

Abstract
An efficient streamlined chemoenzymatic approach has been developed for gram-scale synthesis of Lewis a angtigen (LeaβProN3) and a library of sialyl Lewis a antigens (sLeaβProN3) containing different sialic acid forms. Intially, commercially available inexpensive N-acetylglucosamine (GlcNAc) was converted to its N′-glycosyl p-toluenesulfonohydrazide in one step. Followed by chemical glycosylation, GlcNAcβProN3 was synthesized using this protecting group-free method in high yield (82%). Sequential one-pot multienzyme (OPME) β1–3-galactosylation of GlcNAcβProN3 followed by OPME α1–4-fucosylation reactions produced target LeaβProN3 in gram-scale. Structurally diverse sialic acid forms was successfully introduced using a OPME sialylation reaction containing a CMP-sialic acid synthetase and Pasteurella multocida α2–3-sialyltransferase 1 (PmST1) mutant PmST1 M144D with or without a sialic acid aldolase to form sLeaβProN3 containing naturally occurring or non-natural sialic acid forms in preparative scales.
Keywords: chemoenzymatic synthesis, glycosyltransferase, Lewis a, sialyl Lewis a, sialic acid, protecting group-free glycosylation
Graphical abstract
1. Introduction
Lewis a (Lea) antigen is a blood group antigen found in glycolipids and glycoproteins on the surface of erythrocytes as well as normal glandular and epithelial cells [1, 2]. Its sialylated form, sialyl Lewis a (sLea), is expressed on the cells of some normal tissues such as normal fibroblasts, the luminal side of ductal epithelial cells, and some parenchymatous cells [3]. Nevertheless, overexpression of sLea has been found on various types of cancer cells with increased levels of fucosylation [4], including colon cancer, pancreatic cancer, gallbladder/bile duct cancer, breast cancer, cholangiocarcinoma, and small cell lung cancer (SCLC) [3, 5, 6]. In fact, sLea was named as carbohydrate antigen 19–9 (CA19–9) [7] and was the first reported tumor-associated carbohydrate antigen (TACA) [8]. It was also discovered in the serum of patients with colon or pancreatic cancers [9] and has been used as a serum biomarker of pancreatic cancer [10] and other gastrointestinal cancers in clinic for prognosis determination, postoperative surveillance, and monitoring clinical responses to therapy [4, 7, 11–14]. Nevertheless, false negative and false positive results are possible and precautions are needed in interpreting the data [7, 10, 11].
sLea oligosaccharides and analogs have been synthesized chemically [15–20] or chemoenzymatically [21–24]. Chemoenzymatic synthesis of sLea antigens by sialidase (e.g. Salmonella typhimurium LT2 sialidase)-catalyzed transglycosylation reaction using p-nitrophenyl α-N-acetylneuraminide (Neu5AcαpNP) as the glycosyl donor and Lea trisaccharide as the acceptor was reported but with poor glycosylation yields [25]. All reported glycosyltransferase-catalyzed chemoenzymatic synthesis of sLea antigens were carried out using recombinant mammalian enzymes or human milk enzymes including human α1–4-fucosyltransferase (FucT III) [26, 27] or human milk [28, 29] α1–3/4-fucosyltransferase, rat liver [22, 23, 26] or porcine submaxillary [28] α2–3-sialyltransferase, and/or human β1–3galactosyltransferase [26]. In general, the reactions were carried out by sialyltransferase-catalyzed sialylation of β1–3-linked galactosides followed by fucosylation in the last step [23]. sLea derivatives with variations on the aglycon and the N-acyl group on the D-GlcNAc or the LFuc residue have been synthesized. However, modified sialic acid forms have not been introduced to sLea although variations at C-5 and C-9 positions of sialic acid have been introduced to sialyl Lewis x (sLex) structures [22, 23].
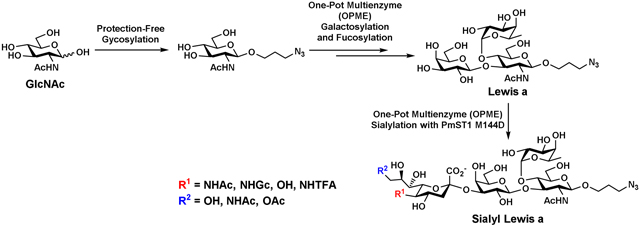
We have been interested in developing an efficient approach for systematically synthesizing sLea antigens containing different sialic acid forms which can be used as important probes to understand the structure-function relationship of sLea and the important roles of sialic acid modification. Unlike the common glycosylation sequence of sialylation followed by fucosylation for the formation of sLea in nature by glycosyltransferase-catalyzed reactions, the most efficient strategy to obtain sLea containing different sialic acid forms is to alter the glycosylation sequence by obtaining Lea followed by α2–3-sialylation using a suitable α2–3-sialyltransferase-catalyzed reaction. This strategy will streamline the synthetic scheme and simplify the purification process as demonstrated previously for the synthesis of a library of sialyl Lewis x (sLex) antigens containing different sialic acid forms [30]. We show here that Lea trisaccharide Galβ1–3(Fucα1–4)GlcNAcβProN3 can be readily assembled in gram-scale by a bacterial disaccharide phosphorylase-containing one-pot multienzyme (OPME) galactosylation reaction followed by a bacterial α1–3/4-fucosyltransferase-containing OPME fucosylation reaction. A previously constructed bacterial α2–3-sialyltransferse mutant is shown to be efficient in tolerating Lea as the acceptor substrate to introduce different sialic acid forms with modifications at C-5, C-7, and/or C-9 positions for the synthesis of desired sLea antigens in good yields.
2. Results and discussion
2.1. Gram-scale chemoenzymatic synthesis of Lea antigen Galβ1–3(Fucα1–4)GlcNAcβProN3
To synthesize Lea trisaccharide which could be potentially used as an acceptor substrate for sialyltransferase-catalyzed sialylation reaction for the production of desired sLea tetrasaccharides containing different sialic acid forms, monosaccharide glycoside GlcNAcβProN3 was chemically synthesized. Comparing different strategies for obtaining the desired compound, a protecting group-free glycosylation strategy using N′-glycosyl p-toluenesulfonohydrazides (GSHs) as glycosyl donors reported by Gudmundsdottir and Nitz [31] was identified as an excellent choice. This method was shown to be especially useful for 2-acetamido-sugars such as free N-acetylglucosamine (GlcNAc) and unprotected disaccharides containing a free GlcNAc at the reducing end. The reactions resulted in good yields and excellent β-selectivity [32]. The GSH donors can be easily prepared from unprotected sugars and p-toluenesulfonyl hydrazide under mild acidic conditions in high yields. They can then be readily activated by N-bromosuccinimide (NBS) to couple with various alcohols to form glycosides [31]. We applied this glycosylation method for efficient synthesis of GlcNAcβProN3 from commercially available and inexpensive GlcNAc. The azido functional group introduced can be used as a versatile chemical handle for bio-conjugation with alkyne-containing molecules [33]. It can also be readily reduced to an amine for immobilization or conjugation [34, 35].
As shown in Scheme 1, starting from GlcNAc (1) and p-toluenesulfonyl hydrazide (1.2 equiv.) in the presence of a catalytic amount of acetic acid in dimethylformamide (DMF), the GSH donor (2) was synthesized on a multi-gram (26.35 g) scale in a quantitative yield. The product was easily isolated by precipitation using diethyl ether. Glycosylation of 3-chloro-1propanol using the GSH donor (2) in the presence of NBS formed glycoside GlcNAcβProCl which was subsequently converted to the desired GlcNAcβProN3 (3) by treating with NaN3 in DMF. Compared to a previously reported method for synthesizing GlcNAcβProN3 (3) which involved converting GlcNAc to peracetylated GlcNAc trichloroacetimidate followed by glycosylation and deprotection (5 steps, 28% yield) [36, 37], the current route (3 steps, 82% yield) is a much more efficient method which allows easy scaling up.
Scheme 1.

Gram-scale chemoenzymatic synthesis of LeaβProN3 (5). Enzyme abbreviations: SpGalK, Streptococcus pneumoniae galactokinase; BiGalHexNAcP, Bifidobacterium infantis Dgalactosyl-β1–3-N-acetyl-D-hexosamine phosphorylase; BfFKP, Bacteroides fragilis strain NCTC9343 bifunctional L-fucokinase/GDP-fucose pyrophosphorylase; PmPpA, Pasteurella multocida inorganic pyrophosphatase; Hp3/4FT, Helicobacter pylori α1–3/4-fucosyltransferase.
The obtained GlcNAcβProN3 (3) was used for gram-scale enzymatic synthesis of disaccharide Galβ1–3GlcNAcβProN3 (4) in the presence of galactose (Gal) and adenosine 5′triphosphate (ATP) using a one-pot two-enzyme galactosylation system containing Streptococcus pneumoniae (SpGalK) [38] and Bifidobacterium infantis D-galactosyl-β1–3-N-acetyl-D-hexosamine phosphorylase (BiGalHexNAcP) [39]. An excellent yield (90%) was achieved after product purification by silica gel column followed by BioGel P-2 gel filtration column chromatography. Compared to β1–3-galactosyltransferase-catalyzed reactions, the strategy using BiGalHexNAcP avoided the need to produce uridine 5’-diphosphate-galactose (UDP-Gal) from uridine 5’-triphosphsate (UTP). Instead, galactose-1-phosphate formed by galactokinase-catalyzed reaction was directly used for the production of the disaccharide product. In addition, BiGalHexNAcP was able to be expressed at a level of 55 mg/L in Escherichia coli culture [39], providing an ample amount of enzyme for large-scale synthesis.
For the synthesis of Lea trisaccharide from the resulting disaccharide Galβ1–3GlcNAcβProN3 (4), a highly efficient fucosyltransferase was needed. We reported recently the cloning and application of a recombinant Helicobacter pylori α1–3/4-fucosyltransferase (Hp3/4FT) in a highly efficient one-pot three-enzyme (OP3E) fucosylation system for the preparative-scale (up to 100 mg) synthesis of diverse α1–3- and α1–4-linked fucosides [40]. The Hp3/4FT-containing OP3E fucosylation system was applied for gram scale synthesis of Lea trisaccharide Galβ1–3(Fucα1–4)GlcNAcβProN3 (LeaβProN3, 5) which was achieve in high-yield (91%) in the presence of L-fucose, adenosine 5’-triphosphate (ATP), and guanosine 5’triphosphate (GTP). In this system, fucosyltransferase donor substrate guanosine 5’-diphosphateL-fucose (GDP-Fuc) was generated in situ from L-fucose, ATP, and GTP using a bifunctional Bacteroides fragilis enzyme (BfFKP) with both L-fucokinase and GDP-Fuc pyrophosphorylase activities [41]. To degrade the by-product pyrophosphate formed in the GDP-fucose formation process to inorganic phosphate, Pasteurella multocida inorganic pyrophosphatase (PmPpA) [37] was used. Hp3/4FT-catalyzed fucosylation of disaccharide Galβ1–3GlcNAcβProN3 (4) using in situ generated GDP-fucose as the donor substrate produced the desired fucoside LeaβProN3 (5). The OPME fucosylation approach generated high cost GDP-Fuc in situ from inexpensive materials without the purification of intermediates, making the gram-scale synthesis of Lea an economic and highly efficient process.
2.2. Enzymatic synthesis of sLea tetrasacchrides containing different sialic acid forms
The most efficient route for systematic synthesis of sLea antigens containing different sialic acid forms would be sialylating Lea using a one-pot multienzyme (OPME) α2–3sialylation system [42]. To do this, the sialyltransferase used in the system has to tolerate fucosylated galactosides (e.g. LeaβProN3, 5) as acceptor substrates. Previously, we reported that a single mutant of Pasteurella multocida multifunctional α2–3sialyltransferase 1 [43], PmST1 M144D, was able to tolerate fucosylated galactoside LexβProN3 as the acceptor substrates for the synthesis of sLex structures containing different sialic acid forms [30]. To our delight, LeaβProN3 (5) that differs from LexβProN3 on both galactosidic and fucosidic linkages was also a suitable acceptor substrates for PmST1 M144D. Therefore, the OPME sialylation system was readily used for the construction of the desired library of sLea antigens.
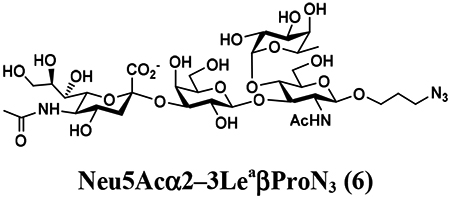
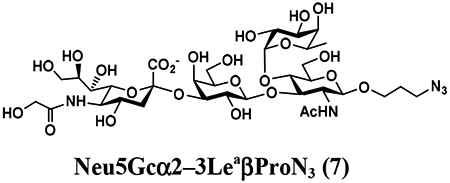
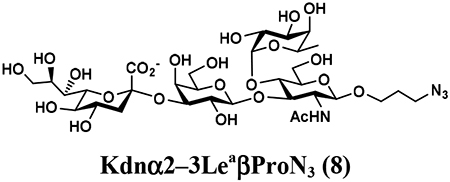
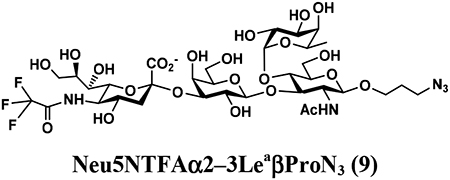
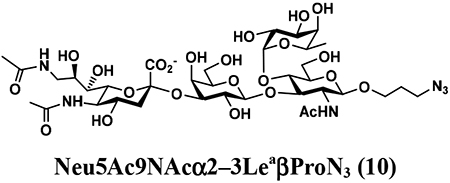
An shown in Scheme 2, the OPME sialylation system containing Neisseria meningitidis CMP-sialic acid synthetase (NmCSS) and PmST1 M144D with (A) or without (B) Pasteurella multocida sialic acid aldolase (PmNanA), allowed the synthesis of α2–3-linked sialosides containing diverse sialic acid forms from LeaβProN3 (5) and sialic acid precursors with in situ generation of a diverse array of CMP-sialic acids. sLeaβProN3 tetrasaccharides (6–12) containing N-acetylneuraminic acid (Neu5Ac), N-glycolylneuraminic acid (Neu5Gc), 3-deoxy-2-ketononulosonic acid (Kdn), N-trifluoroacetylneuraminic acid (Neu5NTFA), 9-N-acetyl-9-deoxyNeu5Ac (Neu5Ac9NAc), 9-O-acety-Neu5Ac (Neu5,9Ac2), or 9-O-acety-Neu5Gc (Neu5Gc9Ac) were successfully synthesized from the corresponding six-carbon (Man/ManNAc derivatives) or 9-carbon (Neu5Ac derivatives) precursors, respectively.
Scheme 2.

Synthesizing sLeaβProN3 containing different sialic acid forms using one-pot multienzyme (OPME) α2–3-sialylation system containing NmCSS and PmST1 M144D with (A) or without (B) PmNanA.
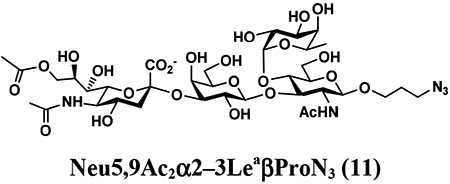
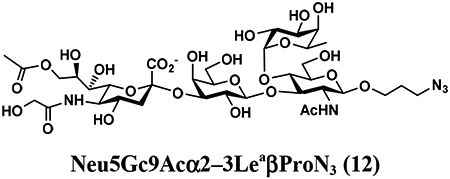
The synthesis of sLea tetrasaccharides 6–10 was carried out in the one-pot three-enzyme system at pH 8.5. As shown in Table 1, sLeaβProN3 containing naturally occurring sialic acid forms Neu5Acα2–3LeaβProN3 (6), Neu5Gcα2–3LeaβProN3 (7), and Kdnα2–3LeaβProN3 (8) were obtained in 85%, 82%, and 86% yields, respectively, from N-acetylmannosamine (ManNAc), N-glycolylmannosamine (ManNGc) [44], and mannose (Man) as sialic acid precursors. sLeaβProN3 containing non-natural sialic acid forms Neu5NTFAα2–3LeaβProN3 (9) and Neu5Ac9NAcα2–3LeaβProN3 (10) were obtained in good yields (80% and 76%, respectively) from C2- or C6-modified ManNAc derivatives N-trifluoroacetylmannosamine (ManNTFA) [45] or 6-acetamido-6-deoxy-N-acetylmannosamine (ManNAc6NAc) [46, 47], respectively. sLeaβProN3 containing naturally occurring 9-O-acetylated sialic acid forms Neu5,9Ac2α2–3LeaβProN3 (11) and Neu5Gc9Acα2–3LeaβProN3 (12) were obtained in 62% and 51% yields from Neu5,9Ac2 and Neu5Gc9Ac [48, 49], respectively, in a one-pot two-enzyme reaction containing NmCSS and PmST1 M144D at pH 7.5. The lower yields for the synthesis of 11 and 12 were due to partial de-O-acetylation during the reaction and purification process.
Table 1:
One-pot multienzyme (OPME) preparative-scale synthesis of sLeaβProN3 (6–12).
| Entry | Sialic acid or precursor | Product | Yield (%) |
|---|---|---|---|
| a | 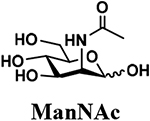 |
 |
85 |
| b | 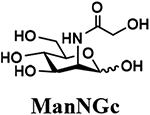 |
 |
82 |
| c | 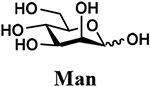 |
 |
86 |
| d | 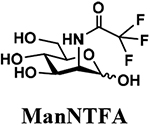 |
 |
80 |
| e | 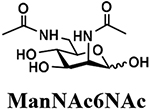 |
 |
76 |
| f | 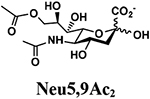 |
 |
62 |
| g | 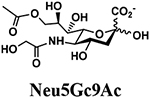 |
 |
51 |
3. Conclusions
In conclusion, an efficient chemoenzymatic method for systematic synthesis of synthetically challenging sialyl Lewis a antigens (sLeaβProN3, 6–12) containing different sialic acid forms was developed successfully. The protecting group-free chemial glycosylation reaction combed with BiGalHexNAcP-catalyzed OPME β1–3-galactosylation and Hp3/4FT-catalyzed OPME fucosylation reactions succeffully produced Lewis a angtigen (LeaβProN3, 5) in a gram scale. PmST1 M144D was proven to be an efficient catalyst for sialylating Lea with different sialic acid forms. The study presented here provides a facile chemoenzymatic route for synthesizing Lea antigen in gram-scale in high yield and for systematic synthesis of sLea antigens containing different sialic acid forms.
4. Experimental
4.1. Materials and general methods
Chemicals were purchased and used without further purification. 1H NMR and 13C NMR spectra were recorded on Bruker Avance-800 NMR spectrometer. High resolution electrospray ionization (ESI) mass spectra were obtained using Thermo Electron LTQOrbitrap Hybrid MS at the Mass Spectrometry Facility in the University of California, Davis. Silica gel 60 Å (230–400 mesh, Sorbent Technologies) was used for flash column chromatography. Thin-layer chromatography (Sorbent Technologies) was performed on silica gel plates using p-anisaldehyde sugar staining with 5% sulfuric acid in ethanol stain for detection. Gel filtration chromatography was performed with a column (100 cm × 2.5 cm) packed with Bio-Gel P-2 Fine resins (Bio-Rad). Recombinant enzymes Streptococcus pneumoniae (SpGalK) [38], Bifidobacterium infantis D-galactosyl-β1–3-N-acetyl-Dhexosamine phosphorylase (BiGalHexNAcP) [39], Bacteroides fragilis strain NCTC9343 bifunctional L-fucokinase/GDP-fucose pyrophosphorylase (BfFKP) [41], Pasteurella multocida inorganic pyrophosphatase [37], Helicobacter pylori α1–3/4-fucosyltransferase (Hp3/4FT) [40], Pasteurella multocida sialic acid aldolase (PmNanA) [50], Neisseria meningitidis CMP-sialic acid synthetase (NmCSS) [44], and Pasteurella multocida α2–3sialyltransferase 1 M144D mutant (PmST1 M144D) [30] were expressed and purified as described previously.
4.2. Gram-scale synthesis of trisaccharide LeaβProN3 (5)
4.2.1. N’-(2-Acetamido-2-deoxy-β-D-glucopyranosyl)-p-toluenesulfono-hydrazide (2)
GlcNAc (15.0 g, 67.8 mmol) and p-toluenesulfonylhydrazide (13.9 g, 74.6 mmol) were dissolved in DMF (30 mL) and H2O (15 mL), 3 mL of acetic acid was added. The mixture was incubated at 37 °C without stirring for 2 days. After all solids were dissolved, the resulting solution was poured into diethyl ether (1 L) and the mixture was stirred vigorously for overnight. The precipitate was collected to produce compound 2 (26.35 g, 99.8%) as a light yellow solid. 1H NMR (800 MHz, D2O) δ 7.71 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 3.91 (d, J = 8.8 Hz, 1H, H-1), 3.86 (dd, J = 1.6 and 12.0 Hz, 1H), 3.68 (dd, J = 5.6 and 12.0 Hz, 1H), 3.49 (t, J = 9.6 Hz, 1H), 3.45 (t, J = 9.6 Hz, 1H), 3.34 (t, J = 9.6 Hz, 1H), 3.30–3.27 (m, 1H), 2.44 (s, 3H), 2.02 (s, 3H); 13C NMR (200 MHz, D2O) δ 174.10, 145.48, 132.93, 129.85, 129.74, 127.80, 127.68, 89.64 (C-1), 76.50, 74.06, 69.68, 60.63, 53.09, 22.07, 20.63. HRMS (ESI) m/z calcd for C15H24N3O7S (M+H) 390.1335, found 390.1366.
4.2.2. Synthesis of GlcNAcβProN3 (3)
To a solution of compound 2 (2.0 g) in anhydrous DMF (10 mL), 3-chloropropanol (8.6 mL) was added. NBS (2.2 g) was then added at room temperature. After stirring the mixture for 10 minutes, Dowex (OH-) resins were added to quench the reaction. The mixture was stirred until the yellow color disappeared. After filtration, the filtrate was concentrated and purified by flash chromatography (EtOAc: MeOH = 4:1, by volume) to produce 3-chloropropyl GlcNAc as a syrup residue. The obtained product was dissolved in DMF (10 mL) and NaN3 (1.6 g) was then added. The reaction mixture was stirred at 65 °C for overnight before it was concentrated. The residue was purified by flash chromatography (EtOAc:MeOH = 4:1, by volume) to provide pure GlcNAcβProN3 (3, 1.28 g, 82%) as a white solid. 1H NMR (800 MHz, D2O) δ 4.50 (d, J = 8.8Hz, 1H, H-1), 3.98–3.95 (m, 1H, H-5), 3.92 (dd, J = 2.4 and 12.8 Hz, 1H),), 3.74 (dd, J = 5.6 and 12.8 Hz, 1H), 3.69–3.65 (m, 2H, OCH2CH2CH2N3), 3.53 (t, J = 8.0 Hz, 1H), 3.46–3.41 (m, 2H), 3.40–3.34 (m, 2H, OCH2CH2CH2N3), 2.04 (s, 3H, NHCOCH3), 1.86–1.80 (m, 2H, OCH2CH2CH2N3). 13C NMR (200 MHz, D2O) δ 174.42, 101.06 (C-1), 75.73, 73.63, 69.80, 66.96, 60.61, 55.46, 47.67, 27.99, 22.03. HRMS (ESI) m/z calcd for C11H21N4O6 (M+H) 305.1461, found 305.1456.
4.2.3. One-pot two-enzyme chemoenzymatic synthesis of Galβ1–3GlcNAcβProN3 (4)
A reaction mixture with 30 mM acceptor GlcNAcβProN3 (3, 1.04 g, 3.42 mmol), containing Tris-HCl buffer (100 mM, pH 6.55), galactose (1.23 g, 6.84 mmol), ATP (3.77 g, 6.84 mmol), MgCl2 (20 mM), SpGalK (4.65 mg), and BiGalHexNAcP (5.74 mg) was incubated in a water bath at 37 °C. The reaction progress was monitored by thin-layer chromatography (TLC) and mass spectrometry analyses. When an optimal yield was achieved (after 2 day), the enzyme reaction was terminated by adding same volume of methanol as the reaction mixture and incubating at 4°C for 30 minutes. The mixture was centrifuged and the precipitates were removed. The supernatant was concentrated and passed through a silica gel column (EtOAc:MeOH:H2O = 6:2:1, by volume) followed by a Bio-Gel P-2 gel filtration column purification to produce disaccharide Galβ1–3GlcNAcβProN3 (4, 1.43 g, 90%). 1H NMR (800 MHz, D2O) δ 4.53 (d, J = 8.0 Hz, 1H, HGlcNAc-1), 4.41 (d, J = 8.0 Hz, 1H, HGal-1), 3.97–3.88 (m, 3H), 3.81 (t, J = 10.4 Hz, 1H), 3.78–3.61 (m, 7H), 3.52 (t, J = 10.4 Hz, 1H), 3.51–3.45 (m, 2H), 3.38–3.28 (m, 2H, OCH2CH2CH2N3), 2.02 (s, 3H, NHCOCH3), 1.84–1.81 (m, 2H, OCH2CH2CH2N3); 13C NMR (200 MHz, D2O) δ 174.46, 103.40 (CGal-1), 100.80 (CGlcNAc-1), 82.25, 75.22, 75.14, 72.34, 70.53, 68.58, 68.39, 67.03, 60.89, 60.58, 54.44, 47.65, 27.97, 22.09. HRMS (ESI) m/z calcd for C17H31N4O11 (M+H) 467.1989, found 467.1986.
4.2.4. One-pot three-enzyme chemoenzymatic synthesis of LeaβProN3 (5)
A reaction mixture containing Tris-HCl buffer (100 mM, pH 7.5), Galβ1–3GlcNAcβProN3 (4, 1.18 g, 30 mM, 2.53 mmol), L-fucose (0.62 g, 3.80 mmol), ATP (2.10 g, 3.80 mmol), GTP (2.15 g, 3.80 mmol), MgCl2 (20 mM), BfFKP (4.49 mg), PmPpA (1.35 mg), and Hp3/4FT (10.55 mg) was incubated in a water bath at 37 °C. The reaction was monitored by TLC and mass spectrometry analyses. When an optimal yield was achieved (after 2 days), the same volume (115 mL) of methanol was added to the reaction mixture. This mixture was incubated at 4 °C for 30 min and then centrifuged. The supernatant was concentrated and passed through a silica gel column (EtOAc:MeOH:H2O = 5:2:1, by volume) followed by a Bio-Gel P-2 gel filtration column purification to produce LeaβProN3 (5, 1.41g, 91%) as a white powder. 1H NMR (800 MHz, D2O) δ 5.01 (d, J = 4.0 Hz, 1H, HFuc-1), 4.88–4.85 (m, 1H, HFuc-5), 4.52 (d, J = 8.8 Hz, 1H, HGlcNAc-1), 4.48 (d, J = 8.0 Hz, 1H, HGal-1), 4.07–3.34 (m, 19H), 2.04 (s, 3H, NHCOCH3), 1.86–1.82 (m, 2H, OCH2CH2CH2N3), 1.17 (d, J = 6.4 Hz, 3H, HFuc-6); 13C NMR (200 MHz, D2O) δ 174.24, 102.77 (CGal-1), 100.93 (CGlcNAc-1), 97.96 (CFuc-1), 75.96, 75.29, 74.70, 72.31, 72.18, 71.83, 70.35, 69.01, 68.24, 67.66, 67.08, 66.75, 61.55, 59.57, 55.65, 47.66, 28.01, 22.16, 15.25. HRMS (ESI) m/z calculated for C23H40N4NaO15 (M+Na) 635.2388, found 635.2353.
4.3. General procedure of one-pot multienzyme synthesis of sLea containing different sialic acid forms (6–12):
LeaβProN3 (5, 10–15 mM, 1 equiv.), a sialic acid precursor (mannose, ManNAc, or their derivatives, 1.5 equiv.), sodium pyruvate (5–7.5 equiv. when six-carbon sugars used as substrates), CTP (1.3–2.0 equiv.) were dissolved in water in a 50 mL centrifugal tube. The pH of this mixture was adjusted to neutral. MgCl2 (20 mM) and Tris-HCl buffer (100 mM, pH 8.5 for reactions using ManNAc, Man, or derivatives as the sialyltransferase donor precursor; or pH 7.5 for reactions using Neu5,9Ac2 or Neu5Gc9Ac as the sialyltransferase donor precursor) were then added. After adding NmCSS (1–3 mg) and PmST1 M144D (1–3.5 mg) with (1–3 mg, for reactions using ManNAc, Man, or derivatives as the sialyltransferase donor precursor) or without PmNanA, water was added to bring the concentration of the acceptor to the range of 10–15 mM. The reactions were carried out for 12–36 hours before they were stopped by adding the same volume of ice-cold methanol followed by incubation at 4 °C for 30 min. After centrifugation, the supernatant was concentrated and purified by a BioGel P-2 gel filtration column chromatography with or without an additional C18-column chromatography in a high-performance liquid chromatography (HPLC) system to obtain the desired products.
4.3.1. Neu5Acα2–3LeaβProN3 (6),
203 mg, 85% yield, white powder: 1H NMR (800 MHz, D2O) δ 5.00 (d, J = 4.0 Hz, 1H, HFuc-1), 4.88–4.84 (m, 1H, HFuc-5), 4.52 (d, J = 8.0 Hz, 2H, HGal-1 and HGlcNAc-1), 4.06 (t, J = 9.6 Hz, 1H), 4.04 (dd, J = 3.2 and 9.6 Hz, 1H), 3.98–3.94 (m, 2H), 3.89 (d, J = 3.2 Hz, 1H), 3.88–3.51 (m, 18H), 3.49 (t, J = 8.0 Hz, 1H), 3.40–3.34 (m, 2H, OCH2CH2CH2N3), 2.76 (dd, J = 4.8 and 12.8 Hz, 1H, HNeu5Ac_eq-3), 2.04 (s, 3H, NHCOCH3), 2.02 (s, 3H, NHCOCH3), 1.85–1.81 (m, 2H, OCH2CH2CH2N3), 1.76 (t, J = 12.0 Hz, 1H, HNeu5Ac_ax-3), 1.16 (d, J =6.4 Hz, 3H, HFuc-6); 13C NMR (200 MHz, D2O) δ 174.81, 174.11, 173.81, 102.68 (CGal-1), 100.86 (CGlcNAc-1), 99.24 (CNeu5Ac2), 97.93 (CFuc-1), 75.93, 75.48, 75.33, 74.63, 72.62, 72.22, 71.82, 71.74, 68.97, 68.66, 68.31, 67.90, 67.66, 67.07, 66.80, 66.72, 62.17, 61.55, 59.58, 51.55, 47.66, 39.89, 28.00, 22.29, 21.92, 15.21, HRMS (ESI) m/z calculated for C34H57N5O23 (M-H) 902.3372, found 902.3366.
4.3.2. Neu5Gcα2–3LeaβProN3 (7),
37 mg, 82% yield, white powder: 1H NMR (800 MHz, D2O) δ 5.00 (d, J = 4.0 Hz, 1H, HFuc-1), 4.88–4.85 (m, 1H, HFuc-2), 4.53 (d, J = 8.0 Hz, 2H, HGal-1 and HGlcNAc-1), 4.11 (s, 2H, NHCOCH2OH), 4.08–4.04 (m, 2H), 3.98–3.95 (m, 2H), 3.93 (t, J = 10.4 Hz, 1H), 3.91 (d, J = 3.2 Hz, 1H), 3.89–3.52 (m, 17H), 3.50 (t, J = 8.0 Hz, 1H), 3.40–3.34 (m, 2H, OCH2CH2CH2N3), 2.78 (dd, J = 4.8 and 12.8 Hz, 1H, HNeu5Ac_eq-3), 2.05 (s, 3H, NHCOCH3), 1.86–1.82 (m, 2H, OCH2CH2CH2N3), 1.78 (t, J = 12.8 Hz, 1H, HNeu5Ac_ax-3), 1.17 (d, J = 6.4 Hz, 3H, HFuc-6); 13C NMR (200 MHz, D2O) δ 175.58, 174.11, 173.85, 102.70 (CGal-1), 100.87 (CGlcNAc-1), 99.25 (CNeu5Ac2), 97.94 (CFuc-1), 75.94, 75.47, 75.34, 74.64, 72.35, 72.22, 71.82, 68.98, 68.67, 68.04, 67.84, 67.67, 67.07, 66.79, 66.73, 62.14, 61.56, 60.86, 59.58, 55.60, 51.26, 47.66, 39.98, 28.01, 22.30, 15.22. HRMS (ESI) m/z calculated for C34H57N5O24 (M-H) 918.3321, found 918.3300.
4.3.3. Kdnα2–3LeaβProN3 (8),
122 mg, 86% yield, white powder: 1H NMR (800 MHz, D2O) δ 4.99 (d, J = 4.0 Hz, 1H, HFuc-1), 4.87–4.84 (m, 1H, HFuc-5), 4.51 (d, J = 7.2 Hz, 2H, HGal-1 and HGlcNAc-1), 4.04 (t, J = 9.9 Hz, 1H), 4.02 (dd, J = 3.2 and 10.4 Hz, 1H), 3.98–3.51 (m, 21H), 3.48 (t, J = 8.0 Hz, 1H), 3.40–3.34 (m, 2H, OCH2CH2CH2N3), 2.71 (dd, J = 4.8 and 12.8 Hz, 1H, HNeu5Ac_eq-3), 2.05 (s, 3H, NHCOCH3), 1.85–1.81 (m, 2H, OCH2CH2CH2N3), 1.70 (t, J = 12.0 Hz, 1H, HNeu5Ac_ax-3), 1.15 (d, J = 6.4 Hz, 3H, HFuc6); 13C NMR (200 MHz, D2O) δ 174.09, 173.98, 102.74 (CGal-1), 100.88 (CGlcNAc-1), 99.10 (CNeu5Ac-2), 97.94 (CFuc-1), 75.97, 75.38, 75.32, 74.64, 73.65, 72.21, 72.04, 71.82, 70.15, 69.61, 68.97, 68.64, 67.66, 67.56, 67.07, 66.72, 66.69, 62.22, 61.56, 59.56, 55.57, 47.66, 39.62, 28.00, 22.29, 15.21. HRMS (ESI) m/z calculated for C32H54N4O23 (M-H) 861.3106, found 861.3115.
4.3.4. Neu5NTFAα2–3LeaβProN3 (9),
64 mg, 80% yield, white powder: 1H NMR (800 MHz, D2O) δ 5.00 (d, J = 4.0 Hz, 1H, HFuc-1), 4.88–4.85 (m, 1H, HFuc-5), 4.52 (d, J = 8.0 Hz, 1H, HGlcNAc-1), 4.51 (d, J = 8.0 Hz, 1H, HGal-1), 4.06–4.04 (m, 2H), 4.00 (t, J = 9.6 Hz, 1H), 3.98–3.94 (m, 2H), 3.90 (d, J = 3.2 Hz, 1H), 3.88–3.51 (m, 17H), 3.50 (t, J = 8.0 Hz, 1H), 3.39–3.34 (m, 2H, OCH2CH2CH2N3), 2.79 (dd, J = 4.8 and 12.8 Hz, 1H, HNeu5Ac_eq-3), 2.04 (s, 3H, NHCOCH3), 1.85–1.81 (m, 2H, OCH2CH2CH2N3), 1.78 (t, J = 12.8 Hz, 1H, HNeu5Ac_ax-3), 1.16 (d, J = 6.4 Hz, 3H, HFuc-6); 13C NMR (200 MHz, D2O) δ 174.11, 173.71, 102.71 (CGal-1), 100.88 (CGlcNAc-1), 99.22 (CNeu5Ac-2), 97.94 (CFuc-1), 75.98, 75.43, 75.33, 74.62, 72.21, 71.93, 71.82, 68.97, 68.64, 67.96, 67.84, 67.66, 67.07, 66.72, 62.11, 61.55, 59.57, 52.10, 47.66, 39.97, 28.00, 22.29, 15.21. HRMS (ESI) m/z calculated for C34H53F3N5O23 (M-H) 956.3089, found 956.3084.
4.3.5. Neu5Ac9NAcα2–3LeaβProN3 (10),
35 mg, 76% yield, white powder: 1H NMR (800 MHz, D2O) δ 5.00 (d, J = 4.0 Hz, 1H, HFuc-1), 4.88–4.83 (m, 1H, HFuc-5), 4.54–4.47 (m, 2H, HGal-1 and HGlcNAc-1), 4.06 (t, J = 9.6 Hz, 1H), 4.01 (dd, J = 3.2 and 9.6 Hz, 1H), 3.98–3.95 (m, 2H), 3.90 (d, J = 3.2 Hz, 1H), 3.88–3.47 (m, 18H), 3.40–3.34 (m, 2H, OCH2CH2CH2N3), 3.25 (dd, J = 8.0 and 14.4 Hz, 1H), 2.76 (dd, J = 4.8 and 12.8 Hz, 1H, HNeu5Ac_eq-3), 2.02 (s, 3H, NHCOCH3), 2.01 (s, 6H, 2xNHCOCH3), 1.87–1.80 (m, 2H, OCH2CH2CH2N3), 1.77 (t, J = 12.0 Hz, 1H, HNeu5Ac_ax-3), 1.16 (d, J =6.4 Hz, 3H, HFuc-6); 13C NMR (200 MHz, D2O) δ 174.75, 174.19, 173.92, 173.80, 102.62 (CGal-1), 100.86 (CGlcNAc-1), 99.47 (CNeu5Ac-2), 97.93 (CFuc-1), 75.94, 75.57, 75.33, 74.63, 72.50, 72.22, 71.82, 69.89, 69.45, 68.98, 68.66, 68.27, 67.67, 67.04, 66.88, 66.73, 61.55, 59.59, 55.60, 51.55, 47.66, 41.92, 39.84, 28.01, 22.18, 21.93, 21.66, 15.21. HRMS (ESI) m/z calculated for C36H60N6O23 (M-H) 943.3637, found 943.3641.
4.3.6. Neu5,9Ac2α2–3LeaβProN3 (11),
29 mg, 62% yield, white powder: 1H NMR (800 MHz, D2O) δ 5.00 (d, J = 3.2 Hz, 1H, HFuc-1), 4.87–4.84 (m, 1H, HFuc-5), 4.53 (d, J = 8.0 Hz, 2H, HGal1 and HGlcNAc-1), 4.38 (dd, J = 2.4 and 11.2 Hz, 1H), 4.18 (dd, J = 7.2 and 12.0 Hz, 1H), 4.083.52 (m, 21H), 3.51 (t, J = 8.0 and 14.4 Hz, 1H), 3.41–3.34 (m, 2H, OCH2CH2CH2N3), 2.76 (dd, J = 4.8 and 12.8 Hz, 1H, HNeu5Ac_eq-3), 2.13 (s, 3H, OCOCH3), 2.04 (s, 3H, NHCOCH3), 2.03 (s, 3H, NHCOCH3), 1.87–1.80 (m, 3H, OCH2CH2CH2N3 and HNeu5Ac_ax-3), 1.17 (d, J = 7.2 Hz, 3H, HFuc-6); 13C NMR (200 MHz, D2O) δ 174.76, 174.17, 174.01, 172.66, 102.64 (CGal-1), 100.85 (CGlcNAc-1), 98.61 (CNeu5Ac-2), 97.92 (CFuc-1), 75.98, 75.39, 75.34, 74.50, 72.55, 72.20, 71.83, 69.08, 69.00, 68.68, 68.12, 67.85, 67.67, 67.06, 66.74, 66.72, 65.77, 61.44, 59.59, 51.52, 47.67, 39.47, 28.01, 22.27, 21.97, 20.16, 15.21. HRMS (ESI) m/z calculated for C36H58N5O24 (M-H) 944.3477, found 944.3430.
4.3.7. Neu5Gc9Acα2–3LeaβProN3 (12),
12 mg, 51% yield, white powder: 1H NMR (800 MHz, D2O) δ 5.00 (d, J = 4.0 Hz, 1H, HFuc-1), 4.88–4.85 (m, 1H, HFuc-2), 4.53 (d, J = 8.0 Hz, 1H, HGlcNAc-1), 4.52 (d, J = 8.0 Hz, 1H, HGal-1), 4.35 (dd, J = 2.4 and 11.2 Hz, 1H), 4.18 (dd, J = 6.4 and 12.0 Hz, 1H), 4.11 (s, 2H, OCH2OCOCH3), 4.07–3.51 (m, 21H), 3.50 (t, J = 8.0 and 14.4 Hz, 1H), 3.40–3.34 (m, 2H, OCH2CH2CH2N3), 2.78 (dd, J = 4.8 and 12.8 Hz, 1H, HNeu5Ac_eq-3), 2.13 (s, 3H, OCOCH3), 2.04 (s, 3H, NHCOCH3), 1.85–1.81 (m, 2H, OCH2CH2CH2N3), 1.78 (t, J = 12.0 Hz, 1H, HNeu5Ac_ax-3), 1.16 (d, J =6.4 Hz, 3H, HFuc-6); 13C NMR (200 MHz, D2O) δ 175.53, 174.25, 174.17, 173.99, 102.77(CGal-1), 102.69(CGlcNAc-1), 100.87(CNeu5Ac-1), 97.94(CFuc-1), 76.00, 75.40, 75.35, 75.30, 74.71, 74.54, 72.32, 72.22, 71.83, 70.36, 69.26, 69.00, 68.68, 68.24, 68.05, 67.67, 67.07, 66.73, 65.67, 61.55, 61.49, 60.88, 59.59, 55.59, 51.20, 47.67, 39.70, 28.01, 22.28, 22.16, 20.17, 15.25, 15.21. HRMS (ESI) m/z calculated for C36H58N5O25 (M-H) 960.3426, found 960.3420.
Supplementary Material
Highlights:
High-yield protection group-free glycosylation for synthesizing GlcNAcβProN3
Gram-scale chemoenzymatic synthesis of Lewis a antigen was efficiently achieved
LeaβProN3 was shown to be a suitable acceptor for PmST1 M144D
Seven sLea antigens containing different sialic acid forms were synthesized
A high-yield chemoenzymatic method for Lea and sLea synthesis was demonstrated
ACKNOWLEDGMENT
This work was supported by the United States National Institutes of Health under Award Numbers U01GM120419 and R01AI130684. The content is solely the responsibility of the authors and does not necessarily represent the official views of the United States National Institutes of Health. The Bruker Avance-800 NMR spectrometer was funded by the United States National Science Foundation grant DBIO-722538.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information. 1H and 13C NMR spectra of synthesized glycans.
Reference
- [1].Blaszczyk M, Pak KY, Herlyn M, Sears HF, Steplewski Z, Characterization of Lewis antigens in normal colon and gastrointestinal adenocarcinomas, Proc. Natl. Acad. Sci. U. S. A 82 (1985) 3552–3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Green C, The ABO, Lewis and related blood group antigens; a review of structure and biosynthesis, FEMS Microbiol. Immunol 1 (1989) 321–330. [DOI] [PubMed] [Google Scholar]
- [3].Zhang S, Zhang HS, Cordon-Cardo C, Reuter VE, Singhal AK, Lloyd KO, Livingston PO, Selection of tumor antigens as targets for immune attack using immunohistochemistry: II. Blood group-related antigens, Int. J. Cancer 73 (1997) 50–56. [DOI] [PubMed] [Google Scholar]
- [4].Blanas A, Sahasrabudhe NM, Rodriguez E, van Kooyk Y, van Vliet SJ, Fucosylated antigens in cancer: An alliance toward tumor progression, metastasis, and resistance to chemotherapy, Front. Oncol 8 (2018) 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ugorski M, Laskowska A, Sialyl Lewis(a): a tumor-associated carbohydrate antigen involved in adhesion and metastatic potential of cancer cells, Acta Biochim. Pol 49 (2002) 303–311. [PubMed] [Google Scholar]
- [6].Narita T, Funahashi H, Satoh Y, Watanabe T, Sakamoto J, Takagi H, Association of expression of blood group-related carbohydrate antigens with prognosis in breast cancer, Cancer 71 (1993) 3044–3053. [DOI] [PubMed] [Google Scholar]
- [7].Scara S, Bottoni P, Scatena R, CA 19–9: Biochemical and clinical aspects, Adv. Exp. Med. Biol 867 (2015) 247–260. [DOI] [PubMed] [Google Scholar]
- [8].Gong E, Hirohashi S, Shimosato Y, Expression of carbohydrate antigen 19–9 and stage-specific embryonic antigen 1 in non-tumorous and tumorous epithelia of the human colon and rectum, J. Natl. Cancer Inst 75 (1985) 447–454. [PubMed] [Google Scholar]
- [9].Koprowski H, Herlyn M, Steplewski Z, Sears HF, Specific antigen in serum of patients with colon carcinoma, Science 212 (1981) 53–55. [DOI] [PubMed] [Google Scholar]
- [10].Duffy MJ, Sturgeon C, Lamerz R, Haglund C, Holubec VL, Klapdor R, Nicolini A, Topolcan O, Heinemann V, Tumor markers in pancreatic cancer: a European Group on Tumor Markers (EGTM) status report, Ann. Oncol 21 (2010) 441–447. [DOI] [PubMed] [Google Scholar]
- [11].Galli C, Basso D, Plebani M, CA 19–9: handle with care, Clin. Chem. Lab. Med 51 (2013) 1369–1383. [DOI] [PubMed] [Google Scholar]
- [12].Trinchera M, Aronica A, Dall’Olio F, Selectin ligands sialyl-Lewis a and sialylLewis x in gastrointestinal cancers, Biology (Basel) 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Reis CA, Osorio H, Silva L, Gomes C, David L, Alterations in glycosylation as biomarkers for cancer detection, J. Clin. Pathol 63 (2010) 322–329. [DOI] [PubMed] [Google Scholar]
- [14].Pinho SS, Reis CA, Glycosylation in cancer: mechanisms and clinical implications, Nat. Rev. Cancer 15 (2015) 540–555. [DOI] [PubMed] [Google Scholar]
- [15].Peilstöcker K, Kunz H, A sialyl-Lewis a-asparagine building block for glycopeptide-synthesis, Synlett (2000) 820–822. [Google Scholar]
- [16].Ragupathi G, Damani P, Srivastava G, Srivastava O, Sucheck SJ, Ichikawa Y, Livingston PO, Synthesis of sialyl Lewis(a) (sLe (a), CA19–9) and construction of an immunogenic sLe(a) vaccine, Cancer Immunol. Immunother 58 (2009) 13971405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Toepfer A, Schmidt RR, An efficient synthesis of the Lewis A (Lea) antigen pentasaccharide moiety, J. Carbohydr. Chem 12 (1993) 809–822. [Google Scholar]
- [18].Manzoni L, Castelli R, Synthesis of the Lewis a trisaccharide based on an anomeric silyl fluorous tag, Org. Lett 6 (2004) 4195–4198. [DOI] [PubMed] [Google Scholar]
- [19].Kobayashi D, Ueki A, Yamaji T, Nagao K, Imamura A, Ando H, Kiso M, Ishida H, Efficient synthesis of the Lewis a tandem repeat, Molecules (Basel, Switzerland) 21 (2016) pii: E614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kiso M, Furui H, Ando K, Ishida H, Hasegawa A, Systematic synthesis of N-methyl-1-deoxynojirimycin-containing, Le(x), Le(a), sialyl-Le(x) and sialyl-Le(a) epitopes recognized by selectins, Bioorg. Med. Chem 2 (1994) 1295–1308. [DOI] [PubMed] [Google Scholar]
- [21].Palcic MM, Venot AP, Ratcliffe RM, Hindsgaul O, Enzymic synthesis of oligosaccharides terminating in the tumor-associated sialyl-Lewis-a determinant, Carbohydr. Res 190 (1989) 1–11. [DOI] [PubMed] [Google Scholar]
- [22].Baisch G, Ohrlein R, Glycosyltransferase catalyzed assemblage of sialyl-Lewis(a)saccharopeptides, Carbohydr. Res 312 (1998) 61–72. [DOI] [PubMed] [Google Scholar]
- [23].Ernst B, Oehrlein R, Substrate and donor specificity of glycosyl transferases, Glycoconj. J 16 (1999) 161–170. [DOI] [PubMed] [Google Scholar]
- [24].Barstrom M, Bengtsson M, Blixt O, Norberg T, New derivatives of reducing oligosaccharides and their use in enzymatic reactions: efficient synthesis of sialyl Lewis a and sialyl dimeric Lewis x glycoconjugates, Carbohydr. Res 328 (2000) 525–531. [DOI] [PubMed] [Google Scholar]
- [25].Makimura Y, Ishida H, Kondo A, Hasegawa A, Kiso M, Regioselective α(2→3)sialylation of LeX and Lea by sialidase-catalyzed transglycosylation, J. Carbohydr. Chem 17 (1998) 975–979. [Google Scholar]
- [26].Baisch G, Ohrlein R, Streiff M, Kolbinger F, On the preparative use of recombinant beta(1–3)galactosyl-transferase, Bioorg. Med. Chem. Lett 8 (1998) 751754. [DOI] [PubMed] [Google Scholar]
- [27].Baisch G, Ohrlein R, Streiff M, Kolbinger F, Enzymatic synthesis of sialylLewis(a)-libraries with two non-natural monosaccharide units, Bioorg. Med. Chem. Lett 8 (1998) 755–758. [DOI] [PubMed] [Google Scholar]
- [28].Palcic MM, Venot AP, Ratcliffe RM, Hindsgaul O, Enzymic synthesis of oligosaccharides terminating in the tumor-associated sialyl-Lewis-a determinant, Carbohydr. Res 190 (1989) 1–11. [DOI] [PubMed] [Google Scholar]
- [29].Nikrad PV, Kashem MA, Wlasichuk KB, Alton G, Venot AP, Use of humanmilk fucosyltransferase in the chemoenzymic synthesis of analogues of the sialyl Lewis(a) and sialyl Lewis(x) tetrasaccharides modified at the C-2 position of the reducing unit, Carbohydr. Res 250 (1993) 145–160. [DOI] [PubMed] [Google Scholar]
- [30].Sugiarto G, Lau K, Qu J, Li Y, Lim S, Mu S, Ames JB, Fisher AJ, Chen X, A sialyltransferase mutant with decreased donor hydrolysis and reduced sialidase activities for directly sialylating LewisX, ACS Chem. Biol 7 (2012) 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gudmundsdottir AV, Nitz M, Protecting group free glycosidations using ptoluenesulfonohydrazide donors, Org. Lett 10 (2008) 3461–3463. [DOI] [PubMed] [Google Scholar]
- [32].Hernandez Armada D, Santos JT, Richards MR, Cairo CW, Protecting groupfree immobilization of glycans for affinity chromatography using glycosylsulfonohydrazide donors, Carbohydr. Res 417 (2015) 109–116. [DOI] [PubMed] [Google Scholar]
- [33].Tiwari VK, Mishra BB, Mishra KB, Mishra N, Singh AS, Chen X, Cu-catalyzed click reaction in carbohydrate chemistry. Chem. Rev 116 (2016) 3086–3240. [DOI] [PubMed] [Google Scholar]
- [34].Deng L, Chen X, Varki A, Exploration of sialic acid diversity and biology using sialoglycan microarrays. Biopolymers 99 (2013) 650–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chokhawala HA, Huang S, Lau K, Yu H, Cheng J, Thon V, Hurtado-Ziola N, Guerrero JA, Varki A, Chen X, Combinatorial chemoenzymatic synthesis and high-throughput screening of sialosides. ACS Chem. Biol 3 (2008) 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sudibya HG, Ma J, Dong X, Ng S, Li L-J, Liu X-W, Chen P, Interfacing glycosylated carbon-nanotube-network devices with living cells to detect dynamic secretion of biomolecules, Angew. Chem. Int. Ed 48 (2009) 2723–2726. [DOI] [PubMed] [Google Scholar]
- [37].Lau K, Thon V, Yu H, Ding L, Chen Y, Muthana MM, Wong D, Huang R, Chen X, Highly efficient chemoenzymatic synthesis of beta1–4-linked galactosides with promiscuous bacterial beta1–4-galactosyltransferases, Chem. Commun 46 (2010) 6066–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chen M, Chen LL, Zou Y, Xue M, Liang M, Jin L, Guan WY, Shen J, Wang W, Wang L, Liu J, Wang PG, Wide sugar substrate specificity of galactokinase from Streptococcus pneumoniae TIGR4, Carbohydr. Res 346 (2011) 2421–2425. [DOI] [PubMed] [Google Scholar]
- [39].Yu H, Thon V, Lau K, Cai L, Chen Y, Mu S, Li Y, Wang PG, Chen X, Highly efficient chemoenzymatic synthesis of beta1–3-linked galactosides, Chem. Commun 46 (2010) 7507–7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yu H, Li Y, Wu Z, Li L, Zeng J, Zhao C, Wu Y, Tasnima N, Wang J, Liu H, Gadi MR, Guan W, Wang PG, Chen X, H. pylori alpha1–3/4-fucosyltransferase (Hp3/4FT)-catalyzed one-pot multienzyme (OPME) synthesis of Lewis antigens and human milk fucosides, Chem. Commun 53 (2017) 11012–11015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yi W, Liu X, Li Y, Li J, Xia C, Zhou G, Zhang W, Zhao W, Chen X, Wang PG, Remodeling bacterial polysaccharides by metabolic pathway engineering, Proc. Natl. Acad. Sci. U. S. A 106 (2009) 4207–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yu H, Chokhawala HA, Huang S, Chen X, One-pot three-enzyme chemoenzymatic approach to the synthesis of sialosides containing natural and nonnatural functionalities, Nat. Protoc 1 (2006) 2485–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yu H, Chokhawala H, Karpel R, Yu H, Wu B, Zhang J, Zhang Y, Jia Q, Chen X, A multifunctional Pasteurella multocida sialyltransferase: a powerful tool for the synthesis of sialoside libraries, J. Am. Chem. Soc 127 (2005) 17618–17619. [DOI] [PubMed] [Google Scholar]
- [44].Yu H, Yu H, Karpel R, Chen X, Chemoenzymatic synthesis of CMP-sialic acid derivatives by a one-pot two-enzyme system: comparison of substrate flexibility of three microbial CMP-sialic acid synthetases, Bioorg. Med. Chem 12 (2004) 64276435. [DOI] [PubMed] [Google Scholar]
- [45].Humphrey AJ, Fremann C, Critchley P, Malykh Y, Schauer R, Bugg TD, Biological properties of N-acyl and N-haloacetyl neuraminic acids: processing by enzymes of sialic acid metabolism, and interaction with influenza virus, Bioorg. Med. Chem 10 (2002) 3175–3185. [DOI] [PubMed] [Google Scholar]
- [46].Khedri Z, Xiao A, Yu H, Landig CS, Li W, Diaz S, Wasik BR, Parrish CR, Wang L-P, Varki A, Chen X, A chemical biology solution to problems with studying biologically important but unstable 9-O-acetyl sialic acids, ACS Chem. Biol 12 (2017) 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Li W, Xiao A, Li Y, Yu H, Chen X, Chemoenzymatic synthesis of Neu5Ac9NAccontaining alpha2–3- and alpha2–6-linked sialosides and their use for sialidase substrate specificity studies, Carbohydr. Res 451 (2017) 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yu H, Cheng J, Ding L, Khedri Z, Chen Y, Chin S, Lau K, Tiwari VK, Chen X, Chemoenzymatic synthesis of GD3 oligosaccharides and other disialyl glycans containing natural and non-natural sialic acids, J. Am. Chem. Soc 131 (2009) 18467–18477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ogura H, Furuhata K, Sato S, Anazawa K, Itoh M, Shitori Y, Synthesis of 9-Oacyl- and 4-O-acetyl-sialic acids, Carbohydr. Res 167 (1987) 77–86. [DOI] [PubMed] [Google Scholar]
- [50].Li Y, Yu H, Cao H, Lau K, Muthana S, Tiwari VK, Son B, Chen X, Pasteurella multocida sialic acid aldolase: a promising biocatalyst, Appl. Microbiol. Biotechnol 79 (2008) 963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.