

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is the most common renal genetic disorder, however it still lacks a cure. The discovery of new therapies heavily depends on understanding key signalling pathways that lead to ADPKD. The JAnus Kinase and Signal Transducers and Activators of Transcription (JAK/STAT) pathway is aberrantly activated and contributes to ADPKD pathogenesis via enhancing epithelial proliferation. Yet the mechanisms underlying the upregulation of JAK/STAT activity in this disease context is completely unknown. Here, we investigate the role of JAK2 in ADPKD using a murine model of ADPKD (Pkd1nl/nl). In normal kidneys, JAK2 expression is limited to tubular epithelial and vascular cells with lesser staining in bowman’s capsule and remains below detection level in the interstitium. By contrast, in kidneys of mice with ADPKD, JAK2 is higher in cyst-lining cells when compared to normal tubules and critically, it is ectopically expressed in the interstitium, suggesting that ectopic JAK2 may contribute to ADPKD. JAK2 activity was inhibited using either curcumin, a natural compound with strong JAK2 inhibitor activity, or Tofacitinib, a clinically used selective JAK small molecule inhibitor. JAK2 inhibition led to significantly reduced tyrosine phosphorylation of STAT3 and markedly reduced cystic growth of human and mouse ADPKD-derived cells in cystogenesis assays. Taken together, our results indicate that blockade of JAK2 shows promise as a novel therapeutic target in ADPKD.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a devastating multi-organ disease lacking a cure. ADPKD accounts for approximately 5–10% of patients with renal failure. Genetically, it arises predominately due to mutations in the Pkd1 or Pkd2 genes, encoding for the polycystin-1 and polycystin-2 proteins. The disease is characterised by the progressive growth and enlargement of renal cysts. Patients with ADPKD also exhibit a vascular phenotype, presenting with hypertension and aneurysms1,2 which together with cysts in kidneys and other organs, shows that ADPKD is a systemic disease. Currently Tolvaptan, a V2-vasopressin receptor antagonist, is the only approved therapy; however, it carries significant side effects3 and in is available to some, but not all, patients with ADPKD. Therefore, a major biomedical challenge is to identify central druggable pathways for the treatment of ADPKD.
Curcumin (diferuloylmethane)4 is a phytochemical known to exhibit strong JAK/STAT inhibitory activity. Consistent with being a JAK/STAT inhibitor, curcumin exhibits robust antioxidant, anti-tumour and anti-inflammatory effects5. Importantly, no appreciable side-effects have been reported making it an attractive drug for long-term inhibition of this pathway. Because of its low toxicity but potency, curcumin has been tested as a therapeutic in ADPKD murine models, where it limites cystic growth6 and is currently being trialled in young patients and children with ADPKD (clinicaltrials.gov - NCT02494141).
We have recently shown that JAK2/STAT5 activity is abnormally activated and contributes to ADPKD7–11. Others have also shown that additional components of the JAK/STAT pathway are dysregulated in ADPKD, previously reviewed11. The mechanisms underlying JAK/STAT hyperactivity in ADPKD are however unknown. STAT transcription factors become activated by a series of phosphorylation events on conserved tyrosine residues facilitated by one of four JAK kinases, namely JAK1-JAK3 or Tyk2. Because of their involvement in disease, JAK small-molecule inhibitors have been developed, including the approved Tofacitinib, Ruxolitinib, and Baracitinib12–15. Moreover, additional JAK inhibitors are in clinical trials, making JAK-STAT a therapeutically tractable pathway. Evidence exists that STAT1, STAT3, STAT5 and STAT6 are involved in ADPKD pathogenesis (7–9,11,16–23), while on the contrary the involvement of JAK kinases, which are druggable, has not been previously studied in ADPKD. Combining our previous and current work with that of other groups we predict that JAK-STAT inhibition may be of therapeutic benefit in ADPKD. We therefore investigated the expression of JAK2 in kidneys of mice with and without ADPKD and inhibited JAK2 activity to examine its role in cystogenesis.
Results
JAK2 is highly expressed by both cystic and normal renal epithelial cells in vivo
To investigate whether JAK2 is involved in the development of cysts in vivo, we stained kidney sections from the Pkd1nl/nl mouse model24 and associated wild-type littermate controls with anti-JAK2 antibodies. Kidneys from 5 weeks old mice, with intermediate disease, and 10 weeks old with advance disease, were studied as previously described7. We found that JAK2 is diffusely expressed by cortical renal epithelial cells and its expression appears predominately cytoplasmic with no detectable expression by fibroblasts or the interstitium (Fig. 1A and A’). During early stages of ADPKD, JAK2 is strongly expressed by renal cystic tubules, and to a lesser extent by non-cystic, otherwise normal, tubular epithelial cells (Fig. 1B). At 10 weeks of age, as the disease advances evident by increased number of larger cysts and diffuse interstitial expansion, JAK2 is strongly expressed in cystic epithelial cells and this expression appears to be stronger in cystic than in healthy non-cystic tubules. Quantification of JAK2 staining intensity indicated that mice with intermediate and late stages of disease (5 and 10 weeks) have stronger JAK2 in cystic when compared to non-cystic tubules and compared to wild-type littermate animals (Fig. 1D). Some JAK2 expression is detected in interstitial cells in mice with ADPKD (Fig. 1C’) which is absent in healthy kidneys (Fig. 1A’), suggesting that JAK2 expression in the interstitium is ectopic. JAK2 is expressed in vessels with a lesser expression by bowman’s capsule cells (Fig. 1C’). Expression of JAK2 in vessels remains strong as disease progresses (Fig. 1C). Hence, JAK2 expression is temporally and spatially coincident with cyst growth in the Pkd1nl/nl mouse model of ADPKD.
Figure 1.

JAK2 is highly expressed by cystic renal epithelial cells in vivo. JAK2 levels were studied in kidneys sections of the Pkd1nl/nl model at 5 and 10 weeks of age (5 mice in each group), representative images at 20x magnification are shown. Kidneys of Pkd1 wild type littermates exhibit JAK2 staining through the kidney parenchyma with strong expression in tubules (A). 5 weeks of age Pkd1nl/nl mice exhibit JAK2 expression in both cystic and non-cystic tubules (c, denotes cysts) (B). While at 10 weeks of age the cysts of the Pkd1nl/nl are larger and JAK2 is highly expressed in epithelial (C and C’) but also ectopically expressed in the interstitium (i). Expression of JAK2 is also found in vessels (v). JAK2 quantification revealed stronger expression in cystic epithelial when compared with non-cystic and with wild-type normal tubules (D).
STAT3 tyrosine phosphorylation is potently suppressed by curcumin
Since JAK2 is highly expressed in cystic epithelial cells in vivo (Fig. 1) we considered whether blocking JAK2 activity may offer protection against cystogenesis and set out to explore this biochemically in vitro. Studies in non-renal systems, with intact Pkd1-triggered signalling, have suggested that curcumin acts as a strong JAK-STAT inhibitor25–27, an observation confirmed by our studies (Supplementry Fig. 1A). We used curcumin to inhibit JAK/STAT activity in three independent ADPKD-derived epithelial cell lines; three cell lines were used to avoid cell-line specific artefacts. All ADPKD lines tested here did not show signs of constitutive activation of JAK/STAT, hence to activate the pathway, we used a defined amount of oncostatin M (OSM)28. 50μΜ of curcumin for 6 hours potently inhibited the OSM-induced STAT3 activity, indicated by a marked reduction in the level of tyrosine phosphorylation (Fig. 2A), without affecting total STAT3 protein levels. We next performed a time-response curve, which showed that one-hour of curcumin treatment is sufficient to cause nearly 50% reduction in STAT3 phosphorylation while 3 and 6 hours result in 100% reduction. Total STAT3 protein levels were unaltered by the treatment (Fig. 2B). To study the amount of curcumin needed to cause 50% decrease in phosphorylation of STAT3, cells were stimulated for three hours with 100, 50 or 25 μM of curcumin. Treatment with 100 μM and 50 μM caused 100% reduction in tyrosine phosphorylation, while 25 μM only reduced it by 50%. These time- and dose-dependent data show that curcumin potently blocks STAT3 activity in ADPKD-derived epithelial lines.
Figure 2.
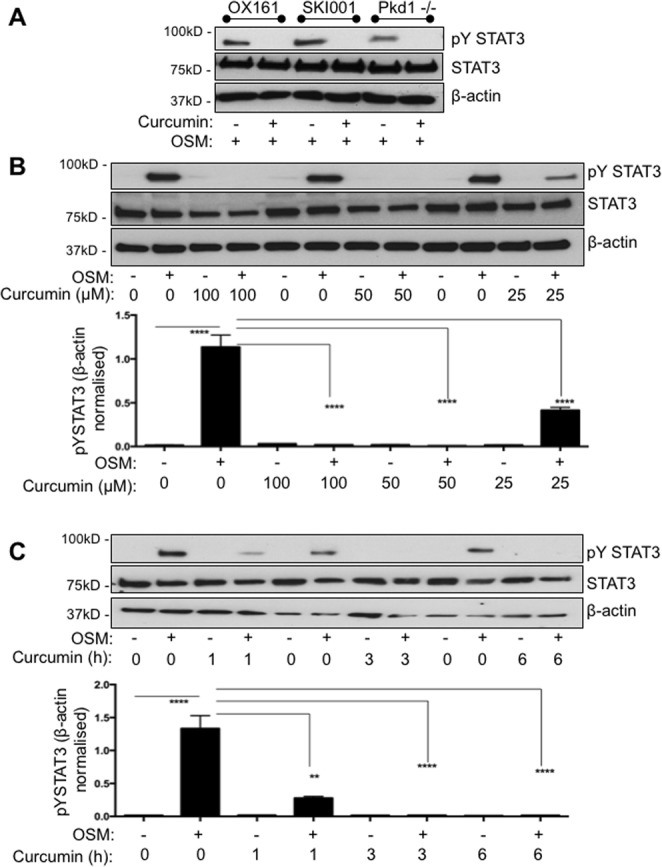
Curcumin is a potent STAT3 inhibitor in renal ADPKD-derived epithelial cells. (A) Oncostatin M (OSM, 10 ng/ml) was used to stimulate phosphorylation of STAT3. Western blotting was performed with total cell lysates from human ADPKD-derived epithelial cells OX161-c1, SKI-001 and mouse F1 Pkd1−/−. Anti- STAT3 phospho-tyrosine 705 antibody (pY STAT3), total STAT3 (STAT3) and β-actin, which served as an internal loading control, were used. In all three cell lines curcumin treatment inhibited OSM-induced pY STAT3. (B) SKI001 cells treated with either 100, 50 or 25 μM of curcumin were subjected to blotting for STAT3, pYSTAT3 and β-actin control. Densitometric quantification of three independent blots was carried out. One-way Anova with Bonferroni corrections was performed, and values lower that 0.05 were considered statistically significant. (C) SKI001 cells were treated with 100 μM of curcumin for 1 or 3 or 6 hours and stimulated either with water vehicle control or 10 ng/ml of OSM. pYSTAT3, STAT3 and β-actin were investigated by blotting. Quantification of three independent blots was carried out. One-way Anova with Bonferroni corrections was performed, and values lower that 0.05 were considered statistically significant. Symbol meaning: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Curcumin blocks STAT3 activity via JAK2
To find out how curcumin blocks STAT3 activity we studied JAK2, which is highly expressed by cyst-lining cells in vivo (Fig. 1) and is known to activate STAT3 by phosphorylation. We found that 100 μM and 50 μM of curcumin caused approximately 100% reduction in JAK2 levels, while 25μΜ resulted in a modest 50% decrease (Fig. 3). Curcumin reduced JAK2 levels in both OSM-treated and control-treated cells, hence showing that it affects JAK2 in an OSM-independent manner. Curcumin’s effect on JAK2 activity was consistent with the blockade of STAT3 phosphorylation observed above (Fig. 2). To test if this effect was not cell line specific, we tested the effect of curcumin in mouse F1-Pkd1−/− cells and found that curcumin reduced JAK2 levels (Supplementry Fig. 2B). Therefore, curcumin blocks JAK2 and this may explain the reduced STAT3 activity.
Figure 3.
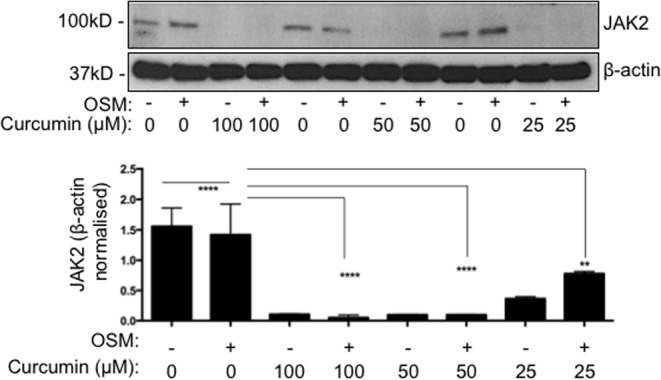
Curcumin inhibits JAK2. SKI001 cells were exposed to 100 or 50 or 25 μM of curcumin and immunoblot was carried out using anti-JAK2 and β-actin antibodies. Total JAK2 is found in both OSM stimulated and unstimulated cells. Quantification of three independent immunoblots was carried out. One-way Anova with Bonferroni corrections was carried out and values lower that 0.05 were considered statistically significant. Symbol meaning: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Curcumin does not change JAK2 levels but makes JAK2 insoluble
We next wished to understand how curcumin causes the apparent alteration of the levels of JAK2 in immunoblots. Firstly, we explored the option that curcumin may cause JAK2 degradation via proteasomal processing of JAK2. To assess this, we treated cells with MG132, a proteasome inhibitor. MG132 caused the expected increase in ubiquitinated proteins, thus confirming adequate level of proteasomal blockade (Fig. 4A). Yet, the effect of curcumin on JAK2 did not depend on the proteosome, as MG132 was unable to restore JAK2 levels following curcumin treatment (Fig. 4A compare lane 3 with 7 and 8). These data show that curcumin-induced JAK2 reduction is not though the proteasome. Given that the proteasome is not responsible for the decreased JAK2 levels we decided to perform immunocytochemistry to visualise JAK2 and test whether curcumin treatment affects its subcellular localisation. Interestingly, we found that JAK2 is found throughout the cell body with some localisation in the plasma membrane in DMSO-vehicle treated cells, however curcumin treatment causes JAK2 to become strongly punctate (Fig. 4B). Previous studies have suggested that curcumin can cause protein precipitation into aggresomes29,30, we therefore suggest that these punctate structures may be JAK2 aggregates. This could explain how JAK2 becomes insoluble and provide an explanation as to why we were unable to detect JAK2 in the detergent-soluble fraction by immunoblotting. Taken together, our data show that curcumin causes JAK2 inactivation without involving the proteasome.
Figure 4.
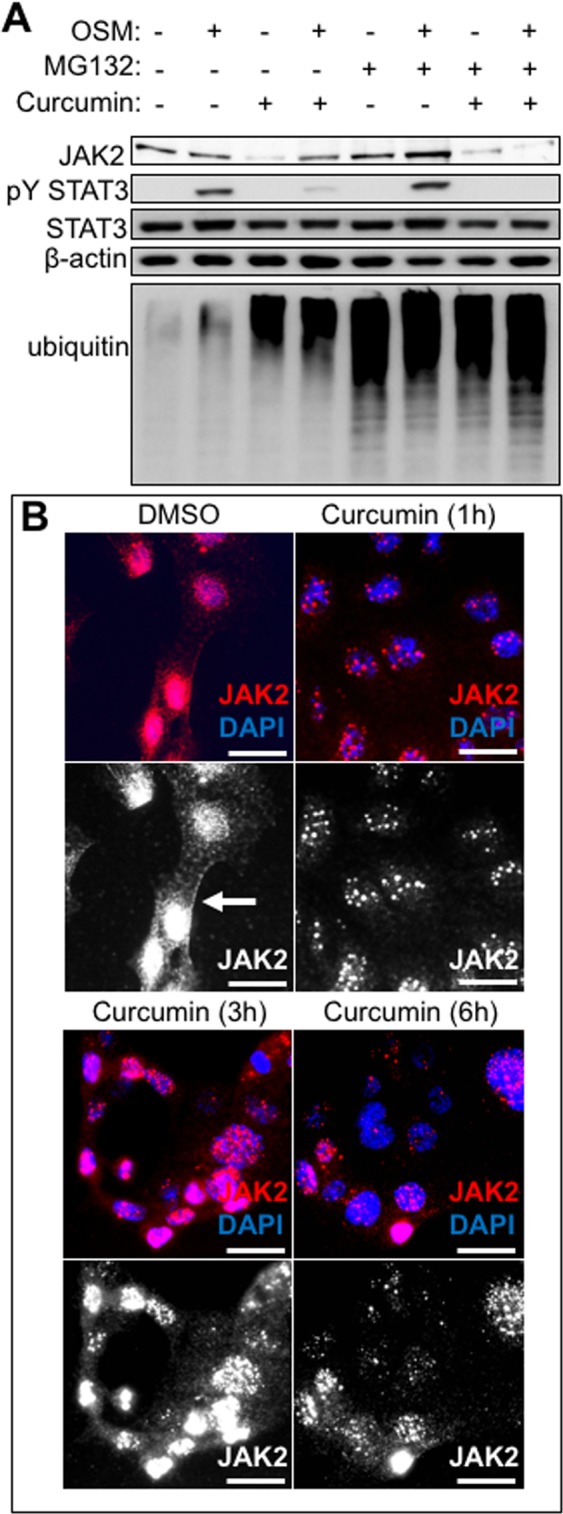
Curcumin controls JAK2 localisation. (A) SKI001 cells were treated with OSM (10 ng/ml), or MG132 (50 μM) or curcumin (50 μM) for 3 hours and lysates were subjected to immunoblotting. Antibodies against JAK2 pYSTAT3, STAT3, ubiquitin (indicator of proteasome inhibition control) and β-actin (internal loading control) were studied. (B) SKI001 cells were either treated with DMSO vehicle control for 3 hours (3 h) or curcumin for 1 h or 3 h or 6 h, they were then snap frozen on ice-cold methanol, stained with anti-JAK2 rabbit antibody, followed by an anti-rabbit AF594 and imaged in an inverted fluorescent microscope. Red is anti-JAK2 (also in greyscale in lower panel), blue is nuclear counterstain (DAPI). Scale bars are 25 μm.
Curcumin-induced JAK2 blockade reduces cystic growth in vitro
To study the ability of the JAK2/STAT3 pathway to drive cystogenesis we performed cystogenesis assays in vitro using a monoculture grown in three dimensions (3D cyst assays). We used three independent cell lines to ensure the robustness of results and avoid cell-type specific artefacts. In these assays microscopic cysts formed, from each independent cell line, which grew in diameter over time, as previously shown by our group7. We found that curcumin-induced JAK2 blockade led to a clear and reliable reduction in the size of cysts in all three independent cell lines tested and, in a time and dose-dependent manner (Fig. 5). Critically, two of the three cell lines used are human derived cells, therefore highlighting human relevance. Curcumin reduced the growth of cysts at day 2, with the effect persisting to day 6 and 9. Curcumin at 50 and 25μΜ also clearly reduced cyst size at day 9 in OX161-c1 cells, which are human-derived ADPKD cells (Fig. 5A). A similar pattern was seen in SKI-001, an additional independent human ADPKD-derived line, which showed reduced cyst size at day 6 and 9 with 50 and 25μΜ of curcumin (Fig. 5B). We also studied a mouse line which has a Pkd1 deletion and is therefore considered as a mouse ADPKD line, as expected curcumin blocked the growth of cysts at day 2 with 100 μM of curcumin, while day 6 and day 9 50 and 100μΜ were able to cause a block in the growth of cysts (Fig. 5C). Pkd1 wild-type F1 mouse renal cells also responded by exhibiting reduced cystic growth (data not shown), suggesting that the strong anti-cystogenic effect of curcumin is Pkd1-independent. To study whether JAK/STAT in indeed activated in cells grown in 3D cyst assay and investigate if curcumin is able to block STAT3 phosphorylation, we performed immunoblotting from cyst assays. STAT3 is phosphorylated within the cysts and curcumin potently blocks this (Fig. 5D), as well as reducing JAK2 levels (Supplementry Fig. 1C). These data suggest that the previously reported anti-cystic actions of curcumin may be via blockade of JAK/STAT. Given that curcumin is pleiotropic and can block pathways other than just JAK/STAT we also used a selective JAK small-molecule inhibitor, namely tofacitinib, which is currently in clinical use for rheumatoid arthritis. We found that tofacitinib led to a significant reduction in cyst size (Fig. 4E), thus suggesting that JAK inhibition alone is sufficient to block cystic growth of ADPKD cells.
Figure 5.
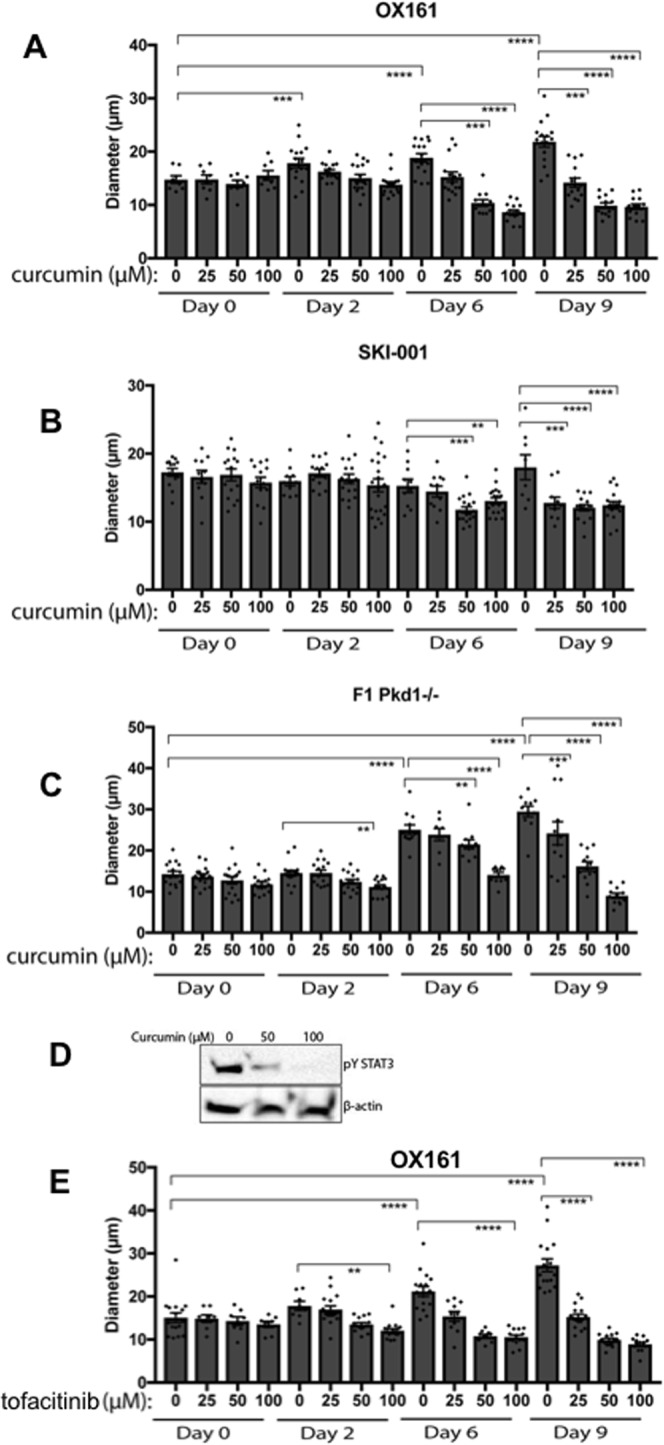
Curcumin slows down cyst growth in vitro. ADPKD-derived human epithelial cells were grown in BD-Matrigel to allow the formation of cysts to grow over time. All cysts formed were photographed at day 0, 2, 6 and 9; using an Olympus inverted microscope. Curcumin at 100, 50, 25 and 10 μM or DMSO vehicle-control was added and cyst growth measured in (A) human OX161 (B) human SKI-001 and (C) mouse derived Pkd1−/− cell lines. (D) Immunoblots were performed from cell lysates obtained from cells grown in cysts at day 9. Matrigel was melted and pYSTAT3 was measured, β-actin was also assessed. Cells were treated with either 0, 50 or 100 μM of curcumin to test whether the latter can reduce JAK/STAT activity in the cysts. One-way Anova with Bonferroni corrections was carried out and values lower that 0.05 were considered statistically significant. Symbol meaning: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Discussion
We show that JAK2 levels are higher in polycystic when compared with wild-type kidneys a finding that may be therapeutically relevant given that we identified that JAK blockade, using either curcumin or a selective JAK inhibitor, potently reduces cystic growth in human ADPKD-derived lines. These data are important because multiple components of the JAK/STAT pathway contribute to cystogenesis, and JAK activity is therapeutically amenable, therefore providing a realistic potential for intervention studies. Our data suggest that JAK2 is a key kinase promoting cystic growth in ADPKD, since inhibiting it, blocks cystic growth of ADPKD cells. JAK2 may contribute to the development of cystic growth likely via canonical activation of one, or multiple, of the downstream STAT transcription factors. Indeed, a number of groups, including our own, have shown that STAT3, STAT5 and STAT6, which are normally latent transcription factors, are activated in ADPKD7,10,11,18–21,23,31. JAK2 is involved in tyrosine phosphorylation of all of these STATs32–34, which is an essential step for the dimerization and activity of these transcription factors. Therefore, the inhibition of cystic growth observed under JAK blockade may be via suppression of canonical JAK/STAT activity. Alternately, STAT transcription factors can also be activated, non-canonically, by other kinases such as Src, which was found to be responsible for activating STAT3 in MDCKII renal cells31. Indeed, curcumin can the biochemical activity of the Src kinase35 in addition to blocking JAK/STAT activity. If STATs are activated by Src or other kinases, then it follows that highlt selective small-molecule inhibitors of JAKs would not be effective in reducing JAK/STAT activity and lowering cystic growth. However, our work shows that tofacitinib, which is a selective JAK inhibitor, used at concentrations found in plasma of treated individuals, resulted in reduced cyst growth. Therefore, we conclude that JAK2 is a major kinase driving cystogenesis of ADPKD cells.
The choice of JAK inhibitor is based on a number of considerations, including how effective a given drug is and balancing this with its side effects. Over the years, phytochemicals, such as curcumin, have been used to inhibit, prevent or reverse disease processes. Curcumin for example has strong anti-cystic effects in pre-clinical models of ADPKD6. One advantage of curcumin is that it can be used long term, due to lack of apparent side effects36. However, the bioavailability of curcumin is low37,38, making it less attractive when compared with small molecule inhibitors, such as tofacitinib, the JAK inhibitor used in our study. Indeed, here we showed that both curcumin and tofacitinib were capable of slowing down the rate of growth of cysts in cystogenesis assays. A comparison of tofacitinib (and other JAK inhibitors) with curcumin in a pre-clinical setting is required to assess effectiveness and measure their side effects in murine models of ADPKD.
Previous studies used systemic administration of curcumin in a murine model of ADPKD6, where curcumin showed promising results; however, whether curcumin acted primarily on renal epithelial or vascular cells was uncertain from these studies. Here, we find that JAK2 is expressed in both vascular and epithelial cells in the ADPKD affected kidney. To gain a deeper understanding we used monoculture assays and found that curcumin reduces cystogenesis of renal epithelial cells without requiring interactions with vascular cells. As such, if, and how, curcumin affects vessels is currently unknown. However, curcumin is currently under clinical investigation (phase 4 clinical trial) for its potential to modify the vascular phenotype in children and young individuals with ADPKD. The results of this study may determine whether curcumin can protect the vasculature of young individuals affected with against ADPKD, which will provide an additional protective mechanism for this drug.
Curcumin causes JAK2 to move into an insoluble fraction, and we observe the generation of large JAK2 positive puncta in cells treated with curcumin. We do not know the nature of these puncta, however, curcumin has been previously shown to cause protein aggregation29, therefore we propose that these structures may be aggresomes. The observation that these JAK2 positive puncta are not dependent on proteasomal processing of JAK2 is evidence that JAK2 has not been degraded. Likewise, others have reported that curcumin-treated cancer cells respond by a temporal blockade of STAT3, which is fully reversible within 24 hours39, this is consistent with our observations. Therefore, we propose a model whereby curcumin takes JAK2 briefly out of action by causing it to aggregate, without leading to JAK2 degradation, which is an irreversible action. It should be noted that the effects of tofacitinib are also reversible but it requires up to 14 days for reversibility. This is important because it may explain the lack of significant side effects in response to systemic treatment with curcumin.
In summary, we show that JAK2 is highly expressed in polycystic kidneys and its blockade reduces cystic growth. Understanding how key signalling pathways, such as the JAK/STAT, are misregulated in ADPKD will help identify suitable molecules to target for therapy. Based on our in vivo immunohistochemical analysis an in vitro cyst assays in human cells, we propose that JAK inhibitors may be of therapeutic benefit for patients with ADPKD. More studies are required to explore the mechanisms employed by JAK-STAT to regulate cystic growth and to identify the most suitable point in the pathway to intervene. In conclusion, our study provides the basis for further testing of JAK inhibitors as a novel therapeutic avenue for patients with polycystic kidney disease.
Methods
Cell lines
Conditionally-immortalized ADPKD-derived lines (OX161-c1 and SKI-001)40 are tubular epithelial cells isolated from human kidneys and immortalized by transduction at an early passage (P1-4) with a retroviral vector containing a temperature-sensitive large T antigen and the catalytic subunit of human telomerase41. The F1-Pkd1 WT renal epithelial cells were previously isolated from kidney papillae of the Pkd1fl/fl mouse (B6.129S4-Pkd1tm2Ggg/J; the Jackson Laboratory) and were immortalized with the lentiviral vector VVPW/mTert expressing the mTert; to delete Pkd1 and produce F1/Pkd1−/− cells they were subsequently transfected with VIRHD/HY/Silntβ1/2363 lentivectors followed by hydromycin selection42.
Cyst assays
Cyst assays were performed as previously described7. In brief, for ADPKD cystic cell lines, 2 human lines namely OX161-c1, SKI-001, and one mouse line F1-Pkd1−/− were used. OX161 and SKI-001 were kept at 33 °C F1-Pkd1−/− were at 37 °C. Healthily growing cells were trypsinised and re-suspended in DMEM media containing 10% FBS (no antibiotics) and counted using a haemocytometer. Subsequently 2 × 104 cells per well were mixed with basement Matrigel (354230, BD biosciences). Prior to use, the Matrigel was left to thaw on ice and once cells were added it was immediately loaded onto the 96 well plate, allowing it to polymerise. 100 μl of DMEM media supplemented with 10% FBS was added to each well and cells returned to the incubator for 24 hours to allow them to recover. After 24 hours, the media was aspirated and replaced with fresh media (DMEM + 10% FBS) supplemented with either vehicle (DMSO) or curcumin (Sigma). The cells were photographed before every media change and every two days, the media was replaced with fresh media containing either curcumin or DMSO.
Immunohistochemistry/immunofluorescence
Cells grown on coverslips were fixed with ice-cold methanol prior to blocking in 2% milk/TBST for 30 minutes and incubated with primary antibodies overnight at 4 °C. Antibody used was anti-rabbit JAK2 (3230S, cell Signalling, D2E12). Cells were washed and incubated with secondary antibody, anti-rabbit AF594 (A-11037, Invitrogen (1:250). Microscopy was carried out using a confocal microscope. Immunohistochemistry using a JAK2 antibody (3230S, Cell Signalling) was performed as previously described7. Quantification of JAK2 staining intensity was performed using Fiji freeware. Image was first colour-convoluted using the H -DAB function, image 2 (DAB) was converted to 8-bit and using the freehand selection tool, an area of interest was manually drawn around each tubule, mean grey intensity, maximum intensity (which is 255 for an 8-bit image) were obtained for each measured tubule. Data are presented as optical density (mean grey value) and were calculated following the formula Optical density = log(max intensity/mean intensity). To minimize the biases, which can be introduced in areas where there is no tissue (middle of a cyst), the non-cell compartment was excluded by drawing around the cysts.
Western blotting and curcumin treatment
Cells were analysed by Western blotting with previously reported protocols43–46. In brief, cells were lysed using ice-cold Lysis Buffer (50 mM Tris (pH 7.4) 250 mM NaCl, 0.3% Triton X-100, 1 mM EDTA) supplemented with protease inhibitor cocktail (Roche), freeze-thawed and sonicated. Whole cell lysates were boiled in 2xLaemmeli sample buffer for 5 minutes. Samples were resolved by SDS-PAGE and transferred using the Mini-PROTEAN system (Bio-Rad). Primary antibodies were anti-β-actin (ab8226, Abcam), anti-phosphorylated STAT3 (p-STAT3) (9145, Cell Signalling), anti-STAT3 (12640, Cell Signalling). Curcumin (08511-10MG, Sigma) was resuspended in DMSO and used at stated doses, DMSO served as vehicle control. For immunoblotting in cyst assays, Matrigel was first dissolved using cultrex organoid harvesting solution, then cysts were lysed directly in 2xLaemmeli sample buffer and immunoblots carried out as described above.
Animals
Pkd1nl/nl, harboring an intronic neomycin-selectable marker, or wild type littermate controls were used7,47. Pkd1nl/nl or control mice were sacrificed at 5 or 10 weeks of age and kidneys collected, formalin-fixed and paraffin embedded. 5-micron section were used for histological examination. All mouse experiments were done under the authority of a U.K. Home Office license (license holder Dr Maria Fragiadaki, license number PPL7008968).
Statistical analysis
Data were analysed using Prism GraphPad and Non-parametric, two-tailed, Mann-Whitney T-tests or one-way ANOVA were performed. Results with a P value of 0.05 or lower were considered statistically significant24.
Supplementary information
Acknowledgements
We would like to thank Ms Fiona Wright, Ms Monica Neilan and Dr Barbora Ndreca for tissue processing and genotyping; Prof AC Ong and Dr Andrew Streets for providing advice and cell lines. Prof DJ Peters for the gift of the Pkd1nl/nl animal model and Dr Luca Gusella for the collecting duct Pkd1−/− F1 cells. This work was supported by a Kidney Research UK Intermediate Fellowship and a Springboard Award by the Academy of Medical Sciences awarded to Dr Fragiadaki.
Author Contributions
M.F., F.P., A.M.C. and Z.H. performed experiments, revised and approved manuscript, M.F. conceived and supervised project, attracted funded, directed the research and wrote & revised manuscript.
Data Availability
All data including supporting datasets are made available as main figures or supplementary information files.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-41106-3.
References
- 1.Azurmendi PJ, et al. Early renal and vascular changes in ADPKD patients with low-grade albumin excretion and normal renal function. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2009;24:2458–2463. doi: 10.1093/ndt/gfp136. [DOI] [PubMed] [Google Scholar]
- 2.Carney EF. Polycystic kidney disease: TGF-beta signalling and vascular complications in ADPKD. Nat Rev Nephrol. 2013;9:694. doi: 10.1038/nrneph.2013.214. [DOI] [PubMed] [Google Scholar]
- 3.Makabe S, et al. Elevation of the serum liver enzyme levels during tolvaptan treatment in patients with autosomal dominant polycystic kidney disease (ADPKD) Clin Exp Nephrol. 2018 doi: 10.1007/s10157-018-1545-7. [DOI] [PubMed] [Google Scholar]
- 4.Jurenka JS. Anti-inflammatory properties of curcumin, a major constituent of Curcuma longa: a review of preclinical and clinical research. Altern Med Rev. 2009;14:141–153. [PubMed] [Google Scholar]
- 5.Boyanapalli SSS, et al. Pharmacokinetics and Pharmacodynamics of Curcumin in regulating anti-inflammatory and epigenetic gene expression. Biopharm Drug Dispos. 2018 doi: 10.1002/bdd.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leonhard WN, et al. Curcumin inhibits cystogenesis by simultaneous interference of multiple signaling pathways: in vivo evidence from a Pkd1-deletion model. American journal of physiology. Renal physiology. 2011;300:F1193–1202. doi: 10.1152/ajprenal.00419.2010. [DOI] [PubMed] [Google Scholar]
- 7.Fragiadaki M, et al. STAT5 drives abnormal proliferation in autosomal dominant polycystic kidney disease. Kidney international. 2017;91:575–586. doi: 10.1016/j.kint.2016.10.039. [DOI] [PubMed] [Google Scholar]
- 8.Bhunia AK, et al. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell. 2002;109:157–168. doi: 10.1016/S0092-8674(02)00716-X. [DOI] [PubMed] [Google Scholar]
- 9.Talbot JJ, et al. Polycystin-1 regulates STAT activity by a dual mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7985–7990. doi: 10.1073/pnas.1103816108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weimbs T, Olsan EE, Talbot JJ. Regulation of STATs by polycystin-1 and their role in polycystic kidney disease. Jak-Stat. 2013;2:e23650. doi: 10.4161/jkst.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weimbs T, Talbot JJ. STAT3 Signaling in Polycystic Kidney Disease. Drug Discov Today Dis Mech. 2013;10:e113–e118. doi: 10.1016/j.ddmec.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goll GL, Kvien TK. New-generation JAK inhibitors: how selective can they be. Lancet. 2018 doi: 10.1016/S0140-6736(18)31325-4. [DOI] [PubMed] [Google Scholar]
- 13.Hoffman HM, Broderick L. JAK inhibitors in autoinflammation. J Clin Invest. 2018 doi: 10.1172/JCI121526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang EH, Liao KP, Kim SC. Cardiovascular Safety of Biologics and JAK Inhibitors in Patients with Rheumatoid Arthritis. Curr Rheumatol Rep. 2018;20:42. doi: 10.1007/s11926-018-0752-2. [DOI] [PubMed] [Google Scholar]
- 15.Musumeci F, et al. An Update on JAK Inhibitors. Curr Med Chem. 2018 doi: 10.2174/0929867325666180327093502. [DOI] [PubMed] [Google Scholar]
- 16.Deltas C, Felekkis K. Is suppression of cyst growth in PKD enough to preserve renal function?: STAT6 inhibition is a novel promising target. Jak-Stat. 2012;1:216–218. doi: 10.4161/jkst.21634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felekkis KN, et al. Mutant polycystin-2 induces proliferation in primary rat tubular epithelial cells in a STAT-1/p21-independent fashion accompanied instead by alterations in expression of p57KIP2 and Cdk2. BMC nephrology. 2008;9:10. doi: 10.1186/1471-2369-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jing Y, et al. Triptolide delays disease progression in an adult rat model of polycystic kidney disease through the JAK2/STAT3 pathway. American journal of physiology. Renal physiology. 2018 doi: 10.1152/ajprenal.00329.2017. [DOI] [PubMed] [Google Scholar]
- 19.Low SH, et al. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev Cell. 2006;10:57–69. doi: 10.1016/j.devcel.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 20.Olsan EE, et al. Signal transducer and activator of transcription-6 (STAT6) inhibition suppresses renal cyst growth in polycystic kidney disease. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18067–18072. doi: 10.1073/pnas.1111966108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olsan EE, West JD, Torres JA, Doerr N, Weimbs T. Identification of targets of IL-13 and STAT6 signaling in polycystic kidney disease. American journal of physiology. Renal physiology. 2018;315:F86–F96. doi: 10.1152/ajprenal.00346.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peda JD, et al. Autocrine IL-10 activation of the STAT3 pathway is required for pathological macrophage differentiation in polycystic kidney disease. Dis Model Mech. 2016;9:1051–1061. doi: 10.1242/dmm.024745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takakura A, et al. Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Human molecular genetics. 2011;20:4143–4154. doi: 10.1093/hmg/ddr338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lantinga-van Leeuwen IS, et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Human molecular genetics. 2004;13:3069–3077. doi: 10.1093/hmg/ddh336. [DOI] [PubMed] [Google Scholar]
- 25.Rajasingh J, Raikwar HP, Muthian G, Johnson C, Bright JJ. Curcumin induces growth-arrest and apoptosis in association with the inhibition of constitutively active JAK-STAT pathway in T cell leukemia. Biochem Biophys Res Commun. 2006;340:359–368. doi: 10.1016/j.bbrc.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 26.Zhao HM, et al. Curcumin Suppressed Activation of Dendritic Cells via JAK/STAT/SOCS Signal in Mice with Experimental Colitis. Front Pharmacol. 2016;7:455. doi: 10.3389/fphar.2016.00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zu J, et al. Curcumin improves the recovery of motor function and reduces spinal cord edema in a rat acute spinal cord injury model by inhibiting the JAK/STAT signaling pathway. Acta Histochem. 2014;116:1331–1336. doi: 10.1016/j.acthis.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 28.Zhang F, Li C, Halfter H, Liu J. Delineating an oncostatin M-activated STAT3 signaling pathway that coordinates the expression of genes involved in cell cycle regulation and extracellular matrix deposition of MCF-7 cells. Oncogene. 2003;22:894–905. doi: 10.1038/sj.onc.1206158. [DOI] [PubMed] [Google Scholar]
- 29.Krishnamoorthy A, et al. Effect of curcumin on amyloid-like aggregates generated from methionine-oxidized apolipoprotein A-I. FEBS Open Bio. 2018;8:302–310. doi: 10.1002/2211-5463.12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mithu VS, et al. Curcumin alters the salt bridge-containing turn region in amyloid beta(1-42) aggregates. J Biol Chem. 2014;289:11122–11131. doi: 10.1074/jbc.M113.519447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Talbot JJ, et al. The cleaved cytoplasmic tail of polycystin-1 regulates Src-dependent STAT3 activation. J Am Soc Nephrol. 2014;25:1737–1748. doi: 10.1681/ASN.2013091026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu X, Kennedy RH, Liu SJ. JAK2/STAT3, not ERK1/2, mediates interleukin-6-induced activation of inducible nitric-oxide synthase and decrease in contractility of adult ventricular myocytes. J Biol Chem. 2003;278:16304–16309. doi: 10.1074/jbc.M212321200. [DOI] [PubMed] [Google Scholar]
- 33.Jin H, Lanning NJ, Carter-Su C. JAK2, but not Src family kinases, is required for STAT, ERK, and Akt signaling in response to growth hormone in preadipocytes and hepatoma cells. Mol Endocrinol. 2008;22:1825–1841. doi: 10.1210/me.2008-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guiter C, et al. Constitutive STAT6 activation in primary mediastinal large B-cell lymphoma. Blood. 2004;104:543–549. doi: 10.1182/blood-2003-10-3545. [DOI] [PubMed] [Google Scholar]
- 35.Leu TH, Su SL, Chuang YC, Maa MC. Direct inhibitory effect of curcumin on Src and focal adhesion kinase activity. Biochem Pharmacol. 2003;66:2323–2331. doi: 10.1016/j.bcp.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 36.Aggarwal BB, Kumar A, Bharti AC. Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res. 2003;23:363–398. [PubMed] [Google Scholar]
- 37.Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4:807–818. doi: 10.1021/mp700113r. [DOI] [PubMed] [Google Scholar]
- 38.Liu W, et al. Oral bioavailability of curcumin: problems and advancements. J Drug Target. 2016;24:694–702. doi: 10.3109/1061186X.2016.1157883. [DOI] [PubMed] [Google Scholar]
- 39.Bharti AC, Donato N, Aggarwal BB. Curcumin (diferuloylmethane) inhibits constitutive and IL-6-inducible STAT3 phosphorylation in human multiple myeloma cells. Journal of immunology. 2003;171:3863–3871. doi: 10.4049/jimmunol.171.7.3863. [DOI] [PubMed] [Google Scholar]
- 40.Parker E, et al. Hyperproliferation of PKD1 cystic cells is induced by insulin-like growth factor-1 activation of the Ras/Raf signalling system. Kidney international. 2007;72:157–165. doi: 10.1038/sj.ki.5002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Hare MJ, et al. Conditional immortalization of freshly isolated human mammary fibroblasts and endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:646–651. doi: 10.1073/pnas.98.2.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee K, Boctor S, Barisoni LM, Gusella GL. Inactivation of integrin-beta1 prevents the development of polycystic kidney disease after the loss of polycystin-1. J Am Soc Nephrol. 2015;26:888–895. doi: 10.1681/ASN.2013111179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fisher KH, et al. A genome-wide RNAi screen identifies MASK as a positive regulator of cytokine receptor stability. J Cell Sci. 2018 doi: 10.1242/jcs.209551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fragiadaki M, et al. Hyperglycemia causes renal cell damage via CCN2-induced activation of the TrkA receptor: implications for diabetic nephropathy. Diabetes. 2012;61:2280–2288. doi: 10.2337/db11-1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fragiadaki M, et al. High doses of TGF-beta potently suppress type I collagen via the transcription factor CUX1. Mol Biol Cell. 2011;22:1836–1844. doi: 10.1091/mbc.E10-08-0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fragiadaki M, et al. Interstitial fibrosis is associated with increased COL1A2 transcription in AA-injured renal tubular epithelial cells in vivo. Matrix Biol. 2011;30:396–403. doi: 10.1016/j.matbio.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 47.Happe H, et al. Cyst expansion and regression in a mouse model of polycystic kidney disease. Kidney international. 2013;83:1099–1108. doi: 10.1038/ki.2013.13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data including supporting datasets are made available as main figures or supplementary information files.