

Abstract
Members of the Mycobacterium chelonae-abscessus complex (MCAC) are close to the mycobacterial ancestor and includes both human, animal and fish pathogens. We present the genomes of 14 members of this complex: the complete genomes of Mycobacterium salmoniphilum and Mycobacterium chelonae type strains, seven M. salmoniphilum isolates, and five M. salmoniphilum-like strains including strains isolated during an outbreak in an animal facility at Uppsala University. Average nucleotide identity (ANI) analysis and core gene phylogeny revealed that the M. salmoniphilum-like strains are variants of the human pathogen Mycobacterium franklinii and phylogenetically close to Mycobacterium abscessus. Our data further suggested that M. salmoniphilum separates into three branches named group I, II and III with the M. salmoniphilum type strain belonging to group II. Among predicted virulence factors, the presence of phospholipase C (plcC), which is a major virulence factor that makes M. abscessus highly cytotoxic to mouse macrophages, and that M. franklinii originally was isolated from infected humans make it plausible that the outbreak in the animal facility was caused by a M. salmoniphilum-like strain. Interestingly, M. salmoniphilum-like was isolated from tap water suggesting that it can be present in the environment. Moreover, we predicted the presence of mutational hotspots in the M. salmoniphilum isolates and 26% of these hotspots overlap with genes categorized as having roles in virulence, disease and defense. We also provide data about key genes involved in transcription and translation such as sigma factor, ribosomal protein and tRNA genes.
Introduction
Mycobacteria occupy various ecological niches and can be isolated from soil, tap water and ground water and they are divided into slow (SGM) and rapid (RGM) growing mycobacteria. Several cause diseases both in humans and animals (land and aquatic). Among these, Mycobacterium tuberculosis (Mtb) and Mycobacterium leprae, the causative agents of tuberculosis and leprosy, respectively, are well-known pathogenic mycobacteria. Mycobacterium piscium was the first Mycobacterium spp. to be isolated from fish1, however, it has been lost. Many different mycobacteria have since been isolated from various infected fish: the fish disease caused by mycobacteria is referred to as mycobacteriosis (fish tuberculosis). Infections are due to three predominant mycobacteria: Mycobacterium marinum (Mma); Mycobacterium chelonae (Mche); and Mycobacterium fortuitum (Mfor). Of these, the SGM Mma seems to be the most important species infecting a wide array of different fish, in particular in warm water systems, while the coldwater pathogen Mche infects predominantly salmonid species2. Several other mycobacteria such as Mycobacterium abscessus (Mabs) and Mycobacterium salmoniphilum (Msal) have emerged as fish pathogens. The RGM Msal belongs to the Mche-Mabs (MCAC) complex3 and, as Mche it causes mycobacteriosis in cold water living fish4–6. Msal was originally identified from salmonids7 but lost its species status in 1980 due to its high biochemical similarity with Mche and Mabs. On the basis of phylogenetic analysis of the 16S rRNA gene, rpoB and hsp65 and mycolic acid composition Msal regained species status 20073,8.
Mycobacterial infections are common among wild fish but it is most problematic in aquaculture and aquarium settings. To prevent and treat bacterial infections in aquaculture settings antimicrobial agents are used in large quantities worldwide as well as the use of medicated fish food9. In addition, the MCAC-complex contains many clinically relevant human pathogens but Msal has not been implicated to cause disease in humans10,11. Recently, the clinically isolated human pathogen Mycobacterium franklinii (Mfra) was classified as a mycobacterial species and member of the MCAC-complex10,12. As exemplified by the emerging pathogen Mabs, members of the MCAC-complex display resistance to many clinically relevant antibiotics and hence infections caused by these mycobacteria can be problematic and treatment requires the use of other antibiotics than in treatment of tuberculosis11. Together this imposes a potential risk for selecting antibiotic resistant microbes and thereby constitutes a threat to animal and human health9,13–16.
In 2012, there was an outbreak of a bacterial infection in the animal facility among the mice population at the Biomedical Center, Uppsala University. Subsequently, two different mycobacteria were isolated from the tap water in the animal facility on two different occasions. On the basis of partial 16S rRNA gene sequences and other biochemical tests (see acknowledgments) the single isolate from the first sampling was identified as Msal, while the three isolates from the second sampling were classified as M. salmoniphilum- (Msal-) like. However, it is still not clear whether the sampled mycobacteria caused the outbreak. Neither is it known if the isolates named as Msal-like are Msal strains or represent different species. On the basis of this and together with the importance of this group of mycobacteria with respect to pathogenicity, emerging antibiotic resistance and the phylogenetic closeness of MCAC-complex members to the mycobacterial ancestor17,18 (unpublished) provided the incentives for a comparative genomic analysis of these closely related mycobacterial species.
Here we present the complete genomes of the Msal DSM43276 (MsalT) and Mche DSM43804 (McheT) type strains, seven Msal strains (including outbreak strains), and five Msal-like isolates. Our comparative genomic analysis, where we included the genomes of Mabs, and 36 additional MCAC-complex members, revealed that Msal and Msal-like strains represent two different species. Whole genome average nucleotide identity and core gene phylogeny further suggested that the Msal-like isolates should be referred to as Mfra strains and that they are phylogenetically close to Mabs. Our data further suggest that Msal constitute three separate groups.
Results
Overall description of the genomes
To understand the interrelationship between Msal and Msal-like strains we obtained strains from various sources including McheT (DSM43804; Table 1 and S1). The type strain MsalT (DSM43276) formed both rough (R) and smooth (S) colony morphotypes (the other Msal strains formed R colonies). After re-streaking to obtain homogenous cultures the R type was used for genome sequencing (sequencing the 16S rDNA suggested that both types correspond to Msal; not shown). The collection also included Msal-like strains from the 2012 outbreak at the animal facility at the Uppsala University Biomedical Center and Msal-like strains isolated from tap water at different time points between 2011 and 2013 in Uppsala (Sweden). DNA from the different strains were isolated and subjected to sequencing (see Methods and Supplementary information).
Table 1.
Summary of genome annotation.
| Species | Name tag | Genome size(bp) | (%)GC content | Annotation (number of.) | Bioproject ID | Accession no | Source | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Scaffolds | CDSs | rRNAs (5S;23S;16S) | tRNAs | ncRNAs | signal peptides | |||||||
| M. salmoniphilum CCUG60883 | Msal CCUG60883 | 5076038 | 64 | 15 | 4950 | (1;1;1) | 51 | 38 | 458 | PRJNA414709 | PECM00000000 | CCUG strain* |
| M. salmoniphilum CCUG60884 | Msal CCUG60884 | 4963292 | 64,2 | 16 | 4832 | (1;1;1) | 56 | 36 | 429 | PRJNA414709 | PECL00000000 | CCUG strain* |
| M. salmoniphilum CCUG60885 | Msal CCUG60885 | 5076073 | 64 | 14 | 4953 | (1;1;1) | 51 | 38 | 455 | PRJNA414709 | PECK00000000 | CCUG strain* |
| M. salmoniphilum CCUG62472 | Msal CCUG62472 | 5176285 | 64,2 | 16 | 5061 | (1;1;1) | 57 | 34 | 435 | PRJNA414709 | PECJ00000000 | CCUG strain* |
| M. salmoniphilum DE4585 | Msal DE4585 | 5072347 | 64,1 | 11 | 4867 | (1;1;1) | 61 | 39 | 442 | PRJNA414709 | PECH00000000 | Outbreak strain Ennis |
| M. salmoniphilum DE4586 | Msal DE4586 | 4817070 | 64,5 | 31 | 4645 | (1;1;1) | 55 | 39 | 428 | PRJNA414709 | PECG00000000 | Outbreak strain Ennis |
| M. salmoniphilum DE4587 | Msal DE4587 | 4802997 | 64,2 | 11 | 4629 | (1;1;1) | 55 | 39 | 427 | PRJNA414709 | PECI00000000 | Outbreak strain Ennis |
| M. salmoniphilum DSM43276 | Msal T | 4776625 | 64,3 | 1 | 4652 | (1;1;1) | 56 | 31 | 414 | PRJNA414709 | CP024633 | DSM strain** |
| M. salmoniphilum-like CCUG63695 | Msal-like CCUG63695 | 4998469 | 64,2 | 14 | 4847 | (1;1;1) | 53 | 45 | 430 | PRJNA414709 | PECE00000000 | CCUG strain* |
| M. salmoniphilum-like CCUG63696 | Msal-like CCUG63696 | 4997587 | 64,2 | 9 | 4844 | (1;1;1) | 53 | 44 | 432 | PRJNA414709 | PECD00000000 | CCUG strain* |
| M. salmoniphilum-like CCUG63697 | Msal-like CCUG63697 | 5008405 | 64,2 | 36 | 4856 | (1;1;1) | 53 | 45 | 430 | PRJNA414709 | PECC00000000 | CCUG strain* |
| M. salmoniphilum-like CCUG64054 | Msal-like CCUG64054 | 5011360 | 64,2 | 46 | 4866 | (1;1;1) | 53 | 45 | 432 | PRJNA414709 | PECB00000000 | CCUG strain* |
| M. salmoniphilum-like CCUG64056 | Msal-like CCUG64056 | 4998395 | 64,2 | 8 | 4844 | (1;1;1) | 53 | 45 | 430 | PRJNA414709 | PECF00000000 | CCUG strain* |
| M. franklinii DSM45524 | Mfra DSM45524T | 5408993 | 64,1 | 34 | 5334 | (1;1;1) | 53 | 45 | 474 | PRJNA509866 | RXLR00000000 | DSM strain** |
| M. chelonae subsp. chelonae DSM43804 | Mche T | 5030282 | 63,9 | 1 | 4894 | (1;1;1) | 47 | 34 | 445 | PRJNA508902 | CP034383 | DSM strain** |
| M. abscessus ATCC19977 | Mabs ATCC19977 | 5067172 | 64,1 | 1 | 4955 | (1;1;1) | 47 | 51 | 440 | PRJNA61613 | NC_10397 | NCBI:NC_10397 |
Summary of genome annotation and sources of Msal and Msal-like strains, McheT and MabsATCC19997.
Note: All genomes were annotated using PROKKA pipeline. CDS: Coding Sequences (Prodigal), rRNA: ribosomal RNA (rnammer), tRNA: Transfer RNA (tRNAScanSE), ncRNA: non-coding RNA (Rfam) and signal peptides (signalp); Species highlighted in bold are representative genomes in this article.
*Strains obtained from the CCUG strain collection, Goteborg, Sweden; **Strains obtained from the Deutsche Sammlung von Mikroorganism and Zellkulturen, Germany.
De novo assembly of the long Pac-bio reads (average length 10 kbp) with a coverage of 100x resulted in single scaffolds (one contig for each genome) representing the complete MsalT and McheT genomes (4,776,625 and 5,030,282 bps, respectively; Fig. 1 and Table 1). The average GC-contents were calculated to be 64.3% and 63.9%, respectively. We predicted that the MsalT and McheT genomes encompass 4712 and 4945 genes. Of these, 4652 (4894 for McheT) correspond to coding sequences (CDS), 56 tRNA genes (McheT 47), one rRNA operon (gene order; 16S, 23S, and 5S rRNA) and one transfer-messenger RNA (tmRNA; Table 1). The genome-wide distribution of the tRNA genes is shown in Fig. S1. Of note, MsalT and McheT encode two tRNACys and two (McheT one) tRNAHis isoacceptors as previously observed in other mycobacteria19,20 (see below; Behra et al. unpublished).
Figure 1.

Overview of the MsalT and McheT genomes. (a) Circos plot showing the complete genome sequence of MsalT. From outer to inner circle: Green track represents the complete genome overlapping with scale along the genome length. The next two circles, marked as brown and violet blocks, represent genes in forward (brown) and reverse (violet) strands. The circle with blue (higher than the mean value) and grey (lower than the mean value) “spikes” show the GC-content distribution calculated using a sliding window of 1000 bp, while each grey circle represent variations of the mean GC-content 64.3% in ±10 and ±20 units (i.e. outer grey circle = 84.3% and inner grey circle = 44.3%). The inner track in red (positive) and green (negative) circle shows the GC-skew using a sliding window of 1000 bp. (b) Same as in (a) for McheT where the complete genome overlapping with scale along the genome length (outer circle), which is illustrated in dark yellow. The mean GC-value equals to 63.9%.
For the other strains, seven Msal and five Msal-like strains (Table 1; Fig. S2), the average genome coverage of the Illumina reads ranged from 250x to 600x. The reads were assembled into high quality, near complete genomes (approx. 95% complete) supported by high N50 values and few scaffolds (Table 1; Fig. S2). The sizes of the assembled draft genomes vary from 4,802,997 to 5,176,285 base pairs (Table 1). The average GC-content for these strains was determined to be similar as the GC-content calculated for MsalT and McheT (Table 1). Predicted number of CDS varies between 4629 and 5061 consistent with their genome sizes, while the number of tRNA genes range between 47 and 61 (see below). As for MsalT and McheT, one rRNA operon and one tmRNA gene were detected (Table 1 and S2). The number of predicted non-coding RNAs (ncRNAs) in Msal varies from 31 to 39, while all Msal-like (except Msal-likeCCUG63696) strains were predicted to encode for 45 ncRNAs (Table 1 and S2). The higher number in the Msal-like strains is due to the presence of extra copies of genes encoding Ms_IGR8 (Table S2). For McheT and MabsATCC19977 we predicted 34 and 51 ncRNA genes, respectively (Table 1 and S2).
Whole genome alignment of the complete genomes MsalT, McheT, MabsATCC19977 and the draft genome Msal-likeCCUG64054 revealed high homology. Short inversions were detected in MabsATCC19977 and Msal-likeCCUG64054 compared to MsalT and McheT (Fig. S1a).
Except for MsalT (Table 1), we could not identify any plasmid sequences in either of the genomes including McheT. Presence of a low number (one or two) of incomplete and intact phages, on the other hand, was predicted in all the genomes. The phage sequences constitute less than one percent of the genome size irrespective of strain, except for MsalCCUG62472 where the phage sequences covers approx. 2.2% (Fig. S3; Table S3). A comparison with other MCAC-members (Msal, and Mfra strains; see below) revealed that the fraction of phage sequences for the MfraDSM45524 isolates was higher (≈3%; Fig. S3; Table S3; of note, these MCAC strains were also predicted to carry phages classified as questionable). For McheT and MabsATCC19977 approx. 1.5% of their genomes represent predicted phage sequences.
For MsalT two insertion sequence (IS) elements belonging to the ISAs1 and IS701 families were detected. These two IS elements were also detected in the MsalDE4585–4587 isolates, while the MsalCCUG-isolates were predicted to have additional IS elements belonging to other families (Table S4). For the Msal-likeCCUG strains we also detected ISAs1 and IS701 and the presence of an additional IS element, ISL3 (two copies in Msal-likeCCUG63697). The ISAs1 and IS701 elements are present in other MCAC-members including McheT and MabsATCC19977 (see below). For these isolates, we also detected other IS element families, in particular different Mfra and M. sp. strains (which cluster together with Msal, see below) carry significantly higher numbers compared to our Msal or Msal-like strains. The total number of IS elements varied between two and 54 with McheT having three, while M. sp. D16Q20 carries 54 belonging to 14 different types and MabsATCC19977 a total of six (five types) IS elements (Table S4).
Average nucleotide identity (ANI) analysis reveals that Msal and Msal-like strains cluster into two groups
Unsupervised hierarchical clustering of the “all-versus-all” ANI scores clustered Msal and Msal-like into different groups. The ANI values for the Msal and Msal-like strains varied between 84–87% (Fig. 2a,b), significantly lower than the threshold 95%20,21 to be considered to belong to the same species. Moreover, the Msal strains can be sub-divided into groups (see also below); group I, MsalCCUG60883, MsalCCUG60885, MsalDE4587, and MsalDE4585, group II, MsalT, MsalCCUG62472, and MsalCCUG60884. The ANI scores between “intra-group” members are >95%, while for “inter-group” members it is ≈92% (Fig. 2a,b). The outbreak strain MsalDE4586 could not be referred to any of these two groups (but see below). Moreover, Msal strains are closer (ANI ≈ 87%) to McheT, while the Msal-like strains cluster close to Mabs (ANI >85%; Fig. 2a).
Figure 2.

Clustering of Msal, Msal-like and other MCAC-members based on the average nucleotide scores (ANI) as indicated. (a) Heat map showing ANI values for ‘all-versus-all’ Msal and Msal-like strains including other members of the MCAC. ANI values were clustered based on unsupervised hierarchical clustering (see Methods). The horizontal tree represents the heatmap clustering of column wise dendogram. (b) Dendogram, extracted from the heat map in (a), showing clustering of different strains/isolates based on ANI values.
Expanding the ANI analysis by including 14 MCAC-members, for which the genomes are publicly available (Table S1; ftp://ftp.ncbi.nlm.nih.gov/genomes/; last accessed Aug 2017; see also ref.22), revealed that the Msal-like strains cluster together with Mfra; ANI scores higher than 96% (Fig. 2a,b; of note, MfraCV002, MfraDSM45524 and MfraDSM45524T are the same Mfra strain but represent draft genomes sequenced in different laboratories where the MfraDSM45524T draft genome was sequenced in connection with the present study, see Table S1). These data also suggested that the Mycobacterium sp. D16 strains cluster together with Msal group I (D16R12 and D16R18), Msal group II (D16Q15) or close to these two groups (D16Q13, D16Q14, D16Q16, D16Q20 and D16R24). Hence, this extended analysis suggested that MsalD16Q13, MsalD16Q14, MsalD16Q16, MsalD16Q20 and MsalD16R24 constitute a third Msal group, group III.
To conclude, these data suggested that Msal and Msal-like strains cluster in distinct groups. Moreover, the Msal strains cluster into three groups separating them from McheT as expected.
Core genes and comparative analysis
To further understand the interrelationship between Msal and Msal-like strains (in total 12 strains) and their relation to McheT and MabsATCC19977 we used the complete MsalT genome sequence as a reference genome for identification of orthologous protein coding sequences (CDS). Using amino-acid percentage identity for these CDS clustered Msal and Msal-like strains into two groups in keeping with the ANI data (Fig. 3a; see above). For the Msal strains (relative MsalT) the amino-acid percentage identity for the majority of CDS was ≥95% (Fig. S4a), while for the Msal-like strains it ranged between 85% and 95%. Comparing amino-acid percentage identity for MsalT, McheT and MabsATCC19977 revealed that MsalT and McheT show higher similarity than MsalT and MabsATCC19977 (Fig. S4a).
Figure 3.

Comparative analysis of orthologous genes predicted to be present in Msal and Msal-like strains, McheT and MabsATCC19977. (a) Circos plot showing the presence of protein coding genes in seven Msal, five Msal-like strains, McheT and MabsATCC19977 compared to the reference genome MsalT. The outer track (green) represents the genome for MsalT with a size scale, while the next circle in blue corresponds to the predicted protein coding sequences (CDS) for MsalT. Subsequent circular tracks represent one genome and the number corresponds to the strain name in the legend on the right. Colored radial blocks represent orthologous genes in the corresponding genome and color intensity (see color scale in the middle) indicates percentage identity at the protein level. The white blocks indicate that no orthologs were identified. (b) Heat map showing presence (red) and absence (white) of orthologous genes (excluding core genes) mapped in different Msal and Msal-like strains, McheT and MabsATCC19977, and clustered using hierarchical clustering. The horizontal and vertical trees represent the heat map clustering of the column and row wise dendograms. (c) Venn diagram showing common and unique coding genes for Msal and Msal-like representative strains as indicated. (d) Venn diagram showing common and unique genes in MsalT, Msal-likeCCUG64054, McheT and MabsATCC19977. (e) Heat map showing presence (red) and absence (white) of orthologous ncRNA genes mapped as in (b). The ncRNA genes were identified using Rfam, see main text. The horizontal and vertical trees represent the heat map clustering of the column and row wise dendograms.
To further investigate the variation in gene content across the Msal and Msal-like strains, we identified core and unique genes where core genes are the set of genes present in all genomes. This analysis also revealed sets of genes predicted to be present in either Msal or Msal-like strains (Fig. 3b). In total 3817 CDS were present in both MsalT and Msal-likeCCUG64054, while 834 and 1048 unique CDS were identified in MsalT and Msal-likeCCUG64054, respectively (Fig. 3c). One may argue that this analysis was biased since we compared the MsalT complete genome with the draft Msal-likeCCUG64054 genome. But, since the Msal-likeCCUG64054 genome is estimated to be 95% complete the numbers of unique genes that are false positive are probably few. Including MabsATCC19977 and McheT in this analysis predicted that 3494 core CDS are present in MsalT, Msal-likeCCUG64054, MabsATCC19977 and McheT (Fig. 3d). For functional classification of CDS (including core and unique genes) in selected species see below.
Considering ncRNAs, while the majority of the Rfam annotated ncRNA genes were predicted to be present in these MCAC-members, the T-box category appears to be missing in Msal strains and McheT (Fig. 3e; T-boxes are riboswitches present in the leader region of genes/operons in bacteria influencing their expression23). Sequence alignment of the ileS gene revealed a putative T-box upstream of ileS in all MCAC-members (Fig. S4b). However, we did detect structural variations comparing ileS T-boxes originating from MCAC-members and Mycobacterium smegmatis (Msmeg) MC2155, in particular with respect to the S-turn in stem II (Fig. S4c)24,25.
Phylogenetic analysis
The data presented above suggest that Msal and Msal-like strains cluster into two groups. Hence, we generated a core gene phylogenetic tree where we used 937 core genes (see above) present in the 13 Msal and Msal-like strains, McheT, MabsATCC19977, and the 14 MCAC-members for which genomes are available (see above and Table S1). As outgroups, we used MforDSM46621, Mma (M strain26) and Mycobacterium ulcerans Agy99 (MulcAgy99). The resulting tree clustered the Msal and Msal-like strains in separate branches and suggested that the Msal strains share a common ancestor with McheT, whereas the Msal-like strains are more closely related to MabsATCC19977 and clustered together with Mfra strains [Fig. 4a; of note, a tree based on complete 16S rRNA gene sequences displayed two main branches and it did not discriminate the Msal-like strains and McheT (Fig. S5a)]. This clustering into two separate branches is in keeping with the ANI data (see above) as is the tree based on 3623 core genes present in 12 Msal and Msal-like strains, and MsalT (Fig. 4b). Moreover, consistent with the ANI data the Msal strains cluster together with Mycobacterium sp. D16 isolates into at least three groups close to McheT (Fig. 4a; for further details with respect to M. sp. D isolates, see ref.22).
Figure 4.
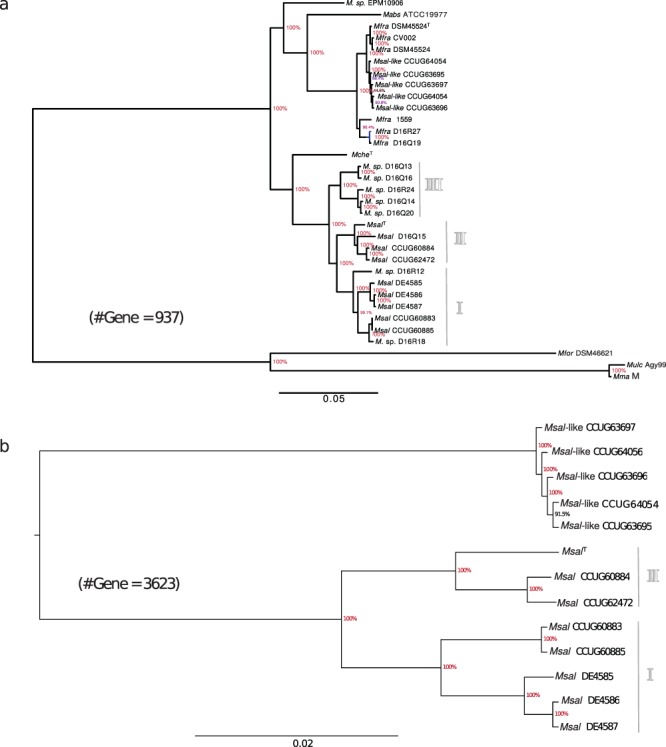
Phylogenetic relationship of Msal and Msal-like strains, McheT and MabsATCC19977. (a) Phylogenetic tree based on 937 core genes present in Msal, Mfra (Msal-like) strains, McheT, MabsATCC19977, MforDSM46621, MulcAgy99 and Mma (M strain). For details see Methods. (b) Phylogenetic tree based on 3623 core genes present in Msal and Msal-like strains as indicated.
Another member of MCAC is Mycobacterium immunogenum (Mimm) and recently the genome sequences for several Mimm and Mycobacterium spp. isolates were published (Table S1; ftp://ftp.ncbi.nlm.nih.gov/genomes/; last accessed Aug 2017). We therefore expanded our analysis by including these genomes and generated a core gene phylogenetic tree based on 623 genes present in these strains, McheT, Mabs subsp. bolletii, Mabs subsp. massiliense, Msal and Msal-like strains (using the same outgroups as above). This tree displayed high bootstrap values and revealed that Mimm and Mabs shared a common ancestor and again clusters Msal and Msal-like strains in separate branches. In addition, this analysis suggested that McheT and several of the Mycobacterium spp. (M. sp. strains) H-strains shared a common ancestor and are grouped into three branches. The other H strains cluster together with Mimm (Fig. S5b). We conclude that Msal-like strains diverged before the separation of Mimm and Mabs.
Functional classification of core and unique genes in MsalT, Msal-likeCCUG64054, McheT and MabsATCC19977
We used the RAST subsystem and classified the function of 3494 core genes, present in MsalT, Msal-likeCCUG64054, McheT and MabsATCC19977, and unique genes, which range between 532 and 810 genes (Fig. 3d; Table S5). This analysis revealed that 62.2% (2173) of the core genes could be classified into different subsystems (Fig. 5a). Considering the total number of CDS in these mycobacteria, the fraction in percentage of functionally classified genes was lower (ranging between approx. 52% and 57%), while the fraction of hypothetical genes was ≈30%. For genes classified into subsystems, the distribution of genes belonging to different categories was similar comparing MsalT, Msal-likeCCUG64054, McheT and MabsATCC19977 (Fig. S6a).
Figure 5.

Functional classification of genes in MsalT, Msal-likeCCUG64054, McheT and MabsATCC19977 into subsystem as indicated. (a) Subsystem classification of 2173 core genes using MsalT. Of note, that a gene can be classified in more than one subsystem. (b) Subsystem classification of specific genes present in MfraCCUG64054 and MabsATCC19977, and present in MsalT and McheT as indictated. (c) Classification of unique genes present in MabsATCC19977, McheT, MsalT and Msal-likeCCUG64054 in the subsystem “Virulence, Disease and Defence”. (d) Subsystem classification of mutational hotspot genes in Msal, see main text for details.
Comparing functional classification of CDS unique (Fig. 3d) to the MsalT/McheT (162 genes) and Msal-likeCCUG64054/MabsATCC19977 (183 genes) pairs showed that the fraction unique genes belonging to in particular the subsystems “Amino acids and Derivatives” and “Carbohydrates” was higher in Msal-likeCCUG64054/MabsATCC19977, while MsalT/McheT carry higher numbers of genes involved in “Sulfur Metabolism” and “Stress Response” (Fig. 5b). Of note, Msal-likeCCUG64054 and MabsATCC19977 also have four unique genes involved in “Regulation and Cell signaling”; classified as transcriptional regulator whiD, hca operon transcriptional activator, HTH-type transcriptional regulator cynR, and carbonic anhydrase 1 gene. However, MsalT/McheT carry other genes annotated as the three former genes whereas no carbonic anhydrase 1 gene could be detected in MsalT/McheT (see Discussion). Moreover, Msal-likeCCUG64054/MabsATCC19977 carry three unique copper homeostasis genes (copB, copZ and copA homologs) belonging to the “Virulence, Disease and Defence” subsystem, while MsalT/McheT appears to have no unique genes in these two latter categories. But analyzing the four species separately, unique genes involved in copper homeostasis were predicted to be present in all species except for MsalT (Fig. 5c).
Furthermore, functional classification of unique genes revealed that all four species carry genes in almost all subsystems (Fig. S6b; Table S5b–e). For example, MabsATCC19977 has a high number of unique genes in the “Amino acid and Derivatives” subsystem, while Msal-likeCCUG64054 has higher numbers in the “Carbohydrate” subsystem. In these two subsystems, we noted the presence of unique genes in these four mycobacteria that are involved in the metabolism of specific amino acids and carbohydrates (Fig. S6c,d). In the “Fatty Acids, Lipids, and Isoprenoids” subsystem we observed differences in the presence/absence and copy number variation of genes comparing MabsATCC19977, McheT, MsalT, and Msal-likeCCUG64054 genomes (Fig. S6e). For example, the long-chain fatty acid CoA ligase fad13 gene, required for maintaining the appropriate mycolic acid composition and permeability of the cell wall27,28, was predicted to be present in nine copies in Msal-likeCCUG64054. Of these nine copies, five were also predicted to be present in MsalT. Interestingly, the number of unique genes belonging to “Virulence, Disease and Defence” appears to be higher in McheT and Msal-likeCCUG64054 relative to MabsATCC19977 and MsalT (Fig. S6b).
Identification of SNVs and mutational hotspots in M. salmoniphilum strains
Single nucleotide variations (SNVs) for the Msal genomes were predicted using MsalT as reference and the program MUMmer29. The number of SNVs ranged between 136702 and 291755 for the different Msal strains. Mutational hotspots, which are genomic regions where the SNV frequencies are higher relative to the background, were identified for Msal. Application of the method described by Das et al.30 revealed 69 mutational hotspot regions in Msal with a high average number of SNVs (>150) per region (Fig. S6f). This corresponds to a frequency of 14.7/Mb. 168 genes overlap with the 69 hotspot regions and of these, 49 were annotated as hypothetical genes and 53 genes were classified into different subsystem categories (Fig. 5d; Table S6). Of the classified genes, >25% were predicted to belong to the category “Virulence, Disease and Defence” with several categorized as mce related (mammalian cell entry; Table S6). Interestingly, the ESX-1 associated gene espR is among the genes that overlap with the hotspot regions (see Discussion).
Horizontal gene transfer, HGT
The total number of putative horizontally transferred genes ranged from 251 (MsalDE4587) to 345 (MfraDSM45524T; Fig. S7a; Table S7). Of these, 66 were predicted to be present in MabsATCC19977, McheT, MsalT and Msal-likeCCUG64054 (Fig. S7b; Table S7). Among possible donors of the HGT genes, members of the order Streptomycetales, Micrococcales, Propionibacteriales, Streptosporangiales and Pseudonocardiales were predicted to be the most likely donors (Fig. S7c). The gene annotations and function of the HGT genes are presented in Table S7. Among the HGT genes one was predicted, the lactate 2-monooxygenase gene, to be of eukaryotic origin and derived from the fungi Ascomycota. Mann-Whitney-Wilcoxon test with respect to GC-content (version R v3.2.231) suggested that the GC-content deviates from the average GC-contents in the majority of the cases (Table S7) supporting the notion that candidate HGT genes have been horizontally transferred.
Virulence genes and ESX genes
Several MCAC-members cause disease and strains belonging to the Mabs branch is of particular interest (see introduction). Mfra (including Msal-like, see above) is phylogenetically close to Mabs, and Msal-like strains were isolated from the water system at BMC (Uppsala University) after an outbreak in the animal facility. Hence, we were interested in to survey the presence of genes encoding for virulence factors (VF) in the Msal and Msal-like strains. For this purpose, we extracted 326 (including homologs) VF genes from a selected number of mycobacteria, including MtbH37Rv (Table S8a), from the virulence factor database (VFDB; last accessed Aug 2017) and searched for orthologs in the Msal and Msal-like genomes. The presence (and absence) of VF genes in the Msal and Msal-like strains were similar compared to MabsATCC19977 and McheT. Of 326 VF genes, 53 are common to all selected mycobacterial species and orthologs to roughly 40% of the 326 VF genes were predicted to be present in McheT, MabsATCC19977, and in the Msal and Msal-like strains (Fig. S8; Table S8b). A comparison of Msal and Msal-like strains revealed that certain VF genes are unique to Msal such as fadE14 and fadD33 (also known as mbtM and mbtN, which are involved in mycobactin biosynthesis32). Of note, fadE14 and fadD33 are also absent in MabsATCC19977 (Fig. S8; Table S8b) while Msal-like strains have two mbtE orthologs, which might influence mycobactin synthesis. Moreover, sigL is absent in the Msal-like strains while MabsATCC19977 carries one sigL copy (Fig. S8; Table S8b; see below, Fig. 6b).
Figure 6.

Analysis of ESX, sigma factor and tRNA genes in MCAC-members. (a) ESX related genes. Presence (blue) and absence (white) of ESX related genes in different mycobacteria as indicated in the phylogenetic tree shown to left (see also Fig. 4b). ESX related genes present in M. stephanolepidis NJB0901 is shown below, see main text. (b) Sigma factor genes. Heat map showing presence (red) and absence (white) of sigma factor genes in MabsATCC19977, McheT, MsalT and Msal-likeCCUG64054. The signature for the respective sigma factor genes correlate with the naming for MtbH37Rv sigma factor genes43. The horizontal and vertical trees represent the heat map clustering of the column and row wise dendograms. (c) Predicted presence of additional tRNA genes in MCAC-members. Gene synteny for a tRNA gene cluster encompassing nine genes in MsalT (seven in MfraCCUG64054). The tRNA genes are marked in red and the vertical boxes marked in brown highlight homologous genes. Note the presence of the HNH endonuclease gene (marked in gray) located within tRNA gene clusters (see main text). See also Figs S1a and S9a.
Two Mabs VFs, adhD and plcC, were predicted to be present in the Msal-like strains (Table S8b). The adhD encodes a potential zinc-type alcohol dehydrogenase, while plcC encodes phospholipase C that hydrolyses membrane phospholipids. The presence of plcC makes Mabs highly cytotoxic to mouse macrophages and, as such plcC is a major virulence factor in Mabs33.
In general, MCAC-members lack ESX-1, ESX-2 and ESX-5 genes (Fig. 6a), where ESX-1 (which encode for esxA/ESAT-6 and esxB/Cfp-10) and ESX-5 have an impact on mycobacterial virulence34,35. However, a few ESX-1, ESX-2 and ESX-5 homologs were predicted to be present in some of the species, where MsalCCUG60884 encodes for several ESX-1 genes, including esxA and esxB. We also noted that Mycobacterium stephanolepidis NJB0901 lacks ESX-1, ESX-2 and ESX-5 genes as other MCAC-members (Fig. 6a; see also36). Moreover, homologs of the VF gene espC were predicted to be present in the MCAC-members (Fig. 6a). The EspC protein is localized on the Mtb surface and is co-secreted with EsxA and EsxB37. In contrast to ESX-1, -2 and -5 genes, ESX-3 and ESX-4 genes were predicted to be present38,39 where ESX-4 is considered to be the ancestor of the ESX-systems38. ESX-3 is suggested to be required for mycobactin mediated iron uptake and, as such, have an impact on virulence of Mtb40 while Mabs ESX-4 genes contribute to intracellular survival39. Of note, absence of ESX-3 and ESX-4 genes is possibly due to draft genome status. Moreover, mce genes affect virulence41 and albeit MCAC-members encode several mce genes belonging to mce4 and mce9 they lack mce2, mce3, mce7 and mce8 genes (Fig. S8; Table S7). We also note that none of the MCAC-members carry ctpV, a putative copper exporter and required for full MtbH37Rv virulence42. Together these data imply variation and differences in genes having a role for a successful infection caused by SGM such as Mtb and RGM MCAC-members, e.g. Msal and Msal-like.
Transcription sigma factor genes
In bacteria, initiation of transcription requires sigma factors and, as such they have key roles in regulating gene expression43–46. While MtbH37Rv is equipped with 13 different sigma factors the number varies between 17 and 19 in MCAC-members (Fig. 6b). Collectively, orthologs for almost all MtbH37Rv sigma factor genes (sigA-M) were also predicted to be present in MCAC-members. In addition to that sigL appears to be missing in the Msal-like strains we note the following. The sigma factor C (sigC), which is suggested to have a role for Mtb virulence47, is present in these mycobacteria, while it is absent in the RGM MsmegMC215546. No sigF orthologs could be detected in the Msal-like strains and in MsalDE4585, MsalDE4586, and MsalDE4587. An ortholog to the Mtb sigI gene was predicted to be present in the Msal strains but it is missing in all the other strains. For sigJ, we predicted three orthologs in McheT, the Msal and Msal-like strains, and four in MabsATCC19977 (of note, for other mycobacteria it has been reported that the sigJ transcript level increases in late stationary phase and during intracellular growth48,49). Moreover, the Msal-like strains carry two sigK orthologs, the Msal strains and McheT were predicted to have one, while we were unable to detect any sigK gene in MabsATCC19977. Interestingly, in Mtb SigK influences expression of the MPT70 and MPT80 antigens and it has been inferred that the SigK/anti-SigK regulatory system is conserved among mycobacteria50. Hence, it seems that MabsATCC19977 consititutes an exception. Together this suggested variation in sigma factor occurrence within MCAC with probable consequences in gene expression patterns in these species.
Genes related to translation
Prediction of ribosomal protein (RP) genes revealed that MsalT encodes 38 large subunit RPs, L1-L36, with two genes encoding L28, L31 and L33, respectively (Table S9a). These genes are also present in McheT, MabsATCC19977 and Msal-likeCCUG64054, albeit with some variations; two copies of the L30 gene is present in Msal-likeCCUG64054, while the L36 gene was not detected in MabsATCC19977 and Msal-likeCCUG64054. With respect to small subunit RPs, 23 genes were predicted, S1-S20 with two paralogs encoding for S1, S14 and S18 in MsalT, McheT, MabsATCC19977, and Msal-likeCCUG64054, while two S5 paralogs were also detected in Msal-likeCCUG64054 (Table S9). Compared to MtbH37Rv, we noted some differences; MtbH37Rv encodes three L28 genes, and it lacks second copies of the L31, L33, and S1. As MsalT, McheT, MabsATCC19977, MtbH37Rv lacks the L36 gene and it does not carry an extra copy of the S5 gene. The presence of extra RP paralogs has been discussed to play a role in adaption to stress and for S18 data suggest that it has a role in zinc homeostasis in MtbH37Rv51,52.
All MCAC-members encode for a complete set of translation factor genes with the exception of prfC (release factor 3; Table S9b). This is the case also for MtbH37Rv and other SGM, while RGM such as MsmegMC2155 and Mycobacterium phlei have prfC homologs20,53,54. Moreover, while MtbH37Rv carries two fusA genes, fusA1 (EF-G) and fusA2 (extra EF-G), MsalT and MCAC-members encode for only one fusA gene corresponding to fusA1 in MtbH37Rv.
Variations in the number of tRNA genes
MCAC-members carry between 47 and 80 tRNA genes and some also encode for pseudo tRNAs, e.g., MsalDE4585 and MsalD16Q15 carry two and three, respectively (Fig. 6c, S1 and S9a–f). Among the tRNA genes, 38 (“core tRNAs”) are present in all strains covering all amino acids except SelCys for which no tRNA gene could be identified. For MabsATCC19977, McheT, MsalT and Msal-likeCCUG64054 the number of tRNA genes vary with 47 for the two formers and 56, and 53 for MsalT and Msal-likeCCUG64054, respectively (Fig. S9a). Their locations on the chromosome in these species are similar (Fig. S9b–d). Several of the extra tRNA genes in MsalT cluster together and were predicted to be present in several Msal strains belonging in particular to Msal group I and II, while those present in Msal-likeCCUG64054 (and Mfra strains; all draft genomes) likely cluster at roughly the same location on the chromosome as in MsalT (Fig. 6c; Fig. S9a,e). Sequence alignments suggest that these extra tRNA genes probably are of different origins (Fig. S9f). Interestingly, the group III Msal strain, MspD16Q14 was predicted to carry 80 tRNA genes where 34 appears to cluster (Fig. S9a). This tRNA gene cluster shows striking similarities, including the presence of the GOLLD RNA gene and an HNH endonuclease gene (not shown), with that detected in e.g., Mabs M24 and the Mycobacterium aubagnense type strain55,56 (Behra et al. unpublished).
To conclude, based on that the extra tRNA show differences in their structure compared to the common tRNAs it is conceivable that they have been acquired through horizontal gene transfer after divergence of Msal and Mche, and Mabs and Msal-like strains (and Mfra). In this context, we note that an HNH endonuclease gene is predicted to be present in close proximity to the extra tRNA gene clusters (Fig. S9e). Moreover, given that MCAC-members are the closest mycobacteria to the mycobacterial ancestor17,18 (unpublished) our data suggest that at least 38 tRNA genes were present before mycobacteria diverged into separate species that constitute the genus.
Discussion
We present the genomes for 14 mycobacteria, including the complete genomes for MsalT and McheT (type strains), belonging to MCAC. The size of the genomes range between 4.8 and 5.2 Mbp with MsalT having the smallest genome. Our comparative genomic analysis, ANI, CDS amino-acid percentage identity and core gene based phylogeny, suggested that Msal and Msal-like strains are representatives of different species and close to Mabs. Including Mfra10,12, Mycobacterium sp. “D16” strains and MabsATCC19977 suggest that the Msal-like isolates should be referred to as Mfra strains (Fig. 4). The data further suggested that the Msal strains clustered into three separate groups, where the Mycobacterium sp. D16 (Q13, Q14, Q16, Q20 and R24) constitute one group. Together, our findings expand and provide insight into the phylogenetic and evolutionary relationships within the MCAC and the Mycobacterium genus and clarify species identity17–20,57.
The number and type of IS elements in Msal, Msal-like, Mfra, McheT and MabsATCC19977 genomes vary as does the presence of phage sequences, while sequences of plasmid origins were only detected in MsalT. Thus, IS elements and phages appear to have contributed to the evolution of these MCAC members. Differences with respect to IS elements and phage sequences are also observed comparing strains of other mycobacteria, as exemplified by our comparative genomic studies of M. phlei and Mma strains20,57. Moreover, the number of SNVs for the Msal strains ranged between 136702 and 291755. This is significantly higher compared to the situation in Mma where the number of SNVs in different strains relative to the Mma M strain varies between 56000 and 8900057. But, we emphasize that Mma display a higher frequency of mutational hotspot regions relative to Msal (26.5/Mb vs. 14.7/Mb, respectively). In this context, some mycobacteria use Distributive Conjugal Transfer (DCT) to transfer DNA and ESX-1 and ESX-4 have been suggested to play a key role in this process58–60. Analysis of Mabs isolates implicates that DCT is in operation in this RGM61. Together this makes it plausible that Msal strain variation and clustering into three groups is at least partly the result of DCT.
As other members of MCAC, Msal and Mfra (Msal-like) lack the majority of the ESX-1, ESX-2, ESX-5 and ESX-6 (duplication of ESX-1 in Mma26,57) genes, while genes belonging to ESX-3 and ESX-4 are present. However, some ESX genes such as esxA, esxB and espI (associated with ESX-1) were predicted to be present in some of the strains, while the ESX-1 associated espR is present in all strains (see below). Interestingly, MsalCCUG60884 encodes for several of the ESX-1 genes including esxA and esxB (Fig. 6a). Possibly, these ESX genes have been aquired through horizontal gene transfer. The ESX systems are involved in transport and secretion and available data suggest that ESX-1, ESX-3 and ESX-5 affect virulence for several mycobacteria including Mtb and Mma34,35. That ESX-5 is missing is consistent with that it is present in SGM and has not been detected in RGM35 (unpublished data).
The transcriptional regulator EspR (espR) is involved in controlling Mtb virulence and expression of ESX-1 genes62 and espR is present in MCAC-members. Apart from being a regulator of the ESX-1 system, EspR is regulating the expression of genes involved in cell wall synthesis. EspR also operates together with PhoP, which is part of a two-component system, regulating the expression of many VF genes. This provides a rationale for its presence in MCAC-members and given that espR constitutes a hotspot region in Msal raises the possibility that this has an impact on the pathogenicity for the different Msal strains. In this context, we note that espR is not essential in MtbH37Rv62.
An intriguing question is whether other genes/systems functionally compensate for the absence of these ESX systems. For example, there appears to be a coupling between the transporter Mce1 family proteins and the ESX-1 system35. Understanding this and other questions that relates to mycobacterial infections will have an impact on our understanding of the biology of mycobacteria. In this context, the PhoPR regulon and ESX-1 secretion in a MtbCDC1551 derivative is inhibited by ethoxzolamide, which is a known carbonic anhydrase inhibitor. As a consequence, virulence is attenuated. Together, this raises the possibility of coupling between carbonic anhydrase activity and signaling mediated by the two-component PhoPR regulatory system in MtbCDC155163. A carbonic anhydrase 1 gene was predicted to be present in MabsATCC19977 and Msal-likeCCUG64054 but absent in MsalT and McheT. Hence, it would be interesting to study whether this coupling is present in Mabs (and Msal-like i.e., Mfra) and if so, does ethoxzolamide also influence the virulence for these two mycobacteria.
As other MCAC-members, Msal and Msal-like belong to RGM but they only harbor one rRNA operon supporting the notion that the number of rRNA operons does not explain the difference in growth rate comparing SGM and RGM57,64–66. Moreover, as Mtb and other SGM, MsalT (complete genome) and other MCAC-members lack the gene encoding the translational release factor RF3, prfC. RF3 is suggested to assist in the dissociation of class I translational release factors from the ribosome, and to abolish competition between the release factors and ribosome recycling factor, RRF, for binding to the ribosome67–69. Inactivation of RF3 in Escherichia coli results in lower growth rates67,70,71. Together this suggests that the absence or presence of prfC does not dictate whether mycobacteria should be classified as SGM or RGM. Moreover, phylogeny based on whole genome sequencing suggests that MCAC is the earliest diverging mycobacterial lineage17,18,36 (unpublished). Hence, acquisition of prfC in other RGM happened after they diverged from MCAC.
MCAC-members, except Msal, have been implicated to be associated with human diseases. Mfra, which belongs to MCAC, causes symptoms similar to those observed in patients infected with Mabs10,12. We cannot conclusively state that the Msal-like strains isolated from the tap water caused the outbreak at the animal facility among the mice population at Uppsala University. To do this the pathogen has to be isolated from infected mice and study whether exposure to the bacteria indeed cause disease. However, on the basis that Mfra causes disease in humans we consider it plausible that Msal-like (i.e. Mfra) also infects and causes disease in animals, such as mice. In this context, Msal-like (and Mfra) strains share several genes coding for virulence factors (VF) with Mabs such as the major Mabs VF plcC, which makes Mabs highly cytotoxic to mouse macrophages33. Nonetheless, its presence in tap water suggest that it can be present in the environment. Moreover, we note that neither of the strains analyzed here carry mutations at positions 1408 (16S rDNA; E. coli numbering) or 2058 (or 2059; 23S rDNA). Mutations at these positions in Mabs isolates results in resistance to amikacin and macrolides, respectively72.
To conclude, understanding the genome composition of mycobacteria will be instrumental to understand not only their evolution but also provide insight into mycobacterial physiology and pathogenicity, and clarify species identity. This knowledge will be instrumental for treatment of infections caused by mycobacteria such as MCAC-members.
Methods
Strains, cultivation and DNA isolation
We collected eight Msal and five Msal-like strains isolated from different sources, MfraDSM45524T and McheT where MsalT and McheT represent the type strains Msal DSM43276 and Mche DSM43804, respectively. MsalT, McheT and MfraDSM45524T were obtained from the Deutsche Sammlung von Mikrooganismen und Zellkulturen, Germany (Table 1 and S1; we refer to the strains such that e.g., strain MsalDE4585 corresponds to M. salmoniphilum DE4585). The strains were grown under conditions as recommended by the supplier (for the outbreak Msal “DE-strains”, we followed the recommendation from DSM); aliquots of −80 °C stocks were plated on Middlebrook 7H10 media and incubated at 30 °C. Genomic DNA was isolated as previously described73 (see also Supplementary information). Prior to submission for genome sequencing we PCR amplified and sequenced 16S rDNA to ensure that the cultures were free from contaminations.
Genome sequencing, assembly and annotation
The MsalT and McheT type strains were sequenced using the Pacific Biosciences (PacBio) platform at the NGI-Uppsala Genome Center, while sequencing of the other 12 strains (and MfraDSM45524T) were performed using Illumnia short read technology (at the SNP@SEQ Technology Platform, Uppsala University). Genome assembly, annotation, plasmid, phage, identification of IS elements, horizontal gene transfer (HGT) analysis and identification of SNV and mutational hotspots were done as previously described19,20,30,57 (see Supplementary information and refs74–83).
Average nucleotide identity
The evolutionary distance between two species can be measured as average nucleotide identity (ANI) of homologous genomic regions84. ANI values were calculated for all the sequenced genomes in a pairwise manner using the Jspecies tool21. The ANI values were clustered using an unsupervised hierarchical clustering algorithm and plotted using “R” environment85.
Identification and analysis of core genes
To identify core and unique genes, predicted CDS from the genomes were used for “all-vs-all” BLAST search. Based on the BLAST results orthologous genes were identified using PanOCT with minimum 45% identity and 65% query coverage86, see also refs19,20,57.
Phylogenetic analysis based on single and multiple genes
We extracted 16S ribosomal RNA (rRNA) gene sequences from the genomes and homologous gene sequences from other mycobacteria as indicated were downloaded from the NCBI database and aligned using MAFFT (version 587). Phylogenetic trees, 16S rDNA and core gene based trees, based on the multiple sequence alignment were computed using the FastTree along with default settings, which infers approximately-maximum-likelihood phylogenetic trees from alignments of nucleotide or protein sequences (Jukes-Cantor + CAT model for nucleotide sequences and Jones-Taylor-Thorton + CAT models of amino acid sequences) and 1000 cycles of bootstrapping88. The figures were generated using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Ethics statement
All methods were carried out in accordance with relevant guidelines and regulations.
Data deposition
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the projects PRJNA414709, PRJNA508902 and PRJNA509866.
Supplementary information
Acknowledgements
We thank our colleagues for discussions, Ms Caroline Sciarrillo is acknowledged for preliminary cultivation of type and outbreak M. salmoniphlium strains in the laboratory of DGE, and Dr Santanu Dasgupta for critical reading the manuscript. We acknowledge Ms S. Cardew, CCUG Laboratory, Göteborg, Sweden (email: elisabeth.inganas@vgregion.se) who classified the Msal and Msal-like isolates obtained from the CCUG strain collection. Sequencing was performed by the SNP&SEQ Technology Platform in Uppsala, which is part of the Science for Life Laboratory at Uppsala University and supported as a national infrastructure by the Swedish Research Council. The computations were performed on resources provided by SNIC through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under Project b2011072. This work was funded by the Swedish Research Council (M and N/T), the Swedish Research Council for Environment, Agricultural Sciences, and Spatial Planning (FORMAS), and Uppsala RNA Research Center (Swedish Research Council Linneus support) to LAK. Ms L. Shirreff was funded by a Louisiana Graduate Fellowship award and funds for supplies and travel by the University of Louisiana Graduate Organization to DGE.
Author Contributions
L.A.K. and D.G.E. conceived the study. P.R.K.B. and S.D. designed and performed the bioinformatics computations. P.R.K.B., S.D., B.M.F.P., D.G.E. and L.A.K. analyzed and interpreted the data. B.M.F.P. and D.G.E. generated culture extracts and DNA isolation. B.M.F.P., L.S. and T.D. maintained, cultivated and prepared DNA from different Msal and Msal-like strains. K.-G.J. isolated the samples from the tap water and analyzed the outbreak at the animal facility (Biomedical Center, Uppsala Unversity). P.R.K.B., S.D., B.M.F.P., D.G.E. and L.A.K. wrote the manuscript. All authors read and approved the final version of the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Phani Rama Krishna Behra and Sarbashis Das contributed equally.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-40922-x.
References
- 1.Bataillon E, Dubard TL. Un nouveau type de tuberculose. Comptes rendus des Sceances de la Societe Biologie. 1897;49:446–449. [Google Scholar]
- 2.Austin, B. & Austin, D. A. Bacterial Fish Pathogens (SpringerDordrecht Heidelberg New York London) (2013).
- 3.Whitman, W. B. et al. Bergey’s Manual® of Systematic Bacteriology. 2nd ed. Springer New York: New York, NY (2012).
- 4.Righetti M, et al. Mycobacterium salmoniphilum infection in a farmed Russian sturgeon, Acipenser gueldenstaedtii (Brandt & Ratzeburg) J Fish Dis. 2014;37:671–674. doi: 10.1111/jfd.12143. [DOI] [PubMed] [Google Scholar]
- 5.Zerihun MA, Nilsen H, Hodneland S, Colquhoun DJ. Mycobacterium salmoniphilum infection in farmed Atlantic salmon, Salmo salar L. J Fish Dis. 2011;34:769–781. doi: 10.1111/j.1365-2761.2011.01293.x. [DOI] [PubMed] [Google Scholar]
- 6.Zerihun MA, Colquhoun DJ, Poppe TT. Experimental mycobacteriosis in Atlantic cod, Gadus morhua L. J Fish Dis. 2012;35:365–377. doi: 10.1111/j.1365-2761.2012.01349.x. [DOI] [PubMed] [Google Scholar]
- 7.Ross AJ. Mycobacterium salmoniphilium sp. nov. from salmonoid fishes. Am Rev Respir Dis. 1960;81:241–250. doi: 10.1164/arrd.1960.81.2.241. [DOI] [PubMed] [Google Scholar]
- 8.Whipps CM, Butler WR, Pourahmad F, Watral VG, Kent ML. Molecular systematics support the revival of Mycobacterium salmoniphilum (ex Ross 1960) sp. nov., nom. rev., a species closely related to Mycobacterium chelonae. Int J Syst Evol Microbiol. 2007;57:2525–2531. doi: 10.1099/ijs.0.64841-0. [DOI] [PubMed] [Google Scholar]
- 9.Watts EM, Schreier HJ, Lanska L, Hale MS. The rising tide of antimicrobial resistance in aquaculture: Sources, sinks and solutions. Mar Drugs. 2017;15:158. doi: 10.3390/md15060158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simmon KE, et al. Mycobacterium chelonae-abscessus complex associated with sinopulmonary disease, northeastern USA. Emerg Inf Dis. 2011;17:1692–1700. doi: 10.3201/eid1709.101667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown-Elliott BA, Philley JV. Rapidly growing mycobacteria. Microbiol Spectrum. 2017;5(1):TNMI7-0027–2016. doi: 10.1128/microbiolspec.tnmi7-0027-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nogueira CL, et al. Mycobacterium saopaulense sp. nov. a rapidly growing mycobacterium closely related to members of the Mycobacterium chelonae-Mycobacterium abscessus group. Int J Syst Evol Microbiol. 2015;65:4403–4409. doi: 10.1099/ijsem.0.000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cabello FC, et al. Antimicrobial use in aquaculture re-examined: its relevance to antimicrobial resistance and to animal and human health. Environ Microbiol. 2013;15:1917–1942. doi: 10.1111/1462-2920.12134. [DOI] [PubMed] [Google Scholar]
- 14.Cabello FC, Godfrey HP, Buschmann AH, Dölz HJ. Aquaculture as yet another environmental gateway to the development and globalisation of antimicrobial resistance. Lancet Infect Dis. 2016;16:e127–33. doi: 10.1016/S1473-3099(16)00100-6. [DOI] [PubMed] [Google Scholar]
- 15.Marti E, Variatza E, Balcazar JL. The role of aquatic ecosystems as reservoirs of antibiotic resistance. Trends Microbiol. 2014;22:36–41. doi: 10.1016/j.tim.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Chang CT, Whipps CM. Activity of antibiotics against Mycobacterium species commonly found in laboratory zebrafish. J Aquat Anim Health. 2015;27:88–95. doi: 10.1080/08997659.2015.1007176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fedrizzi T, et al. Genomic characterization of nontuberculosis mycobacteria. Sci Rep. 2017;7:45258. doi: 10.1038/srep45258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta RS, Lo B, Son J. Phylogenomics and comparative genomic studies robustly support division of the genus Mycobacterium into an emended genus Mycobacterium and four novel genera. Front Microbiol. 2018;9:article 67. doi: 10.3389/fmicb.2018.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das S, et al. Characterization of Three Mycobacterium spp. with Potential Use in Bioremediation by Genome Sequencing and Comparative Genomics. Genome Biol Evol. 2015;7:1871–1886. doi: 10.1093/gbe/evv111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das S, et al. The Mycobacterium phlei Genome: Expectations and Surprises. Genome Biol Evol. 2016;8:975–985. doi: 10.1093/gbe/evw049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA. 2009;106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nogueria CL, et al. Characterization of Mycobacterium chelonae-like strains by comparative genomics. Front Microbiol. 2017;8:789. doi: 10.3389/fmicb.2017.00789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henkin T. The T box riboswitch: A novel regulatory RNA that utilizes tRNA as its ligand. Biochim Biophys Acta. 2014;1839:959–963. doi: 10.1016/j.bbagrm.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherwood AV, Grundy FJ, Henkin TM. T box riboswitches in actinobacteria: translational regulation via a novel tRNA interactions. Proc Natl Acad Sci USA. 2014;112:1113–1118. doi: 10.1073/pnas.1424175112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sherwood AV, Frandsen JK, Grundy FJ, Henkin TM. New tRNA contacts facilitate ligand binding in a Mycobacterium smegmatis T box riboswitch. Proc Natl Acad Sci USA. 2018;115:3894–3899. doi: 10.1073/pnas.1721254115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stinear TP, et al. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 2008;18:729–741. doi: 10.1101/gr.075069.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh A, et al. Requirement of the mymA operon for appropriate cell wall ultrastructure and persistence of Mycobacterium tuberculosis in the spleens of guinea pigs. J Bacteriol. 2005;187:4173–4186. doi: 10.1128/JB.187.12.4173-4186.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersson CS, et al. The Mycobacterium tuberculosis very-long-chain fatty acyl-CoA synthetase: structural basis for housing lipid substrates longer than the enzyme. Structure. 2012;20:1062–1070. doi: 10.1016/j.str.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 29.Delcher AL, et al. Alignment of whole genomes. Nucl Acids Res. 1999;27:2369–2376. doi: 10.1093/nar/27.11.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das S, et al. Identification of hot and cold spots in genome of Mycobacterium tuberculosis using Shewhart Control Charts. Sci Rep. 2012;2:297. doi: 10.1038/srep00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.R Development Core Team: R: A Language and Environment for Statistical Computing. R. Vienna, Austria: R Foundation for Statistical Computing (2015).
- 32.Sritharan M. Iron homeostatis in Mycobacterium tuberculosis: Mechanistic insights into siderophore-mediated iron uptake. J Bacteriol. 2016;198:2399–2409. doi: 10.1128/JB.00359-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.N’Goma JCB, et al. Mycobacterium abscessus Phospholipase C Expression Is Induced during Coculture within Amoebae and Enhances M. abscessus Virulence in Mice. Infect Immun. 2015;83:780–791. doi: 10.1128/IAI.02032-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gröschel MI, Sayes F, Simeone R, Majlessi L, Brosch R. ESX secretion systems: mycobacterial evolution to counter host immunity. Nat Rev Mircro. 2016;14:677–691. doi: 10.1038/nrmicro.2016.131. [DOI] [PubMed] [Google Scholar]
- 35.Bosserman RE, Champion PA. Esx systems and the mycobacterial cell envelope: What’s the connection? J Bacteriol. 2017;199:e00131–17. doi: 10.1128/JB.00131-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tortoli E, et al. The new phylogeny of the genus Mycobacterium: The old and the news. Inf Gene Evol. 2017;56:19–25. doi: 10.1016/j.meegid.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 37.Lou Y, Rybniker J, Sala C, Cole ST. EspC forms a filamentous structure in thecell envelope of Mycobacterium tuberculosis and impacts ESX-1 secretion. Mol Microbiol. 2017;103:26–38. doi: 10.1111/mmi.13575. [DOI] [PubMed] [Google Scholar]
- 38.Dumas E, et al. Mycobacterial pan-genome analysis suggests important role of plasmids in the radiation of type VII secretion systems. Genome Biol Evol. 2016;8:387–402. doi: 10.1093/gbe/evw001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laencina L, et al. Identification of genes required for Mycobacterium abscessus growth in vivo with a prominent role of the ESX-4 locus. Proc Natl Acad Sci USA. 2018;115:e1002–e1011. doi: 10.1073/pnas.1713195115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siegrist MS, et al. Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc Natl Acad Sci USA. 2009;106:18792–18797. doi: 10.1073/pnas.0900589106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casali N, Riley LW. A phylogenomic analysis of the Actinomycetales mce operons. BMC Genomics. 2007;8:60. doi: 10.1186/1471-2164-8-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ward SK, Abomoelak B, Hoye EA, Steinberg H, Talaat AM. CtpV: a putative copper exporter required for full virulence of Mycobacterium tuberculosis. Mol Microbiol. 2010;77:1096–1110. doi: 10.1111/j.1365-2958.2010.07273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manganelli R, et al. Sigma factors and global gene regulation in Mycobacterium tuberculosis. J Bacteriol. 2004;186:715–724. doi: 10.1128/JB.186.4.895-902.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodrigue S, Provvedi R, Jacques PÉ, Gaudreau L, Manganelli R. The σ factors of Mycobacterium tuberculosis. FEMS Microbiol Rev. 2006;30:926–941. doi: 10.1111/j.1574-6976.2006.00040.x. [DOI] [PubMed] [Google Scholar]
- 45.Sachdeva P, Misra R, Tyagi AK, Singh Y. The sigma factors of Mycobacterium tuberculosis: Regulation of the regulators. FEBS J. 2010;277:605–626. doi: 10.1111/j.1742-4658.2009.07479.x. [DOI] [PubMed] [Google Scholar]
- 46.Pettersson BMF, et al. Comparative sigma factor-mRNA levels in Mycobacterium marinum under stress conditions and during host infection. PLOS One. 2015;10:e0139823. doi: 10.1371/journal.pone.0139823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang A, Smollett KL, Gopaul KK, Chan BH, Davis EO. Mycobacterium tuberculosis H37Rv sigC is expressed from two promoters but is not auto-regulatory. Tuberculosis. 2012;92:48–55. doi: 10.1016/j.tube.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 48.Hulten K, et al. In situ hybridization method for studies of cell wall deficient M. paratuberculosis in tissue samples. Vet Microbiol. 2000;77:513–518. doi: 10.1016/s0378-1135(00)00336-9. [DOI] [PubMed] [Google Scholar]
- 49.Sechi LA, et al. Genome and transcriptome scale portrait of sigma factors in Mycobacterium avium subsp. paratuberculosis. Infect Genet Evol. 2007;7:424–432. doi: 10.1016/j.meegid.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 50.Veyrier F, Said-Salim B, Behr MA. Evolution of the mycobacterial SigK regulon. J Bacteriol. 2008;190:1891–1899. doi: 10.1128/JB.01452-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore CM, Helmann JD. Metal ion homeostasis in Bacillus subtilis. Curr Opin Microbiol. 2005;8:188–195. doi: 10.1016/j.mib.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 52.Prisic S, et al. Zinc regulates a switch between primary and alternative S18 ribosomal proteins in Mycobacterium tuberculosis. Mol Microbiol. 2015;97:263–280. doi: 10.1111/mmi.13022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cole ST, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 54.Mohan A, Padiadpu J, Baloni P, Chandra N. Complete genome sequences of a Mycobacterium smegmatis laboratory strain (MC2155) and isoniazid-resistant (4XR1/R2) mutant strains. Genome Announc. 2015;3:e01520–14. doi: 10.1128/genomeA.01520-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weinberg Z, Perreault J, Meyer MM, Breaker RR. Exceptional structured noncoding RNAs revealed by bacterial metagenome analysis. Nature. 2009;462:656–659. doi: 10.1038/nature08586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choo SW, et al. Genomic reconnaissance of clinical isolates of emerging human pathogen Mycobacterium abscessus reveals high evolutionary potential. Sci Rep. 2014;4:4061. doi: 10.1038/srep04061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Das S, et al. Extensive genomic diversity among Mycobacterium marinum strains revealed by whole genome sequencing. Sci Rep. 2018;8:12040. doi: 10.1038/s41598-018-30152-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gray TA, Krywy JA, Harold J, Palumbo MJ, Derbyshire KM. Distributive conjugal transfer in mycobacteria generates progeny with meiotic-like genome-wide mosaicism, allowing mapping of a mating identity locus. PLoS Biol. 2013;11:e1001602. doi: 10.1371/journal.pbio.1001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gray TA, et al. Intercellular communictation and conjugation are mediated by ESX secretion systems in mycobacteria. Science. 2016;354:347–350. doi: 10.1126/science.aag0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gray TA, Derbyshire KM. Blending genomes: distributive conjugal transfer in mycobacteria, a sexier form of HGT. Mol Microbiol. 2018;108:601–613. doi: 10.1111/mmi.13971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sapriel G, et al. Genome-wide mosaicism within Mycobacterium abscessus: evolutionary and epidemiological implications. BMC Genomics. 2016;17:118. doi: 10.1186/s12864-016-2448-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Raghavan S, Manzanillo P, Chan K, Dovey C, Cox JS. Secreted transcription factor controls Mycobacterium tuberculosis virulence. Nature. 2008;454:717–721. doi: 10.1038/nature07219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnson BK, et al. The carbonic anhydrase inhibitor ethoxzolamide inhibits the Mycobacterium tuberculosis PhoPR regulon and Esx-1 secretion and attenuates virulence. Antimicrob Agents Chemother. 2015;59:4436–45. doi: 10.1128/AAC.00719-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gonzalelz-y-Merchand JA, Colston MJ, Cox RA. Roles of multiple promoters in transcription of ribosomal DNA: effects of growth conditions on precursor rRNA synthesis in mycobacteria. J Bacteriol. 1998;180:5756–5761. doi: 10.1128/jb.180.21.5756-5761.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gonzalelz-y-Merchand JA, Colston MJ, Cox RA. Effects of growth conditions on expression of mycobacterial murA and tyrS genes and contributions of their transcripts to precursor rRNA synthesis. J Bacteriol. 1999;181:4617–4627. doi: 10.1128/jb.181.15.4617-4627.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Menedez Mdel C, Rebollo MJ, Núnez Mdel C, Cox RA, Garcia MJ. Analysis of the precursor rRNA fractions of rapidly growing mycobacteria: quantification by methods that include the use of a promoter (rrnAP1) as a novel standard. J Bacteriol. 2005;187:534–543. doi: 10.1128/JB.187.2.534-543.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pavlov MY, Freistroffer DV, Heurgué-Hamard V, Buckingham RH, Ehrenberg M. Release factor RF3 abolishes competition between release factor RF1 and ribosome recycling factor (RRF) for a robosome binding site. J Mol Biol. 1997;273:389–401. doi: 10.1006/jmbi.1997.1324. [DOI] [PubMed] [Google Scholar]
- 68.Mora L, Zavialov A, Ehrenberg M, Buckingham RH. Stop codon recognition and interactions with peptide release factor RF3 of truncated and chimeric RF1 and RF2 from Escherichia coli. Mol Microbiol. 2003;50:1467–1476. doi: 10.1046/j.1365-2958.2003.03799.x. [DOI] [PubMed] [Google Scholar]
- 69.Gao H, et al. RF3 induces ribosomal conformational changes responsible for dissociation of class I release factors. Cell. 2007;129:929–941. doi: 10.1016/j.cell.2007.03.050. [DOI] [PubMed] [Google Scholar]
- 70.Grentzmann G, Brecheimier-Baey D, Heurgue V, Mora L, Buckingham RH. Localization and characterization of the gene encoding release factor RF3 in Escherichia coli. Proc Natl Acad Sci USA. 1994;91:5848–5852. doi: 10.1073/pnas.91.13.5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mikuni O, et al. Identification of the prfC gene, which encodes peptide-chain-release factor 3 of Escherichia coli. Proc Natl Acad Sci USA. 1994;91:5798–5802. doi: 10.1073/pnas.91.13.5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bryant JM, et al. Emergence and spread of a human-transmissible multidrug-resistant nontuberculous mycobacterium. Science. 2016;354:751–357. doi: 10.1126/science.aaf8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pettersson BMF, et al. Draft Genome Sequence of Saccharopolyspora rectivirgula. Genome Announc. 2014;2:e01117. doi: 10.1128/genomeA.01117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lagesen K, et al. RNAmmer: consistent and rapid annotation of ribosomal RNA. genes. Nucl Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guy L, Kultima JR, Andersson SG. E. genoPlotR: comparative gene and genome visualization in R. Bioinformatics (Oxford, England) 2010;26:2334–2335. doi: 10.1093/bioinformatics/btq413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Aziz RK, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Varani AM, Siguier P, Gourbeyre E, Charneau V, Chandler M. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 2011;12:R30. doi: 10.1186/gb-2011-12-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucl Acids Res. 2011;39:W347–52. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tritt A, Eisen JA, Facciotti MT, Darling AE. An integrated pipeline for de novo assembly of microbial genomes. PLoS ONE. 2012;7:e42304. doi: 10.1371/journal.pone.0042304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chin C-S, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Meth. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 82.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics (Oxford, England) 2014;30:2068–2069. [Google Scholar]
- 83.Hawkey J, et al. ISMapper: identifying transposase insertion sites in bacterial genomes from short read sequence data. BMC Genomics. 2015;16:667. doi: 10.1186/s12864-015-1860-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA. 2005;102:2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria (2008).
- 86.Fouts DE, Brinkac L, Beck E, Inman J, Sutton G. PanOCT: automated clustering of orthologs using conserved gene neighborhood for pan-genomic analysis of bacterial strains and closely related species. Nucl Acids Res. 2012;40:e172–e172. doi: 10.1093/nar/gks757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Katoh K, Standley DM. MAFFT: iterative refinement and additional methods. Methods Mol Biol (Clifton, N. J.) 2014;1079:131–146. doi: 10.1007/978-1-62703-646-7_8. [DOI] [PubMed] [Google Scholar]
- 88.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.