

Abstract
Epidermal cancer stem cells (ECS cells) comprise a limited population of cells that form aggressive, rapidly growing, and highly vascularized tumors. VEGF-A/NRP-1 signaling is a key driver of the ECS cell phenotype and aggressive tumor formation. However, relatively less is known regarding the downstream events following VEGF-A/NRP-1 interaction. In the present study, we show that VEGF-A/NRP-1, GIPC1, and Syx interact to increase RhoA-dependent p38 MAPK activity to enhance ECS cell spheroid formation, invasion, migration, and angiogenic potential. Inhibition or knockdown of NRP-1, GIPC1 or Syx attenuates RhoA and p38 activity to reduce the ECS cell phenotype, and NRP-1 knockout, or pharmacologic inhibition of VEGF-A/NRP-1 interaction or RhoA activity, reduces p38 MAPK activity and tumor growth. Moreover, expression of wild-type or constitutively-active RhoA, or p38, in NRP1-knockout cells, restores p38 activity and the ECS cell phenotype. These findings suggest that NRP-1 forms a complex with GIPC1 and Syx to activate RhoA/ROCK-dependent p38 activity to enhance the ECS cell phenotype and tumor formation.
Keywords: angiogenesis, cancer stem cells, GIPC1, NRP-1, p38 MAPK, squamous cell carcinoma, Syx, vascularization
1 |. INTRODUCTION
Squamous cell carcinoma is a highly aggressive and invasive disease.1 Angiogenesis is essential for squamous cell carcinoma tumor growth2,3 and is stimulated by a host of proteins including vascular endothelial growth factor (VEGF).4–7 We recently identified epidermal cancer stem cells (ECS cells) as a small subpopulation (0.15%) of the tumor cells in squamous cell carcinoma.8 ECS cells, enriched by growth as non-attached spheroids, undergo epithelial to mesenchymal transition and form aggressive, invasive, and highly vascularized tumors. Moreover, injection of as few as 100 ECS cells into immunocompromised mice drives formation of highly vascularized and aggressive tumors.8 VEGF-A knockdown, or treatment with VEGF-A inactivating antibody, reduces ECS cell survival and tumor formation.9 This response is interesting in that ECS cells lack VEGFR1 and VEGFR2.9 Instead, VEGF-A stimulates ECS cell survival via interaction with neuropilin-1 (NRP-1).9 VEGF-A/NRP-1 interaction triggers intracellular changes to enhance formation of aggressive, invasive, and highly vascularized tumors. We recently reported that this is mediated by an α6/β4-integrin, FAK, Src, PI3 K/PDK1, Hippo signaling cascade that regulates YAP1/ΔNp63α.10 We have also shown that NRP-1 interacts with GIPC1 (GAIP-interacting protein) to stimulate the α6/β4-integrin/YAP1/ΔNp63α signaling cascade.11
Because GIPC1 is a PDZ domain protein that serves to scaffold multiple signaling pathways,12 VEGF-A/NRP-1 interaction with GIPC1 can potentially trigger multiple signaling outcomes. For example, a recent study indicated that the GIPC1/Syx complex stimulates RhoA activity in DJM-1 skin cancer cells.13 We therefore sought to determine whether VEGF-A/NRP-1/GIPC1 interaction triggers activity in additional cascades. Our present study suggests that VEGF-A/NRP-1 forms a complex with GIPC1 and Syx (synectin-binding RhoA exchange factor) to activate RhoA/ROCK, MEK3/6 and p38. Pharmacology inhibition of VEGF-A/NRP-1 interaction, or VEGF-A, NRP-1, GIPC1 or Syx knockdown reduces p38 activity to attenuate the ECS cell phenotype. In addition, p38 knockdown, reduces spheroid formation, matrigel invasion and migration, and forced expression of RhoA or p38 restores the cancer stem cell phenotype in NRP-1 knockout cells. These findings suggest that VEGF-A/NRP-1 interaction triggers a GIPC1, Syx, RhoA/ROCK, MEK3/6 cascade to activate p38 MAPK and drive the ECS cell phenotype.
2 |. MATERIALS AND METHODS
2.1 |. Chemicals and reagents
Trypsin and Dulbecco’s modified Eagle’s medium were purchased from Invitrogen (Frederick, MD). β-actin (A5441), and pan-p38 (p38T) (M0800) antibodies were obtained from Sigma (St. Louis, MO). Mouse monoclonal antibody to VEGF-A (MAB293) was purchased from R&D Systems (Minneapolis, MN). Antibodies to NRP-1 (ab81321), and MEK6 (ab33866) were obtained from Abcam (Cambridge, MA). Antibodies to total p38-P (9216), ERK1/2 (9102), and ERK1/2-P (9101) were obtained from Cell Signaling Technologies (Danvers, MA). Antibodies to GIPC1 (sc-9648), MEK3 (sc-961), Syx (PLEKHG5, sc-130100), mouse IgG (sc-2025), rabbit IgG (sc-2028), and MEK3/6-P (sc-8407) were obtained from Santa Cruz (Dallas, TX). Rabbit IgG was purchased from Millipore (NI-01, Temecula, CA). Anti-RhoA was from Cytoskeleton (ARH04, Denver, CO). Horseradish peroxidase-conjugated sheep anti-mouse IgG (NXA931) and donkey anti-rabbit IgG (NA934V) were obtained from GE Healthcare (Buckinghamshire, UK) and used at a 1:5000 dilution. SB203580 (A8254) and Y27632 (A3008) were purchased from APExBIO (Houston, TX). EG00229, an inhibitor of VEGF-A/NRP-1 interaction, was obtained from R&D Systems (4931, Minneapolis, MN).
2.2 |. RhoA activity assay
RhoA activity was assessed by measuring interaction of GTP-bound RhoA with the Rho binding domain of rhotekin by immunoprecipitation. Protein extract was prepared in 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 0.5 M NaCl and 2% Igepal from Cytoskeleton, Inc. (BK036), and 400 μg of protein was incubated with Rho binding domain followed by precipitation and assay by immunoblot. Increased RhoA/Rho binding domain interaction indicates enhanced activity. The RhoA activity assay was purchased from Cytoskeleton, Inc. (BK036, Denver, CO).
2.3 |. Cell culture
Monolayer cell cultures were maintained in a Dulbecco’s modified Eagle’s medium containing 5% fetal bovine serum (FBS), 2 mM l- glutamine and 1 mM sodium pyruvate and appropriate antibiotics.8 ECS cell spheroids were grown by plating 40 000 monolayer cells/well in ultra-low attachment six well cluster dishes and growing for 0–10 d in spheroid medium [DMEM/F12 (1:1) (DMT-10–090-CV, Mediatech Inc, Manassa, VA) containing 2% B27 serum-free supplement (17504–044, Invitrogen, Frederick, MD), 20 ng/mL EGF (E4269, Sigma, St. Louis), 0.4% bovine serum albumin (B4287, Sigma) and 4 μg/mL insulin (#19278, Sigma Chemical, St. Louis, MO).8 SCC-13 cells are derived from epidermal squamous cell carcinoma.14 Production of the NRP-1 knockdown cell line, SCC13-NRP1-shRNA1, was previously described.11 SCC13-NRP1-KOc8 cells are NRP-1 knockout cells created using CRISPR/Cas9 vectors purchased from Biocytogen (Worcester, MA). Each figure panel indicates the cell source as “SCC-13 or HaCaT spheroid” to indicate that the ECS cells used were derived from cultures grown as non-attached spheroids.8
2.4 |. Electroporation
For electroporation, 1 × 106 cells were electroporated with 3 μg of control- (D-001206–13-05, Dharmacon), VEGF-A (sc-29520, Santa Cruz), NRP-1 (sc-36038, Santa Cruz), GIPC1 (M-019997–02-005, Dharmacon), p38α (sc-29433, Santa Cruz), p38β (sc-39116, Santa Cruz), p38γ (sc-39118, Santa Cruz), p38δ (sc-36456, Santa Cruz), MEKK1 (sc-35898, Santa Cruz), MEK3 (sc-35907, Santa Cruz), MEK6 (sc-35913, Santa Cruz), Syx (Dharmacon, M-013873–01-0005) or RhoA (Santa Cruz, sc-29471) siRNA using the Amaxa electroporator and the VPD-1002 nucleofection kit (Germany). The cells were plated, permitted to recover overnight, and then harvested and the electroporation was repeated using the same siRNA.15 A similar protocol was used for electroporation of expression plasmids. These included pcDNA3-EGFP-RhoA-wt (RhoA-wild type) obtained from Addgene (Cambridge, MA),16 and pcDNA3-Flag-MKK6.17,18 After a 24 h recovery, the cells were utilized for experiments. Reduced level of the siRNA targeted protein was confirmed by immunoblot. Immunoblot analysis and HUVEC cell angiogenesis assays, were performed as previously described.9
2.5 |. Human umbilical vein endothelial cells (HUVEC) cell angiogenesis assay
HUVEC cell tube formation assay was performed as previously described.9 Briefly, HUVEC cells (1.2 × 105 cells) are plated in EBM basal medium (Lonza, CC-3121) supplemented with human epidermal growth factor, hydrocortisone, bovine brain extract, ascorbic acid, fetal bovine serum, and gentamicin/amphotericin-B (Lonza, CC-4133) in wells pre-coated with a 10 mg/mL solution of BD Matrigel (354234, BD Biosciences). For tube formation assay, this medium was supplemented with 0 or 300 μg of ECS cell extract and the wells were incubated for 18 h at 37°C and microscopic images were collected for analysis of junction using ImageJ (http://imagej.nih.gov/ij/macros/toolsets/).19
2.6 |. Cell invasion and migration assays
To measure cell invasion, BioCoat Millicell inserts (1 cm diameter, 8 µm pore size) were coated with 120 μL of 250 μg/mL Matrigel. ECS cells (20 000) were seeded atop the matrigel in 500 μL of serum-free RPMI1640 containing 1% FCS. The lower chamber contained the identical medium containing 10% FCS. When appropriate, pharmacologic agents were added to the bottom chamber and the cells were placed in an incubator. After 18 h the membrane was washed, fixed with 4% paraformaldehyde and cells on the membrane inner surface were visualized by staining with 4′, 6-diamidino-2-phenylindole (DAPI) for fluorescence detection of nuclei. For the migration assay, ECS cells were plated on plastic and the confluent cultures were wounded by scraping with a 10 μL pipette tip. Migration of ECS cell wound closure was monitored at 0–18 h.
2.7 |. Tumor growth assay
ECS cells, selected by growth as spheroids, were prepared as single cell suspensions and 100 000 cells were injected subcutaneously into each front flank in NOD/scid/IL2 receptor gamma chain knockout mice (NSG mice) (5 mice/treatment group).8 Tumor experiments were repeated three separate times. The studies were approved by the University of Maryland-Baltimore Animal Care and Use Committee and meet all national and international standards. The NRP-1 inhibitor, EG00229, was dissolved at 100 mM in dimethyl sulfoxide and diluted to 2 mM with captisol11 for IP injection of 200 μL (10 mg/kg body weight) three times per week (M/W/F). The ROCK inhibitor, Y27632, was dissolved at 310 mM in dimethyl sulfoxide, diluted to 3.1 mM in phosphate buffered saline for IP injection of 200 μL (10 mg/kg body weight) three times per week (M/W/F). The values are errors of the mean and significant differences were determined using the student’s t-test.
3 |. RESULTS
Epidermal cancer stem cells (ECS cell) comprise a highly aggressive cell subpopulation in epidermal squamous cell carcinoma.8,10,20,21 Our previous studies show that a VEGF-A/NRP-1 signaling complex plays a major role in driving this aggressive ECS cell phenotype.9,11 In this paper we explore the mechanisms downstream of VEGF-A/NRP-1 that mediate these events. NRP-1 knockdown (Figure 1A) reduces ECS cell spheroid formation, matrigel invasion and wound closure (Figure 1B–D), and also attenuates HUVEC cell vessel formation (Figure 1E). NRP-1 has no catalytic activity and it is thought that the c-terminal NRP-1 PDZ binding domain interacts with other factors that mediate downstream events.22 To identify NRP-1 coupled signaling cascades in ECS cells we immunoprecipitated NRP-1 and assayed for co-precipitating proteins. Figure 1F shows that NRP-1, GIPC1, Syx, MEKK1, and p38 are expressed in ECS cells, and that GIPC1, Syx, and p38 co-precipitate with NRP-1. GIPC1 is a PDZ domain protein that serves to scaffold multiple signaling pathways12 and Syx is a PDZ binding domain protein that interacts with and activates RhoA/ROCK signaling to drive angiogenesis.23,24
FIGURE 1.
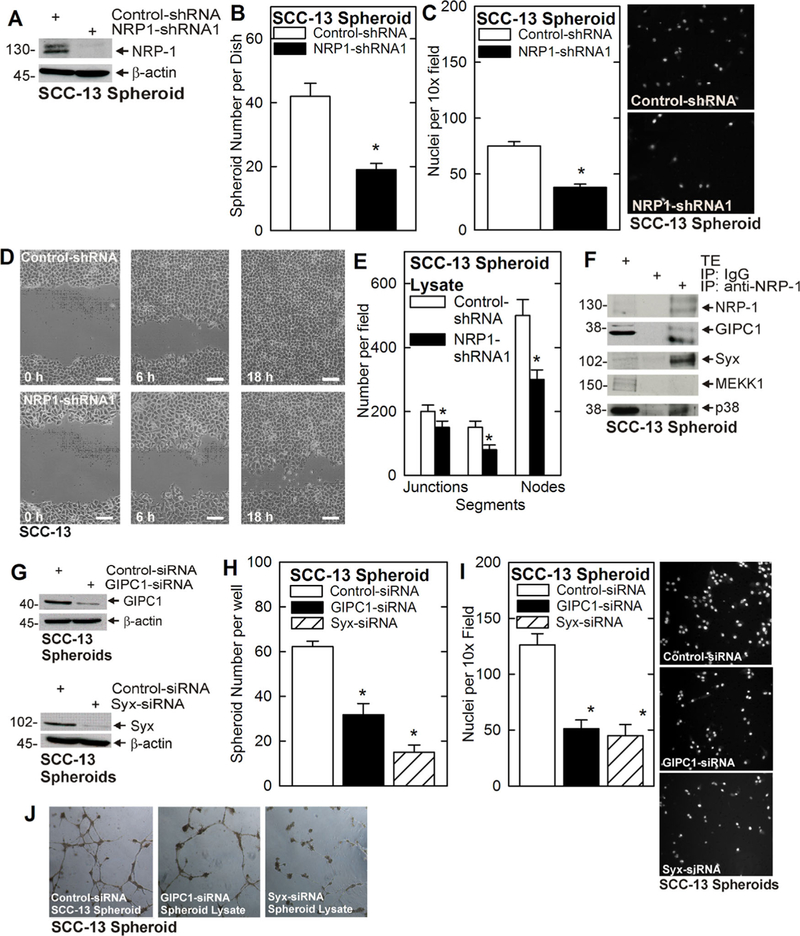
NRP-1 is required for ECS cell survival. A–D) Wild-type and NRP-1 knockdown SCC-13 derived ECS cells were assayed for ability to form spheroids, invade matrigel and migrate. Cells were seeded and grown as spheroids in ultra-low attachment plates. Spheroid diameter and number were measured at 5 d. For invasion, 25 000 cells were seeded atop matrigel per transwell chamber, and cell migration to the lower chamber was monitored at 24 h. For migration, cells were permitted to attach to conventional plates at confluence, wounded and wound closure was monitored. Bars = 100 μm. E) ECS cell lysates were prepared from 5 d spheroids and 300 μg of protein extract was tested for ability to enhanced HUVEC tube formation. The plot shows the number of junctions, segments, and nodes measured using ImageJ analysis.9 The asterisks indicate a significant change (n = 3, P ≤ 0.005). F) NRP-1 interacts with GIPC1, Syx and p38. Extracts were prepared from SCC-13-derived ECS cells and 200 μg of extract was immunoprecipitated with anti-NRP-1 followed by immunoblot to detect the indicated epitopes. Total extract (TE) was electrophoresed as a loading control. G-I) GIPC1 and Syx knockdown reduces spheroid formation and invasion. Cells were electroporated with the indicated siRNA and spheroid number (5 d) and matrigel invasion (24 h) were monitored. J) Cell lysates were prepared from 5 d spheroids and 300 μg protein was tested for ability to enhance HUVEC cell tube formation. The number of junctions, segments, and nodes was assessed using ImageJ.9 All graphical data is presented as the mean ± SEM and the asterisks indicate a significant change, n = 3, P ≤ 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
To assess the importance of these factors in mediating NRP-1 action, we knocked down each protein and monitored the effect on the ECS cell phenotype. Knockdown of GIPC1 or Syx (Figure 1G) reduces ECS cell spheroid formation, matrigel invasion and HUVEC cell vessel formation (Figure 1H–J). As Syx interacts with and activates RhoA, the finding that Syx co-precipitates with NRP-1 implies that RhoA is a signaling mediator. We therefore determined whether suppressing RhoA or ROCK function could attenuate downstream responses. As shown in Figure 2A and 2B, RhoA knockdown, or treatment with ROCK inhibitor (Y27632) (Figure 2C and 2D), reduces spheroid formation and invasion. These findings suggest that GIPC1, Syx, and RhoA have a role in driving the NRP-1 dependent ECS cell phenotype.
FIGURE 2.
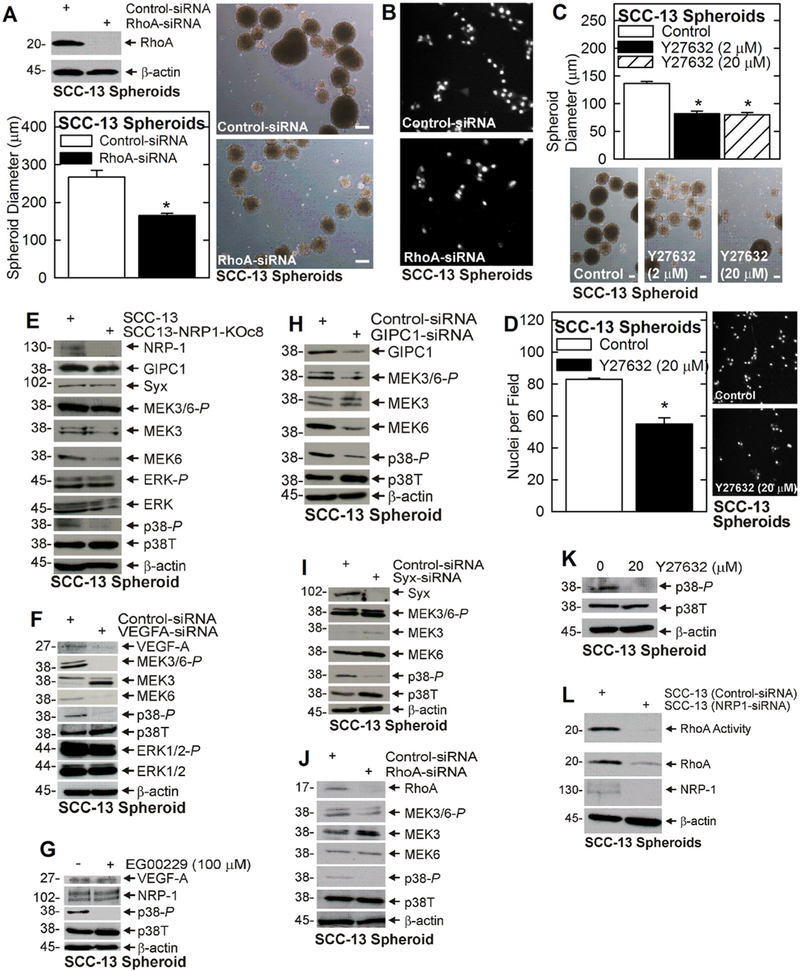
RhoA is required for the ECS cell phenotype. A and B) SCC-13 cells were electroporated with 3 μg of control- or RhoA-siRNA and tested for spheroid formation and invasive potential. C and D) Spheroid formation and invasive potential were monitored after treatment with 0 or 20 μM Y27632, a ROCK inhibitor. E) Wild-type and NRP-1 knockout ECS cell extracts were monitored for signaling protein activity. F) SCC-13 cells were electroporated with 3 µg of control- or VEGFA-siRNA, grown as spheroids, and at 5 d assayed for the indicated proteins. G) SCC13 cells were grown as spheroids for 3 d and then treated for 48 h with 0 or 100 μM EG00229 before extracts were prepared for immunoblot. H-J) SCC-13 ECS cells were electroporated with 3 µg of control-, GIPC1-, Syx-, or RhoA-siRNA and impact on MAPK signaling was assayed. K) SCC13 ECS cells were grown as spheroids for 3 d prior to treatment for 48 h with 0 or 20 μM Y27632 and p38 activity was measured. L) SCC-13 ECS cells were electroporated with control- or NRP-1-siRNA and after 48 h extracts were prepared to monitor RhoA level and activity. Similar results were observed in three experiments. The bars = 100 μm. All graphical data is presented as mean ± SEM and the asterisks indicate a significant change, n = 3, P ≤ 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
3.1 |. VEGF-A/NRP-1 signaling and p38 activity
To characterize the role of these proteins in mediating NRP-1 action, we knocked down or inhibited NRP-1 in ECS cells, and monitored the impact on GIPC1, Syx, and RhoA. We also monitored ERK1/2 and p38 signaling, as these are highly elevated in ECS cells and we suspected they may be VEGF-A/NRP-1 responsive. Figure 2E shows that NRP-1 knockdown does not impact GIPC1, Syx, or ERK1/2-P level, but does reduce MEK3/6-P and p38-P. Consistent with the NRP-1 knockdown findings, VEGF-A knockdown does not suppress ERK1/2-P, but does reduce MEK3/6-P and p38-P (Figure 2F). Moreover, treatment with EG00229, an inhibitor of VEGF-A/NRP-1 interaction, also reduces p38-P (Figure 2G). Thus, these findings implicate p38 as a downstream VEGF-A/NRP-1 target.
3.2 |. GIPC1, Syx, and RhoA link VEGF-A/NRP-A signaling to p38 activation
Since, GIPC1 and Syx interact with NRP-1 (Figure 1F), and Syx interacts with and regulates RhoA,13,25 we assessed if GIPC1, Syx, and RhoA link VEGF-A/NRP-1 to p38 signaling. Figure 2H–J shows that loss of GIPC1, Syx, or RhoA reduces p38 activity. Rho-associated protein kinase (ROCK) is required for RhoA signaling.26,27 Figure 2K shows that treatment with Y27632, a ROCK inhibitor, reduces p38 activity. Finally, we demonstrate that NRP-1 regulates RhoA, as NRP-1 knockdown reduces RhoA activity (Figure 2L). These findings suggest that GIPC1, Syx, and RhoA, serve to link VEGF-A/ NRP-1 to p38 signaling.
3.3 |. Role of MEK3/6 and p38 MAPK in regulating ECS cell phenotype
Figure 2 shows that p38 activity is consistently reduced in VEGF-A, NRP-1, GIPC1, Syx, or RhoA knockdown cells. As MEK3/6 activity is regarded as the canonical direct upstream controller of p38 activity,28 we anticipated that MEK3/6 and p38 activity would be reduced in tandem. However, this is not always the case. For example, although p38 activity is reduced in GIPC1, Syx or RhoA knockdown ECS cells, MEK3/6-P is not (Figure 2H–J). We therefore assessed the role of MEK3/6 in regulating p38 activity and the ECS cell phenotype. Our findings show that MEK3/MEK6 knockdown reduces p38 activity (Figure 3A) and ECS cell spheroid formation, matrigel invasion, and HUVEC cell tube/vessel formation (Figure 3B–D). However, although these studies show that MEK3 and MEK6 can regulate ECS cell p38 activity and phenotype, our evidence (Figure 2H–J) suggests that MEK3/6 do not necessarily assume this role in response to NRP-1 signaling. We think it likely that MEK3/6 regulates p38 in a context specific manner, and that other factors which have been reported to regulate p38 activity,29 may also have a role in mediating VEGF-A/ NRP-1 action in ECS cells. Further studies will be necessary to assess this possibility.
FIGURE 3.
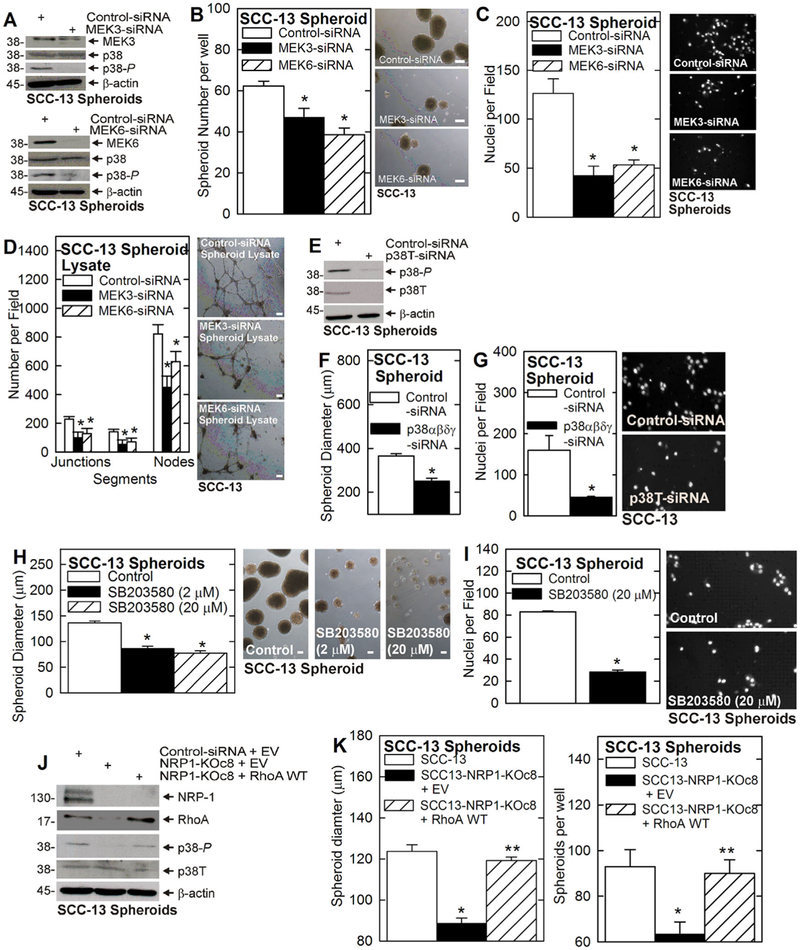
The ECS cell phenotype and MEK3/6 and p38 signaling. A-C) Cells were electroporated with 3 μg of Control-, MEK3-, or MEK6-siRNA and the impact on p38 activity, and spheroid formation and invasive, were measured. D) Cells were electroporated with 3 μg of Control-, MEK3-, or MEK6-siRNA, grown for 5 d as spheroids, and cell extracts (300 μg protein) were assayed for ability to enhance HUVEC tube formation as measured by junction, segment and node formation. E) Cells were electroporated with 3 μg of the indicated siRNA and p38 level and activity were measured at 48 h. F and G) Impact of p38 knockdown on spheroid formation and matrigel invasion. H and I) Treatment with SB203580, a p38α/β inhibitor, suppresses ECS cell spheroid formation and invasive potential. J and K) RhoA restores the ECS cell phenotype in NRP-1 knockdown cells. SCC-13 derived NRP-1 knockout cells were electroporated with 3 μg of empty vector or RhoA WT expression plasmid, and plated for spheroid formation (5 d). The immunoblot monitors NRP-1 knockdown, RhoA overexpression, and the impact on p38T and p38-P level. SCC-13 and SCC13-NRP-1-KOc8 cells were electroporated with 3 μg of the indicated plasmids and ability to form spheroids was monitored at 5 d. The asterisk indicates a significant reduction in spheroid formation in SCC13-NRP1-KOc8 cells versus SCC-13. The double asterisk indicates an increase in spheroid formation in RhoA WT expression plasmid-electroporated versus empty vector-electroporated SCC13-NRP1-KOc8 cells, n = 3, P ≤ 0.005. All bars = 100 μm. [Color figure can be viewed at wileyonlinelibrary.com]
To confirm that p38 at an is a NRP-1 signaling mediator, we used both knockdown and pharmacologic strategies. p38 knockdown (Figure 3E) reduces ECS cell spheroid formation and invasion (Figure 3F and 3G). In addition, treatment with the p38α/β selective inhibitor, SB203580, also reduces spheroid formation and invasion (Figure 3H and 3I).
3.4 |. RhoA overexpression restores p38 activity and the ECS cell phenotype
Our findings suggest that VEGF-A/NRP-1 stimulates RhoA which stimulates p38 signaling to maintain the ECS cell phenotype. To test this prediction, we forced RhoA overexpression in NRP-1 knockout cells and assessed the impact on p38 activity and the ECS cell phenotype. NRP-1 knockout results in reduced p38 activity and ECS cell spheroid formation and expression of wild-type (RhoA WT) protein restores p38 activity (Figure 3J) and ECS cell spheroid formation (Figure 3K). These findings suggest that RhoA acts downstream of VEGF-A/NRP-1 and upstream of p38.
3.5 |. VEGF-A/NRP-1, RhoA, and tumor formation
The above studies predict that NRP-1 triggers a cascade, which includes RhoA, to enhance p38 activity and tumor formation. To assess whether this cascade is active in tumors, we injected wild-type and NRP-1 knockout ECS cells and monitored tumor growth, and MEK3/6 and p38 signaling. Figure 4A and 4B shows that NRP-1 knockout tumors grow more slowly and display reduced MEK3/6 and p38 activity. As a second approach, we treated with EG00229, an inhibitor of VEGF-A/NRP-1 interaction.30,31 Consistent with this finding EG00229, which inhibits VEGF-A/NRP-1 interaction,32 reduces tumor formation and p38 activity (Figure 4C and 4D). Finally, we investigated the role of RhoA/ROCK, by studying the impact of a ROCK inhibitor (Y27632) on signaling and tumor formation. Figure 4E–G shows that treatment with Y27632 reduces tumor formation, and MEK3/6 and p38 signaling.
FIGURE 4.
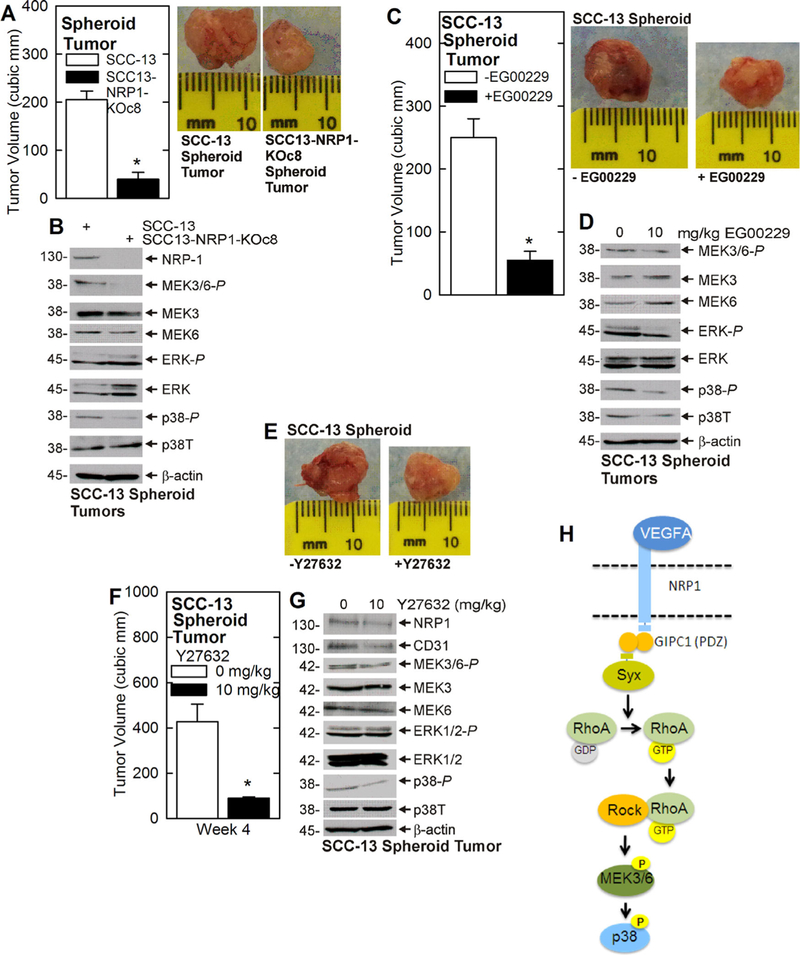
Impact of NRP-1 and RhoA in tumor formation and p38 signaling. A and B) Spheroid-derived SCC-13 and SCC-13-NRP-1-KOc8 cells were injected (100 000 cells per each front flank) into NSG mice. Tumors were harvested at 4 wk and extracts prepared for immunoblot. C and D) Spheroid-derived SCC-13 cells were injected (100 000 per each front flank) into NSG mice and treated with 0 or 10 mg EG00229/kg body weight. Four week tumors were harvested, photographed and extracts were assayed by immunoblot. E–G) Spheroid-derived SCC-13 cells were injected (100 000 per each front flank) into NSG mice and treated with 0 or 10 mg Y27632/kg body weight. Tumors were harvested at 4 wk, photographed, and assayed by immunoblot. K) Proposed VEGF-A/NRP-1/RhoA/p38 signal transduction pathway. The VEGF-A/NRP-1 complex formation causes the NRP-1 PDZ binding domain to engage the PDZ domain of one subunit of the GIPC1 dimer while the second GIPC1 subunit engages the Syx PDZ binding domain. Syx then activates RhoA which interacts with ROCK to stimulate MAPK signaling. Although MEK3/6 is shown as an activator of p38 activity, this role appears to be context-dependent and we cannot rule out a role for other p38 activation mechanisms. The plotted values are mean ± SEM. The asterisks in each plot indicate a significant reducing in tumor volume, n = 10 tumors/group, P ≤ 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
3.6 |. Role of proposed signaling cascade in HaCaT cells
To assess whether the proposed VEGF-A/NRP-1 cascade can be generalized, we determined whether GIPC1 and RhoA are required for NRP-1 signaling in HaCaT cell-derived ECS cells. HaCaT cells are immortal but non-tumorigenic cells derived from epidermis.33 HaCaT cell-derived ECS cells were treated with control-, NRP-1-, GIPC1-, or RhoA-siRNA and the impact on spheroid formation and invasion, and p38 signaling, was monitored. Knockdown of NRP-1 reduces spheroid size (Figure 5A), MEK3/6 and p38 activity (Figure 5B) and RhoA activity (Figure 5C). We also examined the impact of manipulating GIPC1, and RhoA level on the ECS cell phenotype. GIPC1 knockdown reduces ECS cell spheroid size, matrigel invasion, and MEK3/6 and p38 activity (Figure 5D–F). RhoA knockdown also reduces ECS cell spheroid formation, invasion, and MEK3/6 and p38 signaling (Figure 5G–I). Finally, RhoA overexpression restores spheroid formation in NRP-1 knockdown cells and this is associated with a partial restoration of p38 activity (Figure 5J–L).
FIGURE 5.
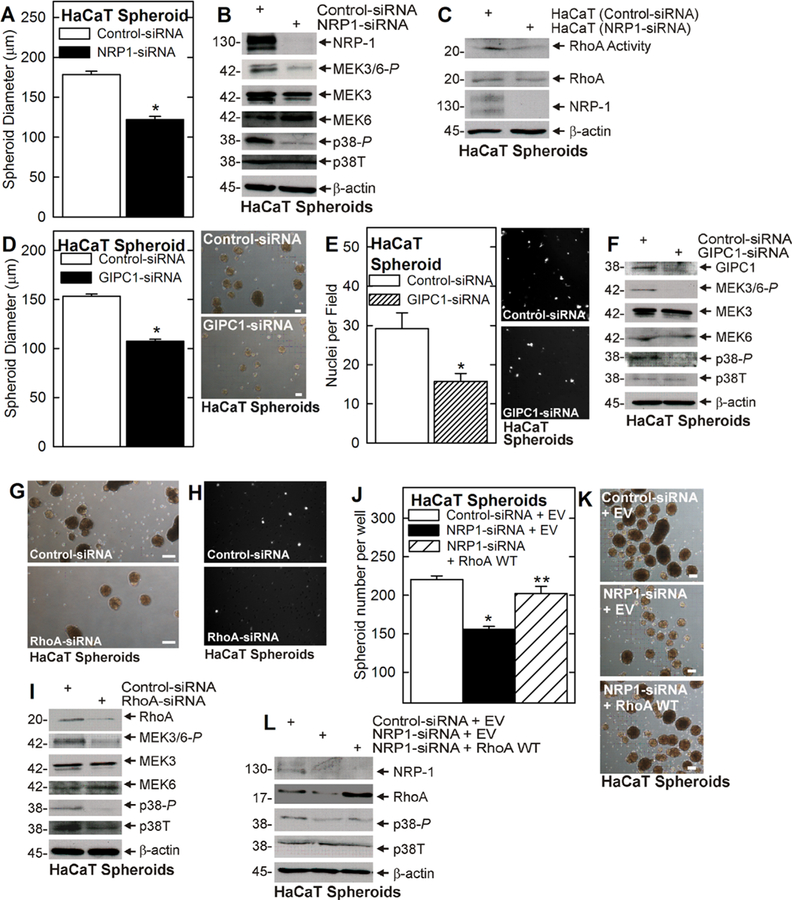
Role of NRP-1 and RhoA in HaCaT ECS cells. A and B) Cells, electroporated with 3 μg control- or NRP-1-siRNA, were tested for spheroid formation and extracts were prepared to assay NRP-1 and MEK3/6 and p38. C) Cells were electroporated with control- or NRP-1- siRNA. After 48 h, extracts were prepared to monitor RhoA level and RhoA activity. D–F) Cells were electroporated with 3 μg of control- or GIPC1-siRNA and tested for spheroid formation, invasion, and MAPK signaling. G–I) Cells were electroporated with 3 μg of control- or RhoA-siRNA and tested for spheroid formation, invasive potential, and MAPK signaling. J–L) Cells were electroporated with 3 μg of control- or NRP1-siRNA in the presence of empty vector (EV) or RhoA WT expression plasmid, and the impact on spheroid formation (3 d) and p38 activity was monitored. All graphical data is presented as the mean ± SEM. In panels A, D, and E, the single asterisk indicates a significant reduction in the treated group compared to control, n = 3, P ≤ 0.005. In panel J the asterisk indicates a significant reduction in spheroid formation in NRP1-siRNA treated versus control. The double asterisk indicates a significant increase in spheroid formation in NRP-1 knockout cells electroporated with RhoA WT expression plasmid versus cells electroporated with empty vector (EV), n = 3, P ≤ 0.005. All bars = 100 μm. [Color figure can be viewed at wileyonlinelibrary.com]
4 |. DISCUSSION
The ECS cell phenotype is characterized by an enhanced ability to form spheroids, invade matrigel, and migrate.8 VEGF-A is required for ECS cell spheroid formation, invasion, and migration.9 However, the absence of VEGFR1/2 suggests that VEGF-A controls these cell responses via an alternate mechanism. Our recent studies reveal that VEGF-A complexes with NRP-1 to activate downstream signaling events.9 The classic role for the NRP-1 is as an adaptor protein to facilitate VEGF-A interaction with VEGFR1/2.34–36 However, recent studies suggest that NRP-1 can mediate VEGF-A action independent of VEGF receptor in a number of systems.9,13,31,37–39 For example, NRP-1 mediates VEGF action in VEGFR1/2-negative pancreatic cancer,37,39 renal cell carcinoma cells,40,41 and DJM-1 skin cancer cells.13
The NRP-1 protein encodes a large extracellular glycoprotein domain, a short transmembrane-spanning region, and a 40 amino acid intracellular segment. The intracellular segment encodes a PDZ binding domain that is required for interaction with PDZ domain proteins.22,42,43 Our recent study indicates that VEGF-A/NRP-1 activates a α6/β4-integrin cascade to increase YAP1/ΔNp63α-dependent ECS cell survival.11 In this cascade, NRP-1 interacts with GIPC1, a PDZ domain scaffold protein, and NRP-1 or GIPC1 knockdown attenuates downstream signaling and reduces ECS cell survival. Because GIPC1 forms a homodimer that encodes a PDZ domain on each subunit, the GIPC1 homodimer can serve as a scaffold for multiple PDZ domain-encoding signaling proteins.12,13,44,45 This, in turn, implies that GIPC1 can funnel signal transduction down alternate signaling cascades depending upon which PDZ domain binding signaling proteins are coupled. For this reason, we suspected that GIPC1 may couple VEGF-A/NRP-1 to multiple alternate signaling cascades in ECS cells. To assess this, we performed NRP-1 immunoprecipitation experiments to identify additional interacting signaling proteins. These experiments revealed NRP-1 interaction with GIPC1 and also identified interaction with Syx. The presence of Syx (synectin-binding RhoA exchange factor), a RhoA guanine exchange factor,25,46 suggested a role for RhoA. RhoA is a small GTPase that is important regulator of cell proliferation and cell migration in cancer cells.27 Our findings show that suppressing VEGF-A/NRP-1, GIPC1, or Syx function reduces RhoA activity and attenuates the ECS cell phenotype. Moreover, suppressing RhoA or ROCK activity, using genetic or pharmacologic intervention, reduces ECS cell survival. Consistent with a role for RhoA, overexpression of wild-type or constitutively-active RhoA restores the ECS cell phenotype, and p38 signaling, in NRP-1 deficient cells, suggesting that RhoA is downstream of NRP-1 and upstream of p38. These studies suggest that VEGF-A/NRP-1 interacts with GIPC1 and Syx to activate RhoA/ROCK signaling to activate p38 signaling to maintain the ECS cell phenotype. We observed NRP-1 control of RhoA activity and RhoA control of p38 activity in ECS cells derived from two different epidermis derived cell lines. For example, treatment with Y27632, a ROCK inhibitor, reduced p38 activity and ECS cell spheroid formation in both cell types. The importance of RhoA/ ROCK regulation of p38 kinase is supported by studies in other systems.47–49 However, Yoshida et al. found that RhoA/ROCK regulation of cell survival was not coupled to p38 signaling in DJM-1 human skin cancer cells.13 This interesting difference suggests that the mechanism of survival regulation may differ in various skin cancer cells and tumors.
4.1 |. The importance of MEK3/6 and p38
In the course of the present studies, we observed a reproducible reduction in p38 MAPK activity in VEGF-A or NRP-1 knockdown ECS cells, suggesting that p38 is a VEGF-A/NRP-1 target. Genetic and pharmacologic inhibition of p38 confirmed that it is required for ECS cell spheroid formation, invasion, and migration. Moreover, the observation that SB203580, a p38α/β selective inhibitor, suppresses the ECS cell phenotype, suggests a role for these isoforms.
An interesting finding is that MEK3/6 does not appear to be an obligatory regulator of p38 activity in the VEGF-A/NRP-1 triggered signaling cascade. For example, reducing ECS cell VEGF-A, NRP-1, GIPC1, Syx, or RhoA signaling always produces a reduction in p38 activity, but this is not always associated with a reduction in MEK3/6 activity. This is unanticipated, as MEK3 and MEK6 are canonical regulators of p38,28 but is not particularly surprising as other proteins can also regulate p38 activity.29 Thus, we conclude that p38 is an essential mediator of VEGF-A/NRP-1 action in ECS cells and that MEK3/6 may (or may not) mediate p38 activation in a context-dependent manner.
4.2 |. Role of NRP-1 dependent RhoA/ROCK and p38 signaling in tumor formation
An important test of the relevance of this signaling cascade is the impact on tumor formation. To test this we monitored the impact of NRP-1 knockout, and EG00229 or ROCK inhibitor treatment, on ECS cell tumor formation and p38 activity. EG00229 is an inhibitor of VEGF-A/NRP-1 interaction.30,31 Each treatment reduced p38 activity and tumor formation, suggesting that this signaling cascade has a role in vivo and may be important in human tumor formation.
4.3 |. VEGF-A/NRP-1 control of ECS cell phenotype
Based on the present findings, we propose that GIPC1 couples VEGF-A/NRP-1 to Syx, via PDZ based interaction, and that Syx activates RhoA/ROCK to stimulate MEK3/6 and p38 activity (Figure 4K) and drive tumor formation. This scheme compliments our previous report showing that VEGF-A/NRP-A engages a α6/β4-integrin, FAK, Src, PI3 K/PDK1 pathway that regulates YAP1/ΔNp63α.10 In this cascade, GIPC1 couples VEGF-A/NRP-1 to TG2/α6/β4-integrin to activate FAK/src to stabilize YAP1/ΔNp63α and drive tumor formation.11
Inhibiting activity in either cascade reduces the ECS cell phenotype and reduces VEGF-A/NRP-1 dependent tumor formation. We do not know which of these cascades is more important in driving the cancer stem cell phenotype. However, it is likely that it depends upon the availability of proteins present in each cascade. Moreover, these cascades could act in a synergistic or additive manner to drive ECS cell survival. Moreover, examples of crosstalk between the MAPK and Hippo/YAP1 signaling cascades have been noted,50–52 suggesting that the cascades may be mutually reinforcing. It is likely that GIPC1 couples VEGF-A/NRP-1 to additional, as yet undiscovered cascades, which drive the ECS cell phenotype. It will be interesting in future studies to examine the impact of simultaneously inhibiting the NRP-1 stimulated YAP1/ΔNp63α and the RhoA/p38 pathways to see if this provides a more effective inhibition of tumor formation.
ACKNOWLEDGMENT
This work was supported by the NIH (CA131074 and CA184027) to R.L. Eckert.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Rogers HW, Weinstock MA, Harris AR, et al. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol 2010;146:283–287. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J The role of angiogenesis in tumor growth. Semin Cancer Biol 1992;3:65–71. [PubMed] [Google Scholar]
- 3.Johnson KE, Wilgus TA. Multiple roles for VEGF in non-melanoma skin cancer: angiogenesis and beyond. J Skin Cancer 2012;2012: 483439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Claesson-Welsh L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med 2013;273:114–127. [DOI] [PubMed] [Google Scholar]
- 5.Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J 2011;437:169–183. [DOI] [PubMed] [Google Scholar]
- 6.Larcher F, Murillas R, Bolontrade M, Conti CJ, Jorcano JL. VEGF/VPF overexpression in skin of transgenic mice induces angiogenesis, vascular hyperpermeability and accelerated tumor development. Oncogene 1998;17:303–311. [DOI] [PubMed] [Google Scholar]
- 7.Hirakawa S, Kodama S, Kunstfeld R, et al. VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med 2005;201:1089–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adhikary G, Grun D, Kerr C, et al. Identification of a population of epidermal squamous cell carcinoma cells with enhanced potential for tumor formation. PLoS ONE 2013;8:84324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grun D, Adhikary G, Eckert RL. VEGF-A acts via neuropilin-1 to enhance epidermal cancer stem cell survival and formation of aggressive and highly vascularized tumors. Oncogene 2016;35: 4379–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fisher ML, Kerr C, Adhikary G, et al. Transglutaminase interaction with α6/β4-integrin to stimulates YAP1-dependent ΔNp63α stabilization and leads to enhanced cancer stem cell survival and tumor formation. Cancer Res 2016;76:7265–7276. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 11.Grun D, Adhikary G, Eckert RL. NRP-1 interacts with GIPC1 and alpha6/beta4-integrins to increase YAP1/Np63alpha-dependent epidermal cancer stem cell survival. Oncogene 2018;37:4711–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katoh M Functional proteomics, human genetics and cancer biology of GIPC family members. Exp Mol Med 2013;45:e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida A, Shimizu A, Asano H, et al. VEGF-A/NRP1 stimulates GIPC1 and Syx complex formation to promote RhoA activation and proliferation in skin cancer cells. Biol Open 2015;4:1063–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rheinwald JG, Beckett MA. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultures from human squamous cell carcinomas. Cancer Res 1981;41:1657–1663. [PubMed] [Google Scholar]
- 15.Chew YC, Adhikary G, Xu W, Wilson GM, Eckert RL. Protein kinase C delta increases kruppel-like factor 4 protein, which drives involucrin gene transcription in differentiating keratinocytes. J Biol Chem 2013; 288:17759–17768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subauste MC, Von HM, Benard V, et al. Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and Fas. J Biol Chem 2000;275:9725–9733. [DOI] [PubMed] [Google Scholar]
- 17.Efimova T, LaCelle P, Welter JF, Eckert RL. Regulation of human involucrin promoter activity by a protein kinase C, Ras, MEKK1, MEK3, p38/RK, AP1 signal transduction pathway. J Biol Chem 1998;273: 24387–24395. [DOI] [PubMed] [Google Scholar]
- 18.Efimova T, Broome AM, Eckert RL. A regulatory role for p38 delta MAPK in keratinocyte differentiation. Evidence for p38 delta-ERK1/2 complex formation. J Biol Chem 2003;278:34277–34285. [DOI] [PubMed] [Google Scholar]
- 19.Bao S, Wu Q, Sathornsumetee S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res 2006;66:7843–7848. [DOI] [PubMed] [Google Scholar]
- 20.Fisher ML, Adhikary G, Xu W, et al. Type II transglutaminase stimulates epidermal cancer stem cell epithelial-mesenchymal transition. Oncotarget 2015;6:20525–20539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eckert RL, Fisher ML, Grun D, et al. Transglutaminase is a tumor cell and cancer stem cell survival factor. Mol Carcinog 2015;54:947–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ballmer-Hofer K, Andersson AE, Ratcliffe LE, Berger P. Neuropilin-1 promotes VEGFR-2 trafficking through Rab11 vesicles thereby specifying signal output. Blood 2011;118:816–826. [DOI] [PubMed] [Google Scholar]
- 23.de Toledo M, Coulon V, Schmidt S, Fort P, Blangy A. The gene for a new brain specific RhoA exchange factor maps to the highly unstable chromosomal region 1p36. 2–1p36. 3. Oncogene 2001;20: 7307–7317. [DOI] [PubMed] [Google Scholar]
- 24.Marx R, Henderson J, Wang J, Baraban JM. Tech: a RhoA GEF selectively expressed in hippocampal and cortical neurons. J Neurochem 2005;92:850–858. [DOI] [PubMed] [Google Scholar]
- 25.Garnaas MK, Moodie KL, Liu ML, et al. Syx, a RhoA guanine exchange factor, is essential for angiogenesis in Vivo. Circ Res 2008;103: 710–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bryan BA, Dennstedt E, Mitchell DC, et al. RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. FASEB J 2010;24:3186–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei L, Surma M, Shi S, Lambert-Cheatham N, Shi J. Novel insights into the roles of rho kinase in cancer. Arch Immunol Ther Exp (Warsz) 2016;64:259–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Enslen H, Brancho DM, Davis RJ. Molecular determinants that mediate selective activation of p38 MAP kinase isoforms. EMBO J 2000;19:1301–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang YJ, Seit-Nebi A, Davis RJ, Han J. Multiple activation mechanisms of p38alpha mitogen-activated protein kinase. J Biol Chem 2006;281: 26225–26234. [DOI] [PubMed] [Google Scholar]
- 30.Djordjevic S, Driscoll PC. Targeting VEGF signalling via the neuropilin co-receptor. Drug Discov Today 2013;18:447–455. [DOI] [PubMed] [Google Scholar]
- 31.Jarvis A, Allerston CK, Jia H, et al. Small molecule inhibitors of the neuropilin-1 vascular endothelial growth factor A (VEGF-A) interaction. J Med Chem 2010;53:2215–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borriello L, Montes M, Lepelletier Y, et al. Structure-based discovery of a small non-peptidic Neuropilins antagonist exerting in vitro and in vivo anti-tumor activity on breast cancer model. Cancer Lett 2014;349:120–127. [DOI] [PubMed] [Google Scholar]
- 33.Boukamp P, Petrussevska RT, Breitkreutz D, et al. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 1988;106:761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998;92: 735–745. [DOI] [PubMed] [Google Scholar]
- 35.Migdal M, Huppertz B, Tessler S, et al. Neuropilin-1 is a placenta growth factor-2 receptor. J Biol Chem 1998;273:22272–22278. [DOI] [PubMed] [Google Scholar]
- 36.Mamluk R, Gechtman Z, Kutcher ME, et al. Neuropilin-1 binds vascular endothelial growth factor 165, placenta growth factor-2, and heparin via its b1b2 domain. J Biol Chem 2002;277: 24818–24825. [DOI] [PubMed] [Google Scholar]
- 37.Li M, Yang H, Chai H, et al. Pancreatic carcinoma cells express neuropilins and vascular endothelial growth factor, but not vascular endothelial growth factor receptors. Cancer 2004;101: 2341–2350. [DOI] [PubMed] [Google Scholar]
- 38.Jia H, Cheng L, Tickner M, et al. Neuropilin-1 antagonism in human carcinoma cells inhibits migration and enhances chemosensitivity. Br J Cancer 2010;102:541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gray MJ, Wey JS, Belcheva A, et al. Neuropilin-1 suppresses tumorigenic properties in a human pancreatic adenocarcinoma cell line lacking neuropilin-1 coreceptors. Cancer Res 2005;65: 3664–3670. [DOI] [PubMed] [Google Scholar]
- 40.Cao Y, E G, Wang E, et al. VEGF exerts an angiogenesis-independent function in cancer cells to promote their malignant progression. Cancer Res 2012;72:3912–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lampropoulou A, Ruhrberg C. Neuropilin regulation of angiogenesis. Biochem Soc Trans 2014;42:1623–1628. [DOI] [PubMed] [Google Scholar]
- 42.Cai H Reed RR Cloning and characterization of neuropilin-1-interacting protein: a PSD-95/Dlg/ZO-1 domain-containing protein that interacts with the cytoplasmic domain of neuropilin-1. J Neurosci 1999;19:6519–6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Vries L, Lou X, Zhao G, Zheng B, Farquhar MG. GIPC, a PDZ domain containing protein, interacts specifically with the C terminus of RGS-GAIP. Proc Natl Acad Sci USA 1998;95:12340–12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valdembri D, Caswell PT, Anderson KI, et al. Neuropilin-1/GIPC1 signaling regulates alpha5beta1 integrin traffic and function in endothelial cells. PLoS Biol 2009;7:e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang G, Chen L, Sun K, et al. Neuropilin-1 (NRP-1)/GIPC1 pathway mediates glioma progression. Tumour Biol 2016;37: 13777–13788. [DOI] [PubMed] [Google Scholar]
- 46.Liu M, Horowitz AA. PDZ-binding motif as a critical determinant of Rho guanine exchange factor function and cell phenotype. Mol Biol Cell 2006;17:1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cardone RA, Bagorda A, Bellizzi A, et al. Protein kinase A gating of a pseudopodial-located RhoA/ROCK/p38/NHE1 signal module regulates invasion in breast cancer cell lines. Mol Biol Cell 2005;16: 3117–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peppelenbosch MP, Qiu RG, de Vries-Smits AM, et al. Rac mediates growth factor-induced arachidonic acid release. Cell 1995;81: 849–856. [DOI] [PubMed] [Google Scholar]
- 49.Roberts LA, Glenn H, Hahn CS. Jacobson BS. Cdc42 and RhoA are differentially regulated during arachidonate-mediated HeLa cell adhesion. J Cell Physiol 2003;196:196–205. [DOI] [PubMed] [Google Scholar]
- 50.Liu Z, Wu H, Jiang K, et al. MAPK-Mediated YAP activation controls mechanical-Tension-Induced pulmonary alveolar regeneration. Cell Rep 2016;16:1810–1819. [DOI] [PubMed] [Google Scholar]
- 51.Huang D, Li X, Sun L, et al. Regulation of Hippo signalling by p38 signalling. J Mol Cell Biol 2016;8:328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muranen T, Selfors LM, Hwang J, et al. ERK and p38 MAPK activities determine sensitivity to PI3 K/mTOR inhibition via regulation of MYC and YAP. Cancer Res 2016;76:7168–7180. [DOI] [PMC free article] [PubMed] [Google Scholar]