

Gordon Holmes syndrome (GHS) is the clinical association of ataxia and hypogonadism1 frequently encountered in patients with autosomal‐recessive cerebellar ataxias. Mutations in genes, such as RNF216, OTUD4, STUB1, PNPLA6, and POLR3A/3B/1C, are associated with ataxia and hypogonadism,2, 3, 4, 5, 6 but the patient's observed phenotypes are generally wider than expected. Here, we report the case of 2 Argentinean siblings with GHS caused by a novel homozygous mutation in the RNF216 gene.
Case Report
Two male siblings, born from nonconsanguineous parents (Fig. S1), were referred to our hospital. Patient 1 was examined at age 28 showing clinical signs of hypogonadism, pes cavus, slight gait disturbance, and dysarthria (Fig. S2). Bone densitometry showed low mineral bone density. He was diagnosed with hypogonadotropic hypogonadism (Table S1) and began treatment with testosterone. At age 29, he experienced profound progression of gait and speech disturbances. Further examination revealed appendicular and truncal cerebellar ataxia (Video S1), dysarthria, and brisk tendon reflexes. Montreal Cognitive Assessment (MOCA) score was 12/30. At age 31, he was severely ataxic (Video S1) and demented (MOCA 10/30).
Patient 2 was also diagnosed with hypogonadotropic hypogonadism at age 18 years attributed to poor development of secondary sexual characteristics and low stature (Table S1). He was referred to our hospital 9 years later. Physical examination revealed eunuchoid appearance, gynoid fat distribution, dysarthria, appendicular and truncal cerebellar ataxia, brisk tendon reflexes, and a MOCA score of 17/30. Cerebellar signs were milder than those observed in patient 1. Two years later, the cerebellar ataxia slightly progressed (Video S1) and the MOCA score was 14/30.
Both siblings dropped out of high school because of learning difficulties. Their brain MRI revealed cerebral white matter changes, diffuse brain cortex, and cerebellar atrophy (Fig. 1; Table S1).
Figure 1.
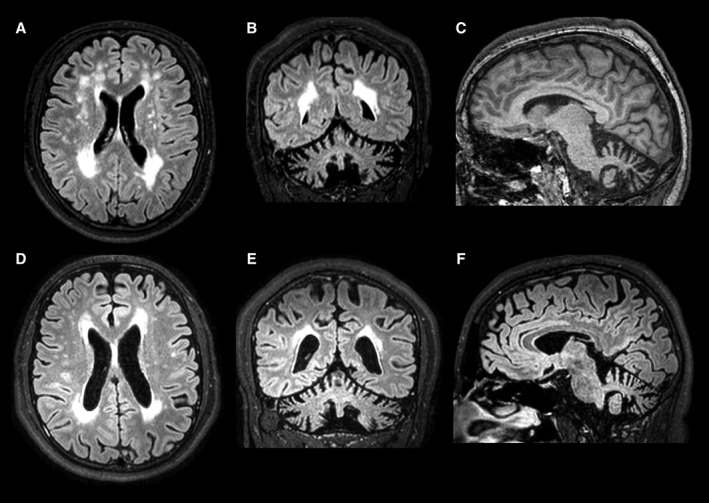
Neuroimaging findings. Multiple cerebral white matter changes, cortical brain, and cerebellar atrophy in axial and coronal FLAIR‐weighted images (A,B, patient 1; D,E, patient 2). Thin posterior segment of corpus callosum and cerebellar atrophy in sagittal T1‐weighted image (C, patient 1) and sagittal FLAIR‐weighted image (F, patient 2).
We performed whole‐exome sequencing (WES) in patient 1. We found a novel homozygous variant in gene RNF216 (NM_207116:c.2042C>T; NM_207111:c.1988C>T), which encodes an E3 ubiquitin ligase, not previously reported and predicted to produce a missense change (NM_207116:P606L), disrupting a highly conserved position in a zinc‐finger IBR domain (Fig. 2). A structural model shows that the proline is adjacent to the zinc coordinating cysteine, in the elbow of a ß‐turn motif. Prolines, because of their imino acid nature, are predominantly found in turns, and mutations to helix prone residues, such as leucine, are thus expected to destabilize the key zinc coordination motif, resulting in a nonfunctional protein. Consistently, numerous pathogenicity prediction softwares classify this variant as likely damaging (Table S2). We define the variant as likely pathogenic according to the American College of Medical Genetics and Genomics Guidelines (PM1, PM2, PM3, PP1, PP3, and PP4).7
Figure 2.
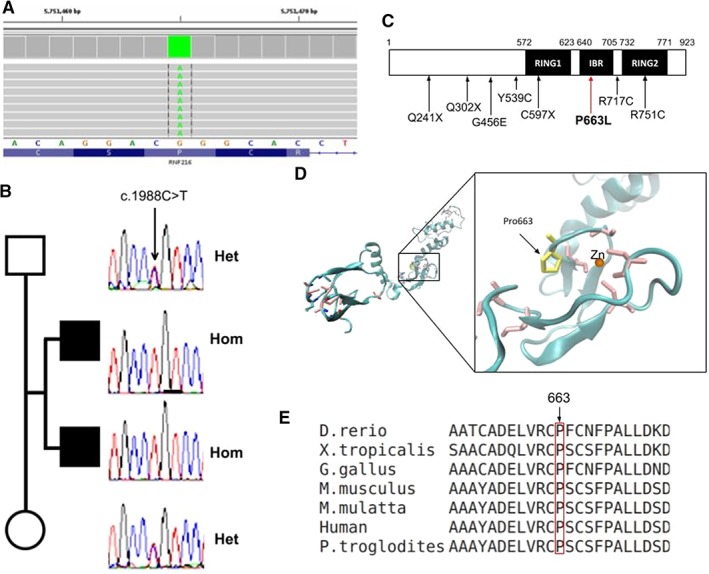
Molecular and bioinformatic results. (A) Exome result of patient 1 in BAM format, the variant is found in the homozygous state. (B) Sanger results of gene RNF216 for the core family. (C) Relative location of the presently found variant together with other reported missense and nonsense pathogenic variants in the RNF216 gene isoform‐2. (D) Model of zinc rings and IBR domain (modeled using 4KBL structure as template). Cystein residues coordinating the zinc ion are shown in pink; mutated proline in yellow. (E) Multiple alignment of proteins homologous to RNF216 isoform‐2 in different species.
Sanger sequencing confirmed the variant and showed that the affected brother (patient 2) is also homozygous, whereas both parents are heterozygous (Fig. 2).
Discussion
The gene RNF216 encodes an E3 ubiquitin ligase which marks different proteins for proteasome‐mediated degradation. Previously, Margolin et al. reported on a consanguineous family with ataxia, dementia, and hypogonadotropism caused by a combination of mutations in RNF216 and OTUD4 genes.2 In the present case, the three variants found in OTUD4 were considered probably benign (Table S3). RNF216‐mediated neurodegeneration has been discovered very recently, with a total of 17 patients, 10 families, and 12 different mutations published to date (including ours; Table S4). Patients with these mutations not only show GHS characteristics, but also other clinical features such as chorea, psychiatric disorders, dementia, dysarthria, and corticospinal signs. Age of onset of the endocrine manifestations might differ among patients. Hormone replacement therapy is of utmost importance. Neuroimaging studies demonstrate cerebral and cerebellar atrophy and focal hyperintense signal changes on T2‐ and fluid‐attenuated inversion recovery (FLAIR)‐weighted images involving the periventricular and subcortical brain white matter, globus pallidus, putamen, thalamus, and pons.2, 8, 9, 10 Patients 1 and 2 showed a thin posterior segment of the corpus callosum, not previously reported. It is important to emphasize that the phenotype of RNF216‐mediated neurodegeneration might have not been yet fully described because of the small number of cases reported on so far.
All previous reported cases with homozygous mutations in RNF216 were from consanguineous families.2, 8, 9, 10 Interestingly, although consanguinity was not reported in the present case, the sibling's parents came from two small nearby towns (only 11 km apart) in the Argentinean countryside. We hypothesize that our findings could be explained by a common ancestor to both families, thus involving some degree of consanguinity unknown by the parents. Supporting this idea, we found a decreased heterozygous to homozygous single‐nucleotide polymorphism ratio in patient 1 after WES (1.31 compared to the observed average of 1.50 in other exomes sequenced by our group).
In summary, we present 2 Argentinean siblings with a GHS as part of the clinical spectrum of the RNF216‐mediated neurodegeneration caused by a novel homozygous mutation.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
C.R.C.: 1A, 1B, 1C, 3A, 3B
Y.M.: 1A, 1B, 1C, 3A, 3B
S.A.V.: 1B, 1C, 3A, 3B
V.T.: 1C, 3A, 3B
J.O.: 1A, 1C, 3A,3B
E.C.C.: 1C, 3A, 3B
G.B.: 1C, 3A, 3B
A.G.T.: 1C, 3A, 3B
M.M.: 1A, 1B, 1C, 3A, 3B
Disclosures
Ethical Compliance Statement: The authors confirm that the approval of an institutional review board was not required for this work. Written informed consent to publish all shown material was obtained from the patients. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for previous 12 months: The authors declare that there are no disclosures to report.
Supporting information
Figure S1. Family pedigree. HH, hypogonadotropic hypogonadism; A, ataxia; D, dementia.
Figure S2. Patient 1. (A) Arm span >5 cm. (B,C) Eunuchoid appearance, gynoid fat distribution, and bilateral lipomastia. (D) Pes cavus.
Video S1. Patient 1, Segment 1: first physical exam. Ataxic gait without assistance. Patient 1, Segment 2: first physical exam. Bilateral upper limb ataxia. Patient 1, Segment 3: last physical exam. Ataxic gait with assistance. Patient 1, Segment 4: last physical exam. Severe cerebellar dysarthria. Patient 2, Segment 5: ataxic gait without assistance. Patient 2, Segment 6: alteration of tandem gait.
Table S1. Clinical manifestations, laboratory, and MRI findings
Table S2. Variant genomic data referred to human genome GRCh37 and pathogenicity prediction analysis
Table S3. Variants in OTUD4 gene
Table S4. RNF216‐mediated neurodegeneration
Acknowledgments
The authors thank all the present case family members for their participation in this study; Dr. Reynaldo M. Gómez at the Endocrinology Department, Hospital de Clínicas José de San Martín, Buenos Aires, Argentina for referral of patients; and Lucas Mac Dougall for edition of figures and videos.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Holmes G. A form of familial degeneration of the cerebellum. Brain 1908;30:466–489. [Google Scholar]
- 2. Margolin DH, Kousi M, Chan YM, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med 2013;368:1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hayer SN, Deconinck T, Bender B, et al. STUB1/CHIP mutations cause Gordon Holmes syndrome as part of a widespread multisystemic neurodegeneration: evidence from four novel mutations. Orphanet J Rare Dis 2017;12:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Synofzik M, Gonzalez MA, Lourenco CM, et al. PNPLA6 mutations cause Boucher‐Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 2014;137:69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bernard G, Vanderver A. POLR3‐related leukodystrophy. In: Adam MP, Ardinger HH, Pagon RA, et al., (eds). GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2018. 2012. Aug 2 [updated 05/11/2017]. Available at: https://www.ncbi.nlm.nih.gov/books/NBK99167/. Accessed on May 1, 2018. [PubMed] [Google Scholar]
- 6. Thiffault I, Wolf NI, Forget D, et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun 2015;6:7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Santens P, Van Damme T, Steyaert W, et al. RNF216 mutations as a novel cause of autosomal recessive Huntington‐like disorder. Neurology 2015;84:1760–1766. [DOI] [PubMed] [Google Scholar]
- 9. Ganos C, Hersheson J, Adams M, Bhatia KP, Houlden H. The 4H syndrome due to RNF216 mutation. Parkinsonism Relat Disord 2015;21:1122–1123. [DOI] [PubMed] [Google Scholar]
- 10. Alqwaifly M, Bohlega S. Ataxia and hypogonadotropic hypogonadism with intrafamilial variability caused by RNF216 mutation. Neurol Int 2016;8:6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Family pedigree. HH, hypogonadotropic hypogonadism; A, ataxia; D, dementia.
Figure S2. Patient 1. (A) Arm span >5 cm. (B,C) Eunuchoid appearance, gynoid fat distribution, and bilateral lipomastia. (D) Pes cavus.
Video S1. Patient 1, Segment 1: first physical exam. Ataxic gait without assistance. Patient 1, Segment 2: first physical exam. Bilateral upper limb ataxia. Patient 1, Segment 3: last physical exam. Ataxic gait with assistance. Patient 1, Segment 4: last physical exam. Severe cerebellar dysarthria. Patient 2, Segment 5: ataxic gait without assistance. Patient 2, Segment 6: alteration of tandem gait.
Table S1. Clinical manifestations, laboratory, and MRI findings
Table S2. Variant genomic data referred to human genome GRCh37 and pathogenicity prediction analysis
Table S3. Variants in OTUD4 gene
Table S4. RNF216‐mediated neurodegeneration