

Abstract
Ligand-dependent oligomerization of receptor tyrosine kinases (RTKs) results in their activation through highly specific conformational changes in the extracellular and intracellular receptor domains. These conformational changes are unique for each RTK sub-family, limiting cross-activation between unrelated RTKs. The proto-oncogene MET receptor tyrosine kinase overcomes these structural constraints and phosphorylates unrelated RTKs in numerous cancer cell lines. The molecular basis for these interactions is unknown. We investigated the mechanism by which MET phosphorylates the human epidermal growth factor receptor-3 (HER3 or ERBB3), a catalytically impaired RTK whose phosphorylation by MET has been described as an essential component of drug resistance to inhibitors targeting EGFR and HER2. We find that in untransformed cells, HER3 is not phosphorylated by MET in response to ligand stimulation, but rather to increasing levels of MET expression, which results in MET activation in a ligand-independent manner. Phosphorylation of HER3 by its canonical dimerization partners, EGFR and HER2, is achieved by engaging an allosteric site on the HER3 kinase domain, but this site is not required when HER3 is phosphorylated by MET. We also observe that HER3 preferentially interacts with MET during its maturation along the secretory pathway, before MET is post-translationally processed by cleavage within its extracellular domain. This results in accumulation of phosphorylated HER3 in the Golgi apparatus. We further show that in addition to HER3, MET phosphorylates other RTKs in the Golgi, suggesting that this mechanism is not limited to HER3 phosphorylation. These data demonstrate a link between MET overexpression and its aberrant activation in the Golgi endomembranes and suggest that non-canonical interactions between MET and unrelated RTKs occur during maturation of receptors. Our study highlights a novel aspect of MET signaling in cancer that would not be accessible to inhibition by therapeutic antibodies.
Keywords: MET, HER3, ERBB3, RTK phosphorylation, Golgi, non-canonical mechanism
INTRODUCTION
Under homeostatic conditions the hepatocyte growth factor (HGF) receptor (HGFR/MET) becomes activated by binding to an extracellular ligand, HGF, which induces receptor dimerization, activation of the intracellular tyrosine kinase domains and subsequent receptor phosphorylation.1–5 The phosphorylated receptor sites then efficiently recruit downstream signaling adaptors to activate signaling cascades that modulate epithelial cell motility and invasion, formation of branched tubules, and cell growth and survival.6–9 In cancer, aberrant signaling by the MET receptor is primarily achieved by overexpression or amplification of genes encoding MET or its ligand, and is associated with tumorigenesis, metastasis, and poor prognosis.10–17 Hyper-activated MET phosphorylates other RTKs, particularly the EGFR/HER family, often as a mechanism of resistance to targeted therapies.
Phosphorylation of one HER receptor, the catalytically impaired HER3 pseudokinase, has been described as an important mechanism of drug resistance.18–21 Under normal conditions, HER3 is phosphorylated by EGFR or HER2, and potently stimulates cell survival through the Akt signaling pathway by direct recruitment of PI3K.22, 23 In lung cancer cells with an activating EGFR mutation and acquired resistance to EGFR inhibitors, MET amplification can restore HER3 phosphorylation and downstream signaling through the PI3K/Akt pathway.18 In numerous other cancer cells lines in which MET is overexpressed, HER3 becomes phosphorylated in a MET-dependent manner19, 24–27 and was shown to interact with MET by co-immunoprecipitation.24, 25, 28 Thus, the ability of MET to phosphorylate HER3 under conditions of overexpression is a well-established phenomenon, however the molecular basis for this non-canonical cross-phosphorylation between RTKs is not understood.
While the mechanisms for activation and phosphorylation remain poorly defined for many RTKs, structural studies on receptors such as EGFR29–33 and the insulin receptor (IR) family34–37 have revealed unique protein-protein interactions that are required to trigger kinase activity. These interactions, promoted by binding of extracellular ligands, are unique for each subfamily of RTKs, but in cancers in which MET efficiently phosphorylates other RTKs, these specific mechanisms no longer seem to apply. At present, it is unknown whether the promiscuity with which MET phosphorylates other RTKs reflects its inherent ability to interact directly with these receptors, or if it is only a consequence of MET overexpression. It is also unclear whether these non-canonical kinase-substrate relationships are mediated by tractable protein-protein interactions that could be explored therapeutically in cancer.
We set out to understand the mechanism of how overexpression of MET leads to phosphorylation of new substrate RTKs by focusing on MET-dependent phosphorylation of HER3. We show that HER3 is a substrate for MET only under conditions of MET overexpression, and that under these circumstances MET phosphorylates HER3 in a ligand-independent manner. HER3 phosphorylation by MET is also independent from its allosteric activator interface which is crucial for HER3 phosphorylation by other HER receptors. Surprisingly, we found that HER3 almost exclusively interacts with and is phosphorylated by MET in endomembranes, primarily the Golgi apparatus, where overexpressed MET accumulates during biosynthesis. Based on these findings, we propose that in MET-amplified cells over-crowding of MET molecules in the secretory pathway facilitates its unspecific interactions with other RTKs, resulting in their premature phosphorylation.
RESULTS
Overexpression of MET drives HER3 phosphorylation independently of EGFR and HER2
To investigate the mechanism by which MET phosphorylates HER3, we used COS7 cells, which do not express detectable levels of endogenous HER3. When HER3 was transfected with its co-receptor HER2 in COS7 cells, HER3 and HER2 phosphorylation could be detected only in the presence of HER3 ligand neuregulin (NRG) (Fig. 1a). In contrast, when HER3 was co-expressed with MET, it was phosphorylated independently of NRG stimulation (Fig. 1a). Under these conditions, MET was also phosphorylated in COS7 cells in a ligand-independent manner (Fig. 1a). To confirm that HER3 phosphorylation is dependent on MET activity, we treated COS7 cells expressing MET and HER3 with a panel of MET inhibitors and found they consistently blocked phosphorylation of both HER3 and MET (Fig. 1b).
Fig. 1.
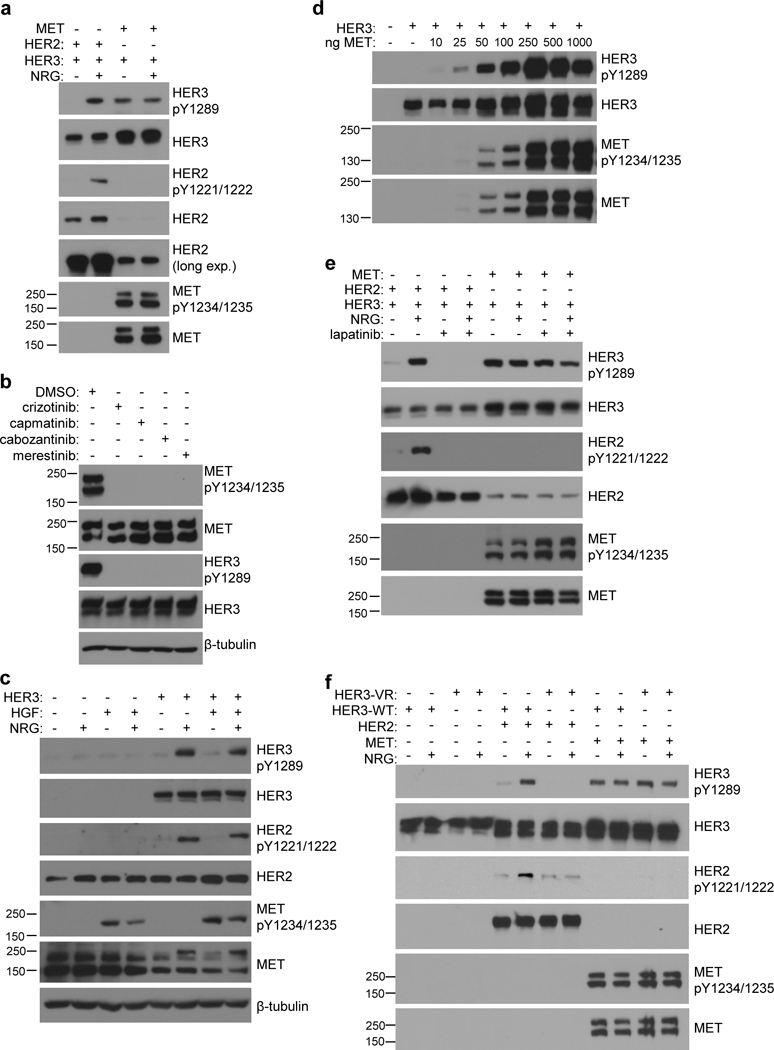
Overexpression of MET drives HER3 phosphorylation independently of EGFR and HER2. (a) COS7 cells expressing either MET or HER2 with HER3 were stimulated −/+ NRG (50ng/mL) and assayed for HER3 phosphorylation by western blot. (b) COS7 cells expressing MET and HER3 were treated with DMSO, or MET inhibitor (1μM crizotinib, 100nM capmatinib, 100nM cabozantinib, or 1μM merestinib) for 6 hours and assayed for MET and HER3 phosphorylation by western blot. (c) COS7 cells transfected with onlyHER3 were stimulated with either NRG (50ng/mL), HGF (50ng/mL), or both, and assayed for HER3 phosphorylation and endogenous levels of MET and HER2 by western blot. (d) COS7 cells were transiently transfected with HER3 and increasing amounts of MET and assayed for HER3 phosphorylation and MET expression by western blot. (e) COS7 cells expressing either HER3 + HER2 or HER3 + MET were treated −/+ NRG (50ng/mL) and −/+ 3μM lapatinib and assayed for HER3 phosphorylation by western blot. (f) COS7 cells expressing either wild-type (WT) or V926R mutant (VR) HER3 together with HER2 or MET were stimulated −/+ NRG (50ng/mL) and assayed for HER3 phosphorylation.
To test if overexpression of MET is necessary for mediating HER3 phosphorylation, we assessed levels of phosphorylated HER3 in COS7 cells as a function of activation of the endogenous MET by HGF. HER3 phosphorylation was not significantly induced by HGF stimulation over the basal level, despite robust activation of endogenous MET (Fig. 1c). We further observed that the extent of HER3 phosphorylation increased linearly with the levels of overexpressed, hyper-activated MET (Fig. 1d). These data suggest that HGF-dependent activation of MET is insufficient to stimulate HER3 phosphorylation, and that MET-dependent HER3 phosphorylation only occurs under conditions of MET overexpression.
Since MET has been reported to interact with other members of the EGFR/HER family24–26, 38 we looked at whether MET-dependent HER3 phosphorylation is a result of cross-activation of EGFR and/or HER2 by MET. We used lapatinib, a highly selective EGFR and HER2 kinase inhibitor. Lapatinib treatment eliminated HER2-dependent HER3 phosphorylation induced by NRG stimulation, but had no effect on HER3 phosphorylation that results from MET overexpression (Fig. 1e). We also examined phosphorylation of the HER3 variant with a mutation in the allosteric site within the kinase domain (V926R). This variant is unable to form functional dimers with other HER receptors and abolishes NRG-dependent HER3 phosphorylation (Fig. 1f).39 Using this mutant, we can test not only whether MET-dependent phosphorylation of HER3 depends on formation of active complexes between HER3 and EGFR or HER2, but also whether HER3 forms structurally similar complexes with MET. As shown in Fig. 1e, the HER3 V926R mutant loses the ability to be phosphorylated by HER2 in a NRG-dependent manner, but is phosphorylated by MET to the same extent as wild-type HER3. Together these results demonstrate that MET overexpression induces HER3 phosphorylation independently from activation of other HER receptors and does not engage the allosteric site of HER3.
HER3 interacts specifically with an intracellular pool of MET
To investigate if MET and HER3 interact under conditions of MET-dependent phosphorylation of HER3 in COS7 cells, we immunoprecipitated FLAG-tagged constructs of MET or HER3, transiently co-expressed in COS7 cells with un-tagged versions of HER3 or MET, respectively. As shown in Fig. 2a and 2b, FLAG-tagged MET co-immunoprecipitated with un-tagged HER3. Likewise, FLAG-tagged HER3 co-immunoprecipitated with un-tagged MET. These results are consistent with previous observations that MET and HER3 interact in lung cancer cells in which MET is amplified.18, 38 This interaction was not significantly affected by capmatinib treatment, despite full inhibition of MET and HER3 phosphorylation (Supplementary Fig. 1).
Fig. 2.

HER3 interacts specifically with an intracellular pool of MET. (a) COS7 cells expressing MET and FLAG-tagged HER3 were immunoprecipitated with anti-FLAG antibody and assayed for HER3 and MET by western blot. (b) COS7 cells expressing HER3 and FLAG-tagged MET were immunoprecipitated with anti-FLAG antibody and assayed for HER3 and MET by western blot. (c) Schematic of cleaved and uncleaved MET protein. COS7 cells expressing MET and FLAG-tagged HER3 were immunoprecipitated for anti-FLAG and assayed for MET by western blot. (d) Membrane and intracellular fractions of COS7 cells expressing MET were isolated by surface biotinylation followed by pull-down with Neutravidin-agarose beads and assayed by western blot.
The MET receptor is typically resolved as a double band on SDS/PAGE due to a post-translational modification that involves cleavage within the extracellular domain of MET.2 This results in an α chain (50 kDa) and β chain (145 kDa) which remain bound together by disulfide linkages.40, 41 When these bonds are broken under reducing SDS/PAGE conditions, MET is typically detected as a doublet composed of the immature uncleaved form (170 kDa) and the β chain (145 kDa) (Fig. 2c).
Our analysis of the co-immunoprecipitates between MET and HER3 led to a consistent observation that HER3 almost exclusively pulls down the upper band of the MET doublet, corresponding to the uncleaved form of MET (Fig. 2c). MET is cleaved by furin or a furin-like protease in the trans-Golgi network during receptor anterograde trafficking to the plasma membrane.42 Uncleaved MET is therefore expected to be located primarily in the endomembranes. We separated the plasma membrane and intracellular pools of MET receptor by biotin-labeling surface proteins in COS7 cells expressing MET, and isolating them from intracellular proteins by affinity purification with Neutravidin-agarose beads. As expected, only mature MET (detected as 145 kDa β chain) is labeled with biotin, whereas the uncleaved form of MET, which we find to interact with HER3, is predominantly protected from labeling (Fig. 2d). This analysis indicates that under conditions of MET overexpression, the interaction between MET and HER3 does not occur at the plasma membrane but rather in the intracellular membranes.
HER3 and MET co-localize in the Golgi under conditions of MET overexpression
To better understand the spatial determinants of the HER3/MET interaction and resulting HER3 phosphorylation, we used immunofluorescence to determine the localization of phosphorylated HER3 in COS7 cells under conditions of MET overexpression. In a control experiment, we examined the localization of phosphorylated HER3 in cells that were co-transfected with HER2, serum-starved, and stimulated with NRG. Under these conditions, in more than 80% of cells HER3 phosphorylation could be detected exclusively at the cell periphery, indicative of localization at the plasma membrane (Fig. 3a). In contrast, in over 80% of cells co-expressing MET and HER3, HER3 phosphorylation was not detected at the cell periphery, but instead within a perinuclear compartment (Fig. 3a). This redistribution of phosphorylated HER3 did not reflect changes in localization of the total HER3 receptor, which was localized both at the cell periphery as well as in the perinuclear compartment under all conditions (Fig. 3a). Notably, in contrast to HER3 and HER2, the overexpressed MET receptor was predominantly detected in the perinuclear compartment (Fig. 3a).
Fig. 3.
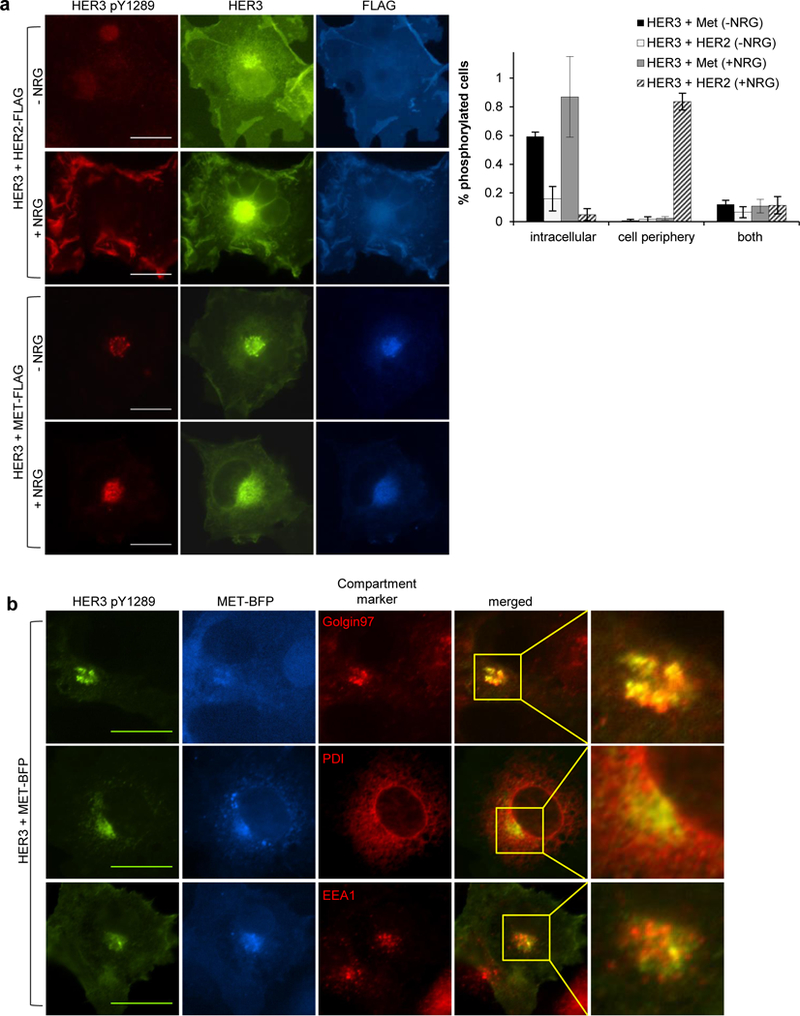
HER3 and MET co-localize in the Golgi under conditions of MET overexpression. (a) COS7 cells expressing HER2-FLAG or MET-FLAG together with HER3-GFP were fixed and stained with anti-FLAG, and anti-phospho-HER3 to visualize HER3 phosphorylation. Localization of HER3 phosphorylation was scored as either membrane, intracellular, or both membrane and intracellular, for 119 cells in the HER3 + MET (-NRG) treatment, 91 cells in the HER3 + MET (+NRG) treatment, 165 cells in the HER3 + HER2 (-NRG) treatment, and 157 cells in the HER3 + HER2 (+NRG) treatment, pooled from a minimum of three experiments and presented as percent of total cells ± SEM. (b) COS7 cells expressing HER3 and MET-BFP were fixed and stained for phospho-HER3 and PDI (ER), EEA1 (early endosome), or Golgin97 (Golgi) markers. Scale bars are 50μm.
To characterize the perinuclear compartment where MET and phosphorylated HER3 are localized, we co-stained COS7 cells expressing MET and HER3 with a set of endomembrane compartment markers, Golgin-97 (Golgi), PDI (endoplasmic reticulum), and EEA1 (early endosomes). The MET and phospho-HER3 signals overlaid most extensively with Golgin-97, with detectable but much less pronounced overlay with PDI and EEA1, demonstrating that a significant pool of phosphorylated HER3 co-localizes with MET in the Golgi (Fig. 3b). These data are consistent with the results of the co-immunoprecipitation and surface biotinylation/fractionation analyses and show that under conditions of MET overexpression, HER3 predominantly interacts with MET in Golgi endomembranes.
HER3 phosphorylation in cancer cells with MET amplification is ligand-independent
To relate our analysis of HER3/MET interactions in the COS7 cell model system of MET overexpression to a physiological scenario of MET amplification, we used a hepatocellular carcinoma cell line, MHCC97-H, in which genomic MET is amplified approximately 15-fold.43, 44 MET protein levels in these cells are ~ 2 fold higher than overexpression of MET that is achieved by transient transfection of MET in our COS7 cell model system (Fig. 1e and Supplementary Fig. 2). In MHCC97-H cells, MET phosphorylation was constitutive, was not further stimulated upon addition of HGF, and was eliminated upon treatment with a MET inhibitor, capmatinib (Fig. 4a). Notably, HER3 was constitutively phosphorylated, and its phosphorylation was significantly diminished by inhibition of MET with capmatinib (Fig. 4b). In contrast, lapatinib treatment resulted only in a minor decrease in HER3 phosphorylation (Fig. 4b). To verify that the effect of capmatinib was not due to off-target inhibition of EGFR-family receptors (primarily EGFR in these cells), we treated MHCC97-H cells with a panel of MET inhibitors and stimulated with EGF or NRG. All MET inhibitors suppressed basal phosphorylation of MET, EGFR, and HER3, but did not affect EGF- or NRG-induced phosphorylation of EGFR and HER3 (Supplementary Fig. 3). These data demonstrate that constitutive phosphorylation of HER3 in MHCC97-H cells is primarily a result of MET activity and that the activity of the canonical HER3 co-receptors, HER2 and EGFR, does not play a major role. To our knowledge, this is the first demonstration of a kinase/substrate relationship between MET and HER3 in cells in which MET amplification has not originated as an acquired mechanism of resistance to EGFR or HER2 inhibitors. Our results strengthen the hypothesis that HER3 phosphorylation is a direct consequence of MET overexpression, and can also occur in cancer cells with no documented dependence on EGFR/HER2 receptor signaling for growth.
Fig. 4.

HER3 phosphorylation in cancer cells with MET amplification is ligand-independent. (a) MHCC97-H cells were treated −/+ MET inhibitor (100nM capmatinib) for 16 hours in serum-free media, stimulated −/+ HGF (50ng/mL) for 5 minutes, and assayed for MET activation by western blotting for phospho-MET. (b) MHCC97-H cells were treated with MET inhibitor (100nM capmatinib), EGFR/HER inhibitor (3μM lapatinib), or both, for 16 hours in serum-free media and assayed for phosphorylation of MET, HER3, EGFR, and HER2 by western blot. (c) MET and HER3 interactions were assessed in MHCC97-H cells via PLA. Red dots were counted in over 40 nuclei for the no primary control, 64 nuclei for the -NRG treatment, and 49 nuclei for the +NRG treatment, from three or four independent fields of view per treatment and data presented are means ± SEM.
An interesting and consistent observation we made was a significant increase in HER3 protein levels upon MET inhibition. This phenomenon has been previously noted in cell lines with constitutively activated MET24, 45 and was determined to occur at the mRNA level in response to prolonged MET inhibition, comparable to the length of treatment in our assay.45 We also observed that EGFR was constitutively phosphorylated in MHCC97-H cells, even in the presence of its own potent inhibitor, lapatinib (Fig. 4b). Treatment with capmatinib blocks EGFR phosphorylation, suggesting that, like HER3, EGFR is a substrate of activated MET in MHCC97-H cells. Remarkably, HER2 is not similarly constitutively phosphorylated by MET in MHCC97-H cells (Fig. 4b).
We observed a significant number of MET and HER3 interactions through proximity ligation assay (PLA) analysis in MHCC97-H cells, which remained unchanged under different conditions, such as steady state growth media, serum starvation, and stimulation with NRG (Fig. 4c). These results are consistent with earlier observations that HER3 phosphorylation by MET is ligand-independent (Fig. 4b).
Phosphorylated MET and HER3 localize to the Golgi in cancer cells with MET amplification
Next, we used immunofluorescence to detect the location of phosphorylated endogenous receptors in MHCC97-H cells. Both the total and the phosphorylated MET signals were detected at the cell periphery as well as concentrated in a perinuclear compartment, consistent with our observations in COS7 cells (Fig. 5a). Treatment with the MET inhibitor, crizotinib, completely eliminated the phosphorylated MET signal (Fig. 5a). The phospho-HER3 signal was much weaker overall than the signal detected for phosphorylated MET (Fig. 5b), and total HER3 levels could only be detected upon prolonged crizotinib treatment which significantly enhances expression of endogenous HER3 in MHCC97-H cells (Fig. 4b and Supplementary Fig. 4). The specificity of the total HER3 staining was confirmed by knocking down HER3 expression in MHCC97-H cells using HER3 siRNA (Supplementary Fig. 4).
Fig. 5.

Phosphorylated MET and HER3 localize to the Golgi in cancer cells with MET amplification. (a) MHCC97-H cells were treated −/+ MET inhibitor (1μM crizotinib) for 4 hours in serum-free media, fixed, and stained for MET or phospho-MET. (b) MHCC97-H cells were treated with EGFR inhibitor (10μM gefitinib), or MET inhibitor (1μM crizotinib) for 4 hours, then fixed and stained for phospho-HER3. (c) MHCC97-H cells were fixed and stained for phospho-MET and PDI (ER), EEA1 (early endosome), or Golgin97 (Golgi) markers. (d) MHCC97-H cells were fixed and stained for phospho-HER3 and Golgin97 (Golgi). Scale bars are 50μm.
Phospho-HER3 and to a lesser extent total HER3 staining could be detected both at the cell periphery and at the perinuclear compartment, similar to the localization of phosphorylated MET (Fig. 5b and Supplementary Fig. 4). This phospho-HER3 staining was eliminated by treatment with crizotinib, but not with the EGFR inhibitor gefitinib (Fig. 5b), consistent with our data in Fig. 4b that HER3 phosphorylation is MET-dependent. Notably, in MHCC97-H cells we detected more significant localization of phosphorylated HER3 at the cell periphery than we did in COS7 cells in which MET was over-expressed.
To identify the nature of the perinuclear compartment in MHCC97-H cells, we used markers specific for Golgi, ER and early endosomes. Similar to our observations in COS7 cells, we observed the most extensive co-localization between phosphorylated receptors and Golgin-97 (Fig. 5c and 5d). These results demonstrate not only that MET is abundantly present in the Golgi apparatus in cancer cells, but also that this population of MET is activated and co-localizes with phosphorylated HER3.
HER3 phosphorylation contributes to proliferation of MHCC97-H cells
In lung cancer cells resistant to EGFR kinase inhibitors, persistent HER3 phosphorylation is essential for cell survival and dependent on MET amplification.18 While HER3 phosphorylation is a potent activator of the PI3K pathway, and to a smaller extent the MAPK pathway, both of these pathways are also efficiently activated by MET. Because MET amplification is the driving oncogenic signal in MHCC97-H cells, we tested whether HER3 phosphorylation is functionally significant in these cells.
As expected, MHCC97-H cells rely on MET signaling, and treatment with capmatinib significantly reduced the proliferation and survival of these cells (Fig. 6a and Supplementary Fig. 5a, 5b). To test the significance of HER3 signaling for MHCC97-H cell proliferation, we used siRNA to knock-down HER3 (Fig. 6b). Our initial analysis showed that loss of HER3 did not have a significant effect on the proliferation and survival of MHCC97-H cells (Fig. 6a, 6d and Supplementary Fig. 5a, 5b). Furthermore, the activation of the PI3K-Akt or MAPK/ERK pathways was not reduced upon HER3 knock-down in MHCC97-H cells (Fig. 6c).
Fig. 6.
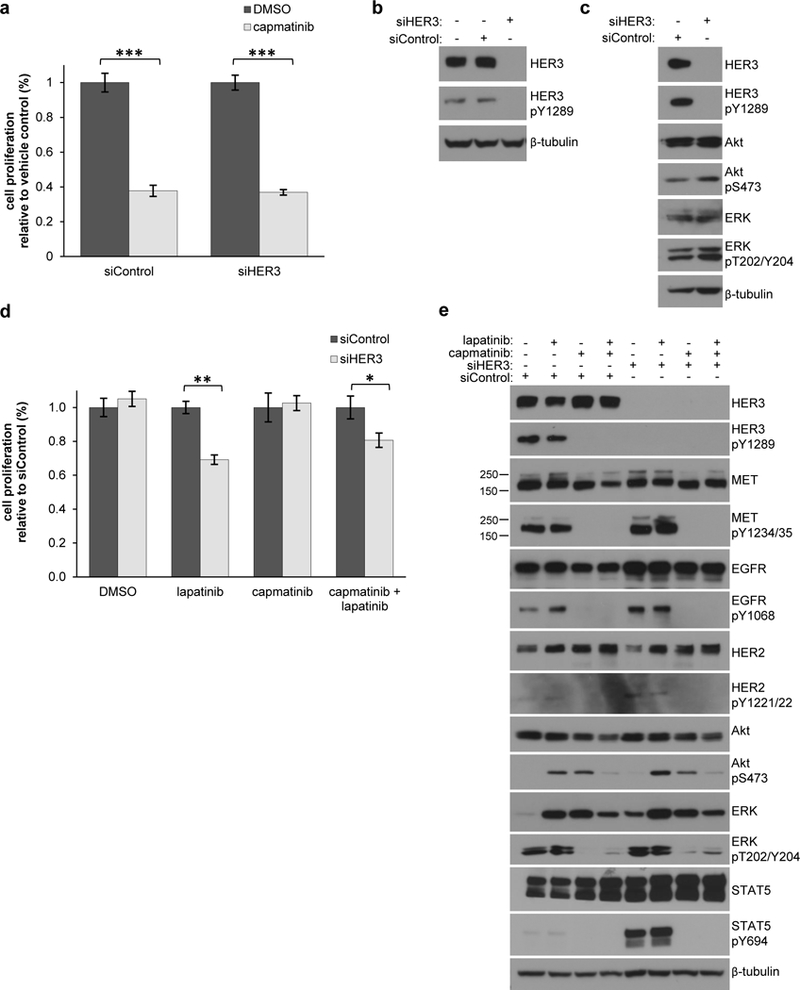
HER3 phosphorylation contributes to proliferation of MHCC97-H cells. (a) MHCC97-H cells were transfected with either non-targeting or HER3 siRNA for 72 hours, treated −/+ 100nM capmatinib for 48 hours in serum-free media, and assayed for cell survival using crystal violet staining. Samples were measured in triplicate for three independent experiments. *** P < 0.0001 (b) MHCC97-H cells were transfected with either non-targeting or HER3 siRNA for 72 hours and assayed for HER3 knock-down by western blot. (c) MHCC97-H cells were transfected with either non-targeting or HER3 siRNA for 72 hours and assayed for activation of phospho-HER3, phospho-Akt and phospho-ERK by western blot. (d) MHCC97-H cells were transfected with either control or HER3 siRNA for 72 hours, treated −/+ 3μM lapatinib, 100nM capmatinib, or both for 48 hours in serum-free media, and assayed for cell proliferation using crystal violet staining. Samples were measured in triplicate for three independent experiments and significance determined by two-tailed t-test.** P < 0.001, * P < 0.05. (e) MHCC97-H cells were transfected with either non-targeting or HER3 siRNA for 72 hours, treated −/+ lapatinib, capmatinib, or both for 48 hours in serum-free media, and assayed for activation of signaling pathways by western blot.
We considered possible redundancy between signaling pathways activated by HER3 and other HER receptors and hypothesized that HER3 phosphorylation may be more critical when its close relatives, EGFR and HER2, can no longer efficiently signal. We therefore used lapatinib or gefitinib to inhibit EGFR and HER2, which are expected to block the ability of EGFR to propagate the signal as the inhibited EGFR will no longer be able to phosphorylate recruited signaling effectors. Indeed, in the presence of lapatinib or gefitinib, HER3 knock-down led to a moderate but significant decrease in cell proliferation and survival to ~ 60–70% of control siRNA treated cells (Fig. 6d & Supplementary Fig. 5a, 5b). Likewise, in control experiments when cells were treated with capmatinib – which is expected to inhibit all MET-dependent phosphorylation events – knock-down of HER3 made a small but significant impact only in the presence of lapatinib (Fig. 6d and Supplementary Fig. 5a). Unexpectedly, analysis of signaling pathway activation under the same serum-starved conditions as the proliferation assays revealed a resurgence of Akt phosphorylation upon sustained treatment with either lapatinib or capmatinib, which was inhibited with the combination of both inhibitors (Fig. 6e). The MAPK/ERK pathway was blocked upon MET inhibition as expected since MET is known to be a strong activator of this pathway. These results suggest that MET-dependent HER3 phosphorylation in MHCC97-H cells plays a functional role in cell proliferation which can be uncovered in the absence of EGFR activation.
EGFR is another RTK substrate of MET phosphorylated in the Golgi
Our data show that MET overexpression in cancer cells results in its premature activation in the Golgi in a ligand-independent manner. Since MET has been previously observed to phosphorylate a broad range of RTKs under conditions of overexpression,19, 20, 26, 38 we hypothesized that this apparent promiscuity reflects encounters between newly synthesized RTKs and hyperactivated MET during receptor trafficking to the plasma membrane. To determine if MET also constitutively phosphorylates a range of RTKs in MET-amplified MHCC97-H cells, we used an RTK array to analyze the phosphorylation status of 49 different RTKs in MHCC97-H cells grown in the presence of DMSO, capmatinib, or crizotinib under serum-starved conditions. In the DMSO-treated sample, MET was highly phosphorylated along with several other receptors, notably EGFR, HER3, Ret, DDR1, and Ryk. Treatment with capmatinib or crizotinib abolished phosphorylation of MET and all other RTKs, demonstrating that hyperactivation of MET in MHCC97-H cells is coupled to phosphorylation of multiple RTKs (Fig. 7a and Supplementary Fig. 6).
Fig. 7.

EGFR is another RTK substrate of MET phosphorylated in the Golgi. (a) MHCC97-H cells were serum starved and treated −/+MET inhibitor for 18 hours (100nM capmatinib) and assayed for RTK phosphorylation using phospho-RTK array. (b) MHCC97-H cells were treated with EGFR inhibitor (10μM gefitinib), or MET inhibitor (100nM capmatinib), for 12 hours in serum-free media then fixed and stained for phospho-EGFR. Scale bars are 50μm.
Among RTK substrates of MET in MHCC97-H cells, EGFR was the most potently phosphorylated (Fig. 7a). Our results in Fig. 4b further show that constitutive EGFR phosphorylation in these cells is largely independent from intrinsic EGFR kinase activity, and is instead dependent on MET. Using immunofluorescence, we also found substantial concentration of phosphorylated EGFR in the Golgi compartment in MHCC97-H cells (Fig. 7b). Phospho-EGFR staining was eliminated with capmatinib treatment, but not when cells were treated with the EGFR inhibitor, gefitinib. These data point to a similar mechanism through which MET phosphorylates both EGFR and HER3.
DISCUSSION
MET-mediated phosphorylation of other RTKs in cancer cells, including EGFR/HER family members, has been reported in numerous malignancies. The mechanisms that allow MET to phosphorylate such a wide spectrum of RTK substrates, which otherwise auto-phosphorylate or are phosphorylated by closely homologous RTK family members, are not understood. Our results describe one mechanism that would allow MET to indiscriminately phosphorylate RTKs through their transient co-localization with hyper-activated MET in the secretory pathway. By focusing our studies on one RTK substrate of MET, HER3, we show that this interaction does not engage molecular interfaces that mediate activation of HER3 by the HER family of receptors.
It is uncertain what leads to intracellular accumulation and activation of overexpressed MET in cancer cells. Following synthesis in the ER, MET undergoes extensive post-translational modification, including N- and O-linked glycosylation, and recently reported palmitoylation.46–48 These modifications are essential for proper MET function, and inhibition of palmitoylation and N-linked glycosylation have both been shown to impair maturation of MET and trafficking to the plasma membrane.46, 47 The efficiency of MET progression through the secretory pathway is also regulated by the functions of acid sphingomyelinase (ASM) and syntaxin 6 (STX6), as well as by the levels of cholesterol.49 Collectively, these regulatory mechanisms are responsible for the maintenance of an intracellular pool of MET in the Golgi that is used to replenish MET at the plasma membrane.49 A perinuclear pool of MET, which primarily consists of de novo synthesized MET, has also been observed in cancer cell models and tumor samples.50.51 In cancer cells with MET overexpression, inefficient post-translational modifications might elicit perinuclear accumulation of immature MET receptors – which nevertheless have a functional kinase domain. Increased local concentration of the kinase domains would then drive receptor activation even in the absence of canonical regulation by ligand binding. One could speculate that cells could mitigate receptor overcrowding by improving the rate of their maturation or alternative mechanisms that would decrease their unspecific interactions. Interestingly, in gastric cancer cells, O-linked glycosylation of MET was shown to negatively regulate MET activation48 which could hypothetically entail preventing receptor interactions in the absence of ligand binding.
During biosynthesis, MET is also proteolytically cleaved in the trans-Golgi-network.42 Interestingly, an uncleaved variant of MET, p190NC is constitutively activated in LoVo cells.52 Although the mechanism through which p190NC is activated is unknown at present, this MET variant is not impaired in trafficking to the plasma membrane. Curiously HER3 is also constitutively phosphorylated in LoVo cells.53 Whether p190NC drives phosphorylation of HER3 and does so through abnormal accumulation in the Golgi, will be an exciting topic for future studies.
Overexpression or mutation of several other RTKs has been shown to impair receptor trafficking to the cell surface resulting in accumulation of an intracellular pool of activated receptors. Specifically, oncogenic mutants of ALK, c-Kit, and Flt3 have all been shown to be retained in the ER and Golgi as a result of their constitutive activation.54–57 Similarly, overexpression of FGFR1 and FGFR2 in COS7 leads to activation of immature receptors at the Golgi apparatus, which still elicit typical downstream signaling responses, such as phosphorylation of downstream effector STAT1.58 In the case of Flt3, change in localization qualitatively changes downstream signaling from its typical membrane-localized downstream effectors to acquisition of new substrates.59 Hence, accumulation of activated RTKs in endomembranes can result in a variety of signaling responses from the intracellular compartment. Based on our data we propose that MET-dependent phosphorylation of other RTKs represents yet another example of aberrant signaling resulting from intracellular RTK activation.
Although our studies focus on the functional interaction between MET and HER3, hyperactivated MET facilitates phosphorylation of many unrelated receptors such as EGFR, HER3, Ret, DDR1, and Ryk, in the hepatocellular carcinoma cell line we tested. Previously published RTK array analyses in cell lines from different cancer types have identified largely overlapping sets of RTKs whose phosphorylation becomes MET-dependent.19, 20, 26, 38 The EGFR/HER family members are consistently identified as substrates of MET in these analyses, as well as Ret, and the MET-family member Ron. The prominence of the EGFR/HER family as substrates of MET is striking and may reflect the fact that these receptors are themselves potent oncogenes. It is possible that cells under physiological stress, such as MET overexpression, compensate by upregulating multiple signaling pathways, and that the EGFR/HER family is particularly good at supporting cell survival in this context. The additional variation of MET substrates observed in these studies might reflect differential receptor expression levels or be indicative of the unique mechanisms through which different cancers support their survival. The lack of clear substrate specificity further argues that functional interactions between MET and other RTKs do not rely on a common molecular mechanism, but rather are driven by MET overexpression, which turns low affinity, unspecific interactions into productive phosphorylation events before the receptors fully progress through the secretory pathway.
Our results increase a body of evidence supporting the propensity of MET to signal intracellularly, which has also been shown to originate from endosomes 60, 61 and the nucleus62, 63. The endosomal pool of MET originates from the plasma membrane following ligand stimulation and results in perinuclear accumulation of activated MET capable of inducing downstream signaling, such as activation of STAT3.60, 61 A truncated and activated form of nuclear MET has been found in both prostate and hepatocellular cancers and linked to activation of a variety of pathways including SRY (sex determining region Y)-box9, β-catenin, and Nanog homeobox, and NF-κB.62, 63 These mechanisms seem to fundamentally differ from how constitutive MET activation is achieved through overexpression during receptor biosynthesis. Collectively, this broad spectrum of intracellular functions of MET has ramifications for antibody-based therapies targeting MET. There are currently numerous antibody therapies targeting MET or its ligand HGF in development and clinical trials,64–69 but these therapies are expected to perform poorly in MET expressing-tumors in which a large population of activated MET is localized intracellularly. Thus far, anti-MET antibody therapies appear to have fallen short of their potential in clinical trials with the absence of a significant clinical benefit to patients.64–67 Our finding that a large population of MET can be activated in the Golgi could help account for these lackluster results and adds weight to the notion that combining small molecule with antibody therapies could more effectively target tumors with this phenotype.
MATERIALS AND METHODS
Cell Culture
MHCC97-H cells were obtained from the Liver Cancer Institute and Zhongshan Hospital of Fudan University, Shanghai. COS7 cells are a monkey kidney fibroblast-like cell line obtained from Dr. John Kuriyan at UC-Berkeley. COS7 and MHCC97-H cells were cultured in DMEM supplemented with 10% FBS and streptomycin/penicillin. Inhibitor assays were treated for 6–24 hours in serum-free media containing capmatinib/INCB28060 (Selleckchem), crizotinib/PF-02341066 (Selleckchem), gefitinib/ZD1839 (Selleckchem), or lapatinib (Selleckchem).
Transfection
COS7 cells were transfected using FuGene6 (Promega) and cultured for 24–48 hours. MHCC97-H cells were reverse transfected with non-targeting (Dharmacon siGENOME Non-Targeting siRNA Pool#1: UAGCGACUAAACACAUCAA, UAAGGCUAUGAAGAGAUAC, AUGUAUUGGCCUGUAUUAG, AUGAACGUGAAUUGCUCAA) or HER3 siRNA (Dharmacon siGENOME Human ERBB3 (2065) siRNA: GCAGUGGAUUCGAGAAGUG, AGAUUGUGCUCACGGGACA, GUGGAUUCGAGAAGUGACA, GCGAUGCUGAGAACCAAUA) using Lipofectamine RNAiMax.
Western blotting
Cells were lysed in lysis buffer (50mM Tris pH 7.5, 150mM NaCl, 1mM EDTA, 1mM Na3VO4, 1mM NaF, 1% Triton X-100), or RIPA buffer (50mM Tris pH 7.5, 150mM NaCl, 1mM EDTA, 1mM Na3VO4, 1mM NaF, 0.1% SDS, 0.1% deochycholate, 1% IGEPAL CA-630) with protease inhibitor cocktail (Roche). Lysates were run on 8 or 10% SDS-PAGE and transferred to PVDF membrane (EMD Millipore). Proteins were detected using: anti-MET (D1C2 XP– Cell Signaling), anti-phospho-MET Tyr1234/Y1235 (Cell Signaling), anti-HER3 (SC-285 – Santa Cruz), anti-HER3 (SC-81455 – Santa Cruz), anti-HER3 (D22C5 XP – Cell Signaling), anti-phospho-HER3 Tyr1289 (21D3 – Cell Signaling), anti-HER2 (SC-284 – Santa Cruz), anti-phospho-HER2 Tyr1221/1222 (Cell Signaling), anti-FLAG M2 (Sigma-Aldrich), anti-EGFR ((1005) SC-03 – Santa Cruz), anti-phospho-EGF-Receptor Tyr1068 (Cell Signaling), β-tubulin (9F3 – Cell Signaling), GAPDH (D4C6R - Cell Signaling). Secondary antibodies were anti-rabbit-IgG HRP-linked antibody (Cell Signaling), or anti-mouse IgG HRP-linked whole antibody (GE Healthcare Biosciences). Blots were developed using ECL/ECL Prime (Thermo Fisher Scientific).
Immunoprecipitation
Cells were lysed, cleared by centrifugation, and incubated with anti-FLAG-M2 antibody (Sigma Aldrich #F1804) for 2–12 hours at 4C followed by incubation with 1:1 protein A-sepharose slurry (Life Technologies). Immunoprecipitates were analyzed by western blotting.
Immunofluorescence
9×104 COS7 cells were plated on coverslips. After 24 hours, cells were transfected and cultured for 24–48 hours. Cells were fixed with 3.7% formaldehyde for 1 hour at room temperature, permeabilized with 0.1% Triton X-100 for 5 minutes, and blocked with 1% BSA for 5 minutes. Cells were incubated with primary antibodies at 37C for 1–2 hours: anti-FLAG-M2 antibody (Sigma Aldrich #F1804), anti-phospho-HER3 Tyr1289 (21D3 – Cell Signaling), anti-MET (D1C2 – Cell Signaling), anti-phospho-MET Tyr1234/Y1235 (Cell Signaling), anti-phospho-EGF-Receptor Tyr1068 (Cell Signaling), anti-Golgin97 (A-21270 – Molecular Probes), anti-PDI (RL90 - Thermo Scientific), or anti-EEA1 (BD Biosciences - 610456), anti-HER3 (D22C5 XP – Cell Signaling). Cells were washed and incubated with fluorescently tagged anti-rabbit or anti-mouse secondary antibodies (Goat Anti-Mouse Alexa-fluor-568 cat#A11031; Alexa-Fluor-488 donkey anti-rabbit cat# A21206 – Life technologies). Images were taken at 60x magnification using a Nikon widefield epifluorescent microscope (Fig. 3a, b, 5a-d, 7b), or at 60x using a Nikon spinning disc confocal microscope Confocal images shown were taken as a stack of 7 slices in the Z-plane and processed as a Z-max projection (Supplementary Fig. 4).
Phospho-RTK Array
Phosphorylated RTKs were identified from MHCC97-H cells using the Proteome Profiler Human Phospho-RTK Array Kit (R&D Systems ARY001B). Cells were treated with DMSO, crizotinib, or capmatinib, for 24 hours in serum-free media. Cells were lysed and protein concentration was standardized by Bradford assay. Samples were incubated on the array membrane overnight at 4C. Membranes were washed and phosphorylated receptors were detected using HRP-conjugated phosphotyrosine antibody.
Proliferation and Cell Survival assays
After 24 hours transfection with siRNA, cells were serum-starved −/+drug and cultured for 48 hours. Cells were washed, incubated with crystal violet solution (0.5% crystal violet, 25% methanol) for 10 minutes, washed, and dried overnight. Dye was solubilized in 1:1 ethanol:sodium citrate solution for 1 hour, transferred to 96-well plate, and absorbance was read at 595nm.
After 24 hours transfection with siRNA, cells were serum starved and treated −/+ inhibitors and cultured for 48 hours. To monitor cell survival, cells were counted at 0, 24, and 48 hours post inhibitor-treatment using an automated Countess hemacytometer and survival curves were plotted.
Proximity Ligation Assay
MHCC97-H cells were grown on four-well chamber slides and processed using the Duolink In Situ Fluorescence kit with red detection reagents (Sigma-Aldrich) per the manufacturer’s instructions. MET (D1C2 XP– Cell Signaling) and HER3 G4 (SC-203) primary antibodies were used.
Statistics
Sample sizes were chosen to be sufficient to obtain reliable results consistent with standard practices within this field. Statistics were determined and displayed using Excel (Microsoft, Redmond, WA). Results represent an average of at least three independent experiments and exact sample size is noted in figure legends. Data are presented as mean ± SEM for co-localization and proximity ligation assay analysis, and as grand mean ± pooled standard deviation for proliferation assay analysis. The variances within each group of compared data were similar in almost all cases, and all were well within one order of magnitude. For proliferation assay analysis, two treatments were compared by unpaired two-tailed t-tests, determined to have normal distributions by Shapiro-Wilk test, and p-values below 0.05 were considered significant.
Supplementary Material
ACKNOWLEDGEMENTS
We thank M. Moasser for insightful discussions and members of the Jura lab for valuable discussions and input on experimental design and execution. This work was supported partially by the grant from the National Institute of General Medical Sciences to N.J. (R01 GM109176), Susan G. Komen Foundation Training Grant to N.J. (CCR14299947), Lung Cancer Research Foundation Grant to N.J. and the grant from National Institute of Dental and Craniofacial Research to J.G. (R01 DE023685).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Park M, Dean M, Kaul K, Braun MJ, Gonda MA, Vande Woude G. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc Natl Acad Sci U S A 1987; 84: 6379–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gherardi E, Youles ME, Miguel RN, Blundell TL, Iamele L, Gough J et al. Functional map and domain structure of MET, the product of the c-met protooncogene and receptor for hepatocyte growth factor/scatter factor. Proc Natl Acad Sci U S A 2003; 100: 12039–12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991; 251: 802–804. [DOI] [PubMed] [Google Scholar]
- 4.Chirgadze DY, Hepple JP, Zhou H, Byrd RA, Blundell TL, Gherardi E. Crystal structure of the NK1 fragment of HGF/SF suggests a novel mode for growth factor dimerization and receptor binding. Nat Struct Biol 1999; 6: 72–79. [DOI] [PubMed] [Google Scholar]
- 5.Rodrigues GA, Park M. Autophosphorylation modulates the kinase activity and oncogenic potential of the Met receptor tyrosine kinase. Oncogene 1994; 9: 2019–2027. [PubMed] [Google Scholar]
- 6.Zhu H, Naujokas MA, Fixman ED, Torossian K, Park M. Tyrosine 1356 in the carboxyl-terminal tail of the HGF/SF receptor is essential for the transduction of signals for cell motility and morphogenesis. J Biol Chem 1994; 269: 29943–29948. [PubMed] [Google Scholar]
- 7.Ponzetto C, Bardelli A, Maina F, Longati P, Panayotou G, Dhand R et al. A novel recognition motif for phosphatidylinositol 3-kinase binding mediates its association with the hepatocyte growth factor/scatter factor receptor. Mol Cell Biol 1993; 13: 4600–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ponzetto C, Bardelli A, Zhen Z, Maina F, dalla Zonca P, Giordano S et al. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994; 77: 261–271. [DOI] [PubMed] [Google Scholar]
- 9.Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995; 376: 768–771. [DOI] [PubMed] [Google Scholar]
- 10.Song Z, Wang X, Zheng Y, Su H, Zhang Y. MET Gene Amplification and Overexpression in Chinese Non-Small-Cell Lung Cancer Patients Without EGFR Mutations. Clin Lung Cancer 2017; 18: 213–219 e212. [DOI] [PubMed] [Google Scholar]
- 11.Christensen JG, Burrows J, Salgia R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett 2005; 225: 1–26. [DOI] [PubMed] [Google Scholar]
- 12.Li A, Niu FY, Han JF, Lou NN, Yang JJ, Zhang XC et al. Predictive and prognostic value of de novo MET expression in patients with advanced non-small-cell lung cancer. Lung Cancer 2015; 90: 375–380. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Li W, He Q, Xu Y, Ren X, Tang X et al. Prognostic value of MET protein overexpression and gene amplification in locoregionally advanced nasopharyngeal carcinoma. Oncotarget 2015; 6: 13309–13319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan S, Jiao X, Zou H, Li K. Prognostic significance of c-Met in breast cancer: a meta-analysis of 6010 cases. Diagn Pathol 2015; 10: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casadevall D, Gimeno J, Clave S, Taus A, Pijuan L, Arumi M et al. MET expression and copy number heterogeneity in nonsquamous non-small cell lung cancer (nsNSCLC). Oncotarget 2015; 6: 16215–16226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dimou A, Non L, Chae YK, Tester WJ, Syrigos KN. MET gene copy number predicts worse overall survival in patients with non-small cell lung cancer (NSCLC); a systematic review and meta-analysis. PLoS One 2014; 9: e107677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seiwert TY, Jagadeeswaran R, Faoro L, Janamanchi V, Nallasura V, El Dinali M et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res 2009; 69: 3021–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–1043. [DOI] [PubMed] [Google Scholar]
- 19.Kataoka Y, Mukohara T, Tomioka H, Funakoshi Y, Kiyota N, Fujiwara Y et al. Foretinib (GSK1363089), a multi-kinase inhibitor of MET and VEGFRs, inhibits growth of gastric cancer cell lines by blocking inter-receptor tyrosine kinase networks. Invest New Drugs 2012; 30: 1352–1360. [DOI] [PubMed] [Google Scholar]
- 20.Kim SM, Kim H, Yun MR, Kang HN, Pyo KH, Park HJ et al. Activation of the Met kinase confers acquired drug resistance in FGFR-targeted lung cancer therapy. Oncogenesis 2016; 5: e241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007; 318: 287–290. [DOI] [PubMed] [Google Scholar]
- 22.Soltoff SP, Carraway KL, 3rd, Prigent SA, Gullick WG, Cantley LC. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol 1994; 14: 3550–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim HH, Sierke SL, Koland JG. Epidermal growth factor-dependent association of phosphatidylinositol 3-kinase with the erbB3 gene product. J Biol Chem 1994; 269: 24747–24755. [PubMed] [Google Scholar]
- 24.Liu X, Wang Q, Yang G, Marando C, Koblish HK, Hall LM et al. A novel kinase inhibitor, INCB28060, blocks c-MET-dependent signaling, neoplastic activities, and cross-talk with EGFR and HER-3. Clin Cancer Res 2011; 17: 7127–7138. [DOI] [PubMed] [Google Scholar]
- 25.Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A 2008; 105: 692–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agarwal S, Zerillo C, Kolmakova J, Christensen JG, Harris LN, Rimm DL et al. Association of constitutively activated hepatocyte growth factor receptor (Met) with resistance to a dual EGFR/Her2 inhibitor in non-small-cell lung cancer cells. Br J Cancer 2009; 100: 941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi P, Oh YT, Zhang G, Yao W, Yue P, Li Y et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett 2016; 380: 494–504. [DOI] [PubMed] [Google Scholar]
- 28.Yun C, Gang L, Rongmin G, Xu W, Xuezhi M, Huanqiu C. Essential role of Her3 in two signaling transduction patterns: Her2/Her3 and MET/Her3 in proliferation of human gastric cancer. Mol Carcinog 2015; 54: 1700–1709. [DOI] [PubMed] [Google Scholar]
- 29.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006; 125: 1137–1149. [DOI] [PubMed] [Google Scholar]
- 30.Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH et al. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 2009; 137: 1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 2002; 110: 763–773. [DOI] [PubMed] [Google Scholar]
- 32.Dawson JP, Berger MB, Lin CC, Schlessinger J, Lemmon MA, Ferguson KM. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Mol Cell Biol 2005; 25: 7734–7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell 2003; 11: 507–517. [DOI] [PubMed] [Google Scholar]
- 34.Kavran JM, McCabe JM, Byrne PO, Connacher MK, Wang Z, Ramek A et al. How IGF-1 activates its receptor. Elife 2014; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Menting JG, Whittaker J, Margetts MB, Whittaker LJ, Kong GK, Smith BJ et al. How insulin engages its primary binding site on the insulin receptor. Nature 2013; 493: 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Favelyukis S, Till JH, Hubbard SR, Miller WT. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat Struct Biol 2001; 8: 1058–1063. [DOI] [PubMed] [Google Scholar]
- 37.McKern NM, Lawrence MC, Streltsov VA, Lou MZ, Adams TE, Lovrecz GO et al. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature 2006; 443: 218–221. [DOI] [PubMed] [Google Scholar]
- 38.Tanizaki J, Okamoto I, Sakai K, Nakagawa K. Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Br J Cancer 2011; 105: 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jura N, Shan Y, Cao X, Shaw DE, Kuriyan J. Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc Natl Acad Sci U S A 2009; 106: 21608–21613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tempest PR, Stratton MR, Cooper CS. Structure of the met protein and variation of met protein kinase activity among human tumour cell lines. Br J Cancer 1988; 58: 3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stamos J, Lazarus RA, Yao X, Kirchhofer D, Wiesmann C. Crystal structure of the HGF beta-chain in complex with the Sema domain of the Met receptor. EMBO J 2004; 23: 2325–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Komada M, Hatsuzawa K, Shibamoto S, Ito F, Nakayama K, Kitamura N. Proteolytic processing of the hepatocyte growth factor/scatter factor receptor by furin. FEBS Lett 1993; 328: 25–29. [DOI] [PubMed] [Google Scholar]
- 43.Tang ZY, Ye SL, Liu YK, Qin LX, Sun HC, Ye QH et al. A decade’s studies on metastasis of hepatocellular carcinoma. J Cancer Res Clin Oncol 2004; 130: 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du Z, Caenepeel S, Shen Y, Rex K, Zhang Y, He Y et al. Preclinical Evaluation of AMG 337, a Highly Selective Small Molecule MET Inhibitor, in Hepatocellular Carcinoma. Mol Cancer Ther 2016; 15: 1227–1237. [DOI] [PubMed] [Google Scholar]
- 45.Steinway SN, Dang H, You H, Rountree CB, Ding W. The EGFR/ErbB3 Pathway Acts as a Compensatory Survival Mechanism upon c-Met Inhibition in Human c-Met+ Hepatocellular Carcinoma. PLoS One 2015; 10: e0128159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen R, Li J, Feng CH, Chen SK, Liu YP, Duan CY et al. c-Met function requires N-linked glycosylation modification of pro-Met. J Cell Biochem 2013; 114: 816–822. [DOI] [PubMed] [Google Scholar]
- 47.Coleman DT, Gray AL, Kridel SJ, Cardelli JA. Palmitoylation regulates the intracellular trafficking and stability of c-Met. Oncotarget 2016; 7: 32664–32677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu SY, Shun CT, Hung KY, Juan HF, Hsu CL, Huang MC et al. Mucin glycosylating enzyme GALNT2 suppresses malignancy in gastric adenocarcinoma by reducing MET phosphorylation. Oncotarget 2016; 7: 11251–11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu L, Xiong X, Kim Y, Okada N, Lu F, Zhang H et al. Acid sphingomyelinase is required for cell surface presentation of Met receptor tyrosine kinase in cancer cells. J Cell Sci 2016; 129: 4238–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruco LP, Ranalli T, Marzullo A, Bianco P, Prat M, Comoglio PM et al. Expression of Met protein in thyroid tumours. J Pathol 1996; 180: 266–270. [DOI] [PubMed] [Google Scholar]
- 51.Kermorgant S, Zicha D, Parker PJ. Protein kinase C controls microtubule-based traffic but not proteasomal degradation of c-Met. J Biol Chem 2003; 278: 28921–28929. [DOI] [PubMed] [Google Scholar]
- 52.Mondino A, Giordano S, Comoglio PM. Defective posttranslational processing activates the tyrosine kinase encoded by the MET proto-oncogene (hepatocyte growth factor receptor). Mol Cell Biol 1991; 11: 6084–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beji A, Horst D, Engel J, Kirchner T, Ullrich A. Toward the prognostic significance and therapeutic potential of HER3 receptor tyrosine kinase in human colon cancer. Clin Cancer Res 2012; 18: 956–968. [DOI] [PubMed] [Google Scholar]
- 54.Mazot P, Cazes A, Boutterin MC, Figueiredo A, Raynal V, Combaret V et al. The constitutive activity of the ALK mutated at positions F1174 or R1275 impairs receptor trafficking. Oncogene 2011; 30: 2017–2025. [DOI] [PubMed] [Google Scholar]
- 55.Bougherara H, Subra F, Crepin R, Tauc P, Auclair C, Poul MA. The aberrant localization of oncogenic kit tyrosine kinase receptor mutants is reversed on specific inhibitory treatment. Mol Cancer Res 2009; 7: 1525–1533. [DOI] [PubMed] [Google Scholar]
- 56.Tabone-Eglinger S, Subra F, El Sayadi H, Alberti L, Tabone E, Michot JP et al. KIT mutations induce intracellular retention and activation of an immature form of the KIT protein in gastrointestinal stromal tumors. Clin Cancer Res 2008; 14: 2285–2294. [DOI] [PubMed] [Google Scholar]
- 57.Schmidt-Arras DE, Bohmer A, Markova B, Choudhary C, Serve H, Bohmer FD. Tyrosine phosphorylation regulates maturation of receptor tyrosine kinases. Mol Cell Biol 2005; 25: 3690–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Citores L, Bai L, Sorensen V, Olsnes S. Fibroblast growth factor receptor-induced phosphorylation of STAT1 at the Golgi apparatus without translocation to the nucleus. J Cell Physiol 2007; 212: 148–156. [DOI] [PubMed] [Google Scholar]
- 59.Choudhary C, Olsen JV, Brandts C, Cox J, Reddy PN, Bohmer FD et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell 2009; 36: 326–339. [DOI] [PubMed] [Google Scholar]
- 60.Kermorgant S, Parker PJ. Receptor trafficking controls weak signal delivery: a strategy used by c-Met for STAT3 nuclear accumulation. J Cell Biol 2008; 182: 855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lefebvre J, Ancot F, Leroy C, Muharram G, Lemiere A, Tulasne D. Met degradation: more than one stone to shoot a receptor down. FASEB J 2012; 26: 1387–1399. [DOI] [PubMed] [Google Scholar]
- 62.Xie Y, Lu W, Liu S, Yang Q, Carver BS, Li E et al. Crosstalk between nuclear MET and SOX9/beta-catenin correlates with castration-resistant prostate cancer. Mol Endocrinol 2014; 28: 1629–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tey SK, Tse EYT, Mao X, Ko FCF, Wong AST, Lo RC et al. Nuclear Met promotes hepatocellular carcinoma tumorigenesis and metastasis by upregulation of TAK1 and activation of NF-kappaB pathway. Cancer Lett 2017; 411: 150–161. [DOI] [PubMed] [Google Scholar]
- 64.Kim KH, Kim H. Progress of antibody-based inhibitors of the HGF-cMET axis in cancer therapy. Exp Mol Med 2017; 49: e307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dieras V, Campone M, Yardley DA, Romieu G, Valero V, Isakoff SJ et al. Randomized, phase II, placebo-controlled trial of onartuzumab and/or bevacizumab in combination with weekly paclitaxel in patients with metastatic triple-negative breast cancer. Ann Oncol 2015; 26: 1904–1910. [DOI] [PubMed] [Google Scholar]
- 66.Shah MA, Bang YJ, Lordick F, Alsina M, Chen M, Hack SP et al. Effect of Fluorouracil, Leucovorin, and Oxaliplatin With or Without Onartuzumab in HER2-Negative, MET-Positive Gastroesophageal Adenocarcinoma: The METGastric Randomized Clinical Trial. JAMA Oncol 2017; 3: 620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shah MA, Cho JY, Tan IB, Tebbutt NC, Yen CJ, Kang A et al. A Randomized Phase II Study of FOLFOX With or Without the MET Inhibitor Onartuzumab in Advanced Adenocarcinoma of the Stomach and Gastroesophageal Junction. Oncologist 2016; 21: 1085–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rosen LS, Goldman JW, Algazi AP, Turner PK, Moser B, Hu T et al. A First-in-Human Phase I Study of a Bivalent MET Antibody, Emibetuzumab (LY2875358), as Monotherapy and in Combination with Erlotinib in Advanced Cancer. Clin Cancer Res 2017; 23: 1910–1919. [DOI] [PubMed] [Google Scholar]
- 69.Cignetto S, Modica C, Chiriaco C, Fontani L, Milla P, Michieli P et al. Dual Constant Domain-Fab: A novel strategy to improve half-life and potency of a Met therapeutic antibody. Mol Oncol 2016; 10: 938–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.