

Abstract
Purpose:
To investigate immune tolerance induction with transient low-dose methotrexate (TLD-MTX) initiated with recombinant human acid α-glucosidase (rhGAA), in treatment-naïve cross-reactive immunologic material (CRIM)-positive infantile-onset Pompe disease (IOPD) patients.
Methods:
Newly-diagnosed IOPD patients received subcutaneous or oral 0.4 mg/kg TLD-MTX for 3 cycles (3 doses/cycle) with the first 3 rhGAA infusions. Anti-rhGAA IgG titers, classified as high-sustained (HSAT; ≥51,200, ≥2 times after 6 months), sustained intermediate (SIT; ≥12,800 and <51,200 within 12 months), or low (LT; ≤6,400 within 12 months), were compared with those of 37 CRIM-positive IOPD historic comparators receiving rhGAA alone.
Results:
Fourteen IOPD TLD-MTX recipients at the median age of 3.8 months (range, 0.7–13.5 months) had a median last titer of 150 (range, 0–51,200) at median rhGAA duration ~83 weeks (range, 36–122 weeks). One IOPD patient (7.1%) developed titers in the SIT range and one patient (7.1%) developed titers in the HSAT range. Twelve of the 14 patients (85.7%) that received TLD-MTX remained LT, versus 5/37 HSAT (peak 51,200–409,600), 7/37 SIT (12,800–51,000), and 23/37 LT (200–12,800) among comparators.
Conclusions:
Results of TLD-MTX co-initiated with rhGAA are encouraging and merit a larger longitudinal study.
Keywords: alglucosidase alfa, anti-drug antibodies, prophylactic immune tolerance induction, methotrexate, Pompe disease
INTRODUCTION
Pompe disease, also known as glycogen storage disease type II (OMIM 232300), is an autosomal recessive lysosomal storage disorder in which deficiency of the lysosomal enzyme acid α-glucosidase (GAA, EC 3.2.1.20) results in a build-up of glycogen in cardiac, skeletal, and smooth muscle of affected individuals.1 Infantile-onset Pompe disease (IOPD) presents in the first days to weeks of life, with symptoms of hypotonia, cardiomyopathy, and respiratory insufficiency. Without treatment, death usually occurs before the age of 2 years.2–4
Alglucosidase alfa (recombinant human GAA; rhGAA) was approved for enzyme replacement therapy (ERT; this abbreviation denotes rhGAA use throughout this paper) in Pompe disease in 2006.5 Its administration has been shown to improve overall and ventilator-free survival in IOPD, with improved clinical outcomes. Today, many long-term survivors have reached adolescence.6–8 However, response to ERT is often affected by an immune response, which may increase infusion-associated reactions (IgE mediated), lead to mistargeting of delivered enzyme (IgG mediated), and/or reduce clinical efficacy (IgG mediated).9 Treatment and the immune response to ERT have been shown to be related to the patient’s endogenous enzyme, commonly referred to as cross-reactive immunologic material (CRIM) status, with CRIM-negative patients developing high and sustained anti-rhGAA immunoglobulin G (IgG) antibody titers (HSAT; defined as titers of ≥51,200 at ≥2 time points at or beyond six months on rhGAA) and having a poor outcome in response to ERT compared with CRIM-positive patients.10 CRIM-negative patients cannot form the native enzyme and usually possess two severe GAA pathogenic variants; their immune systems recognize ERT as foreign and, as a result, form clinically important levels of anti-rhGAA IgG antibodies.10 CRIM-negative patients account for ~25–32% of all patients with IOPD,11,12 and are likely to have a poor clinical outcome when treated with ERT alone. In contrast, CRIM-positive patients have some residual native enzyme, whether functional or non-functional,10 and are more likely to develop lower anti-rhGAA IgG titers or none.13
In a retrospective analysis by Banugaria et al,13 39% (9 of 23) CRIM-positive IOPD patients treated with ERT had high anti-rhGAA IgG antibody titers, and their clinical outcomes were poor, similar to CRIM-negative patients; thus it was established that antibody status affects ERT response. At present, there are no predictive factors to determine which CRIM-positive patients will develop HSAT or sustained intermediate titers (SIT; defined as titers of ≥12,800 and <51,200 within the first year on rhGAA).
Immune tolerance induction (ITI) protocols have been established as an approach to minimize the development of anti-rhGAA IgG antibodies and maintain low or absent antibody titers over time. Protocols for ITI using various immunomodulating drugs have been studied for prophylaxis to preempt immune response in ERT-naïve patients14–16 and for therapy to decrease existing anti-rhGAA antibodies in ERT-treated patients with already established immune responses.14,15,17,18 However, studies in the largest number of CRIM-negative patients naïve to ERT therapy have prophylactically combined rituximab and methotrexate (e.g. one course of rituximab 375 mg/m2 IV weekly four times; methotrexate 0.4 mg/kg SC every 2 weeks), with or without intravenous immunoglobulin, in efforts to preempt an immune response.14,15,17 In the past, there was no success in patients with an entrenched immune response and antibody titers persisted after multiple immune modulating treatment regimens.19,20 Addition of the plasma cell-targeting agent bortezomib21 or other therapies in patients with established HSAT has proven successful in minimizing antibody titers, yet required prolonged immunosuppression arising from the use of maintenance doses of rituximab and methotrexate along with bortezomib.18,22
A short course of prophylactic ITI in CRIM-negative IOPD patients has improved clinical outcomes as compared with ERT monotherapy by preempting the development of HSAT and thus preventing the consequent loss of ERT efficacy. As demonstrated in a prior study by Banugaria et al.,23 a three-drug ITI regimen (methotrexate, rituximab, and intravenous immunoglobulin) initiated concurrently with ERT in 7 CRIM-negative classic IOPD patients – four patients never seroconverted (developed antibodies), 1 patient died of respiratory failure, and 2 patients required additional ITI courses, which left their antibody titers lower than in ERT-treated CRIM-negative infants without ITI. A follow-up study with the same three-drug ITI regimen in 19 CRIM-negative patients, including the 7 patients in Banugaria et al.,23 showed that 15 of 19 patients either did not seroconvert or maintained low antibody titers, in contrast to the natural course of CRIM-negative patients on ERT monotherapy. Only one of these 19 patients broke tolerance and developed HSAT. This patient was subsequently rescued using a bortezomib-based ITI protocol.24 The same prophylactic ITI regimen successfully induced tolerance in the CRIM-positive younger sibling of a CRIM-positive Pompe patient who had developed HSAT on ERT monotherapy.25 Prophylactic ITI protocols concurrent with ERT initiation are used increasingly by treating physicians worldwide and are considered a standard of care for CRIM-negative patients.
A subset of CRIM-positive patients also develop a sustained immune response,13 and it is difficult to predict which CRIM-positive individuals will develop a transient antibody response (seroconvert) or go on to develop HSAT or SIT. Anti-rhGAA antibody titers ≥12,800 were associated with an increase in ERT clearance in a pharmacokinetic study reported in the alglucosidase alfa prescribing information.26 Clinical response to ERT will likely be better conserved by preempting HSAT/SIT than by striving to reduce it once it occurs.25 Indeed, as shown in our studies, once antibodies develop to a significant titer, the intensity of immune suppression to establish tolerance is much greater than that in prophylactic regimens. Moreover, the potential for loss of viable muscle tissue due to loss of ERT activity due to such high titered immune responses is a critical consideration. Thus every effort to prevent the development of these deleterious antibodies is important and study of prophylactic ITI regimens at ERT initiation merits extension into CRIM-positive patients, given that a large subset of them develop significant antibodies likely to have deleterious clinical impact.
Preclinically, a transient low dose of methotrexate alone co-initiated with ERT sustainably reduced anti-rhGAA IgG response in a GAA knock-out mouse model of Pompe disease.27 Studies in wild-type and GAA knock-out mice suggest a mechanism for methotrexate ITI that involves an IL-10 and B regulatory cell-dependent mechanism.28,29 Other immune tolerance-inducing mechanisms may also come into play. In light of these results and given that methotrexate has a favorable safety profile in the doses proposed, is inexpensive and widely accessible, and is administered only during the initial phases of therapy, we undertook the first human protocol incorporating transient use of methotrexate at the initiation phase of ERT in naïve patients with IOPD. For ITI, we investigated a transient, low dose of methotrexate administered only during the first three ERT infusions and then stopped (Figure 1). This is referred to as the transient low-dose methotrexate (TLD-MTX) protocol. We hypothesized that CRIM-positive patients with Pompe disease who receive methotrexate in this manner in the naïve setting will not seroconvert or will have a blunted immune response in comparison with the CRIM-positive patients treated with ERT monotherapy.
Figure 1.
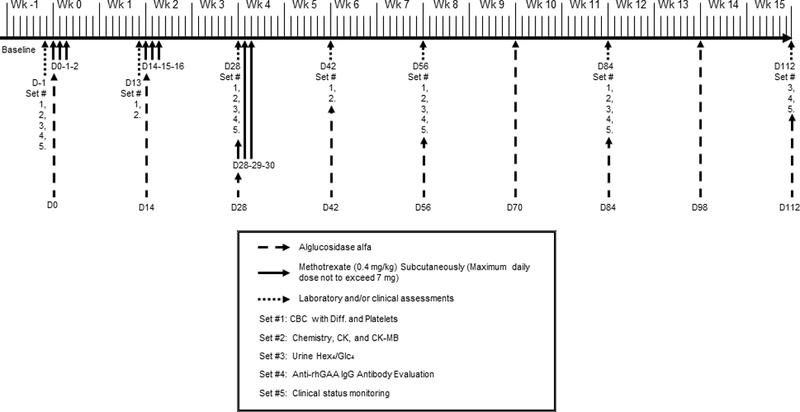
Study timeline for infantile Pompe disease patients receiving ERT infusions every other week.D, day, Wk, week, mg, milligram, kg, kilogram, CBC with diff., complete blood count with differential, CK, creatinine kinase, Hex4/Glc4, glucose tetrasaccharide, rhGAA, recombinant human acid-alpha glucosidase, IgG, immunoglobulin G.
PATIENTS AND METHODS
Patients and inclusion and exclusion criteria
Patients with a confirmed diagnosis of IOPD (two confirmed GAA pathogenic variants) and a low GAA enzyme activity (in blood, muscle, or skin) who were CRIM-positive and ERT-naïve were enrolled in the study. CRIM status was determined by Western blot analysis and confirmed by GAA genotype or was predicted from GAA genotype alone.11 IOPD was defined as symptom onset at ≤12 months of age with cardiomyopathy.
Upon a new diagnosis of Pompe disease, the detailed protocol was shared with the patient’s local treating physician, including the dosing schedule of methotrexate, IgG antibody monitoring guidelines, and laboratory analyses for safety measures. Follow-up data from the local treating team were requested every 3 months. Data collection was completed June 16, 2017 or when at least 6 months of follow-up data were available. If a patient broke tolerance their latest immune responses beyond June 16, 2017 have been presented.
Previously described methods were used for determining CRIM status,11,12 GAA pathogenic variants,7 and anti-rhGAA IgG antibody titers.7 Clinical outcomes used were overall survival and ventilator free survival for patients who received TLD-MTX protocol.
Ethical approval and parent, guardian, or adult patient consent
All patients were enrolled in an Institutional Review Board (IRB)-approved study. Thirteen patients were enrolled in a Duke IRB (Hock Plaza, Suite 405, 2424 Erwin Road, Campus Box #2712, Durham, NC 27705, USA)-approved protocol 00001562 (LDN6709 Site 206; https://clinicaltrials.gov NCT01665326; Determination of Cross-Reactive Immunological Material (CRIM) Status and Longitudinal Follow-up of Individuals with Pompe disease). One patient was enrolled in an IRB-approved study at an institution local to the patient (Shaare Zedek Medical Center, Jerusalem, Israel). Informed consent was obtained in writing from parent(s) or legal guardian(s) of minor patients for their respective studies.
Study design
The study timeline for ERT and methotrexate dosing is shown in Figure 1. Patients received intravenous (IV) infusions of ERT at 20 mg/kg body weight every other week (EOW) based on the package insert or on a different regimen based on decision of the treating clinician.
Methotrexate at 0.4 mg/kg body weight was administered subcutaneously (or orally if subcutaneous administration was not possible) on three consecutive days/MTX cycle/infusion; with the first three ERT infusions (i.e., on Days 0, 14, and 28 for patients infused EOW) for a total of 3 cycles. On the day of ERT infusion, methotrexate was administered 15 minutes (if subcutaneous) or 1 hour (if oral) before infusion initiation and again on the following 2 days. To exemplify the schedule, in a patient receiving ERT infusion EOW, methotrexate was administered on Days 0, 1, and 2 (first cycle); Days 14, 15, and 16 (second cycle), and Days 28, 29, and 30 (third cycle). As stated earlier, the dose and frequency of ERT could be increased at the discretion of the treating physician, based on patients’ clinical status, in which case methotrexate cycles were adjusted accordingly (see Figure S1 for details).
The methotrexate dose was stipulated to be withheld if the absolute neutrophil count (ANC) was <750/mm3 or liver function tests (aspartate aminotransferase [AST] and alanine aminotransferase [ALT]) were >3 times their respective baseline values. Folinic acid supplementation, although not directed in the protocol, could be given at the treating physician’s discretion if there was a concern with methotrexate toxicity.
Immune response
Anti-rhGAA IgG antibodies were determined by Sanofi Genzyme (Framingham, MA, USA) using enzyme-linked immunosorbent assays and confirmed using radioimmunoprecipitation as described previously.7 Antibody analyses were recommended to be performed at Days –1, 28, 56, 84, 112, 140, 168, 196, 224, 252, 280, 308, 336, and 364.
Hematologic and biochemical analyses
On Days –1 (baseline), 13, 28, 42, 56, 84, 168, 252, 336, and 364, blood samples were recommended to be collected for hematologic and biochemical analyses for patients receiving ERT EOW. The sampling schedule was modified for patients receiving ERT weekly (Figure S1). Complete and differential blood counts and platelets were evaluated and monitored for platelet counts <50,000/mm3, ANC <750/mm3, and treatment-resistant infections. Biochemical analyses included ALT, AST, creatine kinase (CK), and CK-MB levels. Post-treatment increases in AST and ALT levels were noted if exceeding three times their respective baseline levels.
Statistical analysis
Patients were classified into HSAT (≥51,200 on ≥2 occasions at or beyond 6 months on ERT13,22), SIT (≥12,800 and <51,200 within the first year of ERT), and low titer (LT; ≤6,400 within the first year of ERT) groups based on the longitudinal anti-rhGAA IgG titers.23 The lower bound of SIT, ≥12,800, was associated with increase in ERT clearance in a pharmacokinetic study reported in the alglucosidase alfa prescribing information; the upper bound of LT, ≤6,400, was 1 dilution level below this. Medians and ranges were determined for baseline, peak, and last titers, although our focus was on longitudinal titers through time because sustained titers affect clinical response.30
Comparator group for CRIM-positive IOPD
A retrospective chart review of 37 CRIM-positive patients with IOPD was conducted from the original rhGAA clinical trials, including patients on ERT for ≥6 months who did not receive immunomodulation.6,7,10,31,32 The GAA variant data and age at ERT initiation were compared with our cohort of TLD-MTX recipients. Immune responses over time were classified into HSAT, SIT, and LT as defined earlier, and time to seroconversion, peak titers, titers at Weeks 12, 24, and 52 were compared with our present cohort of TLD-MTX recipients.
RESULTS
Patient disposition
Fourteen ERT-naïve patients with Pompe disease received TLD-MTX protocol at the median age of 3.8 months (range, 0.7–13.5 months). Longitudinal follow-up data of >6 months were available for 14 patients with IOPD (13 CRIM-positive patients and 1 CRIM-negative patient). Of the 28 total GAA variants in the TLD-MTX group, 18 (64.3%), 3 (10.7%), 3 (10.7%), 3 (10.7%), and 1 (3.6%) were missense, frameshift, initiator codon, nonsense, and splice site variants, respectively. The patients’ baseline demographics are presented in Table 1. The CRIM-negative patient (IOPD9) who received TLD-MTX protocol is an international patient initially thought to be CRIM-positive. No patients in this cohort were diagnosed via newborn screening.
Table 1.
Baseline demographic characteristics of IOPD patients that received TLD-MTX protocol
| Patient | Sex | GAA pathogenic variants | Age at start of ERT | ERT dose and frequency1 | Route of MTX administration | TLD-MTX Protocol deviation | |
|---|---|---|---|---|---|---|---|
| Variant 1 | Variant 2 | ||||||
| IOPD1 | M | c.953T>C | c.1292_1295dupTGCA | 0.9 months | 20 mg/kg weekly | MTX administered SC | Only one dose administered in 2nd cycle of MTX |
| p.Met318Thr | p.Gln433AlafsX74 | ||||||
| IOPD2 | M | c.1004G>A | c.1841C>A | 3.3 months | 20 mg/kg EOW | NA | None |
| p.Gly335Glu | p.Thr614Lys | ||||||
| IOPD3 | M | c.1118T>G | c.1118T>G | 4.0 months | 20 mg/kg EOW | MTX administered SC | Only one dose administered in 2nd & 3rd cycle each and additional 4th cycle was administered |
| p.Leu373Arg | p.Leu373Arg | ||||||
| IOPD4 | F | c.2560C>T | c.1466A>G | 0.8 months | 20 mg/kg EOW | MTX administered SC | 3rd cycle of MTX & ERT was administered on Week 5 instead of Week4 |
| p.Arg854X | p.Asp489Gly | ||||||
| IOPD5 | F | c.665T>G | c.1437+2T>C | 3.5 months | 20 mg/kg weekly | MTX administered SC | None |
| p.Val222Gly | Deletion p.Asp443_Lys479del | ||||||
| IOPD6 | F | c.1114C>T | c. 1979G>A | 11.2 months | 20 mg/kg EOW | MTX administered PO | None |
| p. His372Tyr | p.Arg660His | ||||||
| IOPD7 | F | c.2456G>C | c.2456G>C | 1.2 months | 20 mg/kg EOW | MTX administered SC | None |
| p.Arg819Pro | p.Arg819Pro | ||||||
| IOPD8 | F | c.525delT | c.2297A>C | 4.6 months | 20 mg/kg EOW | MTX administered SC | 3rd cycle of MTX & ERT was administered on Week 5 instead of Week4 |
| p.Glu176Argfs*45 | p.Tyr766Ser | ||||||
| IOPD92 | F | c.1A>G | c.1A>G | 12.9 months | 20 mg/kg EOW | MTX administered SC | None |
| Initiator codon | Initiator codon | ||||||
| IOPD10 | F | c.1942G>A | c.1942G>A | 1.7 months | 20 mg/kg EOW | MTX administered SC | 3rd dose of 3rd cycle of MTX was skipped |
| p.Gly648Ser | p.Gly648Ser | ||||||
| IOPD11 | F | c.1447G>A | c.2560C>T | 13.5 months | 20 mg/kg EOW | 3rd dose of 3rd cycle was given orally, all other MTX administered SC | None |
| p.Gly483Arg | p.Arg854X | ||||||
| IOPD12 | M | c.1A>G | c.2234T>C | 0.7 months | 20 mg/kg EOW | MTX administered SC | None |
| Initiator codon | p.Leu745Pro | ||||||
| IOPD13 | F | c.1979G>A | c.2560C>T | 4.5 months | 20 mg/kg EOW3 | MTX administered SC | None |
| p.Arg660His | p.Arg854X | ||||||
| IOPD14 | F | c.525delT | c.1979G>A | 4.0 months | 20 mg/kg EOW | MTX administered PO | 1st MTX dose of 3rd cycle was skipped |
| p.Glu176Argfs*45 | p.Arg660His | ||||||
TLD-MTX, transient low-dose methotrexate, ANC, absolute neutrophil count; IOPD, infantile-onset Pompe disease, EOW, every other week, PO, orally (per os), SC, subcutaneous, ERT, enzyme replacement therapy.
ERT dose and frequency was determined based on the clinical judgement of the treating physician.
Patient IOPD9 is CRIM-negative.
ERT dose was changed to 40 mg/kg/weekly after two infusions.
ERT-monotherapy treated CRIM-positive patients (Comparator group)
Thirty-seven CRIM-positive patients were identified who received ERT-monotherapy at a median age of 6.9 months (range, 0.5–43.1 months). Age at ERT start was later in ERT-monotherapy group (median, 6.9 months) as compared with TLD-MTX group (median, 3.8 months). Of the 74 total GAA variants in the ERT monotherapy group, 47 (63.5%), 6 (8.1%), 1 (1.4%), 7 (9.5%), 2 (2.7%), 7 (9.5%), and 4 (5.4%) were missense, frameshift, initiator codon, nonsense, splice site, unknown, and in-frame deletion variants, respectively. The GAA variant data in the CRIM-positive patients in the comparator group (ERT-monotherapy) were similar to those in the TLD-MTX group (Figure S3).
Five (13.5%), 7 (18.9%), and 25 (67.6%) of the 37 IOPD monotherapy patients developed HSAT, SIT, and LT, respectively; thus, 32.4% developed SIT or HSAT overall. All HSAT patients seroconverted by 4 weeks; for the SIT and LT patients, median time to seroconversion was 4 weeks (4–8 weeks, n=7) and 8 weeks (4–64 weeks, n=23), respectively; 2 patients did not seroconvert. Group median peak titers were: 204,800 (range, 51,200–409,600) for HSAT (median time of peak, Week 82); 25,600 (range, 12,800–51,200) for SIT (Week 12); and 800 (range, 200–12,800) for LT (Week 38). Group median last titers (at group median times on ERT) were: 102,400 for HSAT (Week 94) (range, 51,200–409,600); 1,600 for SIT (Week 104) (range, 200–25,600); and 400 for LT (Week 130) (range, 0–12,800); individual data are shown in Figure 2. These data were used for comparison with the present TLD-MTX-treated IOPD patients (Figure 2 & Figure S2). Comparison of median anti-rhGAA IgG antibody titers at Weeks 12, 24, and 52, and median peak titers is shown in Table 2.
Figure 2.
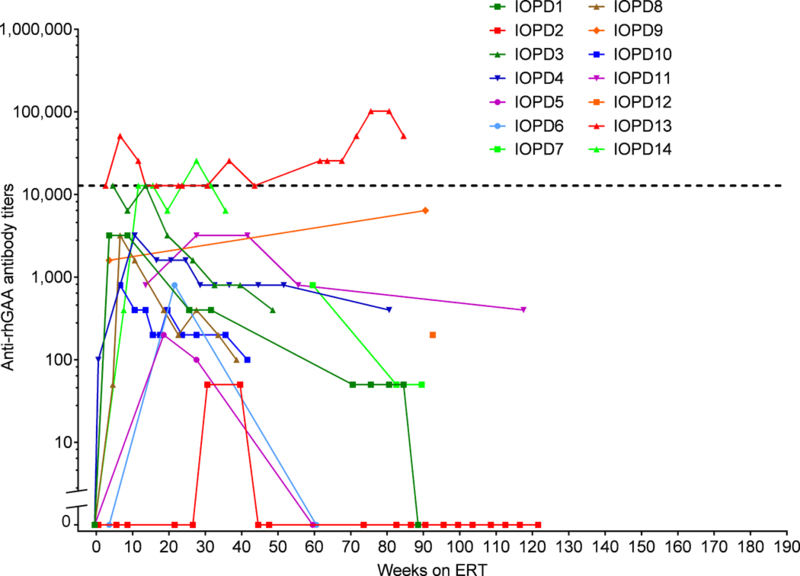
Immune response over time in the current TLD-MTX-treated CRIM-positive IOPD patients. Each patient’s trajectory is graphed individually as a curve. Positive titers <100 are shown as titers of 50.IOPD, infantile-onset Pompe disease, TLD-MTX, transient low-dose methotrexate, CRIM, cross-reactive immunological material, ERT, enzyme replacement therapy, rhGAA, recombinant human acid alpha glucosidase
Table 2.
Comparison of longitudinal immune response between ERT monotherapy and TLD-MTX groups1
| CRIM-positive IOPD on ERT monotherapy (n=37) | |||
| Groups | HSAT | SIT | LT |
| Percent | 13.5% (n=5) | 18.9% (n=7) | 67.6% (n=25) |
|
Median titers (Week 12) |
25,600 (range: 12,800–51,200) |
12,800 (range: 6,400–25,600) |
400 (range: 0–6,400) |
|
Median titers (Week 24) |
25,600 (range: 6,400–204,800) |
12,800 (range: 6,400–51,200) |
400 (range: 0–3,200) |
|
Median titers (Week 52) |
51,200 (range: 12,800–102,400) |
3,200 (range: 400–12,800) |
200 (range: 0–6,400) |
| Median peak titers | 204,800 (range: 51,200–409,600) |
25,600 (range: 12,800–51,200) |
800 (range: 0–12,800) |
| Time since ERT initiation | 82 weeks (range: 24–130) |
12 weeks (range: 8–24) |
38 weeks (range: 8–172) |
| Seroconversion week | 4 weeks (range: 4) |
4 weeks (range: 4–8) |
8 weeks (range: 4–64) |
| IOPD on ERT + TLD-MTX (n=14) | |||
| Groups | HSAT | SIT | LT |
| Percent | 0 | 14.2% (n=2) | 85.7% (n=12)2 |
|
Median titers (Week 12) |
NA | IOPD13: 12,800; IOPD14: 25,600 |
1,600 (range: 0–12,800) |
|
Median titers (Week 24) |
NA | IOPD13: 12,800; IOPD14: 12,800 |
600 (range: 0–3,200) |
| Median titers (Week 52) | NA | IOPD13: 12,800 | 2503 (range: 0–800) |
| Median peak titers | NA | IOPD13: 51,200; IOPD14: 12,800 |
3,200 (range: <100–12,800) |
| Time since ERT initiation | NA | IOPD13: 7 weeks; IOPD14: 12 weeks |
20.5 weeks (range: 4–93) |
| Seroconversion week | NA | IOPD13: 3 weeks; IOPD14: 8 weeks |
6 weeks (range: 4–31) |
HSAT, high and sustained antibody titers, SIT, sustained intermediate titers, LT, low titers, IOPD, infantile-onset Pompe disease, TLD-MTX, transient low-dose methotrexate, CRIM, cross-reactive immunological material, ERT, enzyme replacement therapy, NA, not applicable
All patients in ERT + TLD-MTX group and 35/37 patients in ERT monotherapy group seroconverted.
One patient had peak titers of 12,800 twice (Week 5 and Week 12) on ERT but maintained low titers throughout, so was included in LT group.
Patients who tested seropositive but had titers <100 were deemed to have a titer of 50.
Immune Response—Transient Low-Dose Methotrexate Recipients
The immune response for each TLD-MTX treated patient over time is shown in Figure 2, and summarized in Table 2 and Table 3. The median last titer was 150 (range, 0–51,200) at median time on ERT of ~83 weeks (range, 36–122 weeks). One IOPD patient (IOPD14; 7.1%) developed titers in the SIT range and one patient (IOPD13; 7.1%) developed titers in the HSAT range.). Patient IOPD13 had a titer of 102,400 at Weeks 76 and 81 decreasing to 51,200 at Week 85. Patient IOPD14 had a titer of 25,600 at Week 28 only, and maintained titers of 12,800 at Weeks 12, 16, and 24, and 32 decreasing to 6,400 at week 36. These 2 TLD-MTX recipient patients (IOPD13 and IOPD14) had the highest titers observed in the study; the other 12 TLD-MTX recipients remained LT. One patient (IOPD2) had titers remaining <100 (seropositive on screening assay, but below the limit of titer measurement).
Table 3.
Immune response for IOPD patients that received TLD-MTX protocol with >6 months of follow-up data available
| Patient | Peak antibody titer | Time on ERT (peak titer) (weeks) | Last antibody titer | Time on ERT (last titer) (weeks) |
|---|---|---|---|---|
| IOPD1 | 3,200 | 4 | 0 | 89 |
| IOPD2 | <100 | 31 | 0 | 122 |
| IOPD3 | 12,8001 | 5 | 400 | 49 |
| IOPD4 | 3,200 | 11 | 400 | 81 |
| IOPD5 | 200 | 19 | 0 | 60 |
| IOPD6 | 800 | 22 | 0 | 61 |
| IOPD7 | 800 | 60 | <100 | 90 |
| IOPD8 | 3,200 | 7 | 100 | 39 |
| IOPD9 | 6,400 | 91 | 6,400 | 91 |
| IOPD10 | 800 | 7 | 100 | 42 |
| IOPD11 | 3,200 | 28 | 400 | 118 |
| IOPD12 | 200 | 93 | 200 | 93 |
| IOPD13 | 102,4002 | 76 | 51,200 | 85 |
| IOPD14 | 25,6003 | 28 | 6,400 | 36 |
| Median | 3,200 | 25 | 150 | 83 |
IOPD, infantile-onset Pompe disease, TLD-MTX, transient low-dose methotrexate, ERT, enzyme replacement therapy.
Patient IOPD3 only received a single dose of methotrexate in cycles 2 and 3 instead of 3 doses.
Patient IOPD13 had an increase in dose of ERT from 20 mg/kg/EOW to 40 mg/kg/EOW at Week 4 on ERT.
Patient IOPD14 had skipped a dose of methotrexate on cycle 3.
Safety
Hematologic and biochemical analyses
ANC, AST, and ALT data in the first 6 weeks on ERT were available for all patients except IOPD7 (missing all 3 measures), and IOPD2 and IOPD13 (missing baseline AST and ALT). Patients IOPD1 and IOPD3 developed ANC <750 cells/mm3. Neutropenia in these two patients was transient, and ANC returned to normal levels. Patient IOPD5 developed increases exceeding three times baseline levels of both AST and ALT; patients IOPD3 and IOPD6 had such increases only in ALT. Infection incidence during the time of TLD-MTX administration was available on nine patients. Two patients had hospitalizations related to infections around the time of methotrexate administration. One patient (IOPD8) was hospitalized for rhinovirus infection and the other patient (IOPD13) was hospitalized for concerns of underlying respiratory infections. No infections were noted in the remaining seven patients (IOPD1, IOPD5, IOPD7, IOPD9, IOPD10, IOPD12, and IOPD14).
Clinical outcome
Overall and ventilator-free survival
At baseline, four patients (IOPD2, IOPD3, IOPD5, and IOPD10) were invasively ventilated, two patients (IOPD8 and IOPD13) needed BiPAP support, one patient (IOPD14) needed high flow nasal cannula support, and seven patients required no support. Two of these fourteen patients, IOPD3 and IOPD14, died at the age of 20 months and 15 months, respectively. The cause of death was cardiorespiratory failure secondary to the progression of disease in IOPD3 and respiratory arrest secondary to viral infection during the flu season for IOPD14. For the remaining twelve surviving patients only one patient (IOPD2) needed ventilator support which was only needed during sleep. The remaining eleven patients do not need any support. The median age for the 12 surviving patients was 4.1 years (range: 2.3 – 5.7 years).
DISCUSSION
We have developed an ITI protocol using TLD-MTX, a brief-course (a total of 9 doses) of methotrexate in patients with CRIM-positive IOPD, which may also be considered for CRIM-negative IOPD patients lacking access to rituximab. We demonstrate the feasibility of implementation of this protocol in 13 centers in United States of America, Israel, and India, a collaboration that demonstrates how physicians can work together worldwide to leverage clinical learning in rare diseases and in this case, mitigate the deleterious impact of immune response on ERT therapy. Brief-course, low-dose, single-drug methotrexate administered concurrently with rhGAA initiation in treatment-naïve patients with Pompe disease resulted in the cohort’s median last titer of 150 (range, 0–51,200) at median time of 83 weeks (range, 36–122 weeks) of ERT. One patient (IOPD13) had antibodies in the HSAT range and one patient (IOPD14) had antibodies in the SIT range. All other methotrexate recipients remained LT throughout, unlike the responses in the comparator group. IOPD patients in the comparator group had a similar GAA variant profile. Importantly, all patients in this study were identified clinically and thus were symptomatic at time of diagnosis, again making the two groups very comparable. We collected data on overall and invasive ventilator-free survival on TLD-MTX recipient patients. Two of these fourteen patients (IOPD3 and IOPD14) were deceased and only one patient (IOPD2) needed ventilator support at night. The main focus of this study was ameliorating the deleterious antibody response to rhGAA. The deleterious effect of antibody titers in HSAT and SIT on cardiac and motor response has been previously published. 10,13
Two patients (IOPD13 and IOPD14) developed antibodies in SIT/HSAT range. Patient IOPD13 had an increase in dose of ERT from 20 mg/kg/EOW to 40 mg/kg/EOW at Week 4 on ERT. The bortezomib-based protocol was recommended for this patient with HSAT, but the parents declined. Importantly, patient IOPD14 whose last titer was 6,400 (Week 36 had skipped a dose of methotrexate on cycle 3.
It is important to note that 32.4% of patients in comparator group developed HSAT or SIT vs 14.2% of patients in TLD-MTX group. It appears that, based on expectations from historical data, TLD-MTX possibly blunted the immune response overall as demonstrated by the finding that 86.7% of patients in our cohort maintained low titers. Our patient cohort was younger, which could be more indicative of severe disease or earlier diagnosis. None of the patients was identified via newborn screening and were all clinically identified through clinical features of infantile Pompe disease including cardiomyopathy.
One of the patients was CRIM-negative, suggesting that TLD-MTX can be attempted in CRIM-negative patients in parts of the world where ITI with rituximab is not feasible. A study on a larger CRIM-negative cohort would be needed to establish the efficacy of TLD-MTX in that population for this purpose.
No serious AEs were related to methotrexate. Some patients had methotrexate doses postponed because of their clinical status. Neutropenia (ANC <750 cells/mm3) affected 2 of 13 IOPD patients with ANC data (15.4%; IOPD1 and IOPD3); this was transient, and ANC returned to normal levels. Two of nine patients had hospitalizations related to infections around the time of methotrexate administration with complete resolution. Overall, there was a reasonably good tolerance to the methotrexate protocol in these fragile patients.
Our protocol uses only one-fourth to one-seventh of the methotrexate dose typical for cancer chemotherapy, and its brief time course avoids prolonged immunosuppression. Methotrexate is inexpensive and has a wide geographic availability; its lack of B-cell suppression may confer an advantageous safety profile in CRIM-positive patients as opposed to combination regimens including rituximab.15,23 TLD-MTX is also less expensive than regimens including biologics or lengthy immunosuppression.
Our data show the importance of 3 cycles of methotrexate for clinical efficacy. While 1-cycle methotrexate has not been studied systematically for ITI in humans, our patient who received less than 3-cycle seemed to have a higher immune response than those who followed the 3-cycle protocol. Our study did not formally compare 1 and 3 cycles (which had similar preclinical results); nonetheless, we hypothesize that 3 cycles are beneficial at this low dose to maintain B-cell regulation, without suppressing immunity altogether, as would be expected of high-dose methotrexate.
This report of clinical ITI experience using single-drug methotrexate is confined to CRIM-positive patients, except for IOPD9, who was predicted to be CRIM-negative based on GAA variants. Further study is needed to determine potential applicability of this methotrexate protocol in a larger cohort of CRIM-positive patients and in CRIM-negative patients in regions where rituximab is not available. The TLD-MTX protocol requires specific evaluation before being applied in CRIM-negative patients, the highest-risk group; any empirical clinical use that is attempted will provide valuable experiential data. Further study is warranted in a larger cohort of IOPD patients, and should also evaluate longer-term outcomes such as overall survival, ventilator free survival, reduction in left ventricular mass index, and urinary glucose tetrasaccharide (Glc4).
Preclinical findings33 suggest that with further study, TLD-MTX, which is given as a brief course at enzyme initiation (unlike ITI regimens requiring re-administration over the course of treatment), may be appropriate for prevention of immune responses in other diseases treated by therapeutic enzymes. Other incipient immunomodulation methods are also being studied, such as anti-CD4 monoclonal antibodies,34 Tregitopes (peptides stimulating regulatory T cell expansion and activation),35 rapamycin,16 or tolerogenic nanoparticles containing rapamycin.36 Anti-drug antibody responses and the approaches needed to preempt them may differ among diseases and therapeutic proteins. Further study is needed to understand the mechanism of methotrexate effects, evaluate safety and efficacy in larger CRIM-positive and CRIM-negative cohorts, and monitor long-term outcomes of TLD-MTX-only ITI in Pompe disease.
Supplementary Material
Acknowledgments.
This study was supported in part by Sanofi Genzyme and the Lysosomal Disease Network (U54NS065768). Karen Welch and Christopher Abhulime of Sanofi Genzyme contributed laboratory antibody expertise and maintained validation of the assays used. Dr. Rick Fang of Sanofi Genzyme provided pharmacokinetic and pharmacodynamic modeling. Contributing care givers Ms. Stephanie Dearmey, MHS, PA-C, Dr. Einat Zivi, Dr. Dhanya Yesodharan, Ms. Kathleen Crosby, MS CGC, Ms. Jennifer Goodwin, BS, CCRC, Ms. Jennifer Cook, CPNP, Mr. Matthew Stein, Ms. Valerie Marrero-Stein, CPNP, and late Dr. James Weisfeld-Adams, MB, ChB are acknowledged for patient care and data acquisition. The authors acknowledge editorial assistance of Kim Coleman Healy, PhD, CMPP and Jane M. Gilbert, BSc, CMPP, of Envision Scientific Solutions, which was contracted by Sanofi to provide publication support services.
Footnotes
Conflicts of Interest
Zoheb B. Kazi: Grant support: Lysosomal Disease Network; Sanofi Genzyme Ankit K. Desai: Grant support: Sanofi Genzyme
Bradley Troxler: None
David Kronn: Research funding: Sanofi Genzyme and New York Medical College
Seymour Packman: Recipient of a research grant from Sanofi Genzyme, Member of Sanofi Genzyme speakers’ bureau, Clinical trial of an unrelated product of Sanofi Genzyme
Marta Sabbadini: None
William Rizzo: None
Katalyn Scherer: None
Omar Abdul-Rahman: None
Pranoot Tanpaiboon: None
Sheela Nampoothiri: None
Neerja Gupta: None
Annette Feigenbaum: None
Dmitriy Niyazov: Speaker Bureau of Sanofi Genzyme
Sherry Langston: None
Reeval Seegal: None
Alison McVie-Wylie: Employment: Sanofi Genzyme
Crystal Sung: Employment: Sanofi Genzyme
Alexandra M. Joseph: Employment: Sanofi Genzyme
Susan Richards: Employment: Sanofi Genzyme
Priya S. Kishnani: Research support, honoraria, and Pompe and Gaucher Disease Registries’ advisory board membership: Sanofi Genzyme; grants: Shire Pharmaceuticals, Valerion; Amicus; personal fees: Alexion Pharmaceuticals, Inc., Amicus Therapeutics, Shire Pharmaceuticals; advisory board membership: Baebies, Inc.
REFERENCES
- 1.Hirschhorn R, Reuser AJJ. 135: Glycogen storage disease type II: acid a-glucosidase (acid maltase) deficiency. In: Valle D, Beaudet AL, Vogelstein B, Kinzler K, Antonarakis SE, Ballabio A, Gibson KM, Mitchell G, editors. OMMBID The Online Metabolic & Molecular Bases of Inherited Disease. New York: McGraw-Hill. [Google Scholar]
- 2.Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 2006;148:671–676. [DOI] [PubMed] [Google Scholar]
- 3.van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003;112:332–340. [DOI] [PubMed] [Google Scholar]
- 4.Kishnani PS, Amartino HM, Lindberg C, et al. Timing of diagnosis of patients with Pompe disease: data from the Pompe registry. Am J Med Genet A 2013;161A:2431–2443. [DOI] [PubMed] [Google Scholar]
- 5.Genzyme Corporation. MYOZYME® (alglucosidase alfa) Prescribing information. Available at: https://www.myozyme.com/~/media/MyozymeUS/Files/Documents/mz_pi.pdf. Accessed June 23, 2016.
- 6.Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 2007;68:99–109. [DOI] [PubMed] [Google Scholar]
- 7.Kishnani PS, Nicolino M, Voit T, et al. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr 2006;149:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicolino M, Byrne B, Wraith JE, et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med 2009;11:210–219. [DOI] [PubMed] [Google Scholar]
- 9.Kishnani PS, Dickson PI, Muldowney L, et al. Immune response to enzyme replacement therapies in lysosomal storage diseases and the role of immune tolerance induction. Mol Genet Metab 2016;117:66–83. [DOI] [PubMed] [Google Scholar]
- 10.Kishnani PS, Goldenberg PC, DeArmey SL, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab 2010;99:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bali DS, Goldstein JL, Banugaria S, et al. Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: lessons learned from 10 years of clinical laboratory testing experience. Am J Med Genet C Semin Med Genet 2012;160C:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berrier KL, Kazi ZB, Prater SN, et al. CRIM-negative infantile Pompe disease: characterization of immune responses in patients treated with ERT monotherapy. Genet Med 2015;17:912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banugaria SG, Prater SN, Ng YK, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med 2011;13:729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lacaná E, Yao LP, Pariser AR, Rosenberg AS. The role of immune tolerance induction in restoration of the efficacy of ERT in Pompe disease. Am J Med Genet C Semin Med Genet 2012;160C:30–39. [DOI] [PubMed] [Google Scholar]
- 15.Messinger YH, Mendelsohn NJ, Rhead W, et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet Med 2012;14:135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elder ME, Nayak S, Collins SW, et al. B-Cell depletion and immunomodulation before initiation of enzyme replacement therapy blocks the immune response to acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr 2013;163:847–854 e841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendelsohn NJ, Messinger YH, Rosenberg AS, Kishnani PS. Elimination of antibodies to recombinant enzyme in Pompe’s disease. N Engl J Med 2009;360:194–195. [DOI] [PubMed] [Google Scholar]
- 18.Deodato F, Ginocchio VM, Onofri A, Grutter G, Germani A, Dionisi-Vici C. Immune tolerance induced using plasma exchange and rituximab in an infantile Pompe disease patient. J Child Neurol 2014;29:850–854. [DOI] [PubMed] [Google Scholar]
- 19.Hobson-Webb LD, Proia AD, Thurberg BL, Banugaria S, Prater SN, Kishnani PS. Autopsy findings in late-onset Pompe disease: a case report and systematic review of the literature. Mol Genet Metab 2012;106:462–469. [DOI] [PubMed] [Google Scholar]
- 20.Hunley TE, Corzo D, Dudek M, et al. Nephrotic syndrome complicating alpha-glucosidase replacement therapy for Pompe disease. Pediatrics 2004;114:e532–535. [DOI] [PubMed] [Google Scholar]
- 21.Banugaria SG, Prater SN, McGann JK, et al. Bortezomib in the rapid reduction of high sustained antibody titers in disorders treated with therapeutic protein: lessons learned from Pompe disease. Genet Med 2013;15:123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banugaria SG, Patel TT, Mackey J, et al. Persistence of high sustained antibodies to enzyme replacement therapy despite extensive immunomodulatory therapy in an infant with Pompe disease: need for agents to target antibody-secreting plasma cells. Mol Genet Metab 2012;105:677–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banugaria SG, Prater SN, Patel TT, et al. Algorithm for the early diagnosis and treatment of patients with cross reactive immunologic material-negative classic infantile pompe disease: a step towards improving the efficacy of ERT. PLoS One 2013;8:e67052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kazi ZB, Desai AK, Berrier KL, et al. Sustained immune tolerance induction in enzyme replacement therapy-treated CRIM-negative patients with infantile Pompe disease. JCI Insight 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stenger EO, Kazi Z, Lisi E, Gambello MJ, Kishnani P. Immune tolerance strategies in siblings with infantile Pompe disease-Advantages for a preemptive approach to high-sustained antibody titers. Mol Genet Metab Rep 2015;4:30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Genzyme Corporation. LUMIZYME® (alglucosidase alfa) Prescribing information. Aug 2014. Available at: https://www.lumizyme.com/~/media/LumizymeUS/Files/lumizyme_prescribing_information.pdf. Accessed Nov 24, 2016.
- 27.Joseph A, Munroe K, Housman M, Garman R, Richards S. Immune tolerance induction to enzyme-replacement therapy by co-administration of short-term, low-dose methotrexate in a murine Pompe disease model. Clin Exp Immunol 2008;152:138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joly MS, Martin RP, Mitra-Kaushik S, et al. Transient low-dose methotrexate generates B regulatory cells that mediate antigen-specific tolerance to alglucosidase alfa. J Immunol 2014;193:3947–3958. [DOI] [PubMed] [Google Scholar]
- 29.Mauri C, Menon M. The expanding family of regulatory B cells. Int Immunol 2015;27:479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abbott MA, Prater SN, Banugaria SG, et al. Atypical immunologic response in a patient with CRIM-negative Pompe disease. Mol Genet Metab 2011;104:583–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.CORRECTION. Neurology 2008;71:1748. [Google Scholar]
- 32.Kazi ZB, Desai AK, Erwin A, et al. Prophylactic immune modulation in infantile Pompe disease using low-dose methotrexate induction: A safe, inexpensive, widely accessible, and efficacious strategy. Mol Genet Metab 2016;117:S65–S66. [Google Scholar]
- 33.Garman RD, Munroe K, Richards SM. Methotrexate reduces antibody responses to recombinant human alpha-galactosidase A therapy in a mouse model of Fabry disease. Clin Exp Immunol 2004;137:496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun B, Banugaria SG, Prater SN, et al. Non-depleting anti-CD4 monoclonal antibody induces immune tolerance to ERT in a murine model of Pompe disease. Mol Genet Metab Rep 2014;1:446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cousens LP, Mingozzi F, van der Marel S, et al. Teaching tolerance: New approaches to enzyme replacement therapy for Pompe disease. Hum Vaccin Immunother 2012;8:1459–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lim HH, Yi HQ, Kishimoto TK, Gao FQ, Sun BD, Kishnani PS. Immunomodulation to enzyme replacement therapy with tolerogenic nanoparticles containing rapamycin in a murine model of Pompe disease. Mol Genet Metab 2017;120:S83–S84. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.