

Abstract
Augmented vasoconstriction is a hallmark of hypertension and is mediated partly by hyper-stimulation of G protein couple receptors (GPCRs) and downstream signaling components. Although GPCR blockade is a key component of current anti-hypertensive strategies, whether hypertension is better managed by directly targeting G proteins has not been thoroughly investigated. Here, we tested whether inhibiting Gq/11 proteins in vivo and ex vivo using natural cyclic depsipeptide, FR900359 (FR) from the ornamental plant, Ardisia crenata, and YM-254890 (YM) from Chromobacterium sp. QS3666, or it’s synthetic analog, WU-07047 (WU), was sufficient to reverse hypertension in mice. All three inhibitors blocked G protein-dependent vasoconstriction, but to our surprise YM and WU and not FR inhibited K+-induced Ca2+ transients and vasoconstriction of intact vessels. However, each inhibitor blocked whole-cell L-type Ca2+ channel current in vascular smooth muscle cells. Subcutaneous injection of FR or YM (0.3 mg/kg, s.c.) in normotensive and hypertensive mice elicited bradycardia and marked blood pressure decrease, which was more severe and long lasting after the injection of FR relative to YM (FRt1/2 ≅ 12 hr vs. YMt1/2 ≅ 4 hr). In deoxycorticosterone acetate (DOCA)-salt hypertension mice, chronic injection of FR (0.3 mg/kg, s.c., daily for seven days) reversed hypertension (vehicle SBP: 149 ± 5 vs. FR SBP: 117 ± 7 mmHg), without any effect on heart rate. Our results together support the hypothesis that increased LTCC and Gq/11 activity is involved in the pathogenesis of hypertension, and that dual targeting of both proteins can reverse hypertension and associated cardiovascular disorders.
Keywords: G proteins, cyclic depsipeptides, blood pressure, L-type calcium channel, Gq/11 inhibitor ligands, calcium signaling
Graphical abstarct
Introduction
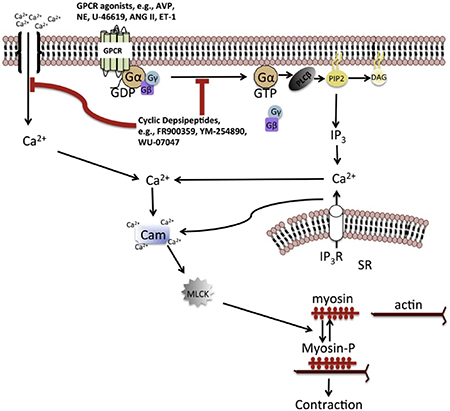
Signaling via heterotrimeric G protein-coupled receptors (GPCRs) plays a critical role in the maintenance of normal blood pressure and in the pathophysiology of hypertension and associated cardiovascular and renal diseases. GPCRs act as signal transducers for vasoactive substances such as angiotensin II, norepinephrine, endothelin-1 and vasopressin, regulating blood pressure largely by two principal mechanisms: modulation of renal control of extracellular fluid volume, and hence cardiac output, and modulation of total peripheral resistance by regulating the balance between constriction and dilatation of the resistance vasculature1, 2. Upon stimulation by cognate agonists, GPCRs evoke a physiological response usually by activating intracellular signaling via heterotrimeric G proteins, thereby increasing intracellular levels of second messengers, including cAMP and calcium. In the vasculature, a rise in intracellular calcium concentration is a critical co-factor for actomyosin cross-bridge formation and contraction of smooth muscle3–5, and for the production of endothelium-derived relaxing factors, including nitric oxide and EDHF6, 7. Most GPCRs often couple to more than one of four classes of G proteins, including Gi/o, Gs, G12/13 and Gq/11, and act in concert with other GPCR/G-protein pairs in vivo to impinge on a physiological process such as vascular tone or the control of electrolyte balance by the kidney. Thus, G proteins act as signaling nexus enabling multiple GPCRs to elicit a common physiological response upon stimulation by their cognate agonists. Abnormal GPCR signaling due to increases in the circulating levels of endogenous agonists, or abnormal activity at the level of the receptor or downstream effectors can lead to augmented vascular resistance, increased sodium retention, or a combination of both, contributing to the development of hypertension2. Accordingly, pharmacological blockade of GPCRs, or the prevention of intracellular calcium rise with calcium channel inhibitors are major therapeutic strategies for controlling blood pressure in human hypertension8. While blocking individual GPCRs alone is an effective anti-hypertensive strategy, particularly in mild cases of human hypertension, this approach has not proven to be sufficient in many other cases, particularly in resistance hypertension. This may be due, at least partly, to the level of complexity of GPCR/G protein pairs and the downstream signaling mechanisms that could facilitate adaptation of the signaling mechanisms to blunt the therapeutic effects of GPCR blockers. Agents that block G proteins could help circumvent such maladaptive mechanisms. However, such agents have been difficult to find. A few years ago, a group of cyclic depsipeptides was shown to inhibit Gq/11 class G proteins with high specificity9–11. These natural cyclic depsipeptides, including YM-254890 (henceforth referred to as YM) and FR900359 (also referred to as FR) inhibit Gq/11 by binding to a hydrophobic pocket in the α-subunit to stabilize the inactive, GDP-bound conformation, thus precluding activation by stimulated GPCRs and subsequent rise in intracellular calcium via Gq/11-mediated calcium release from internal stores9, 11. FR and YM have been shown to have strong effects in the cardiovascular system, including the inhibition of aortic constriction12, blockade of platelet aggregation13 and induction of vasodilation and bronchodilation, when given systemically to anesthetized animals11, 14. Thus FR and YM are versatile tools for in-depth probing of the physiological and pathophysiological roles of Gq/11 class G proteins in the cardiovascular system. Despite the noted cardiovascular effects of the cyclic depsipeptides, very little is known about their therapeutic potential as anti-hypertensive agents.
In this study, we have characterized the hemodynamic effects of YM and FR, as well as the synthetic analog of YM, WU-0704715. We examined the effects of all three inhibitors in an ex vivo assay for Gq/11-mediated reactivity of the resistance vasculature. In the process, we have discovered an additional mechanism by which YM and WU elicit their inhibitory effects in the resistance vasculature. We also demonstrate the mechanism and in vivo consequence of the pseudo-irreversible action of FR and YM on blood pressure. Finally, we use FR to demonstrate the anti-hypertensive potential of chronically blocking Gq/11 in a mouse model of established hypertension.
Our findings demonstrate the potential therapeutic benefits of pharmacological targeting of Gq/11 as a novel approach to treating hypertension and associated cardiovascular disorders. Finally, our results provide a paradigm for future in vivo studies elucidating the biological role of Gq/11 in the cardiovascular and other organ systems.
Material and Methods
Animals
The Institutional Animal Care and Use Committee of Drexel University approved the protocols for all animal experiments performed in this study, in accordance with the U.S. animal welfare act. In all experiments involving animals, we used 2- to 4-month-old male and female mice of the Charles River C57/Bl 6 genetic background. The mice were provided access to food and water ad libitum in our institution’s animal facility at 22°C and a 12-h light/dark cycle.
Reagents
YM-254890 (YM) was purchased from Wako Pure Chemical Industries, Ltd. FR900359 (FR) was a kind gift from Professor Kendall Blumer at Washington University School of Medicine in St. Louis, MO. WU-07047 (WU) was synthesized using our previously described reaction steps15. Phenylephrine (PE), U-46619, endothelin-1 (ET-1), arginine vasopressin (AVP), and Bay K8644 were obtained from Tocris Bioscience. All other reagents for preparing physiological saline solution (PSS) were obtained from Sigma Aldrich.
Vascular reactivity studies
We used small resistance arteries from the mouse mesenteric vascular bed for all the reactivity studies. The mice were euthanized by deep anesthesia with ketamine/xylazine (ketamine; 43 mg/kg, i.p., and xylazine; 6 mg/kg, i.p.) followed by cervical dislocation. The gut was harvested and placed in chilled physiological saline solution (PSS) of the following composition (in mM): 140 NaCl, 5 KC1, 1.2 MgSO4, 2.0 CaC12, 10 NaAcetate, 10 HEPES, 1.2 Na2H2PO4, 5 glucose, and pH adjusted to 7.4 with NaOH. Second-order mesenteric arteries were isolated and transferred into a vessel chamber where they were cannulated at both ends with glass pipettes and secured with nylon ligature. Intraluminal pressure and vessel bath temperature were maintained by servo-controlled pressure pump and temperature control systems, respectively. The vessel lumen and chamber were filled with PSS of the same composition described above. After 30-min equilibration period at 37°C and 60 mmHg, vessel viability was tested with increasing concentrations (20, 40, 60, 80 mM) of high potassium-PSS solution. To evoke vasoconstriction, increasing concentrations of contractile agents (PE, 10−9 – 10−4 M; U-46619, 10−11 – 10−5 M; AVP, 10−11 – 10−5 M; or ET-1, 10−11 – 10−7 M) were applied after 30-minute incubation of the vessel with vehicle (0.02% DMSO in PSS) or a Gq/11 inhibitor ligand (FR or YM applied at 1 μM, or WU 50 μM). To determine the inhibitory profile of the Gq/11 inhibitors, a series of PE-evoked vasoconstriction was performed in the presence of FR (0, 0.001, 0.01, 0.1, 1 μM), YM (0, 0.001, 0.01, 0.1, 1 μM) or WU (0, 1, 5, 10, 20, 50 μM) at various concentrations. To induce L-type calcium channel (LTCC)-mediated vasoconstriction, the artery segments were first incubated in calcium-free PSS containing vehicle or 1 μM of FR or YM, and then treated with PSS containing increasing concentrations of calcium (0, 0.1, 0.2, 0.4, 0.8, 1.6, 3.2 mM) in the presence or absence of the LTCC opener Bay K8644 (0.1 or 1 μM). Percent vasoconstriction was calculated as we have previously described16, 17.
Cell culture and electrophysiological recording of L-type Ca2+ channel currents
Vascular smooth muscle-derived A7r5 cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA). The cells were cultured in DMEM/F-12 (Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12, Gibco) supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin and incubated at 37°C with 5% CO2. L-type Ca2+ channel currents were recorded using a standard whole-cell voltage-clamp technique at room temperature(20–24°C) using an EPC 10 amplifier and PatchMaster software (HEKA Elektronik, Lambrecht, Germany). Currents were recorded from a holding potential of −80 mV with a 500 ms step-pulse from −60 mV to +40 mV and a pulse frequency of 0.1 Hz. Ba2+ was used as the charge carrier and K+ currents were blocked by Cs+ and tetraethylammonium chloride (TEA-Cl). The extracellular solution contained (in mM): 100 NaCl, 10 CsCl, 20 TEA-Cl, 10 BaCl2, 1 MgCl2, 10 Hepes, and 10 glucose. The pH of the solution was adjusted to 7.4 using TEA-OH. The resistance of the patch-glass pipettes was 3–5 MΩ filled with a solution containing (in mM): 140 CsMeSO4, 2 MgCl2, 10 Hepes, 5 EGTA, 3 Na2ATP, 0.3 Na2GTP. The pH of the pipette solution was adjusted to 7.2 with CsOH. L-type Ca2+ channel current recording was verified by application of 1 μM nifedipine. To test the effects of various compounds on L-type Ca2+ channel activity, the cells were pre-incubated with vehicle (0.02% DMSO), YM-254890 (1 μM), FR (1 μM), or WU-07047 (50 μM) for 15 – 60 min prior to the recording of currents.
Blood pressure and heart rate monitoring by radiotelemetry
Mice were instrumented with pressure-sensing catheters connected to radiotelemetry transmitters following well-described procedures17–19. In short, the radiotelemetry catheter was placed in the left carotid artery of isoflurane (3% mixed with 95% oxygen)-anesthetized mice and advanced into the ascending aorta, while the transmitter body (TA11PA-CA10, Data Sciences international) was placed in the intraperitoneal cavity. The mice were allowed to recover for 7 days after surgery, before the beginning of baseline blood pressure and heart rate recordings, and administration of drugs. Baseline and drug-induced changes in diurnal blood pressure and heart rate were recorded and analyzed using Ponemah data acquisition software (DSI). Averages of continuous systolic blood pressure (SBP) and heart rate recordings in one-hour intervals over 24 hours were plotted starting at 12-noon until the same time the following day. Vehicle or drugs were injected subcutaneously after 4 hours of baseline recordings.
Acute in vivo dose-response in conscious, freely moving normotensive and hypertensive mice
After 4 hr of baseline radiotelemetry BP and HR recordings, the mice received a bolus, subcutaneous injection of vehicle (0.01% DMSO in 5% dextrose solution). Twenty-four hours later, the mice were divided into two groups with one group receiving YM-254890 (0.1, 0.3, or 0.5 mg/kg, s.c.) and the other group receiving FR900359 (0.05, 0.1, or 0.3 mg/kg, s.c.). BP and HR were continuously monitored for at least 24 hr post injection. At the end of the 24-hr recording, a washout period of at least 24 hr was allowed before the administration of the next dose of each drug. We determined in preliminary studies that subcutaneous injection of YM and FR above the respective highest dose above caused extreme hypotension and bradycardia, and eventually death within a few hours (data not shown). For acute dose-response in hypertensive state, the mice were first made hypertensive using a previously described method20. Briefly, the mice were provided L-NAME in their drinking water (0.5 g/L,), which caused 20–30 mm Hg increase in SBP by day 10. The mice then received subcutaneous injections of YM or FR, as described above. All injections were performed at 5 pm.
Chronic in vivo dose-response in conscious, freely moving DOCA-salt hypertensive mice
Adult male and female mice were subjected to uninephrectomy, 2 weeks prior to being instrumented with radiotelemetry transmitters, as described above. After one week of recovery, baseline blood pressure and heart rate data were acquired for 3 hours (3–6 pm) every day for 5 days. Then, each mouse received subcutaneous implantation of deoxycorticosterone acetate (DOCA) pellet (50 mg over 21 days) under light isoflurane anesthesia and returned to their home cages, while on 1% drinking salt (NaCl)-water, for more data acquisition as described above. Seven days after DOCA pellet implantation, when systolic blood pressure had plateaued, the mice were divided into two groups; one group received bolus, subcutaneous injection of vehicle (0.01% DMSO in 5% dextrose solution) or FR900359 (0.3 mg/kg, s.c.) every day for seven days. Daily data acquisition session was started 1 hr prior to each injection, administered at 5 p.m., and stopped 1 hour after the injection. After 7 days of injection, blood pressure and heart rate were monitored further for 7 days.
Assessment of arterial Baroreflex in anesthetized mice
Previously described method, with modifications, was used to examine arterial baroreflex response in mice acutely treated with vehicle (0.02% DMSO in normal saline), YM (0.1 mg/kg), or FR (0.1 mg/kg)18. Male and female mice were instrument with arterial and venous catheters in the left carotid artery and right jugular vein, respectively. After 15 min of stabilization after surgery, the mice received bolus intravenous injections of phenylephrine (injected at 1.5 to 24 μg/kg in volumes of 10 μL), followed by injections of the nitric oxide donor, sodium nitroprusside (injected at 1.5 to 24 μg/kg in volumes of 10 μL) via the jugular vein catheter while blood pressure was continuously recorded. A recovery period of 10 min was allowed between injections to allow blood pressure to return to baseline and to avoid the effects of each dose on the subsequent injections. Maximal change in blood pressure and heart rate response to each dose of phenylephrine and sodium nitroprusside were used to plot dose-response curves to compare the effects of YM and FR on arterial baroreflex.
Molecular docking studies
All docking experiments were performed using ICM-Pro™ version 3.8–6a (Molsoft LLC, San Diego, CA). The chemical structure of YM-254890 was extracted from the x-ray crystal structure of Gqαβγ in complex with YM-254890, with Protein Data Bank code 3AH8. The structure file for FR900359 was retrieved from Pubchem chemical database and the structure of WU-07047 was manually built using ICM ligand editor module. The ligands were prepared by using the ligand setup option in ICM-Pro. The crystal structure of bacterial CavAb protein (PDB code 5KMD) was retrieved from the RCSB protein database. The protein was prepared for docking by addition of hydrogen atoms, optimizing side chains and deleting water molecules. The bound amlodipine molecule was removed from the site. A molecular surface of the DHP binding site was generated by selecting residues surrounding the bound amlodipine, and the ligand docking was restricted to this pocket. The docking was performed using the ICM-Pro docking module with effort or thoroughness value of 5. The stacks with various conformations for each docked ligand were selected based on their docking energy scores. The final poses of ligands were chosen based on visual analyses of the validity of the pose and interactions made within the pocket.
Statistics
All data are presented as means ± standard error of the mean. We performed all statistical analysis using Prism software (version 2.0). Repeated-measures ANOVA (Analysis of variance) was used to determine significant interaction within and between groups. Neuman Keuls post hoc analysis was used to determine significance of differences between groups, and results were considered significant if the P value was < 0.05.
Results
Gq/11-inhibitor ligands block vasoconstriction evoked by multiple Gq-coupled receptors in resistance arteries.
Initially, our goal was to compare the inhibitory potencies of three Gq/11 inhibitor ligands, including FR900359, YM-254890 and WU-07047 in the resistance vasculature. We performed ex vivo concentration-response studies using pressurized resistance arteries from the mesenteric vascular bed of C57bl/6 wild type mice of both sexes. We used vasoactive agonists known to evoke vasoconstriction by stimulating GPCRs that couple at least partly to Gq/11 class G proteins, including phenylephrine (α1-adrenergic receptor), the prostaglandin H2 analogue, U-46619 (thromboxane A2 receptor), arginine vasopressin (vasopressin V1 receptor), and endothelin-1 (endothelin ETA receptor). Because the α1-adrenergic receptor couples only to Gq/11 in the vascular smooth muscle, we used the cognate agonist, phenylephrine (PE), to first assess the inhibitory profile of the three Gq/11 inhibitor ligands. All three inhibitor ligands effectively blocked PE-evoked vasoconstriction (Fig 1). As little as 10 nM and FR or YM almost completely blocked PE-evoked constriction, and 1 μM of YM tended to cause dilatation of the vessel from baseline, though the effect was not statistically significant (Fig. 1A and1B). In contrast, WU had the least inhibitory effect on PE-evoked constriction and required 50 times the concentration of FR or YM to elicit a similar level of marked blockade of vasoconstriction (Fig 1A vs. 1C and 1B vs. 1C). In addition to blocking vasoconstriction, all three inhibitors also blocked PE-induced calcium transients in the intact mesenteric artery preparations (supplemental Fig S1). FR, YM and WU showed similar inhibitory effects on vasoconstriction elicited by other Gq/11-coulped receptor agonists. As shown in Figure 1D–1F, FR completely blocked vasoconstriction elicited by U-46619, arginine vasopressin (AVP), and ET-1. YM markedly decreased constriction by U-46619 and completely blocked constriction by AVP and ET-1. In contrast, WU had a weak inhibitory effect on constriction by U-46619, AVP, and ET-1. Taken together, the data showed that FR most potently inhibits vasoconstriction involving Gq/11 activation, whereas WU has the weakest inhibitory effect.
Figure 1.

Inhibition of Gq-coupled GPCR-induced vasoconstriction of small mesenteric arteries by cyclic depsipeptide Gq/11 inhibitor ligands FR900359 (FR), YM-254890 (YM), and WU-07047 (WU). Vasoconstrictor responses to phenylephrine (PE) are expressed as mean percent decrease in diameter (n=3 animals [2 vessels/animal] per group). A – C, Concentration-dependent blockade of PE-induced vasoconstriction by FR, YM, and WU. D, E, F, Effects of the highest concentrations of FR (1 μM), YM (1 μM), and WU (50 μM) on contractile responses to the thromboxane analogue, U-46619, arginine vasopressin (AVP), and endoethelin-1 (ET-1), respectively. Values are mean ± s.e.m. **P< 0,01 vs. control; *, **P< 0.05, 0.01 WU vs. control; #, ##P< 0.05, 0.01 YM vs. control; ++P< 0.01 FR vs. control
YM and WU but not FR block vasoconstriction partly by targeting L-type calcium channels
Calcium influx through voltage-gated L-type calcium channels and receptor-operated calcium channels is critical to vascular smooth muscle contraction21. Previously, it was reported that FR induces vasodilation in part by blocking LTCC in aortic rings12. Therefore, we determined whether the inhibition of vasoconstriction in resistance arteries by FR, YM, and WU was mediated at least partly by blockade of extracellular Ca2+ influx. To this end, we examined the ability of the Gq/11 inhibitors to block LTCC. We used increasing concentrations of K+ to induce plasma membrane depolarization and influx of Ca2+ via LTCC, in the absence or presence of the Gq/11-inhibitor ligands. As shown in Figure 2A and2D, incubation of mesenteric arteries with a concentration of FR (1 μM) that completely blocked PE-induced vasoconstriction (supplemental Fig S1) had no effect on K+-induced calcium transients or vasoconstriction. In contrast, concentrations of YM and WU that had similar inhibitory effects on PE-induced constriction caused a marked reduction in K+-induced calcium transients and vasoconstriction (Fig 2B, 2C, 2E & 2F). To confirm that YM or WU inhibited K+-induced vasoconstriction by targeting LTCC, we developed an ex vivo assay in which vasoconstriction was elicited by pharmacologically opening LTCC and applying increasing concentrations of extracellular Ca2+. Figure 2G shows that abluminal application of increasing concentrations of Ca2+ caused vasoconstriction in the presence of the LTCC opener, Bay K8644, compared to control. However, pre-incubation of the arteries with YM almost completely blocked LTCC-mediated vasoconstriction; interestingly, the inhibitory effect of YM was abolished by incubating the vessel with YM and Bay K simultaneously (Fig 2H). In contrast, FR had no effect on LTCC-mediated vasoconstriction (Fig 2I). Together, these data suggested that YM and WU elicit their inhibitory effect in part by targeting LTCC, whereas FR targets only Gq/11 to block vasoconstriction.
Figure 2.

Effects of Gq/11 inhibitor ligands on G protein-independent, high-potassium and L-type calcium channel (LTCC)-mediated Ca2+ transients and vasoconstriction. Contractile responses were elicited by abluminal application of PSS containing increasing concentrations of potassium to cause membrane depolarization, or the LTCC activator Bay K 8644 (Bay K), opening LTCC and facilitating the influx of Ca2+ into vascular smooth muscle. A-C, High potassium-induced Ca2+ transients in small mesenteric arteries in the absence or presence of FR (1 μM), YM (1 μM), or WU (50 μM). Ca2+ transients were measured as changes in fluorescence intensity of the Ca2+ binding dye, Fluo-4 that was preloaded in the vessel prior to incubation with an inhibitor or the application of high potassium PSS. Values are expressed as percent change in fluorescence from baseline. Vasoconstrictor responses are expressed as mean percent decrease in diameter (n=6 animals [2 vessels/animal] per group). D – F, High potassium-induced vasoconstriction in the absence and presence of FR (1 μM), YM (1 μM), or WU (50 μM). YM and WU but not FR inhibit high potassium-evoked contractile responses. G, Calcium-induced vasoconstriction in the absence and presence of the LTCC opener, Bay K8644. Both low and high concentrations of Bay K have similar facilitative effects on Ca2+-induced vasoconstriction. H, I, Effects of YM (1 μM) and FR (1 μM) on Ca2+-induced, Bay K-facilitated vasoconstriction. YM but not FR blocks LTCC-mediated vasoconstriction. All values are mean ± s.e.m. *,**P<0.05, 0,01 vs. control.
Structural basis for inhibition of LTCC by Gq/11 inhibitor ligands
As previously reported, Gq/11 inhibitors act as GDIs to keep the G-alpha subunit in the inactive GDP-bound state, thereby preventing activation by stimulated GPCRs11, 22. Conversely, 1,4 dihydropyridines (DHPs) are thought to inhibit LTCC by allosterically binding to a hydrophobic groove located on the external, lipid-facing surface at the interface of two pore-forming subunits, inducing an asymmetric conformation of the selectivity filter to block the channel pore23. Therefore, we compared the published amino acid residues that are critical for the binding of YM to the hydrophobic pocket in Gq/11 to the putative, so-called ‘DHP-sensing residues that facilitate the binding of DHPs to the hydrophobic groove in the mammalian LTCC ortholog, the bacterial CavAb22, 24. We found that the key residues in the hydrophobic groove of CavAb that facilitate amlodipine binding (F167, Y168, F171, Y195, I199)23 are also present in the hydrophobic pocket of Gqα subunit, where YM binds (I56, K57, R60, Y67, F75, L78, V184, P185, T186, T187, I189, I190, Y192, P193)22. Therefore, using the published model of the crystal structure of the homotetrameric CavAb channel with amlodipine in the DHP-binding pocket23, we performed molecular docking experiments to determine whether and/or how Gq/11 inhibitors can bind to LTCC. As shown in Figure 3, both FR and YM can form docked complexes by binding to the DHP-binding pocket of CavAb, assuming similar binding modes as amlodipine. Both FR and YM make extensive hydrophobic interactions in the binding pocket, with the phenyl rings oriented towards Y168 as in amlodipine binding (Fig. 3A, B, & C). WU can also bind to CavAb, however, in contrast to amlodipine, FR and YM, the binding mode of WU is inverted, with the phenyl ring pointing downward towards residue F203 (Fig. 3D). Together, these results suggested that Gq/11 inhibitors can compete with DHPs and analogs including Bay K8644, and further supported the hypothesis that Gq/11 inhibitors block vasoconstriction by directly inhibiting LTCC.
Figure 3.

Structural comparison of the binding modes of amlodipine (A, green), YM-254890 (B, pink), FR 900359 (C, yellow), and WU-07047 (D, orange) bound within the 5KMD dihydropyridine binding site of bacterial CaVAb, viewed from the side of the pore module. A, 5KMD x-ray crystal structure with amlodipine bound within the bacterial homotetrameric model CaV channel, CaVAb. B – D, docked complexes of YM, FR and WU compounds, respectively, bound within the amlodipine binding site of CaVAb. Comparison of the binding modes reveal that the phenyl ring of amlodipine, YM and FR orients in the same direction towards amino acid residue Y168. YM and FR make more extensive hydrophobic interactions within the pocket and have the same binding modes. The binding mode of WU is distinct from all others with the phenyl ring pointing down towards the hydrophobic amino acid residue F203. The CaV channel is oriented such that the pore opening is on the top. The structure orientation is similar to what is shown in Figure 1e/f of the reference paper (doi:10.1038/nature19102), which reports details on the 5KMD crystal structure. Residues T138, Y195, I199, P200, F203 are part of the S6 helix as previously reported in the paper23. Residues E165, F167, F171 and Y168 are part of the P helix. E, ribbon and stick structure of CavAb with superimposed poses of docked Gq/11 inhibitor ligands in side view in 5KMD protein.
Gq/11 inhibitor ligands directly block vascular smooth muscle L-type calcium channel activity
As shown by the results in Figure 2, Gq/11 inhibitor ligands block vasoconstriction induced with high potassium, or with the LTCC opener, Bay K8644 in the presence of increasing concentrations of extracellular calcium. This result strongly suggested that G protein inhibitor ligands cause vasodilation in part by blocking Ca2+ influx via voltage-gated Ca2+ channels. To directly test this hypothesis, we used whole-cell electrophysiological recording to examine the effects of the Gq/11 inhibitor ligands on LTCC activity in vascular smooth muscle cells. After establishing a stable whole-cell configuration, Ca2+ current was generated by stepwise application of depolarizing voltages (−60 to +40 mV, in 10 mV increments) to cultured rat vascular smooth muscle A7r5 cells seeded overnight on coverslips. To verify that the current being recorded was from LTCC, after recording ICa-L, bath application of 1 μM of the dihydropyridine, nifedipine, completely blocked the Ca2+ currents, indicating that the source of the recorded currents was through LTCC (Figure 4A). To test the effects of G protein inhibitor ligands on ICa-L, cells were incubated with vehicle (0.02% DMSO in extracellular solution) as control, or 1 μM of FR, YM, or WU-07047 for 15–60 min at room temperature followed by recording of Ca2+ currents as described above. As shown in Figure 4B, all three Gq/11 inhibitor ligands markedly decreased Ca2+ currents. Notably, 1 μM of YM and WU had similar inhibitory effect toward the LTCC at 0 mV (−3.11 ± 0.76 vs. −3.33 ± 0.40 pA/pF, YM vs. WU) and −10 mV (−3.21 ± 0.63 vs. −3.82 ± 0.44 pA/pF, YM vs. WU), while a much greater inhibitory effect by WU was achieved with 50 μM at −10 mV (−3.82 ± 0.44 vs. −1.86 ± 0.44 pA/pF, P<0.01; 1 vs. 50 μM of WU). Taken together, these results are consistent with the hypothesis that the Gq/11 inhibitor ligands elicit their effect partly by blocking the rise in intracellular Ca2+ transients via influx through the LTCC, thereby facilitating vasodilation.
Figure 4.

Inhibition of A7r5 vascular smooth muscle L-type calcium (LCa) current by Gq/11 inhibitor ligands. A, left panel, representative tracings of whole-cell currents (top left panel) recorded with a 500 ms step-pulse from a holding membrane potential of −80 mV to testing potential from −60 mV to +40 V at a pulse frequency of 0.1 Hz (bottom left panel). The A7r5 cells were pre-incubated with vehicle (0.02% DMSO) for 10–60 min prior to establishing whole-cell patch; right panel, whole cell LCa tracings in the presence of the 1,4 dihydropyridine, nifedipine. The cells were pre-incubated with nifedipine (1 μM) for 15 min prior to the recording of LCa. B, Conductance – voltage curves of LTCC derived from peak Lca - voltage relationships in the presence of Gq/11 inhibitors. Prior to LCa current (Ica-L) recordings, the cells were incubated with vehicle (control), YM (1 μM), FR (1 μM), or WU (1 or 50 μM) for 10–60 min. For control and each compound, an average of 10 cells were recorded. All values are mean ± s.e.m. *P<0.05, vs. control.
Subcutaneous administration of FR and YM elicits marked and prolonged depressor response in normotensive mice
Signaling via Gq/11 class G proteins is key to maintaining normal blood pressure and is also involved in the development of hypertension25. Therefore, we determined the hemodynamic effects of systemically blocking Gq/11 with the cyclic depsipeptides, FR and YM, and the YM analogue WU in vivo in conscious mice. To this end, we used normotensive and hypertensive mice instrumented with radiotelemetry devices to monitor changes in conscious blood pressure and heart rate following acute subcutaneous injection of vehicle or a Gq/11 inhibitor ligand. A bolus administration of WU at 30 mg/kg had no effect on blood pressure or heart rate (data not shown). In contrast, a bolus administration of a medium- or high-dose YM or FR caused a rapid drop in blood pressure, reaching a nadir 1–2 hr after injection (Fig 5A & 5B). Whereas low-dose YM prevented the normal nighttime rise in SBP in normotensive mice, a similar dose of FR or vehicle had no effect. To determine the half-life of the depressor effect of FR and YM, we compared the rate of blood pressure return to baseline after injection of the doses of the inhibitors that produced similar decreases in SBP. As shown in Table 1 and Fig. 5C, SBP rise was much slower after injection of FR (t1/2 ~7 hr) compared to YM (t1/2 ~2 hr). Both compounds had minimal effect on heart rate. Only high-dose FR caused bradycardia, whereas both medium- and high-dose YM decreased heart rate (Fig 5D & 5E); however, the bradycardic effect of FR and YM were short-lived, as heart rate returned to baseline 1 hr after drug injection (Fig 5F). Both inhibitors had a dose-dependent depressor effect on SBP but not on heart rate (Fig 6A & 6B). In contrast to reflex tachycardia in response to a fall in blood pressure following systemic administration of Gq/11-coupled GPCR antagonists such as prazosin26, subcutaneous administration of FR and YM in conscious mice caused a fall in blood pressure accompanied by bradycardia, which was visible at 0.3 mg/kg of each inhibitor (Fig 6C & 6D). This result suggested that, in vivo, FR and YM alter arterial baroreflex control of heart rate.
Figure 5.

Summary of the acute effects of systemic administration of FR and YM on overnight systolic blood pressure (SBP) and heart rate (HR) of conscious normotensive mice. Each dose of the inhibitors was administered subcutaneously as a bolus at the same time (5 pm, indicated by the dashed line) of the day, after 3 hr of baseline recording. Hemodynamic parameters were continuously recorded for at least 24 hr. A, FR (n=7) elicits depressor responses only at medium and high doses. B, YM (n=7) elicits depressor responses at all doses. Note the blockade of nighttime rise in SBP by the lowest dose of YM. C, Comparison of the rate of SBP recovery from the nadir following the injection of FR and YM, using the same data as in A and B. SBP returns to control levels 7 and 20 hr after reaching the nadir following the injection of YM and FR, respectively. D, FR causes transient decrease in HR within 2 hr after injection of the highest dose. E, Both the medium and high dose of YM cause a rapid decrease in HR within 1 hr after injection. F, HR slowly returns to baseline after FR injection whereas it rapidly returns to control levels after YM injection. All values are mean ± s.e.m.
Table 1:
Overnight blood pressure response and recovery time in conscious normotensive or hypertensive mice given a bolus subcutaneous injection of the Gq/11 inhibitor, FR900359 or YM-254890.
| Treatment/Measurement | Baseline SBP (mmHg) | Change in SBP (mmHg) | SBP @ 50% recovery (mmHg) | Time to 50% recovery of SBP (hr) | Time to 100% recovery of SBP (hr) | |
|---|---|---|---|---|---|---|
| Normotension | FR900359 (0.3 mg/kg, s.c.) | 105 ± 3 | 37 ± 3 | 87 ± 2 | 6.9 ± 0.7** | 14.5 ± 1.7*** |
| YM-254890 (0.3 mg/kg, s.c.) | 108 ± 2 | 37 ± 3 | 87 ± 2 | 2.4 ± 0.2 | 5.7 ± 0.4 | |
| L-NAME Hypertension | FR900359 (0.3 mg/kg, s.c.) | 120 ± 1 | 49 ± 3 | 96 ± 1 | 10.0 ± 1.0** | 18.0 ± 0*** |
| YM-254890 (0.5 mg/kg, s.c.) | 121 ± 2 | 54 ± 2 | 94 ± 2 | 2.8 ± 0.2 | 6.3 ± 0.7 | |
Values are mean ± s.e.m. from experiments with 3-7 mice under each treatment condition. Statistical differences between paired values (i.e. FR900359 vs. YM-254890) under normotensive or hypertensive condition are indicated.
P<0.001
P<0.0002
SBP - systolic blood pressure; L-NAME - N(ω)-nitro-L-arginine methyl ester.
Figure 6.

Blood pressure and heart rate responses to systemic administration of Gq/11 inhibitors FR and YM in normotensive mice. A, B, maximal depressor response to increasing doses of FR and YM, respectively. Both inhibitors caused a dose-dependent decrease in SBP following a bolus subcutaneous (s.c.) injection in conscious mice. C, D, maximal bradycardic response to increasing doses of FR and YM, respectively. Only the highest dose of FR caused a significant decrease in HR, whereas the medium and high dose YM significantly decrease HR following a bolus s.c. injection. All values are mean ± s.e.m. **P<0.01 vs. control; ##P<0.01, 0.05 vs. 0.1 mg/kg of FR or YM; ++P<0.01, 0.1 vs. 0.3 mg/kg of FR.
FR and YM disrupt arterial baroreflex control of heart rate
To test the hypothesis that both LTCC activity and signaling via Gq/11 are involved in autonomic nervous system control of heart rate, we assessed heart rate control by the arterial baroreflex in the absence and presence of FR or YM. We induced reflex bradycardia and tachycardia by intravenous injection of increasing doses of the α1-adrenergic receptor agonist, phenylephrine (PE), and nitric oxide donor, sodium nitroprusside (SNP), respectively, in anesthetized mice. As shown in Figure 7A and7B, intravenous injection of FR or YM (0.1 mg/kg) caused a marked decrease in blood pressure accompanied by a precipitous drop in heart rate within 2 minutes of injection. Prior to the administration of FR or YM, injection of PE and SNP evoked a dose-dependent rise and fall in blood pressure, respectively (Figure 7C); this was accompanied by corresponding bradycardia and tachycardia, respectively (Figure 7D). However, the bradycardic response to PE was completely abolished after the administration of FR or YM. The tachycardic response to SNP was also abolished following the administration of FR or YM. Together these results indicated that FR and YM elicit inverse heart rate response to changes arterial blood pressure.
Figure 7.

Changes in mean arterial pressure (MAP) and heart rate (HR) in response to intravenous injection of FR900359 (FR) or YM-254890 (YM), or increasing doses of the alpha-adrenergic receptor agonist, phenylephrine (PE), and the nitric oxide donor, sodium nitroprusside (SNP) in the absence or presence of FR or YM in anesthetized mice. A, B, representative time-course tracings of changes in MAP and HR following a bolus administration (0.1 mg/kg, i.v.) of FR (blue tracings) or YM (orange tracings). The arrow indicates the time of drug injection. C, D, maximum changes in BP and HR in response to increasing doses (1.5, 3, 6, 12, 24 μg/kg, i.v.) of phenylephrine (PE) and sodium nitroprusside (SNP), respectively, before (closed squares) or after intravenous injection of 0.1 mg/kg FR (white triangles) or YM (inverted gray triangles). All values are mean ± s.e.m. **P< 0.01 vs. control
The duration of the depressor effect of FR increases in L-NAME hypertension mice
To determine the potential therapeutic effect of Gq/11 blockade with FR and YM, we repeated the acute blood pressure dose-response experiment in two mouse models of hypertension, L-NAME- and DOCA-salt-induced hypertension. The mice were made hypertensive using the chronic Nω-Nitro-L-arginine methyl ester (L-NAME)20 or DOCA-salt regimen as previously described25. In L-NAME hypertensive mice, FR administration elicited similar depressor effects as in normotensive state, though the onset of the blood pressure response was delayed (Fig 8A vs. 5A). Additionally, the blood pressure effects of YM were unchanged in hypertensive mice (Fig 8B vs. 5B), showing a similar half-life and duration of the depressor effects (Table 1). In contrast, the half-life of the effect of FR increased, with SBP returning to pre-injection level ~18 hr after injection of the highest dose (Fig 8C). Under L-NAME hypertension, both FR and YM produced minimal but short-lived bradycardic effect at the highest dose (Fig 8D – 8F). As in normotensive state, both inhibitors produced dose-dependent decreases in SBP; however, these depressor responses were not accompanied by bradycardia (supplemental Fig S2A – D).
Figure 8.

Summary of the acute effects of FR and YM on overnight systolic blood pressure (SBP) and heart rate of conscious hypertensive mice. The mice were made hypertensive by placing them on L-NAME in their drinking water (0.5 g/L) for 10 days. Each dose of FR or YM was administered subcutaneously as a bolus at the same time (5 pm, indicated by the dashed line) of the day, after 3 hr of baseline recording. Hemodynamic parameters were continuously recorded for at least 24 hr. A, B, both FR (n=4) and YM (n=4) elicited depressor responses only at medium and high doses. Note the lack of effect of the lowest dose of YM on nighttime SBP. C, a comparison of the rate of SBP recovery from the nadir following the injection of FR and YM, using the same data as in A and B. Note that a higher dose of YM was required to reduce SBP to the same nadir as 0.3 mg/kg of FR in hypertensive state. SBP returned to control levels 5 hr after YM injection; in contrast, SBP reached control levels 16 hr after FR injection. D, E, F, both FR and YM did not affect HR at any dose in hypertensive mice. All values are mean ± s.e.m.
Chronic administration of FR reverses DOCA-salt hypertension
To examine whether chronic blockade of Gq/11 was sufficient to reverse established hypertension, we performed daily administration of FR in mice that had been made hypertensive with DOCA and high-salt drinking water. We chose this hypertension model because abnormal Gq/11 activity in vascular smooth muscle has been shown to play a causal role in the pathogenesis of this model of salt-induced hypertension25. As shown in figure 8A, acute injection of FR produced a dose dependent depressor effect but did not affect nighttime increases in heart rate (Fig 9C). These effects of FR on SBP in DOCA-salt hypertensive mice were similar to those seen in L-NAME hypertensive mice (Fig. 8A vs. 9A). In a separate group of mice, we initiated daily subcutaneous administration of FR after DOCA-salt hypertension was fully established. Each injection was performed at 5 pm, i.e. just before the beginning of the nighttime rise in blood pressure and heart rate. Blood pressure and heart rate were assessed daily at 20 hr post injection. Systolic blood pressure at 23 hours post injection was similar to pre-DOCA-salt level and remained normalized throughout the drug injection period (Fig 9B, gray-shaded area). After FR injection was terminated, SBP began to rise gradually but remained relatively low compared to vehicle-treated mice. Despite lowering SBP, daily FR administration did not have any effect on heart rate (Fig 9D). Together these data indicated that chronic inhibition of Gq/11 by systemic administration of cyclic depsipeptide inhibitor ligands reverses salt hypertension.
Figure 9.

Effects of FR900359 on systolic blood pressure (SBP) and heart rate (HR) of conscious, deoxycorticosterone acetate (DOCA)-salt hypertensive male and female mice. A, C, 24-hour SBP (A) and HR (C) responses to a single injection of FR (0.1 or 0.3 mg/kg, s.c.). FR was administered subcutaneously as a bolus at the same time (5 pm, indicated by the dashed line) of the day, after 3 hr of baseline recording.
Hemodynamic parameters were continuously recorded for at least 24 hr. B, D, SBP and HR levels before and after DOCA implantation, during daily injection (shaded area) of vehicle (0.01% DMSO in 0.9% saline) or FR (0.3 mg/kg, s.c.), and during post-injection period. All values are mean ± s.e.m. *,**P<0.05, 0,01 vs. control.
Discussion
G proteins, most notably members of the Gq/11 class, are ubiquitously expressed in mammalian organ systems. It is well established that aberrant GPCR activity via Gq/11 signaling is involved in the pathogenesis of hypertension. However, the effects or consequences of post-receptor pharmacological blockade of Gq/11 on cardiovascular function, and importantly the maintenance of blood pressure homeostasis, are unclear. Using natural cyclic depsipeptides and a synthetic analogue that inhibit Gq/11 with relatively high specificity in vitro, in combination with experimental models of human hypertension, we have defined the effects of acute and chronic systemic blockade of Gq/11 on blood pressure and heart rate homeostasis under normotensive and hypertensive conditions. Most importantly, we have shown that chronic administration of FR is sufficient to quickly and effectively reduce blood pressure to normal levels in established salt-hypertension, a commonly diagnosed form of primary human hypertension27. When administered subcutaneously, FR and YM elicit transient bradycardia and a prolonged depressor response. We have found that the blood pressure lowering effect of FR and YM are mediated, largely, by mechanisms involving the inhibition of Gq/11- and LTCC-mediated vasoconstriction of resistance arteries. Altogether, our findings provide direct evidence for dual pharmacological targeting of Gq/11 and LTCC as a potential anti-hypertensive strategy. Moreover, we have established a dosing paradigm for future studies that employ YM and FR as tool compounds to investigate the role of Gq/11 and LTCC in various cardiovascular disease models.
Receptors and ion channels that mediate changes in intracellular Ca2+ fluxes play key roles in the pathogenesis of hypertension. A rise in intracellular Ca2+ concentration is a primary factor for vascular smooth muscle contraction and also for the production of endothelium-derived relaxing factors such as nitric oxide and EDHF28, 29. In the vascular smooth muscle, two processes that mediate intracellular Ca2+ rise are the release from the sarcoplasmic reticulum (SR) and influx through the LTCC. Calcium release from the SR is mediated by the activation of the GPCR-Gq/11-PLC signaling pathway leading to IP3 generation and activation of SR IP3 receptors30. On the other hand, extracellular calcium influx in vascular smooth muscle cells is mediated mainly by the opening of voltage-gated LTCC following membrane depolarization31, 32. Accordingly, blocking GPCRs and LTCC is a major therapeutic strategy and have proven to be effective medications, separately, for managing human hypertension8, 33. However, the use of a single GPCR antagonist may not be sufficient to manage established hypertension. This could be due partly to post-receptor adaptive mechanisms that enable G proteins to pair with many receptor types and function in concert, in vivo, to impinge on a physiological process such as vascular tone regulation and renal control of electrolyte balance. By interacting directly with Gq/11 and locking the G protein in the inactive, GDP-bound state, the natural cyclic depsipeptides FR and YM and their synthetic analogs can be very effective anti-hypertensive agents by reducing LTCC- and Gq/11-mediated intracellular Ca2+ rise in vascular smooth muscle, thereby promoting vasodilation. Indeed, our results show that systemic administration of FR can normalize blood pressure even after hypertension is well established in a mouse model of salt-hypertension.
Findings from this study indicate that the mechanisms mediating the anti-hypertensive effects of cyclic depsipeptides are more complex than previously reported. In addition to the established mechanism whereby YM, FR, and related synthetic analogs reduce vasoconstriction and blood pressure by locking the Gα subunit in the inactive, GDP-bound state10, 11, 22, other studies have suggested that FR reduces vasoconstriction through the inhibition of LTCC in the vasculature12. Our study provides the first line of evidence that Gq/11 inhibitor ligands block vasoconstriction, at least partly, by directly inhibiting LTCC in vascular smooth muscle cells. The bacterial Gq/11 inhibitor ligand, YM and its synthetic analog, WU, block vasoconstriction in part by directly inhibiting calcium influx through the LTCC. Although the plant derived Gq/11 inhibitor, FR, failed to block LTCC-induced vasoconstriction, it showed inhibitory effect on LTCC when applied directly to vascular smooth muscle cells. This discrepancy in the inhibitory effects of FR could be due to high selectivity/preference for Gq/11 relative to LTCC in the intact tissue. The exact binding mechanism that mediates the inhibition of mammalian LTCC by Gq/11 inhibitors is unknown. However, results from our docking experiments, using x-ray crystal structure of the bacterial ortholog, CavAb, gives a good insight into the expected structural basis for the inhibitory effect of depsipeptides and their synthetic analogs on LTCC. Similar to the binding pocket in Gq/11 subunit, the binding of the depsipeptides to LTCC is facilitated by key residues in the dihydropyridine binding site that form hydrophobic contacts with the phenyl ring of the inhibitors. It is noteworthy that WU interacts with CavAb in an inverted pose. This difference could be due to the replacement of the depsipeptide core with a simplified aliphatic ring, which is known to decrease the potency of WU as an inhibitor of Gq/1115.
The blockade of LTCC by cyclic depsipeptides may promote vasodilation and blood pressure decrease by several mechanisms. First, and as previously mentioned, by inhibiting the activity of LTCC, these cyclic depsipeptides and related analogs could prevent the influx of extracellular Ca2+, thereby preventing the rise in intracellular Ca2+ concentration necessary for the activation of the contractile signaling cascade in the vascular smooth muscle. Indeed, we found that YM inhibited potassium-induced vasoconstriction of mesenteric arteries. Second, cyclic depsipeptides that inhibit LTCC could reduce blood pressure by decreasing cardiac output, which is a primary determinant of blood pressure. Because the activity of LTCC is critical to calcium-induced calcium release in cardiomyocytes24, LTCC inhibitors such as YM could reduce cardiac output by decreasing cardiac chronotropic and ionotropic responses to acute changes in blood pressure. Such an effect may underlie the reversal of reflex tachycardia leading to a bradycardic response to acute fall in blood pressure induced with FR or YM.
Non-vascular mechanisms by which Gq/11 inhibition by cyclic depsipeptides reduce blood pressure were not assessed here but need to be examined in detail because of the ubiquitous role of this G protein class in various organ systems that are key to the maintenance of blood pressure homeostasis. Of particular note, sodium reabsorption by the renal tubular system is the primary determinant of extracellular fluid volume and thus cardiac output and blood pressure. This function of the kidney depends largely on the expression and activity of key sodium transporters including Na+/H+ exchangers, Na+/K+ATPase, Na+-Cl− co-transporter, Na+-K+−2Cl− co-transporter, and epithelial Na+ channel (ENaC)34. Several of these sodium transporters are regulated by signaling via Gq/11 class G proteins. For instance, Gq/11-mediated rise in intracellular Ca2+ concentration plays a key role in the insertion of ENaC in the luminal membrane of principal cells of the distal tubule, which is critical for sodium reabsorption at the distal tubule and collecting duct35. In addition, sodium reabsorption via ENaC at the luminal membrane of principal cells is tightly regulated by the ATP/UTP/P2Y2/4 receptor system that couples to the Gq/11 signaling pathway36. Previous studies have established that activation of PLC by Gq/11 upon P2Y2/4 receptor stimulation negatively regulates the open probability (Po) of ENaC to inhibit sodium reabsorption36–38. Accordingly, P2Y2−/− mice have high ENaC Po and are hypertensive38. With the availability of these cyclic depsipeptide inhibitors, a more in-depth understanding of the role that Gq/11 plays in long-term blood pressure control through the regulation of renal epithelial sodium handling by ATP/UTP/P2Y2/4 receptor and other receptor systems that couple to this G protein class can be pursued.
In summary, our study comprehensively reveals the hemodynamic effects of systemic Gq/11 inhibition in normotensive and hypertensive states and is the first in-depth assessment of the potential anti-hypertensive effects of targeting heterotrimeric G proteins. This study also establishes a paradigm for using FR and YM as tool compounds to investigate the pathogenesis and progression of cardiovascular and other diseases such as chronic kidney disease, cardiac arrhythmias, and various forms cancers that involve aberrant activity of Gq/11 proteins and LTCC.
Supplementary Material
Acknowledgements
We thank Dr. Kendall Blumer of Washington University School of Medicine for his generous gift of FR900359. We also thank all members of the Osei-Owusu lab for technical assistance and for comments on the manuscript.
Funding: This study was supported by grants from Pennsylvania Department of Health (CURE), Margaret Q. Landenberger Foundation, American Heart Association (16SDG27260276) and NIH - NHLBI (HL139754) to P. Osei-Owusu, and NIH - MH106912 to O.V. Mortensen. Dr. Shaili Aggarwal is supported by Brody postdoctoral fellowship from the Philadelphia Foundation.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Chemical Compounds
Chemical compounds studied in this article: FR900359 (PubChem CID: 14101198); YM-254890 (PubChem CID: 9919454); WU-07047 (PMID: 25875152); Nifedipine (PubChem CID: 63011); Phenylephrine (PubChem CID: 5284443); Endothelin-1 (PubChem CID: 16212950); U46619 (PubChem CID: 5311493); Arginine vasopressin (PubChem CID: 644077); Bay K8644 (PubChem CID: 2303
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Guyton AC. Blood pressure control--special role of the kidneys and body fluids. Science. 1991;252:1813–6. [DOI] [PubMed] [Google Scholar]
- 2.Hall JE, Granger JP, do Carmo JM, da Silva AA, Dubinion J, George E, Hamza S, Speed J and Hall ME. Hypertension: physiology and pathophysiology. Comprehensive Physiology. 2012;2:2393–442. [DOI] [PubMed] [Google Scholar]
- 3.Kim HR, Appel S, Vetterkind S, Gangopadhyay SS and Morgan KG. Smooth muscle signalling pathways in health and disease. J Cell Mol Med. 2008;12:2165–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hill MA, Zou H, Potocnik SJ, Meininger GA and Davis MJ. Invited review: arteriolar smooth muscle mechanotransduction: Ca(2+) signaling pathways underlying myogenic reactivity. J Appl Physiol (1985). 2001;91:973–83. [DOI] [PubMed] [Google Scholar]
- 5.Bova S, Goldman WF, Yauan XJ and Blaustein MP. Influence of Na+ gradient on Ca2+ transients and contraction in vascular smooth muscle. The American journal of physiology. 1990;259:H409–23. [DOI] [PubMed] [Google Scholar]
- 6.Busse R, Luckhoff A and Mulsch A. Cellular mechanisms controlling EDRF/NO formation in endothelial cells. Basic Res Cardiol. 1991;86 Suppl 2:7–16. [DOI] [PubMed] [Google Scholar]
- 7.Zheng XF, Kwan CY and Daniel EE. Role of intracellular Ca2+ in EDRF release in rat aorta. J Vasc Res. 1994;31:18–24. [DOI] [PubMed] [Google Scholar]
- 8.Bravo EL. Rational drug therapy based on understanding the pathophysiology of hypertension. Cleve Clin J Med. 1989;56:362–8. [DOI] [PubMed] [Google Scholar]
- 9.Takasaki J, Saito T, Taniguchi M, Kawasaki T, Moritani Y, Hayashi K and Kobori M. A novel Galphaq/11-selective inhibitor. The Journal of biological chemistry. 2004;279:47438–45. [DOI] [PubMed] [Google Scholar]
- 10.Taniguchi M, Suzumura K, Nagai K, Kawasaki T, Takasaki J, Sekiguchi M, Moritani Y, Saito T, Hayashi K, Fujita S, Tsukamoto S and Suzuki K. YM-254890 analogues, novel cyclic depsipeptides with Galpha(q/11) inhibitory activity from Chromobacterium sp. QS3666. Bioorg Med Chem. 2004;12:3125–33. [DOI] [PubMed] [Google Scholar]
- 11.Schrage R, Schmitz AL, Gaffal E, Annala S, Kehraus S, Wenzel D, Bullesbach KM, Bald T, Inoue A, Shinjo Y, Galandrin S, Shridhar N, Hesse M, Grundmann M, Merten N, Charpentier TH, Martz M, Butcher AJ, Slodczyk T, Armando S, Effern M, Namkung Y, Jenkins L, Horn V, Stossel A, Dargatz H, Tietze D, Imhof D, Gales C, Drewke C, Muller CE, Holzel M, Milligan G, Tobin AB, Gomeza J, Dohlman HG, Sondek J, Harden TK, Bouvier M, Laporte SA, Aoki J, Fleischmann BK, Mohr K, Konig GM, Tuting T and Kostenis E. The experimental power of FR900359 to study Gq-regulated biological processes. Nat Commun. 2015;6:10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zaima K, Deguchi J, Matsuno Y, Kaneda T, Hirasawa Y and Morita H. Vasorelaxant effect of FR900359 from Ardisia crenata on rat aortic artery. J Nat Med. 2013;67:196–201. [DOI] [PubMed] [Google Scholar]
- 13.Taniguchi M, Nagai K, Arao N, Kawasaki T, Saito T, Moritani Y, Takasaki J, Hayashi K, Fujita S, Suzuki K and Tsukamoto S. YM-254890, a novel platelet aggregation inhibitor produced by Chromobacterium sp. QS3666. J Antibiot (Tokyo). 2003;56:358–63. [DOI] [PubMed] [Google Scholar]
- 14.Matthey M, Roberts R, Seidinger A, Simon A, Schroder R, Kuschak M, Annala S, Konig GM, Muller CE, Hall IP, Kostenis E, Fleischmann BK and Wenzel D. Targeted inhibition of Gq signaling induces airway relaxation in mouse models of asthma. Sci Transl Med. 2017;9. [DOI] [PubMed] [Google Scholar]
- 15.Rensing DT, Uppal S, Blumer KJ and Moeller KD. Toward the Selective Inhibition of G Proteins: Total Synthesis of a Simplified YM-254890 Analog. Org Lett. 2015;17:2270–3. [DOI] [PubMed] [Google Scholar]
- 16.Osei-Owusu P, Knutsen RH, Kozel BA, Dietrich HH, Blumer KJ and Mecham RP. Altered reactivity of resistance vasculature contributes to hypertension in elastin insufficiency. American journal of physiology Heart and circulatory physiology. 2014;306:H654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osei-Owusu P, Sabharwal R, Kaltenbronn KM, Rhee MH, Chapleau MW, Dietrich HH and Blumer KJ. Regulator of G protein signaling 2 deficiency causes endothelial dysfunction and impaired endothelium-derived hyperpolarizing factor-mediated relaxation by dysregulating Gi/o signaling. The Journal of biological chemistry. 2012;287:12541–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gross V, Lippoldt A, Yagil C, Yagil Y and Luft FC. Pressure natriuresis in salt-sensitive and salt-resistant Sabra rats. Hypertension. 1997;29:1252–9. [DOI] [PubMed] [Google Scholar]
- 19.Holobotovskyy V, Manzur M, Tare M, Burchell J, Bolitho E, Viola H, Hool LC, Arnolda LF, McKitrick DJ and Ganss R. Regulator of G-protein signaling 5 controls blood pressure homeostasis and vessel wall remodeling. Circulation research. 2013;112:781–91. [DOI] [PubMed] [Google Scholar]
- 20.Obst M, Tank J, Plehm R, Blumer KJ, Diedrich A, Jordan J, Luft FC and Gross V. NO-dependent blood pressure regulation in RGS2-deficient mice. American journal of physiology Regulatory, integrative and comparative physiology. 2006;290:R1012–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zamponi GW, Striessnig J, Koschak A and Dolphin AC. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacological reviews. 2015;67:821–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishimura A, Kitano K, Takasaki J, Taniguchi M, Mizuno N, Tago K, Hakoshima T and Itoh H. Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang L, Gamal El-Din TM, Swanson TM, Pryde DC, Scheuer T, Zheng N and Catterall WA. Structural basis for inhibition of a voltage-gated Ca(2+) channel by Ca(2+) antagonist drugs. Nature. 2016;537:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu L, Li D, Tao L, Yang Y, Li Y and Hou T. Binding mechanisms of 1,4-dihydropyridine derivatives to L-type calcium channel Cav1.2: a molecular modeling study. Mol Biosyst. 2016;12:379–90. [DOI] [PubMed] [Google Scholar]
- 25.Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS and Offermanns S. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nature medicine. 2008;14:64–8. [DOI] [PubMed] [Google Scholar]
- 26.Atkinson J, Luthi P, Sonnay M and Boillat N. Effect of acute administration of prazosin on blood pressure, heart rate and plasma renin level in the conscious normotensive rat. Clinical and experimental pharmacology & physiology. 1986;13:535–41. [DOI] [PubMed] [Google Scholar]
- 27.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr,, Jones DW, Materson BJ, Oparil S, Wright JT Jr, Roccella EJ, National Heart L, Blood Institute Joint National Committee on Prevention DE, Treatment of High Blood P and National High Blood Pressure Education Program Coordinating C. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. Jama. 2003;289:2560–72. [DOI] [PubMed] [Google Scholar]
- 28.Feletou M and Vanhoutte PM. The alternative: EDHF. Journal of molecular and cellular cardiology. 1999;31:15–22. [DOI] [PubMed] [Google Scholar]
- 29.Jeremy JY, Rowe D, Emsley AM and Newby AC. Nitric oxide and the proliferation of vascular smooth muscle cells. Cardiovascular research. 1999;43:580–94. [DOI] [PubMed] [Google Scholar]
- 30.Fill M Mechanisms that turn-off intracellular calcium release channels. Front Biosci. 2003;8:d46–54. [DOI] [PubMed] [Google Scholar]
- 31.Navedo MF, Amberg GC, Votaw VS and Santana LF. Constitutively active L-type Ca2+ channels. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:11112–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santana LF, Navedo MF, Amberg GC, Nieves-Cintron M, Votaw VS and Ufret-Vincenty CA. Calcium sparklets in arterial smooth muscle. Clinical and experimental pharmacology & physiology. 2008;35:1121–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meggs LG and Kodali P. Emerging concepts in antihypertensive therapy: the benefits of angiotensin II blockade. J Assoc Acad Minor Phys. 1999;10:34–43. [PubMed] [Google Scholar]
- 34.Coffman TM. The inextricable role of the kidney in hypertension. The Journal of clinical investigation. 2014;124:2341–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wildman SS, Marks J, Turner CM, Yew-Booth L, Peppiatt-Wildman CM, King BF, Shirley DG, Wang W and Unwin RJ. Sodium-dependent regulation of renal amiloride-sensitive currents by apical P2 receptors. Journal of the American Society of Nephrology : JASN. 2008;19:731–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vallon V and Rieg T. Regulation of renal NaCl and water transport by the ATP/UTP/P2Y2 receptor system. American journal of physiology Renal physiology. 2011;301:F463–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pochynyuk O, Rieg T, Bugaj V, Schroth J, Fridman A, Boss GR, Insel PA, Stockand JD and Vallon V. Dietary Na+ inhibits the open probability of the epithelial sodium channel in the kidney by enhancing apical P2Y2-receptor tone. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24:2056–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rieg T, Bundey RA, Chen Y, Deschenes G, Junger W, Insel PA and Vallon V. Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na+ and water reabsorption. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21:3717–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.