenotype (10, 67, 70, 71, 79, 91). Metformin may suppress hepatic de novo lipogenesis pathways to reduce plasma TG, but the mechanisms and effects on ApoB-lipoprotein and lipid metabolism in PCOS remain unclear (10, 105, 117). Metformin-flutamide combination therapy has shown improvements in fasting plasma total cholesterol and LDL-C in obese, insulin-resistant women with PCOS treated for 6 mo, but no change in total plasma TG was observed (41). A longer-term intervention of 12 mo in conjunction with a hypocaloric diet showed similar results (42). Although a hypocaloric diet alone improved insulin resistance, plasma lipid profile was improved only in conjunction with metformin-flutamide therapy (42). In young, normal-weight insulin-resistant PCOS women, a low-dose flutamide-metformin-pioglitazone in combination with oral contraceptive (OC) use was shown to improve hyperandrogenemia, HDL-C, and carotid intima media thickness; however, fasting plasma TG concentrations were significantly increased (49, 52, 101). Metformin and statin combination therapy have been shown to improve fasting plasma lipids (TG, total cholesterol, and LDL-C), with no effect on androgens or improved insulin sensitivity (91). To date, there have been limited and inconsistent reports on the effects of metformin and flutamide on insulin-glucose metabolism and fasting blood lipids, and no studies have examined the mechanisms of how these compounds regulate insulin-glucose metabolism or plasma lipid and ApoB-lipoprotein metabolism in PCOS. The aim of this study was to investigate the effects and mechanisms of action of metformin and flutamide, alone and in combination, on plasma lipid and ApoB-lipoprotein metabolism, insulin-glucose metabolism, and endocrine-reproductive indices in a PCOS-prone MetS rodent model. We hypothesized that metformin would improve insulin resistance, flutamide would improve plasma lipid and ApoB-lipoprotein metabolism, and metformin-flutamide in combination would additively improve both insulin resistance and plasma lipid and ApoB-lipoprotein metabolism to improve cardiometabolic risk in the PCOS-prone MetS rodent model. In addition, we hypothesized that treatment with metformin and flutamide independently and in combination would improve reproductive-endocrine outcomes in the PCOS-prone MetS rodent model.
METHODS
Animal Model and Study Design
PCOS-prone and control (lean, normal, non-PCOS) rodents were raised in an established breeding colony at the University of Alberta, as previously described (88, 89, 102). The PCOS-prone model develops hyperandrogenemia, acyclicity, arrested follicular development, and cystic follicles, accompanied by MetS, obesity, and insulin resistance (88, 89). This PCOS-prone genotype spontaneously develops MetS and PCOS-like features without induction using androgen treatments (31, 102). The PCOS-prone MetS model has a recessive spontaneous defect in the stop codon of the Ob gene. The polygenic recessive corpulent (cp) defect is in the leptin receptor, and PCOS-prone MetS rats are homozygous for the recessive cp gene (cp/cp), and control (lean, normal, non-PCOS) animals are either homozygous normal (+/+) or heterozygous (+/?). Therefore, an obese-MetS control genotype that does not develop PCOS-like features or a lean, normal body weight PCOS-prone phenotype is not available, and this is a limitation of this model. However, the control heterozygous genotype without MetS is available and was used in these studies, as previously described (31, 88, 102). Rats were weaned at 21 days of age and housed in the 12:12 h reversed light cycle to allow establishment of a normal diurnal cycle. At 10 wk of age, PCOS-prone rats were randomly assigned to 1 of the 3 treatment groups or a control group. PCOS-prone control animals (n = 12) were fed a standard chow diet, and the treatment groups of PCOS-prone rats were fed a diet supplemented with 1) metformin (300 mg/kg body weight, n = 12); 2) flutamide (30 mg/kg body weight, n = 12); or 3) metformin-flutamide combination (n = 12) for 6 wk. The dose and period of treatment (6 wk) of metformin and flutamide were based on previous rodent studies, and the dose of flutamide in the feed was also based on pilot studies in the PCOS-prone MetS animal model, which has severe dyslipidemia (16, 53, 56, 85, 102). Control and PCOS-prone rats were fed ad libitum their randomly assigned diet. The chow for all animal groups was prepared using standard dry chow powder (5001, PMI Nutrition International, Brentwood, MO). Animals were fed the assigned diets for 6 wk. Animal care and experimental protocols were conducted in accordance with the Canadian Council of Animal Care and approved by Animal Ethics Committee of the University of Alberta.
Metabolic Assessment
Fasting plasma biochemical profile.
Blood samples were collected after a 16-h fast, plasma was separated by 10-min centrifugation (3,000 revolutions/min) at 4°C, and samples were stored at −80°C. Plasma TG, total cholesterol, non-esterified fatty acids, LDL-C, HDL-C, and glucose were measured using commercially available colorimetric kits as previously described (WAKO Chemicals USA, Inc., Richmond, VA) (102). Plasma insulin (ALPCO Diagnostics), free testosterone (free T), total testosterone, serum hormone binding globulin (SHBG), and estradiol were measured using commercially available ELISA kits specific to rodents with variability within assays of ≤5% (CUSABIO, Wuhan, China), as previously described (33, 104). Although testosterone and isomers measured by extraction of unconjugated steroids and liquid or gas chromatography mass spectrophotometric methods are the best option, the use of the rodent-specific ELISA in this study design provides relative testosterone levels for comparison of treatment effects (46). The free androgen index (FAI) was calculated as FAI = 100 (total testosterone/SHBG) (6). Plasma concentration of ApoB48 and ApoB100 were determined using a Western blotting SDS-PAGE and ECL procedure, and a known mass of purified rodent ApoB protein standard was used to quantify plasma ApoB48 and ApoB100, as previously described (102, 103).
Non-fasting plasma insulin-glucose, lipid, and ApoB-lipoprotein metabolism response.
Animals were fasted overnight and given either a 5.0-g meal pellet of chow to determine non-fasting plasma insulin-glucose response or a 5.0-g meal pellet high in fat (47% wt/wt fat) to assess non-fasting plasma lipid and ApoB-lipoprotein response, as previously described using an established tail-snip blood collection procedure (102, 103). Plasma was separated by centrifugation (10 min, 3,000 revolutions/min) at 4°C and stored at −80°C for measurement of insulin and glucose concentrations or lipid (TG and cholesterol) and ApoB-lipoprotein concentrations, respectively, as previously described (102, 103). The magnitude of the postprandial or non-fasting response of plasma glucose and insulin following the meal, and plasma TG, total cholesterol, ApoB48, and ApoB100 following the high-fat meal, were determined by area under the curve (AUC) analysis software (GraphPad Software, San Diego, CA). AUC represents the total plasma levels of the parameter measured in the non-fasting phase, inclusive of fasting and non-fasting response. The incremental AUC (iAUC) was generated by subtracting the fasting plasma concentration of the parameter from the non-fasting plasma concentration to determine the change or response in the parameter following the meal or high-fat meal, as previously described (102, 103).
Intestinal secretion of lipids and chylomicrons.
The animals were fasted overnight before intestinal mesenteric lymph duct and duodenal cannulation surgery, as previously described (102). In brief, saline (representing fasted state) was infused into the duodenum for 5 h, and then Intralipid (representing the fed state) was infused for 5 h, and intestinal lymph chylomicrons were collected, as previously described (102, 103). Lymph chylomicron TG, total cholesterol, ApoB48, and ApoB100 were measured using the Western blot procedure coupled with ECL analysis, as described above.
Measurement of lipidogenic, insulin signaling, AR and estrogen receptor gene mRNA expression and protein analysis.
Quantitative real-time PCR technique was used to measure relative expression of mRNA for AR, estrogen receptor (ER)α, ERβ, liver X receptor (LXR)α, peroxisome proliferator-activated receptor (PPAR)α, SREBP1, SREBP2, LDL receptor/ApoB-ApoE receptor (LDL-R), HMGR, ApoB, microsomal transport protein (MTP), acetyl CoA carboxylase (ACC), fatty acid synthase (FAS), SCAP, diacylglycerol O-acyltransferase (DGAT)1, DGAT2, insulin receptor (IR), mitogen-activated protein kinase-1 (MAPK1), serine/threonine-protein kinase-2 (AKT2), protein-tyrosine phosphatase 1B (PTPN1), Jun N-terminal kinase (JNK), and glycogen synthase kinase-3 (GSK3) in liver and intestinal mucosa. Primer sequences are shown in Table 1 for genes. Total RNA was isolated from tissues using TRIzol (Invitrogen, Canada) as described in the manufacturer’s protocol and reversed transcribed into cDNA (MMLV reverse transcriptase; Applied Biosystems, Burlington, ON, Canada). Target gene copy number was quantified using the comparative Ct (cycle threshold) and normalized to the housekeeping gene, Cyclophilin. Results were expressed as a ratio in mRNA expression relative to housekeeping gene control (2−ΔCt), and all assays were performed in duplicate, as previously described (15).
Table 1.
Primer sequences for quantitative RT-PCR
| Gene | Sequence |
|---|---|
| apoB | F 5′-CGCTGAGTTACTGAAAAAGCTG-3′ |
| R 3′-CCTTAGGTAGGGGCTCACATT-5′ | |
| ar | F 5′-ACTTGATCGCATCATTGCAT-3′ |
| R 3′-GAATTGATGCAGCTCTCTTGC-5′ | |
| er-α | F 5′-TGCTGGTCCTCTGGCAGT-3′ |
| R 3′-CAACCACCAGCAGATGAGAC-5′ | |
| er-β | F 5′-TGCTGGTCCTCTGGCAGT-3′ |
| R 3′-CAACCACCAGCAGATGAGAC-5′ | |
| dgat1 | F 5′-AAAGGCCAGCCTCCCTAAC-3′ |
| R 3′-TCCAAACTAGGGGAGTGTGC-5′ | |
| dgat2 | F 5′-AGGATCTGCCCTGTCACG-3′ |
| R-3′-GTCTTGGAGGGCCGAGAG-5′ | |
| Fasn | F 5′-TCTCAGCCCACAGGACAAG-3′ |
| R 3′-AGGCTGGGAGGAGGATGT-5′ | |
| hmgcr | F 5′-TCTCTGCAGTACCTGCCTTACA-3′ |
| R 3′-CCGATCACGTTCTCACAGC-5′ | |
| ldlr | F 5′-TGCTACTGGCCAAGGACAT-3′ |
| R 3′-CTGGGTGGTCGGTACAGTG-5′ | |
| NR1H3, lxra | F 5′-CAGGAAGAGATGTCCTTGTGG-3′ |
| R 3′-TCTTCCACAACTCCGTTGC-5′ | |
| Mttp | F 5′-AGCCAGGCAGTATGATCCTC-3′ |
| R 3′-GAGGCCAGTTGTGTGACCTT-5′ | |
| ppar-α | F 5′-TGCGGACTACCAGTACTTAGGG-3′ |
| R 3′-GGAAGCTGGAGAGAGGGTGT-5′ | |
| scap | F 5′-TCCATCAGCATGAGCCTAAA-3′ |
| R 3′-GGAACACCGAACAGCAAGTC-5′ | |
| srebf1 | F 5′-ACAAGATTGTGGAGCTCAAGG-3′ |
| R 3′-TGCGCAAGACAGCAGATTTA-5′ | |
| srebf2 | F 5′-ATCAAGTCAGCAGCCAAGGA-3′ |
| R 3′-CTGGTGTACCTGGGCAATG-5′ | |
| IR | F 5′-CAGAAAAACCTCTTCAGGCAAT-3′ |
| R 3′-TTCAAGGGATCTTCGCTTT-5′ | |
| Mapk1 | F 5′-TCTGCACCGTGACCTCAA-3′ |
| R 3′-GCAAGGCCAAAGTCACAGA-5′ | |
| jnk1 | F 5′-GCTTCTCTGGGGAACCTAGTG-3′ |
| R 3′-TTCCCACAGTAGAGTCCAAGG-5′ | |
| akt2 | F 5′-CCGCTATTATGCCATGAAGAT-3′ |
| R 3′-TGTGGGCGACTTCATCCT-5′ | |
| Gsk3a | F 5′-TGGAGCCACAGATTACACCTC-3′ |
| R 3′-CTGGCCAAGAAGCAGCTC-5′ | |
| ptpn1 | F 5′-GGAACAGGTACCGAGATGTCA-3′ |
| R 3′-AGTCATTATCTTCCTGATGCAATTT-5′ |
Immunoblot protein analysis was performed on liver homogenates using SDS-PAGE and ECL, as previously described (60). In brief, proteins were separated using 10% SDS-PAGE and transferred to Immun-Blot PVDF membrane (Bio-Rad). Membranes were incubated with the following antibodies for protein detection (Cell Signaling Technology, Danvers, MA): phospho-(Ser79) ACC (p-ACC) and ACC, FAS, Anti-CideB, MTP, adenosine monophosphate-activated protein kinase (AMPK), and phospho-AMPK (p-AMPK), phospho-AKT (p-AKT), and AKT. Actin or GADPH were used as loading controls. Immunoreactivity was detected by ECL system (Amersham-Pharmacia, ON, Canada) and visualized by G:BOX system 5 (SYNGENE, Cambridge, UK). Immunoblots were quantified by the GeneTools program (SYNGENE). After densitometric analysis, the ratios between p-AMPK/total-AMPK and p-AKT/total AKT were calculated in each group.
Ovary follicular assessment.
Ovaries were longitudinally sectioned (4 μm) and stained with hematoxylin and eosin, and all sections were analyzed using inverted birefringence microscope Axio Observer (Carl Zeiss Microscopy, LLC, Thornwood, NY). The quantitative frequency analysis of ovarian follicles at different stages of development and corpora lutea was assessed at metestrus, as described previously (19, 31, 89). In brief, follicles were defined as primary if they had one layer of cuboidal granulosa cells; secondary follicles had multiple layers of granulosa cells but lacked a fluid-filled antrum; tertiary follicles had a fluid-filled antrum in addition to multiple layers of surrounding granulosa cells; preovulatory follicles had compact multiple-layered granulosa cells and a large fluid filled antrum; and atretic follicles exhibited signs of a degenerated oocyte nucleus, folding or discontinuous oocyte membrane, collapse or involution of the antrum, and detachment of the granulosa cells (89).
Statistical Analysis
Results are expressed as means ± SE. One-way ANOVA was used to test for differences between all groups following treatment with significance set at P < 0.05, and post hoc analysis was performed using the Bonferoni test (SPSS Statistics, IBM Corp., v.20.0.0). A trend in outcome variables was defined as P > 0.05 to P < 0.10.
RESULTS
Body Weight, Food Intake, Tissue Lipids, and Fasting Plasma Biochemical Parameters
The differences in body weight, food intake, and plasma biochemical parameters between control (lean, normal, non-PCOS) and the PCOS-prone control phenotype is consistent with previous established findings demonstrating the PCOS-prone group has features of MetS: obesity, insulin resistance and hypertriglyceridemia (88, 89, 102, 103). There was no difference in the final body weight or total food intake between the PCOS-prone control compared with PCOS-prone metformin, flutamide, or combination metformin-flutamide treatment groups, as shown in Table 1. However, metformin-flutamide combination treatment in PCOS-prone animals tended to lower food intake by ~10% compared with the PCOS-prone control and PCOS-prone metformin treated groups.
Fasting Plasma Glucose, Insulin, and HOMA-IR
There was no significant difference in fasting plasma glucose concentration between the PCOS-prone control and PCOS-prone treatment groups. The PCOS-prone metformin-flutamide combination group was shown to have reduced plasma glucose concentration, and this was not significantly different to the control group. The plasma insulin concentration decreased in the PCOS-prone groups treated with metformin and metformin-flutamide by ~50% compared with the PCOS-prone control group and were shown to be not significantly different compared with the control group. Homeostatic model assessment of insulin resistance (HOMA-IR) was lowered by up to 50% in the PCOS-prone metformin and metformin-flutamide combination treatment groups compared with the PCOS-prone control group, but these differences did not reach statistical significance, as shown in Table 2.
Table 2.
Body weight, fasting biochemical parameters, and tissue lipids in control and PCOS-prone control and treatment groups
| Control | PCOS-Prone Control | PCOS-Prone Metformin | PCOS-Prone Flutamide | PCOS-Prone Met+Flut | |
|---|---|---|---|---|---|
| Body weight, g | 208.2 ± 4.1 | 376.2 ± 6.2a | 377.3 ± 6.1a | 371.2 ± 6.3a | 378.4 ± 7.1a |
| Food intake, g | 585 ± 12 | 947 ± 19a | 977 ± 18a | 892 ± 19a | 864 ± 19a |
| Glucose, mg/dl | 96.9 ± 8.1 | 157.2 ± 10.3a | 164.1 ± 16.8a | 154.4 ± 12.9a | 127.6 ± 7.7a |
| Insulin, ng/ml | 0.32 ± 0.07a | 3.2 ± 0.75b | 1.64 ± 0.35a,b | 2.76 ± 0.77b | 1.89 ± 0.34a,b |
| HOMA-IR | 1.94 ± 0.39a | 35.06 ± 7.82b | 15.53 ± 2.91b | 25.36 ± 6.18b | 15.24 ± 2.90b |
| TG, mg/dl | 19.5 ± 2.8 | 993.6 ± 129.0a | 1,031.0 ± 102.8a | 556.8 ± 43.6b | 594.5 ± 68.6b |
| TC, mg/dl | 58.1 ± 6.0 | 131.0 ± 25.3a | 138.7 ± 14.4a | 145.2 ± 19.6a | 145.1 ± 18.9a |
| NEFA, mmol/l | 0.69 ± 0.08 | 0.78 ± 0.06a | 0.88 ± 0.08a | 1.14 ± 0.19a | 1.10 ± 0.14a |
| LDL-C, mg/dl | 24.4 ± 3.2 | 19.2 ± 4.9 | 24.3 ± 5.6 | 13.9 ± 1.8 | 13.4 ± 1.7 |
| HDL-C, mg/dl | 23.4 ± 1.9 | 15.8 ± 2.9a | 13.3 ± 3.5a | 17.9 ± 2.5a | 17.7 ± 3.5a |
| ApoB48, μg/ml | 30.0 ± 2.0 | 619.6 ± 42.8a | 614.8 ± 49.4a | 435.8 ± 39.7b | 404.7 ± 42.6b |
| ApoB100 (µg/ml) | 572.2 ± 62.8 | 2028.0 ± 316.1a | 1657.0 ± 154.2a,b | 1279.0 ± 159.78b | 1185.0 ± 127.6b |
| Liver TG (mg/mg protein) | 0.39 ± 0.060a | 1.36 ± 0.19b | 1.21 ± 0.13b | 1.01 ± 0.20a,b | 1.18 ± 0.16b |
| Intestinal TG (mg/mg protein) | 1.1 ± 0.04 | 0.69 ± 0.12 | 0.86 ± 0.19 | 1.02 ± 0.19 | 0.95 ± 0.01 |
Values are means ± SE. Values with a common letter are not statistically different (P < 0.05). Apo, apolipoprotein; Flut, flutamide; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; Met, metformin; NEFA, non-esterified fatty acids; PCOS, polycystic ovary syndrome; TC, total cholesterol; TG, triglyceride; HOMA-IR, homeostasis model assessment of insulin resistance.
Fasting Plasma Lipid and ApoB-Lipoproteins
In PCOS-prone animals, flutamide and metformin-flutamide treatments significantly lowered fasting plasma TG and ApoB48 concentrations by 45% and 30%, respectively, compared with the PCOS-prone control and PCOS-prone metformin groups, as shown in Table 2. In PCOS-prone animals, flutamide and metformin-flutamide combination treatment reduced fasting plasma ApoB100 concentrations by >30% compared with the PCOS-prone control group. There was no significant difference in fasting plasma total cholesterol and non-esterified fatty acids between the PCOS-prone control and PCOS-prone treatment groups. HDL-C tended to increase and LDL-C decrease in the PCOS-prone flutamide and metformin-flutamide groups, but these findings did not reach statistical significance.
Hepatic and Intestinal Lipid Content
The hepatic TG content was threefold higher in PCOS-prone animals compared with controls, as shown in Table 2. In PCOS-prone animals, flutamide treatment significantly lowered hepatic TG by 20% compared with the PCOS-prone control group and was not different compared with the control group. There was no significant difference in intestinal TG content between the control and PCOS-prone control and PCOS-prone treatment groups (Table 2).
Endocrine Hormones and Ovarian Follicular Morphology
Endocrine hormones.
Endocrine hormone profiles of PCOS-prone control and PCOS-prone treated animals are shown in Table 3. Free testosterone, total testosterone, and FAI were higher in PCOS-prone control compared with controls. Free testosterone, total testosterone, and FAI were lowered in PCOS-prone animals following treatment with metformin, flutamide, and metformin-flutamide combination, but the levels remained not significantly different from the PCOS-prone control group. SHBG concentration was 15% lower in the PCOS-prone groups compared with the control (lean, normal, non-PCOS) group. Metformin, flutamide, or metformin-flutamide treatments in the PCOS-prone groups had no significant effect on SHBG concentrations.
Table 3.
Endocrine hormone profile and ovary follicular assessment in control, PCOS-prone control, and PCOS-prone treatment groups
| Control | PCOS-Prone Control | PCOS-Prone Metformin | PCOS-Prone Flutamide | PCOS-Prone Met+Flut | |
|---|---|---|---|---|---|
| Free T, pg/ml | 0.62 ± 0.09a | 1.38 ± 0.13b | 0.93 ± 0.13a,b | 1.07 ± 0.20a,b | 0.97 ± 0.23a,b |
| Total T, ng/ml | 0.23 ± 0.01a | 0.35 ± 0.04b | 0.23 ± 0.03a,b | 0.26 ± 0.02a,b | 0.23 ± 0.03a,b |
| SHBG, ng/ml | 0.48 ± 0.01 | 0.41 ± 0.05a | 0.41 ± 0.06a | 0.41 ± 0.03a | 0.41 ± 0.06a |
| Estradiol, ng/ml | 21.6 ± 1.4 | 22.7 ± 1.4 | 28.7 ± 2.5 | 23.1 ± 2.6 | 20.7 ± 1.0 |
| FAI | 47.9 ± 2.1a | 85.0 ± 3.1b | 56.1 ± 2.5a,b | 63.4 ± 3.1a,b | 58.5 ± 2.6a,b |
| Follicles: | |||||
| Primary | 3.4 ± 0.7a | 1.0 ± 0.3b | 1.6 ± 0.5a,b | 2.0 ± 0.5a,b | 2.5 ± 0.6a,b |
| Secondary | 2.0 ± 0.6 | 1.6 ± 0.4 | 1.4 ± 0.5 | 1.7 ± 0.3 | 2.0 ± 0.3 |
| Tertiary | 3.2 ± 0.6 | 3.4 ± 0.5 | 4.2 ± 0.8 | 3.8 ± 1.1 | 5.7 ± 1.6 |
| Preovulatory | 1.2 ± 0.2a,c | 0.2 ± 0.2b | 1.0 ± 0.3a,c | 0.7 ± 0.3a,b | 2.0 ± 0.4c |
| Atretic | 2.6 ± 0.8a | 12.2 ± 2.5b | 5.2 ± 1.2a | 3.2 ± 0.7a | 5.5 ± 1.0a |
| Corpus luteum | 2.4 ± 0.2a | 1.0 ± 0.3b | 1.6 ± 0.4a,b | 1.4. ± 0.3a,b | 1.5 ± 0.5a,b |
| Corpus albicans | 3.0 ± 1.1 | 2.2 ± 0.4 | 2.4 ± 0.5 | 1.8 ± 0.5 | 3.7 ± 0.7 |
PCOS-prone animals were fed metformin (300 mg/kg) and flutamide (30 mg/kg) for 6 wk. Results are means ± SE; n = 12 animals/group. Values with common letter are not statistically different (P < 0.05, ANOVA). FAI, free androgen index; Flut, flutamide; Met, metformin; PCOS, polycystic ovary syndrome; SHBG, serum hormone binding globulin; T, testosterone.
Ovarian follicular assessment.
Ovarian follicular histological assessment is shown in Table 3. PCOS-prone animals had a threefold lower frequency of primary follicles compared with the control (lean, normal, non-PCOS) group, whereas the PCOS-prone treated groups tended to have an increase in primary follicle number compared with the PCOS-control group. There was no difference in secondary or tertiary follicle number between groups. Preovulatory follicles were 6-fold lower in PCOS-prone animals compared with controls. The number of preovulatory follicles was increased by 3- to 10-fold or normalized in PCOS-prone treated groups compared with the PCOS-prone control, and this was significant in the PCOS-prone metformin and metformin-flutamide combination treatment groups. In addition, PCOS-control animals had five times greater the number of atretic-cystic follicles compared with controls, whereas all PCOS-prone treatment groups showed a significant reduction in atretic-cystic follicle number. No significant difference in the frequency of corpus luteum was observed between PCOS-prone control and PCOS-prone treatment groups.
Consistent with previous observations in the PCOS-prone group, we observed lipid accumulation in the ovary stroma that is not present in the ovaries of normal control animals (89). There appeared to be a reduction of ~20%–30% in ovary lipid deposition in the PCOS-prone treatment groups compared with the PCOS-control group (data not shown).
Non-Fasting Insulin and Glucose, Plasma Lipids, and ApoB-Lipoprotein Responses to a Meal
Non-fasting plasma insulin and glucose response following a meal challenge.
PCOS-prone animals were shown to have an elevated non-fasting or postprandial (AUC) response in plasma glucoseAUC and insulinAUC compared with controls (lean, normal, non-PCOS animals), consistent with previous observations in this model (102) (Fig. 1). Treatment with metformin and metformin-flutamide combination in PCOS-prone groups lowered the plasma glucoseAUC response, but there was no significant difference in the incremental glucoseiAUC response between all groups. In PCOS-prone groups, treatment with metformin and flutamide did not modify the non-fasting plasma insulinAUC response; however, the combination of metformin-flutamide was shown to decrease the plasma insuliniAUC response toward that observed in the control group (Fig. 1).
Fig. 1.

The non-fasting plasma glucose and insulin response (AUC and iAUC) to a meal in control, PCOS-prone control, and PCOS-prone treatment groups following an oral meal tolerance test. Results are means ± SE, n = 12 animals/group. *, **Values with a common symbol are not statistically different (P < 0.05, ANOVA). AUC, area under the curve; iAUC, incremental AUC; Flut, flutamide; Met, metformin; PCOS, polycystic ovary syndrome.
Non-fasting lipid and ApoB-lipoprotein response following a high-fat meal.
Non-fasting total plasma TGAUC, cholesterolAUC, and ApoB48AUC following a high-fat meal were elevated in PCOS-prone animals compared with controls, as previously observed in this model (89) (Fig. 2, A–D). In PCOS-prone groups the treatments did not alter total plasma TGAUC, cholesterolAUC, ApoB48AUC, or ApoB100AUC compared with the PCOS-prone control group. However, flutamide and metformin-flutamide combination treatments did lower the postprandial or non-fasting response of TGiAUC, cholesteroliAUC, and Apo100iAUC to levels that were not significantly different from the control (lean, normal, non-PCOS) group. Flutamide and combination treatments significantly lowered ApoB48iAUC in PCOS-prone groups.
Fig. 2.

The non-fasting plasma triglyceride (A), cholesterol (B), ApoB48 (C), and ApoB100 (D) response (AUC and iAUC) to a high-fat meal in control, PCOS-prone control, and PCOS-prone treatment groups. Results are means ± SE, n = 12 animals/group. *, **Values with a common symbol are not statistically different (P < 0.05, ANOVA). Apo, apolipoprotein; AUC, area under the curve; iAUC, incremental AUC; Flut, flutamide; Met, metformin; PCOS, polycystic ovary syndrome.
Intestinal lipid and ApoB48-chylomicron secretion in the fasted and fed state.
Intestinal lymph secretion of TG, cholesterol, and ApoB48 (representing chylomicron particle number) are shown in Fig. 3, A–E. PCOS-prone animals have markedly elevated intestinal secretion of TG, cholesterol, and ApoB48 in the fasting and fed state compared with control (lean, normal, non-PCOS) animals. Intestinal TG secretion in the fasting and fed states was decreased by 75% in the flutamide treatment group compared with the PCOS-prone control group and was also decreased in the metformin-flutamide combination group by >30% (Fig. 3A). Metformin treatment alone had no effect on intestinal secretion of TG (Fig. 3A). Intestinal secretion of cholesterol in the fasted state was 60% and 50% lower in the metformin and flutamide treatment groups, respectively, compared with the PCOS-prone control group, and this was similar to that observed in the control group (lean, normal, non-PCOS) (Fig. 3B). In the fed state, treatments did appear to lower cholesterol secretion, and this was most marked in the flutamide treatment group (Fig. 3B). Despite changes in lipid secretion, there was no difference in intestinal secretion of ApoB48, indicative of chylomicron particle secretion, in the fasting or fed state following treatments in the PCOS-prone groups (Fig. 3C). However, flutamide treatment alone or in combination with metformin reduced or normalized the TG/ApoB48 ratio, which is representative of the TG associated with chylomicron particles, by 75% and 65% in the fasted state, and 85% and 70% in the fed state compared with the PCOS-prone control and metformin alone treatment groups (Fig. 3D). Furthermore, the cholesterol/ApoB48 ratio was reduced by 65%–80% in the fasted state with metformin, flutamide, and combination treatments (Fig. 3E). In addition, in the fed state, flutamide treatment alone or in combination with metformin decreased cholesterol/ApoB48 secretion by 35%–50% compared with both the PCOS-prone and control groups.
Fig. 3.
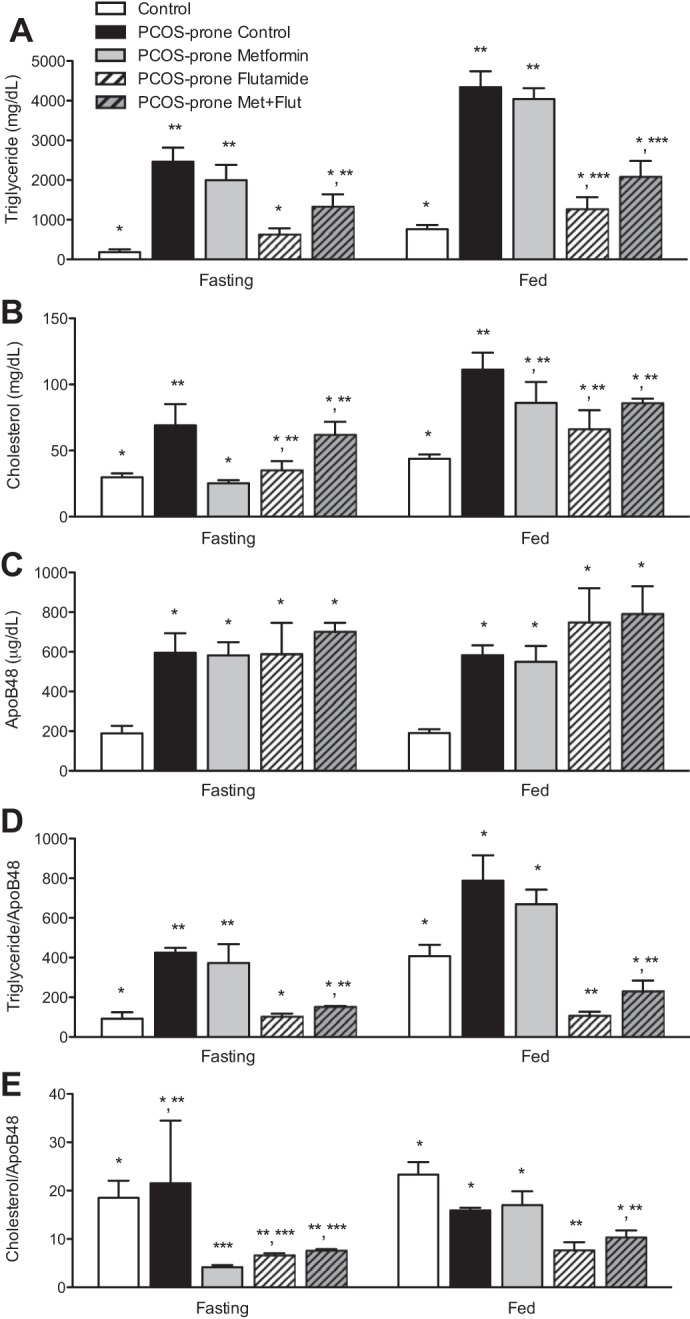
Intestinal lymph chylomicron-ApoB48 and lipid composition. Triglyceride (A), cholesterol (B), ApoB48 (C), TG/ApoB48 (D), and TC/ApoB48 (E) content following saline (fasting state) and Intralipid (fed state) infusion in control, PCOS-prone control, and PCOS-prone treatment groups. Results are means ± SE, n = 12 animals/group. *, **, *** Values with a common symbol are not statistically different (P < 0.05, ANOVA). Apo, apolipoprotein; Flut, flutamide; Met, metformin; PCOS, polycystic ovary syndrome; TC, total cholesterol; TG, triglyceride.
Hepatic and Intestinal Androgen and ER Gene Expression
Hepatic and intestinal expression of nuclear receptors AR and ER are shown in Fig. 4, A–B. Hepatic AR mRNA expression was not different between groups, and ERα mRNA expression was fourfold higher in PCOS-prone animals compared with controls (lean, normal, non-PCOS) (Fig. 4A). Metformin and metformin-flutamide combination treatments increased hepatic ERα mRNA expression by 35% compared with the PCOS-prone control group (Fig. 4A). In the intestine, there was no difference in AR mRNA expression between control and PCOS-prone groups (Fig. 4B). Metformin and flutamide treatments reduced intestinal AR mRNA expression, and this was significant in the metformin-flutamide treatment group. In contrast to the liver, the intestine ERα mRNA expression was twofold higher in control animals compared with the PCOS-prone group (Fig. 4B). ERβ mRNA expression was not different between control and PCOS-prone control groups. Metformin-flutamide treatment significantly decreased intestinal ERβ mRNA expression by >60% compared with the PCOS-prone control and other treatment groups (Fig. 4B).
Fig. 4.

Hepatic (A) and intestinal (B) androgen receptor (AR) and estrogen receptor-α (ERα) and estrogen receptor-β (ERβ) mRNA expression in control, PCOS-prone control, and PCOS-prone treatment groups. Results are expressed as a fold change compared with the control group and are means ± SE, n = 12 animals/group. Groups with a common letter are not statistically different (P < 0.05, ANOVA). Flut, flutamide; Met, metformin; PCOS, polycystic ovary syndrome.
Hepatic and Intestinal Lipogenic Gene and Protein Expression
PCOS-prone animals exhibited increased hepatic mRNA expression of LXRα, SREBP1, LDL-R, HMGCR, ApoB, ACC, FAS, and DGAT1 compared with the control group, as shown in Fig. 5. The combination of metformin-flutamide significantly increased hepatic LXRα and PPARα by 35% and 50% compared with the PCOS-prone control group. In the intestine, the PCOS-prone control group showed increased mRNA expression of SREBP2, LDL-R, and ApoB compared with the control group, as shown in Fig. 6. These results are consistent with the observations of higher plasma and intestinal lipid and ApoB48-chylomicron secretion. Intestinal mRNA expression of LXRα was significantly reduced in PCOS-prone animals compared with control animals by 25% and was restored to control group expression levels with metformin treatment alone. Treatment with flutamide resulted in a significant lowering of ACC mRNA compared with the PCOS-prone control group (Fig. 6). Hepatic expression of select lipidogenic, antilipogenic, and ApoB-lipoprotein assembly proteins was explored based on mRNA results. The hepatic ratio of p-AMPK/AMPK was higher in the control group (lean, normal, non-PCOS) compared with the PCOS-prone control group, reflecting the normal p-AMPK levels inhibiting lipogenesis (Fig. 7). The metformin-flutamide combination was shown to improve the ratio of p-AMPK/AMPK compared with the PCOS-prone control and treated groups (Fig. 7). MTP but not CideB, as indicators of ApoB-lipoprotein assembly, was reduced following flutamide and metformin-flutamide treatments compared with the PCOS-prone control group (Fig. 7). However, the control group had similar MTP and CideB compared with the PCOS-prone control group, suggesting an adaptive lipogenic expression of protein in the PCOS-prone hyperlipidemic phenotype. FAS and ACC proteins were shown to be lower in the control group (lean, normal, non-PCOS) compared with the PCOS-prone groups consistent with plasma lipid data; however, there was no effect of treatments on these markers of TG synthesis in the PCOS-prone groups (Fig. 7).
Fig. 5.

Hepatic lipogenic gene mRNA expression in control, PCOS-prone control, and PCOS-prone treatment groups. Results are expressed as a fold change compared with the control group and are means ± SE, n = 12 animals/group. Groups with a common letter are not statistically different (P < 0.05, ANOVA). ACC, acetyl CoA carboxylase; ApoB, apolipoprotein B; DGAT, diacylglycerol O-acyltransferase; FAS, fatty acid synthase; Flut, flutamide; LDLR, low-density lipoprotein receptor; LXRα, liver X receptor-α; Met, metformin; MTP, microsomal transport protein; PCOS, polycystic ovary syndrome; PPARα, peroxisome proliferator-activated receptor-α; SCAP, SREBP cleavage activating protein; SREBP, sterol regulatory element-binding protein.
Fig. 6.

Intestinal lipogenic gene mRNA expression in control, PCOS-prone control, and PCOS-prone treatment groups. Results are expressed as a fold change compared with the control group and are means ± SE, n = 12 animals/group. Groups with a common letter are not statistically different (P < 0.05, ANOVA). ACC, acetyl CoA carboxylase; ApoB, apolipoprotein B; DGAT, diacylglycerol O-acyltransferase; FAS, fatty acid synthase; Flut, flutamide; LDLR, low-density lipoprotein receptor; LXRα, liver X receptor-α; Met, metformin; MTP, microsomal transport protein; PCOS, polycystic ovary syndrome; PPARα, peroxisome proliferator-activated receptor-α; SCAP, SREBP cleavage activating protein; SREBP, sterol regulatory element-binding protein.
Fig. 7.

Hepatic protein expression in control, PCOS-prone control, and PCOS-prone treatment groups. Results are means ± SE, n = 6 animals/group. Groups with a common letter are not statistically different (P < 0.05, ANOVA). AKT, serine/threonine-protein kinase; AMPK, adenosine monophosphate-activated protein kinase; FAS, fatty acid synthase; Flut, flutamide; Met, metformin; p-ACC, phospho-acetyl CoA carboxylase; pAKT, phospho-AKT; pAMPK, phospho-AMPK; PCOS, polycystic ovary syndrome.
Hepatic and Intestinal Insulin Signaling Gene and Protein Expression
Hepatic and intestinal insulin signaling gene mRNA expression are shown in Fig. 8, A and B. The hepatic mRNA expression of insulin receptor (IR) was not different between control (lean, normal, non-PCOS) and PCOS-prone animals (Fig. 8A). Following metformin treatment, IR mRNA expression was upregulated by 30% and was further increased by twofold in the flutamide and metformin-flutamide combination treatment groups. MAPK1 mRNA expression increased by >30% in the treatment groups compared with the PCOS-control. Protein kinase B (also known as AKT2) mRNA expression was increased by all treatments (40%, 90%, and 80% in the metformin, flutamide, and metformin-flutamide combination, respectively) compared with the PCOS-prone control group. No change in the hepatic mRNA expression of PTPN1, JNK, and GSK3 were observed following treatments (Fig. 8A). The hepatic ratio of p-AKT/AKT protein, a marker of activation in the mediation of insulin signaling, was shown to improve with metformin and metformin-flutamide treatments in PCOS-prone groups, but this did not reach statistical significance (Fig. 7). IR mRNA expression was significantly lower in the intestine of PCOS-prone groups by 50% compared with the control group (lean, normal, non-PCOS), and the treatments in the PCOS-prone groups were observed to have no effect on the mRNA expression of this receptor (Fig. 8B). Similarly, intestinal MAPK1, AKT2, and PTPN1 mRNA levels were found to be lower in PCOS-prone animals compared with controls (lean, normal, non-PCOS). Flutamide treatment significantly lowered MAPK1 expression by 30%, and flutamide-metformin reduced MAPK1 expression by a further 30%. Flutamide and metformin treatments did not have an effect on AKT2 expression independently but in combination reduced AKT2 by 50% (Fig. 8B). PTPN1 and JNK mRNA expression was also significantly reduced with flutamide-metformin combination treatment by 30% and 40%, respectively, compared with the PCOS-prone control group.
Fig. 8.

Hepatic (A) and intestinal (B) insulin signaling gene mRNA expression in control, PCOS-prone control, and PCOS-prone treatment groups. Results are expressed as a fold change compared with the control group and are means ± SE, n = 12 animals/group. Groups with a common letter are not statistically different (P < 0.05, ANOVA). AKT2, serine/threonine-protein kinase-2; Flut, flutamide; GSK3, glycogen synthase kinase-3; IR, insulin receptor; JNK, Jun N-terminal kinase; MAPK1, mitogen-activated protein kinase-1; Met, metformin; PCOS, polycystic ovary syndrome; PTPN1, protein-tyrosine phosphatase 1B.
DISCUSSION
Hyperinsulinemia and hyperandrogenemia have been proposed to contribute to the pathophysiology of cardiometabolic risk in PCOS, including atherogenic dyslipidemia (9, 27, 36, 102). The insulin sensitizer metformin is commonly used in anovulatory infertility and to improve insulin-glucose metabolism in PCOS, but the effects on blood lipid metabolism are inconsistent, and the mechanisms remain unclear (10, 71, 91). The AR antagonist flutamide is used to treat hyperandrogenism and has been shown to lower fasting plasma lipids, but the physiological mechanisms remain unknown in PCOS (29, 42, 67). Exploring how metformin and flutamide regulate metabolic pathways may help us to further understand the mechanisms involved in the pathophysiology of PCOS and cardiometabolic risk. In turn, these mechanisms may facilitate the development of evidence-based research to investigate metabolic targets and new treatments to improve health outcomes in women with PCOS. We have used a PCOS-prone MetS rodent model to examine the physiological and mechanistic effects of metformin and flutamide on insulin-glucose, lipid, and ApoB-lipoprotein metabolism and reproductive-endocrine indices. Metformin and flutamide interventions were shown to mimic the metabolic effects observed in the clinical setting, and we have further demonstrated how these modulate lipid and ApoB-lipoprotein metabolism and steroid, lipidogenic, and insulin signaling pathways. Metformin was shown to decrease fasting insulin and HOMA-IR and androgen indices (free testosterone, total testosterone, and FAI) and to improve ovarian follicular development in PCOS-prone animals. On the other hand, flutamide and metformin-flutamide combination treatment attenuated androgen indices (free testosterone, total testosterone, and FAI), and this was associated with improved fasting plasma TG, ApoB48, and ApoB100-lipoproteins, and reduced intestinal secretion of TG/ApoB48-lipoproteins. These findings were associated with limited modulation of AR, ER, or lipidogenic protein and mRNA expression. Metformin-flutamide combination treatment upregulated hepatic and intestinal insulin signaling pathways, including IR, MAPK1, and AKT2 mRNA, and improved hepatic p-AKT/AKT and p-AMPK/AMPK protein levels in PCOS-prone animals.
Effect of Metformin, Flutamide, and Metformin-Flutamide Combination on Endocrine-Reproductive Indices
Metformin, flutamide, and metformin-flutamide combination treatments appeared to reduce plasma free testosterone, total testosterone, and FAI compared with the PCOS-prone control group and normalized these compared with the control (lean, normal, non-PCOS) group. Plasma SHBG concentrations were significantly lower in PCOS-prone groups compared with the control group, but treatments did not modulate plasma SHBG concentrations. These results are consistent with clinical findings in PCOS that show metformin reduces serum free testosterone concentrations while having limited effects on serum SHBG concentrations (92). Metformin is used in anovulatory PCOS to improve cyclicity and fertility outcomes, and metformin has been reported to downregulate steroidogenesis in the ovary and adrenal gland via inhibition of steroidogenic enzymes to decrease free testosterone, estrogen, dehydroepiandrosterone sulfate (DHEAS), and FAI (34, 73, 77, 107). The enzymes 17,20-lyase and 17α-hydroxylase are essential for testosterone synthesis and have been reported to be dysregulated in the ovaries of PCOS women (74, 84). Metformin’s effect to improve insulin sensitivity is proposed to be mediated by a downregulation of 17,20-lyase, 17α-hydroxylase, 3β-hydroxysteroid dehydrogenase, and StAR enzymes in ovarian androgen synthesis (8, 34, 92, 105). Therefore, metformin and metformin-flutamide combination may mediate effects through insulin regulation of ovarian androgen synthesis. Clinically, flutamide treatment has been demonstrated to lower plasma free testosterone, total testosterone, DHEA, androstenedione, and SHBG concentrations in PCOS after short- (8 wk) and long-term (18 mo) treatment and to increase the number of ovulatory cycles (41, 48, 78). Although not measured in this study, flutamide has also been shown to inhibit 17α-hydroxylase and 17,20-lyase activity in rat testis in vitro (5). In addition, flutamide inhibition of the AR may mediate effects on steroidogenic pathways, in particular decreased ACTH-stimulated adrenal androgen production [DHEAS, free testosterone (free T)] and 17β-hydroxysteroid dehydrogenase enzyme activity (106). It has been shown that atretic follicles have higher testosterone/estrogen ratio, and it has been proposed that androgen-induced follicular atresia opposes estradiol-driven granulosa cell proliferation and follicular development (96). Testosterone via the AR may lead to altered follicular development, reduced ovulation rate, and impaired cyclicity in PCOS (14, 78). Although estrus cyclicity was not measured in this study, the number of atretic follicles was significantly reduced, and preovulatory follicles and corpus luteum were shown to have normalized frequency following metformin, flutamide, and combination treatments.
Beneficial Effect of Metformin and Metformin-Flutamide Combination on Insulin-Glucose Metabolism
Metformin and metformin-flutamide treatments appeared to improve fasting plasma insulin and HOMA-IR; however, these treatments did not reduce postprandial impaired glucose tolerance and insulin secretion (insulinAUC) following a meal. In PCOS, metformin is commonly prescribed as standard of care in those with obesity, impaired glucose tolerance, and hyperinsulinemia with the aim of preventing transition to type 2 diabetes (71, 91, 92). In this study, metformin treatment increased hepatic MAPK1, AKT2, and IR mRNA levels and reduced JNK mRNA compared with the PCOS-prone control group. MAPK1 activation is downregulated in insulin resistance, and activation of this pathway regulates IR expression (105). Metformin has been shown to inhibit mitochondrial respiratory-chain complex-1, resulting in activation of AMPK (105, 117). AMPK phosphorylates IR and tyrosine kinase as well as downstream targets, IR substrate (IRS)-1 and IRS-2, to restore skeletal muscle and hepatic insulin signaling (23, 24). Metformin is proposed to inhibit hepatic gluconeogenesis by promoting AMPK activation, which acts to decrease activity of catabolic pathways utilizing ATP, such as gluconeogenesis and lipogenesis (105, 117). The p-AMPK/AMPK protein ratio, an indicator of activation of AMPK, was improved in metformin-flutamide treated PCOS-prone animals. AKT2 is an important regulator of insulin signaling, and AKT2 activation promotes translocation of glucose transporters to the plasma membrane (43), and the ratio of p-AKT/AKT protein appeared to improve with metformin and metformin-flutamide treatment in PCOS-prone animals. Increase in JNK expression has been shown to increase serine phosphorylation of IRS-1, leading to impaired insulin signaling (113). In turn, AMPK can activate JNK and suppress phosphatase and tension homologue phosphorylation, which is a negative regulator of insulin signaling. Hence, inhibition of phosphatase and tension homologue restores AKT2 activation and is linked to improved insulin signaling in cells and responsiveness to insulin (59, 105). Therefore, metformin appears to improve insulin signaling in the PCOS-prone MetS rodent model via IR, AKT2, MAPK1, and AMPK signaling pathways, and this may contribute to the lowering of fasting plasma glucose, insulin, and HOMA-IR.
Interestingly, PCOS prone animals exhibited elevated hepatic ERα mRNA expression compared with control animals. ERα has been proposed to regulate insulin-glucose homeostasis, and ERα knockout mice develop insulin resistance (17). Metformin, flutamide, and combination treatments were shown to increase ERα mRNA expression in PCOS-prone animals. The ERα can activate PPARγ and GLUT4 genes in skeletal muscle and adipose tissues, resulting in an increase in tissue glucose uptake (11). However, liver-specific ERα knockout mice have been shown to be metabolically normal, suggesting that hepatic ERα may not influence whole-body insulin-glucose metabolism (66). Flutamide treatment alone did not significantly affect insulin-glucose metabolism, whereas flutamide-metformin combination treatment reduced fasting glucose, insulin, and HOMA-IR similarly to metformin alone. These results are consistent with clinical studies, in which flutamide alone does not attenuate impaired insulin-glucose metabolism (42, 86).
It has been shown that plasma insulin concentrations positively correlate with decreased SHBG and increased testosterone concentrations in PCOS (22, 33). Testosterone acts via the AR to elicit metabolic effects and AR polymorphisms, such as shorter (CAG)n repeat length, and these are associated with increased transcriptional activity of the AR (12). Shorter CAG length of the AR and increased circulating plasma testosterone have been positively correlated with insulin concentrations in women with PCOS (69, 87). The mechanism linking AR activation to insulin resistance is not well understood. In the metformin-flutamide treated group, hepatic AKT2 and MAPK1 mRNA expression and the ratio of p-AKT/AKT protein were increased and AR mRNA expression was decreased. AKT2 has been shown to phosphorylate AR at Ser213 and Ser791 residues. The phosphorylation allows AR to be recognized by Mdm2 E3 ligase and undergo further ubiquitination and proteasomal degradation (58). This may lead to a decrease in cytoplasmic AR levels and decreased androgen binding to the AR. Therefore, improved insulin sensitivity observed in animals treated with metformin-flutamide combination may be due to a synergistic effect on insulin-AR signaling pathways.
Beneficial Effect of Flutamide and Metformin-Flutamide Combination on Fasting and Non-Fasting Plasma Lipids and ApoB-Lipoprotein Metabolism
The PCOS-prone model is a well-established model of cardiometabolic risk and demonstrates increased plasma concentrations of ApoB-lipoproteins, TG, and total cholesterol in the fasting and non-fasted state (88, 89, 102). In this study, metformin treatment alone did not impact fasting or postprandial plasma lipids and ApoB-lipoproteins in the PCOS-prone model. These results are consistent with clinical monotherapy with metformin in PCOS, which has been shown to have limited effects on plasma lipids (71). A few studies have shown moderate effects of metformin on fasting plasma lipids, including TG and LDL-C, but this appears to be only associated with significant improvements in glucose-insulin metabolism (67, 71, 91). Flutamide treatment reduced fasting plasma TG by 40%, and decreased ApoB-lipoproteins and LDL-C by ~25%, in the PCOS-prone model. These findings are consistent with clinical studies, which have shown flutamide reduces fasting plasma lipids, TG, total cholesterol, and LDL-C (7, 29, 41, 49, 50). Flutamide was also shown to attenuate the non-fasting lipid response (iAUC) for plasma TG, total cholesterol, ApoB100, and ApoB48-lipoproteins by 30%–40%. Combination metformin-flutamide treatment did not significantly improve fasting or non-fasting lipids, suggesting the effects were primarily associated with flutamide. Metformin-flutamide treatment has been shown to reduce fasting plasma TG after 12 mo treatment, albeit under conditions of dietary caloric restriction (41, 42), and metformin-flutamide combined with an OC reduces LDL-C and increases HDL-C but does not reverse OC-induced hypertriglyceridemia (52). Collectively, these results support the role of the AR to independently modulate lipidogenic pathways in TG and ApoB-lipoprotein metabolism.
Flutamide and metformin-flutamide treatments reduced intestinal lymph secretion of TG, cholesterol, and TG/ApoB48, and total cholesterol/ApoB48 ratios, the latter representing lipid associated with each chylomicron-ApoB48 particle. These findings may represent a downregulation of intestinal synthesis and secretion of lipid in the fasted stated and may reflect decreased absorption of lipid and lipogenesis in the non-fasted state (102). Less lipidated intestinal chylomicrons have been shown to be lipolyzed more readily and cleared at a higher rate from the circulation, which may render the chylomicron-ApoB48-lipoprotein remnant to be less atherogenic (65). Furthermore, it has been demonstrated that atherogenic cholesterol-dense ApoB48-remnants readily permeate the arterial wall, and therefore reduced cholesterol content and increased rate of ApoB48-remnant clearance may contribute to a reduction in atherogenic risk (82, 98). Flutamide inhibits activation of the AR, preventing binding to androgen response elements on target genes. Indeed, activation of the AR, at least in cancer cell lines, has been shown to activate SREBP1, and cholesterol and TG synthesis (47). Our results showed flutamide reduced the expression of hepatic SREBP-1c and ApoB mRNA, and MTP, and this may reflect the decrease in fasting and non-fasting plasma TG and ApoB-lipoproteins. However, no change in hepatic mRNA or protein for FAS or ACC involved in TG synthesis was observed. Total cholesterol was not altered, and LDL-C tended to be lower by 30% in flutamide treated animals. These effects could be due to LXR-mediated degradation of LDL-R, as LXRα mRNA expression was increased with flutamide and metformin-flutamide combination, and LXR activation has been shown to increase LDL-R degradation in hepatocytes and enterocytes, thus reducing LDL-C uptake into cells (35, 115). Decreased intestinal secretion of cholesterol was observed in PCOS-prone animals in response to metformin and flutamide, and this may be related to LXRα regulation of cholesterol homeostasis. Intestine-specific LXRα activation has been shown to lower the intracellular cholesterol pool in the enterocyte, which reduces cholesterol incorporation into chylomicrons, but LXRα mRNA expression was not increased in the enterocytes of metformin-treated animals (57).
Insulin resistance is associated with intestinal oversecretion of chylomicrons-ApoB48 (37, 103), and enterocytes from insulin-resistant hamsters have decreased protein expression and activity of IRS-1 and AKT and increased protein expression and activity of PTPN1 (37). In this study, PCOS-prone animals with insulin resistance had reduced intestinal IR, AKT, MAPK, and PTPN1. Metformin treatment did not attenuate enterocyte mRNA expression of these genes, and there was no reduction in intestinal lipid or chylomicron-ApoB48 secretion in the PCOS-prone group. However, intestinal MAPK1, AKT2, PTPN1, and JNK expression were reduced with metformin-flutamide combination, suggesting a possible role of AR in the regulation of these genes that could mediate reductions in intestinal lipogenesis associated with insulin signaling in PCOS-prone animals. Our investigation of protein expression was done in hepatic tissue but not intestine because of limited tissue availability. Future studies could determine the effects of testosterone and flutamide on protein and/or phosphorylated protein modulation and the protein expression of AR and AR activity to determine the role of AR in regulating intestinal insulin signaling and lipogenic pathways in PCOS. Testosterone may directly regulate insulin action, and AR action is known to be modulated by phosphorylation by AKT2 (58, 94). Metformin has been proposed to activate AMPK, resulting in downregulation of lipogenic genes, such as SREBP1, HMGR, and ACC, to reduce hepatic TG and cholesterol synthesis (68). Our results showed metformin-flutamide but not metformin alone increased p-AMPK/AMPK protein and altered hepatic lipogenic gene expression, which may reflect the improvement in lipids and ApoB48 observed in this PCOS-prone group. Hepatic PPARα expression was significantly increased in metformin-flutamide combination compared with control, suggestive of an increase in fatty acid oxidation, which may limit fatty acid availability for TG synthesis and incorporation into ApoB-lipoproteins. An alternative possibility, although not measured in this study, is that the AR may interact with other nuclear transcription factors such as the glucocorticoid receptor (GR) (25). Genetic studies on GR receptor polymorphism in PCOS have reported GR has an important role in insulin sensitivity and lipid metabolism, suggesting that impaired glucocorticoid signaling and altered hypothalamic-pituitary-adrenocortical axis activity may contribute to metabolic aberrations in PCOS (64).
Intestinal and Hepatic AR and ER Gene Expression Is Altered with Metformin, Flutamide, and Metformin-Flutamide Combination Treatment
Enterocyte ERβ expression was significantly reduced following metformin-flutamide combination treatment, and no effect of treatments on ERα was observed. Little is known regarding ERβ and lipid and insulin-glucose metabolism, particularly in conditions of PCOS associated with hyperandrogenemia and insulin resistance. Ovariectomized ERβ knockout mice have been reported to have restored insulin-glucose metabolism and normalized plasma TG, suggesting that ERβ deficiency may play a cardiometabolic protective role, at least in diet-induced obesity, which has been associated with the obese-MetS PCOS phenotype (38).
Flutamide, metformin, and combination treatments lowered intestinal mRNA AR expression. This is in contrast to other cell culture studies that have shown flutamide upregulates AR mRNA expression (45, 58). Furthermore, our results showed flutamide treatment appeared to lower androgen indices including free T and total T, or FAI. Whole-body AR knockout female mice have been shown to have increased fasting plasma TG, free fatty acids, insulin, and glucose, and increased hepatic TG accumulation (61), whereas our results show flutamide-AR inhibition normalizes an aberrant lipid profile and hepatic TG in the PCOS-prone MetS rodent model. The onset of elevated plasma TG and glucose concentrations and hepatic TG has also been shown to be dependent on neuronal AR expression in dihydrotestosterone-treated mice (18). Reports to date suggest that the presence of functional AR is required for normal energy, insulin-glucose, and lipid metabolism in females, but there is limited knowledge on normal and altered AR expression and activity, androgen modulation of the AR, effects of coactivators or repressors, and downstream interaction with steroidogenic, lipogenic, and insulinotropic genes in PCOS. More research is needed to understand the role of the AR and androgen sensitivity in PCOS, the possible long-term use of low-dose flutamide or other AR-antagonists, and the development of safe selective AR modulators as potential treatments for the endocrine and cardiometabolic dysfunction in PCOS (49, 55, 67, 72).
We did not measure subclinical cardiovascular risk variables such as endothelial and cardiac function, which is a limitation of the study, but clinical studies have shown the use of combination metformin-flutamide-pioglitazone treatment can reduce carotid artery media thickness (49, 101). Flutamide is a teratogen and is contraindicated for use in women of reproductive age; however, very-lose-dose flutamide (62.5 mg/day) may be a potential treatment for improving fasting and non-fasting lipid and ApoB-lipoprotein metabolism (48, 49, 101). In addition, low dose flutamide-metformin and polytherapy with metformin-flutamide-pioglitazone and/or in combination with OC and statin treatments have been reported to improve fasting plasma lipids and insulin-glucose metabolism, and no clinical side effects or hepatoxicity have been reported (49, 67, 101). However, further randomized clinical trial evidence is warranted to test efficacy and safety of interventions to reduce fasting and non-fasting ApoB-lipoprotein dyslipidemia and subclinical CVD indices in women with PCOS.
Conclusion
The insulin-sensitizing agent metformin, the AR antagonist flutamide, and metformin-flutamide combination treatment were shown to selectively improve metabolic and endocrine aberrations in the PCOS-prone MetS rodent model. Metformin improved insulin sensitivity, which was associated with improvement in insulin signaling. Flutamide was shown to improve fasting plasma TG and ApoB-lipoprotein concentrations and intestinal TG secretion, and these results are consistent with the hypolipidemic benefits of flutamide observed in clinical studies. Furthermore, these findings highlight the effect of inhibition of the AR on reducing intestinal lipid secretion associated with ApoB48-chylomicrons. Metformin-flutamide combination treatment upregulated hepatic and insulin signaling pathways, which may contribute to improvements observed in lipid metabolism. In conclusion, these findings highlight the interaction of the AR with insulin signaling and lipogenic pathways and the need for further investigation into potential pharmaceutical and dietary approaches that are safe and target the pathophysiological mechanisms associated with the complex endocrine and cardiometabolic milieu observed in high-risk PCOS patients with MetS.
GRANTS
This research was supported by Natural Sciences and Engineering Research Council of Canada (D. Vine and S. Proctor).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.F.V. conceived and designed research; M.K., A.D., and R.W. performed experiments; M.K., R.W., and D.F.V. analyzed data; R.L., S.D.P., and D.F.V. interpreted results of experiments; M.K. and D.F.V. prepared figures; M.K. and D.F.V. drafted manuscript; R.L., M.G., S.D.P., and D.F.V. edited and revised manuscript; R.L., S.D.P., and D.F.V. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge the important technical support of S. Kelly and S. Sokolik.
REFERENCES
- 1.Abbott DH, Barnett DK, Bruns CM, Dumesic DA. Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update 11: 357–374, 2005. doi: 10.1093/humupd/dmi013. [DOI] [PubMed] [Google Scholar]
- 2.Adeli K, Lewis GF. Intestinal lipoprotein overproduction in insulin-resistant states. Curr Opin Lipidol 19: 221–228, 2008. doi: 10.1097/MOL.0b013e3282ffaf82. [DOI] [PubMed] [Google Scholar]
- 3.Alexander CJ, Tangchitnob EP, Lepor NE. Polycystic ovary syndrome: a major unrecognized cardiovascular risk factor in women. Rev Cardiovasc Med 10: 83–90, 2009. [PubMed] [Google Scholar]
- 4.Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group Consensus on women’s health aspects of polycystic ovary syndrome (PCOS). Hum Reprod 27: 14–24, 2012. doi: 10.1093/humrep/der396. [DOI] [PubMed] [Google Scholar]
- 5.Ayub M, Levell MJ. Inhibition of rat testicular 17 α-hydroxylase and 17,20-lyase activities by anti-androgens (flutamide, hydroxyflutamide, RU23908, cyproterone acetate) in vitro. J Steroid Biochem 28: 43–47, 1987. doi: 10.1016/0022-4731(87)90122-1. [DOI] [PubMed] [Google Scholar]
- 6.Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W, Janssen OE, Legro RS, Norman RJ, Taylor AE, Witchel SF; Task Force on the Phenotype of the Polycystic Ovary Syndrome of The Androgen Excess and PCOS Society . The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: the complete task force report. Fertil Steril 91: 456–488, 2009. doi: 10.1016/j.fertnstert.2008.06.035. [DOI] [PubMed] [Google Scholar]
- 7.Bahceci M, Aydemir M, Tuzcu A. Effects of oral fat and glucose tolerance test on serum lipid profile, apolipoprotein, and CRP concentration, and insulin resistance in patients with polycystic ovary syndrome. Fertil Steril 87: 1363–1368, 2007. doi: 10.1016/j.fertnstert.2006.11.031. [DOI] [PubMed] [Google Scholar]
- 8.Baptiste CG, Battista MC, Trottier A, Baillargeon JP. Insulin and hyperandrogenism in women with polycystic ovary syndrome. J Steroid Biochem Mol Biol 122: 42–52, 2010. doi: 10.1016/j.jsbmb.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barber TM, Dimitriadis GK, Andreou A, Franks S. Polycystic ovary syndrome: insight into pathogenesis and a common association with insulin resistance. Clin Med (Lond) 16: 262–266, 2016. doi: 10.7861/clinmedicine.16-3-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bargiota A, Diamanti-Kandarakis E. The effects of old, new and emerging medicines on metabolic aberrations in PCOS. Ther Adv Endocrinol Metab 3: 27–47, 2012. doi: 10.1177/2042018812437355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barros RP, Gabbi C, Morani A, Warner M, Gustafsson JA. Participation of ERalpha and ERbeta in glucose homeostasis in skeletal muscle and white adipose tissue. Am J Physiol Endocrinol Metab 297: E124–E133, 2009. doi: 10.1152/ajpendo.00189.2009. [DOI] [PubMed] [Google Scholar]
- 12.Beilin J, Ball EM, Favaloro JM, Zajac JD. Effect of the androgen receptor CAG repeat polymorphism on transcriptional activity: specificity in prostate and non-prostate cell lines. J Mol Endocrinol 25: 85–96, 2000. doi: 10.1677/jme.0.0250085. [DOI] [PubMed] [Google Scholar]
- 13.Berneis K, Rizzo M, Hersberger M, Rini GB, Di Fede G, Pepe I, Spinas GA, Carmina E. Atherogenic forms of dyslipidaemia in women with polycystic ovary syndrome. Int J Clin Pract 63: 56–62, 2009. doi: 10.1111/j.1742-1241.2008.01897.x. [DOI] [PubMed] [Google Scholar]
- 14.Blank SK, McCartney CR, Helm KD, Marshall JC. Neuroendocrine effects of androgens in adult polycystic ovary syndrome and female puberty. Semin Reprod Med 25: 352–359, 2007. doi: 10.1055/s-2007-984741. [DOI] [PubMed] [Google Scholar]
- 15.Borthwick F, Mangat R, Warnakula S, Jacome-Sosa M, Vine DF, Proctor SD. Simvastatin treatment upregulates intestinal lipid secretion pathways in a rodent model of the metabolic syndrome. Atherosclerosis 232: 141–148, 2014. doi: 10.1016/j.atherosclerosis.2013.10.031. [DOI] [PubMed] [Google Scholar]
- 16.Brill DS, Moenter SM. Androgen receptor antagonism and an insulin sensitizer block the advancement of vaginal opening by high-fat diet in mice. Biol Reprod 81: 1093–1098, 2009. doi: 10.1095/biolreprod.109.079301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryzgalova G, Gao H, Ahren B, Zierath JR, Galuska D, Steiler TL, Dahlman-Wright K, Nilsson S, Gustafsson JA, Efendic S, Khan A. Evidence that oestrogen receptor-alpha plays an important role in the regulation of glucose homeostasis in mice: insulin sensitivity in the liver. Diabetologia 49: 588–597, 2006. doi: 10.1007/s00125-005-0105-3. [DOI] [PubMed] [Google Scholar]
- 18.Caldwell ASL, Edwards MC, Desai R, Jimenez M, Gilchrist RB, Handelsman DJ, Walters KA. Neuroendocrine androgen action is a key extraovarian mediator in the development of polycystic ovary syndrome. Proc Natl Acad Sci USA 114: E3334–E3343, 2017. doi: 10.1073/pnas.1616467114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caligioni CS. Assessing reproductive status/stages in mice. Curr Protoc Neurosci 48, Appendix: 4I.1–4I.8, 2009. doi: 10.1002/0471142301.nsa04is48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carmina E, Chu MC, Longo RA, Rini GB, Lobo RA. Phenotypic variation in hyperandrogenic women influences the findings of abnormal metabolic and cardiovascular risk parameters. J Clin Endocrinol Metab 90: 2545–2549, 2005. doi: 10.1210/jc.2004-2279. [DOI] [PubMed] [Google Scholar]
- 21.Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Borén J, Catapano AL, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Nordestgaard BG, Ray KK, Reiner Z, Taskinen MR, Tokgözoglu L, Tybjærg-Hansen A, Watts GF; European Atherosclerosis Society Consensus Panel . Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J 32: 1345–1361, 2011. doi: 10.1093/eurheartj/ehr112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen C, Smothers J, Lange A, Nestler JE, Strauss Iii JF, Wickham Iii EP. Sex hormone-binding globulin genetic variation: associations with type 2 diabetes mellitus and polycystic ovary syndrome. Minerva Endocrinol 35: 271–280, 2010. [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng JT, Huang CC, Liu IM, Tzeng TF, Chang CJ. Novel mechanism for plasma glucose-lowering action of metformin in streptozotocin-induced diabetic rats. Diabetes 55: 819–825, 2006. doi: 10.2337/diabetes.55.03.06.db05-0934. [DOI] [PubMed] [Google Scholar]
- 24.Chopra I, Li HF, Wang H, Webster KA. Phosphorylation of the insulin receptor by AMP-activated protein kinase (AMPK) promotes ligand-independent activation of the insulin signalling pathway in rodent muscle. Diabetologia 55: 783–794, 2012. doi: 10.1007/s00125-011-2407-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Claessens F, Joniau S, Helsen C. Comparing the rules of engagement of androgen and glucocorticoid receptors. Cell Mol Life Sci 74: 2217–2228, 2017. doi: 10.1007/s00018-017-2467-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dejager S, Pichard C, Giral P, Bruckert E, Federspield MC, Beucler I, Turpin G. Smaller LDL particle size in women with polycystic ovary syndrome compared to controls. Clin Endocrinol (Oxf) 54: 455–462, 2001. doi: 10.1046/j.1365-2265.2001.01245.x. [DOI] [PubMed] [Google Scholar]
- 27.Diamanti-Kandarakis E. Polycystic ovarian syndrome: pathophysiology, molecular aspects and clinical implications. Expert Rev Mol Med 10: e3, 2008. doi: 10.1017/S1462399408000598. [DOI] [PubMed] [Google Scholar]
- 28.Diamanti-Kandarakis E, Kandaraki E, Christakou C, Panidis D. The effect of pharmaceutical intervention on lipid profile in polycystic ovary syndrome. Obes Rev 10: 431–441, 2009. doi: 10.1111/j.1467-789X.2009.00588.x. [DOI] [PubMed] [Google Scholar]
- 29.Diamanti-Kandarakis E, Mitrakou A, Raptis S, Tolis G, Duleba AJ. The effect of a pure antiandrogen receptor blocker, flutamide, on the lipid profile in the polycystic ovary syndrome. J Clin Endocrinol Metab 83: 2699–2705, 1998. doi: 10.1210/jcem.83.8.5041. [DOI] [PubMed] [Google Scholar]
- 30.Diamanti-Kandarakis E, Papavassiliou AG, Kandarakis SA, Chrousos GP. Pathophysiology and types of dyslipidemia in PCOS. Trends Endocrinol Metab 18: 280–285, 2007. doi: 10.1016/j.tem.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 31.Diane A, Kupreeva M, Borthwick F, Proctor SD, Pierce WD, Vine DF. Cardiometabolic and reproductive benefits of early dietary energy restriction and voluntary exercise in an obese PCOS-prone rodent model. J Endocrinol 226: 193–206, 2015. doi: 10.1530/JOE-14-0711. [DOI] [PubMed] [Google Scholar]
- 32.Duez H, Lamarche B, Uffelman KD, Valero R, Cohn JS, Lewis GF. Hyperinsulinemia is associated with increased production rate of intestinal apolipoprotein B-48-containing lipoproteins in humans. Arterioscler Thromb Vasc Biol 26: 1357–1363, 2006. doi: 10.1161/01.ATV.0000222015.76038.14. [DOI] [PubMed] [Google Scholar]
- 33.Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes 38: 1165–1174, 1989. doi: 10.2337/diab.38.9.1165. [DOI] [PubMed] [Google Scholar]
- 34.Ehrmann DA, Cavaghan MK, Imperial J, Sturis J, Rosenfield RL, Polonsky KS. Effects of metformin on insulin secretion, insulin action, and ovarian steroidogenesis in women with polycystic ovary syndrome. J Clin Endocrinol Metab 82: 524–530, 1997. [DOI] [PubMed] [Google Scholar]
- 35.Engelking LJ, McFarlane MR, Li CK, Liang G. Blockade of cholesterol absorption by ezetimibe reveals a complex homeostatic network in enterocytes. J Lipid Res 53: 1359–1368, 2012. doi: 10.1194/jlr.M027599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fauser BC, Tarlatzis BC, Rebar RW, Legro RS, Balen AH, Lobo R, Carmina E, Chang J, Yildiz BO, Laven JS, Boivin J, Petraglia F, Wijeyeratne CN, Norman RJ, Dunaif A, Franks S, Wild RA, Dumesic D, Barnhart K. Consensus on women's health aspects of polycystic ovary syndrome (PCOS): the Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil Steril 97: 28–38.e25, 2012. doi: 10.1016/j.fertnstert.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 37.Federico LM, Naples M, Taylor D, Adeli K. Intestinal insulin resistance and aberrant production of apolipoprotein B48 lipoproteins in an animal model of insulin resistance and metabolic dyslipidemia: evidence for activation of protein tyrosine phosphatase-1B, extracellular signal-related kinase, and sterol regulatory element-binding protein-1c in the fructose-fed hamster intestine. Diabetes 55: 1316–1326, 2006. doi: 10.2337/db04-1084. [DOI] [PubMed] [Google Scholar]
- 38.Foryst-Ludwig A, Clemenz M, Hohmann S, Hartge M, Sprang C, Frost N, Krikov M, Bhanot S, Barros R, Morani A, Gustafsson JA, Unger T, Kintscher U. Metabolic actions of estrogen receptor beta (ERβ) are mediated by a negative cross-talk with PPARγ. PLoS One 4: e1000108, 2008. doi: 10.1371/journal.pgen.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fruchart JC, Sacks FM, Hermans MP; International Steering Committee of R(3)i . Implications of the ACCORD lipid study: perspective from the Residual Risk Reduction Initiative (R(3)i). Curr Med Res Opin 26: 1793–1797, 2010. doi: 10.1185/03007995.2010.489341. [DOI] [PubMed] [Google Scholar]
- 40.Fruzzetti F, Perini D, Lazzarini V, Parrini D, Genazzani AR. Hyperandrogenemia influences the prevalence of the metabolic syndrome abnormalities in adolescents with the polycystic ovary syndrome. Gynecol Endocrinol 25: 335–343, 2009. doi: 10.1080/09513590802630146. [DOI] [PubMed] [Google Scholar]
- 41.Gambineri A, Pelusi C, Genghini S, Morselli-Labate AM, Cacciari M, Pagotto U, Pasquali R. Effect of flutamide and metformin administered alone or in combination in dieting obese women with polycystic ovary syndrome. Clin Endocrinol (Oxf) 60: 241–249, 2004. doi: 10.1111/j.1365-2265.2004.01973.x. [DOI] [PubMed] [Google Scholar]
- 42.Gambineri A, Patton L, Vaccina A, Cacciari M, Morselli-Labate AM, Cavazza C, Pagotto U, Pasquali R. Treatment with flutamide, metformin, and their combination added to a hypocaloric diet in overweight-obese women with polycystic ovary syndrome: a randomized, 12-month, placebo-controlled study. J Clin Endocrinol Metab 91: 3970–3980, 2006. doi: 10.1210/jc.2005-2250. [DOI] [PubMed] [Google Scholar]
- 43.Gonzalez E, McGraw TE. Insulin signaling diverges into Akt-dependent and -independent signals to regulate the recruitment/docking and the fusion of GLUT4 vesicles to the plasma membrane. Mol Biol Cell 17: 4484–4493, 2006. doi: 10.1091/mbc.e06-07-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goodman NF, Cobin RH, Futterweit W, Glueck JS, Legro RS, Carmina E; American Association of Clinical Endocrinologists (AACE); American College of Endocrinology (ACE); Androgen Excess and PCOS Society (AES) . American Association of Clinical Endocrinologists, American College of Endocrinology, and Androgen Excess and PCOS Society disease state clinical review: guide to the best practices in the evaluation and treatment of polycystic ovary syndrome–Part I. Endocr Pract 21: 1291–1300, 2015. doi: 10.4158/EP15748.DSC. [DOI] [PubMed] [Google Scholar]
- 45.Hackenberg R, Hawighorst T, Filmer A, Slater EP, Bock K, Beato M, Schulz KD. Regulation of androgen receptor mRNA and protein level by steroid hormones in human mammary cancer cells. J Steroid Biochem Mol Biol 43: 599–607, 1992. doi: 10.1016/0960-0760(92)90284-P. [DOI] [PubMed] [Google Scholar]
- 46.Handelsman DJ, Jimenez M, Singh GK, Spaliviero J, Desai R, Walters KA. Measurement of testosterone by immunoassays and mass spectrometry in mouse serum, testicular, and ovarian extracts. Endocrinology 156: 400–405, 2015. doi: 10.1210/en.2014-1664. [DOI] [PubMed] [Google Scholar]
- 47.Heemers HV, Verhoeven G, Swinnen JV. Androgen activation of the sterol regulatory element-binding protein pathway: current insights. Mol Endocrinol 20: 2265–2277, 2006. doi: 10.1210/me.2005-0479. [DOI] [PubMed] [Google Scholar]
- 48.Ibáñez L, de Zegher F. Low-dose flutamide-metformin therapy for hyperinsulinemic hyperandrogenism in non-obese adolescents and women. Hum Reprod Update 12: 243–252, 2006. doi: 10.1093/humupd/dmi054. [DOI] [PubMed] [Google Scholar]
- 49.Ibáñez L, López-Bermejo A, del Rio L, Enríquez G, Valls C, de Zegher F. Combined low-dose pioglitazone, flutamide, and metformin for women with androgen excess. J Clin Endocrinol Metab 92: 1710–1714, 2007. doi: 10.1210/jc.2006-2684. [DOI] [PubMed] [Google Scholar]
- 50.Ibáñez L, López-Bermejo A, Díaz M, Enríquez G, Valls C, de Zegher F. Pioglitazone (7.5 mg/day) added to flutamide-metformin in women with androgen excess: additional increments of visfatin and high molecular weight adiponectin. Clin Endocrinol (Oxf) 68: 317–320, 2008. doi: 10.1111/j.1365-2265.2007.03137.x. [DOI] [PubMed] [Google Scholar]
- 51.Ibáñez L, Oberfield SE, Witchel S, Auchus RJ, Chang RJ, Codner E, Dabadghao P, Darendeliler F, Elbarbary NS, Gambineri A, Garcia Rudaz C, Hoeger KM, López-Bermejo A, Ong K, Peña AS, Reinehr T, Santoro N, Tena-Sempere M, Tao R, Yildiz BO, Alkhayyat H, Deeb A, Joel D, Horikawa R, de Zegher F, Lee PA. An international consortium update: pathophysiology, diagnosis, and treatment of polycystic ovarian syndrome in adolescence. Horm Res Paediatr 88: 371–395, 2017. doi: 10.1159/000479371. [DOI] [PubMed] [Google Scholar]
- 52.Ibáñez L, Valls C, Cabré S, De Zegher F. Flutamide-metformin plus ethinylestradiol-drospirenone for lipolysis and antiatherogenesis in young women with ovarian hyperandrogenism: the key role of early, low-dose flutamide. J Clin Endocrinol Metab 89: 4716–4720, 2004. doi: 10.1210/jc.2004-0047. [DOI] [PubMed] [Google Scholar]
- 53.Jenkins NT, Padilla J, Arce-Esquivel AA, Bayless DS, Martin JS, Leidy HJ, Booth FW, Rector RS, Laughlin MH. Effects of endurance exercise training, metformin, and their combination on adipose tissue leptin and IL-10 secretion in OLETF rats. J Appl Physiol (1985) 113: 1873–1883, 2012. doi: 10.1152/japplphysiol.00936.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jovanovic VP, Carmina E, Lobo RA. Not all women diagnosed with PCOS share the same cardiovascular risk profiles. Fertil Steril 94: 826–832, 2010. doi: 10.1016/j.fertnstert.2009.04.021. [DOI] [PubMed] [Google Scholar]
- 55.Ketefian A, Jones MR, Krauss RM, Chen YD, Legro RS, Azziz R, Goodarzi MO. Association study of androgen signaling pathway genes in polycystic ovary syndrome. Fertil Steril 105: 467–473.e4, 2016. doi: 10.1016/j.fertnstert.2015.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim HS, Shin JH, Moon HJ, Kim TS, Kang IH, Seok JH, Kim IY, Park KL, Han SY. Evaluation of the 20-day pubertal female assay in Sprague-Dawley rats treated with DES, tamoxifen, testosterone, and flutamide. Toxicol Sci 67: 52–62, 2002. doi: 10.1093/toxsci/67.1.52. [DOI] [PubMed] [Google Scholar]
- 57.Kruit JK, Groen AK, van Berkel TJ, Kuipers F. Emerging roles of the intestine in control of cholesterol metabolism. World J Gastroenterol 12: 6429–6439, 2006. doi: 10.3748/wjg.v12.i40.6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee DK, Chang C. Endocrine mechanisms of disease: Expression and degradation of androgen receptor: mechanism and clinical implication. J Clin Endocrinol Metab 88: 4043–4054, 2003. doi: 10.1210/jc.2003-030261. [DOI] [PubMed] [Google Scholar]
- 59.Lee SK, Lee JO, Kim JH, Kim SJ, You GY, Moon JW, Jung JH, Park SH, Uhm KO, Park JM, Suh PG, Kim HS. Metformin sensitizes insulin signaling through AMPK-mediated PTEN down-regulation in preadipocyte 3T3-L1 cells. J Cell Biochem 112: 1259–1267, 2011. doi: 10.1002/jcb.23000. [DOI] [PubMed] [Google Scholar]
- 60.Lian J, Wei E, Groenendyk J, Das SK, Hermansson M, Li L, Watts R, Thiesen A, Oudit GY, Michalak M, Lehner R. Ces3/TGH deficiency attenuates steatohepatitis. Sci Rep 6: 25747, 2015. doi: 10.1038/srep25747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin HY, Xu Q, Yeh S, Wang RS, Sparks JD, Chang C. Insulin and leptin resistance with hyperleptinemia in mice lacking androgen receptor. Diabetes 54: 1717–1725, 2005. doi: 10.2337/diabetes.54.6.1717. [DOI] [PubMed] [Google Scholar]
- 62.Lin LH, Baracat MC, Maciel GA, Soares JM Jr, Baracat EC. Androgen receptor gene polymorphism and polycystic ovary syndrome. Int J Gynaecol Obstet 120: 115–118, 2013. doi: 10.1016/j.ijgo.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 63.Luque-Ramírez M, Mendieta-Azcona C, Alvarez-Blasco F, Escobar-Morreale HF. Androgen excess is associated with the increased carotid intima-media thickness observed in young women with polycystic ovary syndrome. Hum Reprod 22: 3197–3203, 2007. doi: 10.1093/humrep/dem324. [DOI] [PubMed] [Google Scholar]
- 64.Maciel GA, Moreira RP, Bugano DD, Hayashida SA, Marcondes JA, Gomes LG, Mendonça BB, Bachega TA, Baracat EC. Association of glucocorticoid receptor polymorphisms with clinical and metabolic profiles in polycystic ovary syndrome. Clinics (São Paulo) 69: 179–184, 2014. doi: 10.6061/clinics/2014(03)06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martins IJ, Mortimer BC, Miller J, Redgrave TG. Effects of particle size and number on the plasma clearance of chylomicrons and remnants. J Lipid Res 37: 2696–2705, 1996. [PubMed] [Google Scholar]
- 66.Matic M, Bryzgalova G, Gao H, Antonson P, Humire P, Omoto Y, Portwood N, Pramfalk C, Efendic S, Berggren PO, Gustafsson JÅ, Dahlman-Wright K. Estrogen signalling and the metabolic syndrome: targeting the hepatic estrogen receptor alpha action. PLoS One 8: e57458, 2013. doi: 10.1371/journal.pone.0057458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mehrabian F, Ghasemi-Tehrani H, Mohamadkhani M, Moeinoddini M, Karimzadeh P. Comparison of the effects of metformin, flutamide plus oral contraceptives, and simvastatin on the metabolic consequences of polycystic ovary syndrome. J Res Med Sci 21: 7, 2016. doi: 10.4103/1735-1995.177354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13: 1016–1023, 2011. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Möhlig M, Jürgens A, Spranger J, Hoffmann K, Weickert MO, Schlösser HW, Schill T, Brabant G, Schüring A, Pfeiffer AF, Gromoll J, Schöfl C. The androgen receptor CAG repeat modifies the impact of testosterone on insulin resistance in women with polycystic ovary syndrome. Eur J Endocrinol 155: 127–130, 2006. doi: 10.1530/eje.1.02195. [DOI] [PubMed] [Google Scholar]
- 70.Moran LJ, Hutchison SK, Norman RJ, Teede HJ. Lifestyle changes in women with polycystic ovary syndrome. Cochrane Database Syst Rev CD007506, 2011. doi: 10.1002/14651858.CD007506.pub3. [DOI] [PubMed] [Google Scholar]
- 71.Naderpoor N, Shorakae S, de Courten B, Misso ML, Moran LJ, Teede HJ. Metformin and lifestyle modification in polycystic ovary syndrome: systematic review and meta-analysis. Hum Reprod Update 21: 560–574, 2015. doi: 10.1093/humupd/dmv025. [DOI] [PubMed] [Google Scholar]
- 72.Narayanan R, Coss CC, Dalton JT. Development of selective androgen receptor modulators (SARMs). Mol Cell Endocrinol 465: 134–142, 2018. doi: 10.1016/j.mce.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nestler JE, Jakubowicz DJ. Decreases in ovarian cytochrome P450c17 alpha activity and serum free testosterone after reduction of insulin secretion in polycystic ovary syndrome. N Engl J Med 335: 617–623, 1996. doi: 10.1056/NEJM199608293350902. [DOI] [PubMed] [Google Scholar]
- 74.Nestler JE, Jakubowicz DJ. Lean women with polycystic ovary syndrome respond to insulin reduction with decreases in ovarian P450c17 alpha activity and serum androgens. J Clin Endocrinol Metab 82: 4075–4079, 1997. [DOI] [PubMed] [Google Scholar]
- 75.Orio F Jr, Palomba S, Cascella T, De Simone B, Di Biase S, Russo T, Labella D, Zullo F, Lombardi G, Colao A. Early impairment of endothelial structure and function in young normal-weight women with polycystic ovary syndrome. J Clin Endocrinol Metab 89: 4588–4593, 2004. doi: 10.1210/jc.2003-031867. [DOI] [PubMed] [Google Scholar]
- 76.Padmanabhan V, Manikkam M, Recabarren S, Foster D. Prenatal testosterone excess programs reproductive and metabolic dysfunction in the female. Mol Cell Endocrinol 246: 165–174, 2006. doi: 10.1016/j.mce.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 77.Palomba S, Falbo A, Zullo F, Orio F Jr. Evidence-based and potential benefits of metformin in the polycystic ovary syndrome: a comprehensive review. Endocr Rev 30: 1–50, 2009. doi: 10.1210/er.2008-0030. [DOI] [PubMed] [Google Scholar]
- 78.Paradisi R, Fabbri R, Battaglia C, Venturoli S. Ovulatory effects of flutamide in the polycystic ovary syndrome. Gynecol Endocrinol 29: 391–395, 2013. doi: 10.3109/09513590.2012.754876. [DOI] [PubMed] [Google Scholar]
- 79.Pasquali R, Gambineri A, Biscotti D, Vicennati V, Gagliardi L, Colitta D, Fiorini S, Cognigni GE, Filicori M, Morselli-Labate AM. Effect of long-term treatment with metformin added to hypocaloric diet on body composition, fat distribution, and androgen and insulin levels in abdominally obese women with and without the polycystic ovary syndrome. J Clin Endocrinol Metab 85: 2767–2774, 2000. doi: 10.1210/jcem.85.8.6738. [DOI] [PubMed] [Google Scholar]
- 80.Patel SS, Truong U, King M, Ferland A, Moreau KL, Dorosz J, Hokanson JE, Wang H, Kinney GL, Maahs DM, Eckel RH, Nadeau KJ, Cree-Green M. Obese adolescents with polycystic ovarian syndrome have elevated cardiovascular disease risk markers. Vasc Med 22: 85–95, 2017. doi: 10.1177/1358863X16682107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Phillips C, Murugasu G, Owens D, Collins P, Johnson A, Tomkin GH. Improved metabolic control reduces the number of postprandial apolipoprotein B-48-containing particles in type 2 diabetes. Atherosclerosis 148: 283–291, 2000. doi: 10.1016/S0021-9150(99)00275-0. [DOI] [PubMed] [Google Scholar]
- 82.Proctor SD, Vine DF, Mamo JC. Arterial permeability and efflux of apolipoprotein B-containing lipoproteins assessed by in situ perfusion and three-dimensional quantitative confocal microscopy. Arterioscler Thromb Vasc Biol 24: 2162–2167, 2004. doi: 10.1161/01.ATV.0000143859.75035.5a. [DOI] [PubMed] [Google Scholar]
- 83.Rocha MP, Marcondes JA, Barcellos CR, Hayashida SA, Curi DD, da Fonseca ÂM, Bagnoli VR, Baracat EC. Dyslipidemia in women with polycystic ovary syndrome: incidence, pattern and predictors. Gynecol Endocrinol 27: 814–819, 2011. doi: 10.3109/09513590.2010.508852. [DOI] [PubMed] [Google Scholar]
- 84.Rosenfield RL. Ovarian and adrenal function in polycystic ovary syndrome. Endocrinol Metab Clin North Am 28: 265–293, 1999. doi: 10.1016/S0889-8529(05)70070-0. [DOI] [PubMed] [Google Scholar]
- 85.Russell JC, Ravel D, Pégorier JP, Delrat P, Jochemsen R, O’Brien SF, Kelly SE, Davidge ST, Brindley DN. Beneficial insulin-sensitizing and vascular effects of S15261 in the insulin-resistant JCR:LA-cp rat. J Pharmacol Exp Ther 295: 753–760, 2000. [PubMed] [Google Scholar]
- 86.Sahin I, Serter R, Karakurt F, Demirbas B, Culha C, Taskapan C, Kosar F, Aral Y. Metformin versus flutamide in the treatment of metabolic consequences of non-obese young women with polycystic ovary syndrome: a randomized prospective study. Gynecol Endocrinol 19: 115–124, 2004. doi: 10.1080/09513590400004736. [DOI] [PubMed] [Google Scholar]
- 87.Schüring AN, Welp A, Gromoll J, Zitzmann M, Sonntag B, Nieschlag E, Greb RR, Kiesel L. Role of the CAG repeat polymorphism of the androgen receptor gene in polycystic ovary syndrome (PCOS). Exp Clin Endocrinol Diabetes 120: 73–79, 2012. doi: 10.1055/s-0031-1291343. [DOI] [PubMed] [Google Scholar]
- 88.Shi D, Dyck MK, Uwiera RR, Russell JC, Proctor SD, Vine DF. A unique rodent model of cardiometabolic risk associated with the metabolic syndrome and polycystic ovary syndrome. Endocrinology 150: 4425–4436, 2009. doi: 10.1210/en.2008-1612. [DOI] [PubMed] [Google Scholar]
- 89.Shi D, Vine DF. Animal models of polycystic ovary syndrome: a focused review of rodent models in relationship to clinical phenotypes and cardiometabolic risk. Fertil Steril 98: 185–193.e2, 2012. doi: 10.1016/j.fertnstert.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 90.Sidhwani S, Scoccia B, Sunghay S, Stephens-Archer CN, Mazzone T, Sam S. Polycystic ovary syndrome is associated with atherogenic changes in lipoprotein particle number and size independent of body weight. Clin Endocrinol (Oxf) 75: 76–82, 2011. doi: 10.1111/j.1365-2265.2011.04015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sun J, Yuan Y, Cai R, Sun H, Zhou Y, Wang P, Huang R, Xia W, Wang S. An investigation into the therapeutic effects of statins with metformin on polycystic ovary syndrome: a meta-analysis of randomised controlled trials. BMJ Open 5: e007280, 2015. doi: 10.1136/bmjopen-2014-007280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tang T, Lord JM, Norman RJ, Yasmin E, Balen AH. Insulin-sensitising drugs (metformin, rosiglitazone, pioglitazone, D-chiro-inositol) for women with polycystic ovary syndrome, oligo amenorrhoea and subfertility. Cochrane Database Syst Rev CD003053, 2012. doi: 10.1002/14651858.CD003053.pub5. [DOI] [PubMed] [Google Scholar]
- 93.Tarkun I, Arslan BC, Cantürk Z, Türemen E, Sahin T, Duman C. Endothelial dysfunction in young women with polycystic ovary syndrome: relationship with insulin resistance and low-grade chronic inflammation. J Clin Endocrinol Metab 89: 5592–5596, 2004. doi: 10.1210/jc.2004-0751. [DOI] [PubMed] [Google Scholar]
- 94.Tepavčević S, Vojnović Milutinović D, Macut D, Žakula Z, Nikolić M, Božić-Antić I, Romić S, Bjekić-Macut J, Matić G, Korićanac G. Dihydrotestosterone deteriorates cardiac insulin signaling and glucose transport in the rat model of polycystic ovary syndrome. J Steroid Biochem Mol Biol 141: 71–76, 2014. doi: 10.1016/j.jsbmb.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 95.Twickler T, Dallinga-Thie GM, Chapman MJ, Cohn JS. Remnant lipoproteins and atherosclerosis. Curr Atheroscler Rep 7: 140–147, 2005. doi: 10.1007/s11883-005-0037-x. [DOI] [PubMed] [Google Scholar]
- 96.Uilenbroek JT, Woutersen PJ, van der Schoot P. Atresia of preovulatory follicles: gonadotropin binding and steroidogenic activity. Biol Reprod 23: 219–229, 1980. doi: 10.1095/biolreprod23.1.219. [DOI] [PubMed] [Google Scholar]
- 97.Valkenburg O, Steegers-Theunissen RP, Smedts HP, Dallinga-Thie GM, Fauser BC, Westerveld EH, Laven JS. A more atherogenic serum lipoprotein profile is present in women with polycystic ovary syndrome: a case-control study. J Clin Endocrinol Metab 93: 470–476, 2008. doi: 10.1210/jc.2007-1756. [DOI] [PubMed] [Google Scholar]
- 98.Varbo A, Benn M, Tybjærg-Hansen A, Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol 61: 427–436, 2013. doi: 10.1016/j.jacc.2012.08.1026. [DOI] [PubMed] [Google Scholar]
- 99.Varbo A, Nordestgaard BG, Tybjaerg-Hansen A, Schnohr P, Jensen GB, Benn M. Nonfasting triglycerides, cholesterol, and ischemic stroke in the general population. Ann Neurol 69: 628–634, 2011. doi: 10.1002/ana.22384. [DOI] [PubMed] [Google Scholar]
- 100.Velázquez M E, Bellabarba GA, Mendoza S, Sánchez L. Postprandial triglyceride response in patients with polycystic ovary syndrome: relationship with waist-to-hip ratio and insulin. Fertil Steril 74: 1159–1163, 2000. doi: 10.1016/S0015-0282(00)01601-0. [DOI] [PubMed] [Google Scholar]
- 101.Vinaixa M, Rodriguez MA, Samino S, Díaz M, Beltran A, Mallol R, Bladé C, Ibañez L, Correig X, Yanes O. Metabolomics reveals reduction of metabolic oxidation in women with polycystic ovary syndrome after pioglitazone-flutamide-metformin polytherapy. PLoS One 6: e29052, 2011. doi: 10.1371/journal.pone.0029052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vine DF, Shi D, Proctor SD. Insulin and testosterone are associated with elevated intestinal secretion of lipids and lipoproteins in a rodent model of the metabolic syndrome and polycystic ovary syndrome. J Diabetes Metab 5: 391, 2014. doi: 10.4172/2155-6156.1000391. [DOI] [Google Scholar]
- 103.Vine DF, Takechi R, Russell JC, Proctor SD. Impaired postprandial apolipoprotein-B48 metabolism in the obese, insulin-resistant JCR:LA-cp rat: increased atherogenicity for the metabolic syndrome. Atherosclerosis 190: 282–290, 2007. doi: 10.1016/j.atherosclerosis.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 104.Vine DF, Wang Y, Jetha MM, Ball GD, Proctor SD. Impaired ApoB-lipoprotein and triglyceride metabolism in obese adolescents with polycystic ovary syndrome. J Clin Endocrinol Metab 102: 970–982, 2017. doi: 10.1210/jc.2016-2854. [DOI] [PubMed] [Google Scholar]
- 105.Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 122: 253–270, 2012. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vrbíková J, Hill M, Dvoráková K, Stanická S, Vondra K, Stárka L. Flutamide suppresses adrenal steroidogenesis but has no effect on insulin resistance and secretion and lipid levels in overweight women with polycystic ovary syndrome. Gynecol Obstet Invest 58: 36–41, 2004. doi: 10.1159/000077827. [DOI] [PubMed] [Google Scholar]
- 107.Vrbíková J, Hill M, Stárka L, Cibula D, Bendlová B, Vondra K, Sulcová J, Snajderová M. The effects of long-term metformin treatment on adrenal and ovarian steroidogenesis in women with polycystic ovary syndrome. Eur J Endocrinol 144: 619–628, 2001. doi: 10.1530/eje.0.1440619. [DOI] [PubMed] [Google Scholar]
- 108.Vryonidou A, Papatheodorou A, Tavridou A, Terzi T, Loi V, Vatalas IA, Batakis N, Phenekos C, Dionyssiou-Asteriou A. Association of hyperandrogenemic and metabolic phenotype with carotid intima-media thickness in young women with polycystic ovary syndrome. J Clin Endocrinol Metab 90: 2740–2746, 2005. doi: 10.1210/jc.2004-2363. [DOI] [PubMed] [Google Scholar]
- 109.Vural B, Caliskan E, Turkoz E, Kilic T, Demirci A. Evaluation of metabolic syndrome frequency and premature carotid atherosclerosis in young women with polycystic ovary syndrome. Hum Reprod 20: 2409–2413, 2005. doi: 10.1093/humrep/dei100. [DOI] [PubMed] [Google Scholar]
- 110.Wild RA. Dyslipidemia in PCOS. Steroids 77: 295–299, 2012. doi: 10.1016/j.steroids.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 111.Wild RA, Rizzo M, Clifton S, Carmina E. Lipid levels in polycystic ovary syndrome: systematic review and meta-analysis. Fertil Steril 95: 1073–1079.e11, 2011. doi: 10.1016/j.fertnstert.2010.12.027. [DOI] [PubMed] [Google Scholar]
- 112.Wild S, Pierpoint T, McKeigue P, Jacobs H. Cardiovascular disease in women with polycystic ovary syndrome at long-term follow-up: a retrospective cohort study. Clin Endocrinol (Oxf) 52: 595–600, 2000. doi: 10.1046/j.1365-2265.2000.01000.x. [DOI] [PubMed] [Google Scholar]
- 113.Yang R, Trevillyan JM. c-Jun N-terminal kinase pathways in diabetes. Int J Biochem Cell Biol 40: 2702–2706, 2008. doi: 10.1016/j.biocel.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 114.Yildir IC, Kutluturk F, Tasliyurt T, Yelken BM, Acu B, Beyhan M, Erkorkmaz U, Yilmaz A. Insulin resistance and cardiovascular risk factors in women with PCOS who have normal glucose tolerance test. Gynecol Endocrinol 29: 148–151, 2013. doi: 10.3109/09513590.2012.730573. [DOI] [PubMed] [Google Scholar]
- 115.Zhang L, Reue K, Fong LG, Young SG, Tontonoz P. Feedback regulation of cholesterol uptake by the LXR-IDOL-LDLR axis. Arterioscler Thromb Vasc Biol 32: 2541–2546, 2012. doi: 10.1161/ATVBAHA.112.250571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang T, Liang W, Fang M, Yu J, Ni Y, Li Z. Association of the CAG repeat polymorphisms in androgen receptor gene with polycystic ovary syndrome: a systemic review and meta-analysis. Gene 524: 161–167, 2013. doi: 10.1016/j.gene.2013.04.040. [DOI] [PubMed] [Google Scholar]
- 117.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108: 1167–1174, 2001b. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]