

Abstract
Background
The mutation of TANK‐binding kinase 1 (TBK1) gene has been regarded as a causative gene of frontotemporal dementia (FTD)‐amyotrophic lateral sclerosis (ALS) spectrum disease in recent years. So far, more than 70 TBK1 variants have been identified in patients with FTD‐ALS spectrum.
Methods
We reported a Chinese FTD patient carrying TBK1 p.Ile334Thr variant detected by target sequencing and Sanger sequencing. The patient's clinical materials were collected. The transcription and translation levels of TBK1 mutant were investigated in fibroblast by qPCR and western blot. The effects of TBK1 mutant in inflammation pathway and autophagy were detected by luciferase reporter assay and GST pull‐down assay.
Results
The patient was diagnosed as behavioral variant FTD (bvFTD) and displayed progressively severe cognitive impairment especially in executive function. A pattern of frontotemporal atrophy and hypometabolism was shown through MRI and PET‐CT. In vitro functional experiments of TBK1 p.Ile334Thr variant demonstrated reduced transcription and translation levels, decreased kinase activity but maintenance of interaction with optineurin. The variant was classified as likely pathogenic according to American College of Medical Genetics and Genomics guideline.
Conclusion
We proposed the TBK1 mutation p.Ile334Thr as a likely pathogenic variant in bvFTD which also expanded the clinical spectrum of this variant. It can partially abrogate TBK1 functions and be responsible for FTD‐ALS spectrum diseases through neuroinflammatory pathway.
Keywords: frontotemporal dementia, gene, TBK1, variant
1. INTRODUCTION
Frontotemporal dementia (FTD) is a progressive neurodegenerative syndrome with the clinical manifestations of executive dysfunction, language deficits, and behavior as well as personality changes (Perry & Miller, 2013). It was the second common in young‐onset dementia, accounting for 20% dementia in 45–64 age‐group (Mercy, Hodges, Dawson, Barker, & Brayne, 2008). Its clinical subtypes include behavioral variant FTD (bvFTD) and two primary progressive aphasias: nonfluent aphasia and semantic dementia. Extrapyramidal symptoms and/or amyotrophic lateral sclerosis (ALS) could also be presented in FTD patients (Fujioka & Wszolek, 2011).
Frontotemporal dementia is a syndrome of genetic heterogeneity. Since MAPT was first identified as the causative gene of FTD in 1998, an increasing number of pathogenic genes have been reported associated with FTD including GRN, C9orf72, VCP, SQSTM1, CHMP2B, TBK1, OPTN, TARDBP, CHCHD10, UBQLN2, and DCTN1 (Pottier, Ravenscroft, Sanchez‐Contreras, & Rademakers, 2016). Most of them were also the causative genes of ALS except for GRN (Mackenzie & Neumann, 2016; Renton et al., 2011; Van Mossevelde, van der Zee, Cruts, & Van Broeckhoven, 2017). Besides, approximately 15% FTD patients develop clinical features of ALS (Lomen‐Hoerth, Anderson, & Miller, 2002) and up to 15% ALS patients finally reach the criteria of FTD, ALS, and FTD are now considered as ALS‐FTD spectrum, other than two separate diseases.
TBK1 (OMIM:604834) is the causative genes of FTD‐ALS recognized recently based on a large‐scale whole‐exome sequencing study and the following variant analysis (Cirulli et al., 2015; Le Ber et al., 2015). Up till now, more than 70 TBK1 variants have been reported in ALS, FTD, or FTD‐ALS patients and most were Caucasian (Ahmad, Zhang, Casanova, & Sancho‐Shimizu, 2016). Its encoding protein, tumor necrosis factor receptor‐associated factor NF‐kB activator‐binding kinase 1 (TBK1), was a serine/threonine protein kinase involved in multiple cellular pathways. It has functions in neuroinflammation through NF‐κB pathway and can interact with downstream proteins such as optineurin and p62, which are involved in selective autophagy degradation. In vitro functional assessments showed that mutations of TBK1 were associated with disruption of inflammation or autophagy pathways for the impairment of optineurin binding or IFN‐β signaling inducement, respectively (Freischmidt et al., 2015).
In this study, we reported a Chinese FTD patient carrying mutation of p.Ile334Thr in TBK1. This mutation was firstly reported by Shu et al. in a Chinese ALS patient with no symptoms of FTD (Shu et al., 2016). We also performed the in vitro functional studies to investigate its potential pathogenesis.
2. METHODS
2.1. Ethical compliance
This research was approved by the Institutional Ethics Committee of Huashan Hospital Fudan University. The study was conducted after receiving written informed consent from the patient.
2.2. Clinical materials
The patient came from central China. The clinical materials were collected. Blood biochemical analysis (serum creatine kinase, electrolytes, glucose, retinal and liver function, thyroxine, thyroid‐stimulating hormone, and sedimentation rate) and examinations including cranial magnetic resonance imaging (MRI), positron emission computed tomography (PET/CT), electromyography plus nerve conduction velocity (EMG+NCV), electroencephalogram (EEG), and neuropsychological assessments were carried out.
The diagnosis of bvFTD was made according to the Rascovsky criteria (Rascovsky et al., 2011).
2.3. Genetic test
Genomic DNA was extracted from peripheral blood through a standard method (Qiagen, German). A panel containing over 4,000 known virulence genes was performed by target sequencing of the exons. In brief, all exons and their corresponding flanking regions of these genes were selected as target regions. Paired‐end sequencing was performed on Illumina HiSeq X‐ten platform. All variants different from the reference sequence were further screened by allele frequency <1% according to 1,000 Genomes Project (http://www.internationalgenome.org/data), Inhouse database, ESP6500 (evs.gs.washington.edu/EVS/), and ExAC (exac.broadinstitute.org). The synonymous variants were excluded. The phenotypes of the screened genes were compared with the clinical manifestations of the patient, and the inherited modes were considered to further exclude irrelevant genes. Then, the mutations left were confirmed by Sanger sequencing. The databases including Mutation Taster and MUpro were used to predict the pathogenicity of the variants.
The GenBank version was CM000674.1. The NCBI Reference Sequence of TBK1 gene in this study was NC_000012.11, with a transcript ID of ENST00000331710.5. The NCBI Reference Sequence of TBK1 mRNA was NM_013254.4, which encodes a protein of 729 amino acids (NP_037386.1). Interpretation of the variants was based on the American College of Medical Genetics and Genomics (ACMG) recommended standards (Richards et al., 2015).
2.4. In vitro functional studies
2.4.1. Cell culture and transfection
Fibroblasts were obtained from this patient and healthy controls. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 20% FBS (Gibco, Carlsbad, CA, USA), nonessential amino acids (Gibco), sodium bicarbonate (Sigma‐Aldrich, St. Louis, MO, USA), and 1% (vol/vol) penicillin/streptomycin/fungizone (Cellgro, Manassas, VA, USA) in an incubator at 37°C under 5% CO2.
HEK293T cells were cultured in DMEM with 10% FBS in an incubator at 37°C under 5% CO2. For transient overexpression, cells were transfected with Lipofectamine 2000 (Life Technologies, Grand Island, NY, USA) according to the manufacturer's instructions.
2.4.2. TBK1 and phosphorylated IRF3 protein analysis
Fibroblasts from both healthy control and the patient were washed twice with phosphate‐buffered saline (PBS) and lysed in RIPA buffer with protease and phosphatase inhibitor cocktails on ice. Proteins were resolved by SDS‐PAGE and transferred to a PVDF membrane (GE Healthcare, Little Chalfont, UK). After being blocked with 5% skim milk, the membrane was then incubated with primary antibodies overnight, followed by HRP‐conjugated secondary antibodies (Santa Cruz Biotechnology, Dallas, TX, USA). Detection was made with a West‐Q Chemiluminescent Substrate Plus Kit (GenDEPOT, Barker, TX, USA).
2.4.3. TBK1 quantitative real‐time polymerase chain reaction (qRT‐PCR)
Total RNA was isolated with RNA isoplus (Takara, Japan) from the fibroblast, and cDNA was synthesized via Toyobo cDNA kit (Toyobo, Japan). Power SYBR Green PCR Master Mix and primers were used to amplifying cDNA. The relative quantities (RQs) of TATA‐binding protein (TBP) were calculated as internal control in the method. The following primers were used in this study: TBK1, 5′‐CGAGATGTGGTGGGTGGAATG‐3′ and 5′‐CACAGACTGTCCATCTTCCCC‐3′; TBP, 5′‐CCCATGACTCCCATGACC‐3′ and 5′‐TTTACAACCAAGATTCACTGTGG‐3′.
2.4.4. Plasmids and constructs
N‐terminally GFP‐tagged wild‐type human TBK1 cDNA was cloned into the pReceiver vector (GeneCopoeia, Rockville, MD, USA). The missense variants (c.1001T>C, p.Ile334Thr) were introduced into wild‐type GFP‐TBK1 using the EZchange™ site‐directed mutagenesis kit (Enzynomics, Daejeon, Korea) according to the manufacturer's protocol. For expression in bacteria, OPTN cDNA fragments were amplified by PCR and subcloned into pGEX6P1 (GE Healthcare). All constructs were verified by Sanger sequencing.
2.4.5. GST pull‐down assay
Recombinant GST fusion protein GST‐OPTN was produced in E. coli BL‐21 cells. HEK293T cells were transfected with plasmids of wild‐type human TBK1 and p.Ile334Thr TBK1 separately. Both cells were lysed in lysis buffer of GST Protein Interaction Pull‐Down Kit (Thermo Scientific™, #21516). Following experiments were performed according to the manufacturer's protocol.
2.4.6. Luciferase reporter assay
For luciferase reporter assays, HEK293T cells were cultured on 24‐well plates and cotransfected with a plasmid encoding an IFN‐β luciferase reporter (NanoLuc luciferase; 500 ng), pGL4.54 (firefly luciferase plasmid; 50 ng), and 1,000 ng of a plasmid‐encoding GFP‐tagged wild‐type or p.Ile334Thr TBK1. Transfection was performed with Lipofectamine 2000 (Invitrogen). At 24 hr post‐transfection, the cells were washed with PBS. The levels of luciferase activity (NanoLuc luciferase and firefly luciferase) were measured according to the manufacturer's protocol (Promega, Mannheim, Germany). Three independent experiments were performed using triplicate samples in each experiment.
2.4.7. Antibodies
Anti‐TBK1 (1:1,000; Thermo Fisher Scientific, Rockford, IL, PA5‐17478), anti‐IRF3 (1:1,000; Abcam, MA, #ab25950), anti‐pIRF3 (Ser396) (1:500 Cell Signaling, MA, 4D4G, #4947), anti‐OPTN(1:1,000, Proteintech, 108371‐AP), and anti‐β‐actin (1:10,000; Sigma, #A5060) antibodies were used. The following secondary HRP‐conjugated antibodies were used for immunoblotting: goat anti‐mouse IgG (1:1,000; Santa Cruz, CA, #sc‐2005) and goat anti‐rabbit IgG (1:1,000; Santa Cruz, #sc‐2004).
3. RESULTS
3.1. Clinical evaluation
The patient was a 38‐year‐old female who referred to Huashan Hospital after a one‐year history of slowness in reaction, changes in personality, slow movement, and stiffness in her extremities under no predisposing cause. She has no past medical history. She had a suspicious family history as her father had “mental problem” and got lost in his thirties. During her last physical examination, two years from disease onset, the patient showed less cooperation, hyperactivity, and communication difficulties. She also showed stereotyped movement which was repeatedly rubbing her left thigh with her left hand several times and then touching her mouse or hair. Forced grasping was induced in her left hand. Her right hand presented stiffness of all fingers and could not perform fine operations. There was no muscle atrophy. Muscle strength and deep tendon reflexes were normal, while the muscle tone of her upper limbs was elevated with the right one more obvious. Bilateral Babinski signs, Hoffmann signs, and palmomental reflex were negative. The patient scored 12 in Neuropsychiatry Inventory Assessment, 39 in Dysexecutive Questionnaire, 33 in Frontal Behavioral Inventory, and 10.5 in Clinical Dementia Rating (sum of box), suggesting severe cognitive impairment particularly in executive dysfunction, attention deficit, and loss of empathy (Table 1).
Table 1.
Neuropsychological assessments of the FTD patient carrying TBK1 variant of p.Ile334Thr
| Test | Score/total score | Cutoff score | Reference |
|---|---|---|---|
| Mini‐mental state examination | 19/30a | ≥24 | Strong et al. (2009) |
| Boston naming test | 12/30a | ≥24 | Jefferson et al. (2007) |
| Stroop test | Fail to Conduct | – | Scarpina and Tagini (2017) |
| Auditory verbal learning test | |||
| AVLT‐I | 5/36a | ≥12 | Crossen and Wiens (1994) |
| AVLT‐T | 6/60a | ≥23 | |
| Neuropsychiatry inventory | 12/36 | – | Lai (2014) |
| Dysexecutive questionnaire | 39/80a | <10 | Pedrero‐Perez et al. (2011) |
| Frontal behavioral inventory | 33/72a | <27 | Kertesz, Davidson, and Fox (1997) |
| Clinical dementia rating sum of box | 10.5/18a | <2.5 | O'Bryant et al. (2010) |
FTD: frontotemporal dementia.
Score outside the normal value.
Cranial MRI indicated bilateral frontotemporal atrophy (Figure 1a). Her PET‐CT findings also suggested severe bilateral frontotemporal hypometabolism (Figure 1b). Both EMG and EEG were normal. She was diagnosed as bvFTD and donepezil, amantadine and clonidine were prescribed.
Figure 1.
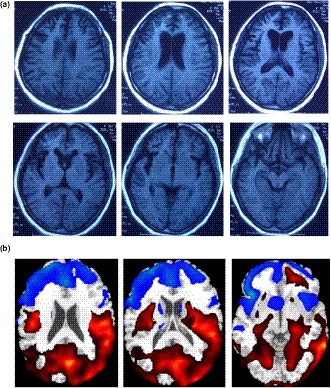
Imaging findings of the patient. (a) Cerebral MRI showed bilateral atrophy of frontal and temporal lobes. (b) FDG‐PET showed severe hypometabolism in frontal temporal areas
3.2. Mutation analysis
The mean depth of target sequencing of the patient was 247.96X. The percentage of the target region with mean depth over 20X was 99.7%. According to the criteria mentioned above, only one heterozygous mutation of c.1001T>C in TBK1 was found, leading to a p.Ile334Thr change in exon 9 (Figure 2a). The MAF of the variant TBK1 p.Ile334Thr (12:64878091 T/C) was about C = 2/102,266 in the total population. The variant was not detected in 200 Chinese seniors without medical history of neurological diseases from the community by Sanger sequencing. The evaluation by Mutation Taster (Schwarz, Cooper, Schuelke, & Seelow, 2014) and MUpro (Cheng, Randall, Sweredoski, & Baldi, 2005) suggested the variant be deleterious: Mutation Taster‐Disease causing, with a score of 89; MUpro‐Decrease stability, with a score of −1.85. Furthermore, according to the protein analysis on a website (http://www.cmbi.ru.nl/hope/), the mutant residue became smaller and less hydrophobic than the wild type, which might lead to a possible loss of interaction with other molecules on the surface of the protein (Figure 2b,c). The wild‐type residue was not conserved at this position (Figure 2d).
Figure 2.
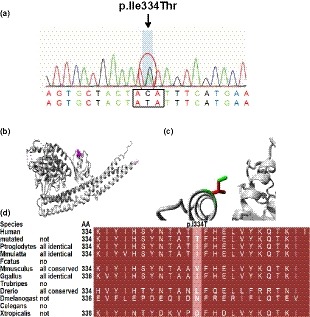
Detection of p.Ile334Thr in TBK1 (NP_037386.1). (a) Sanger sequencing showed the heterozygous of the mutation. (b) The overview of protein. The protein is colored gray, and the side chain of the mutant residue is colored magenta. (c) Close‐up of the mutation. The protein is colored gray, and both the wild‐type (green) and mutant (red) residue are shown. (d) Conservation among multiple species at position 334
3.3. Expression and function analysis
Compared with control fibroblasts, the mRNA level of TBK1 in the fibroblasts with p.Ile334Thr variant was decreased by about 50% (Figure 3a), and the same was found in the protein expression (Figure 3b,c).
Figure 3.
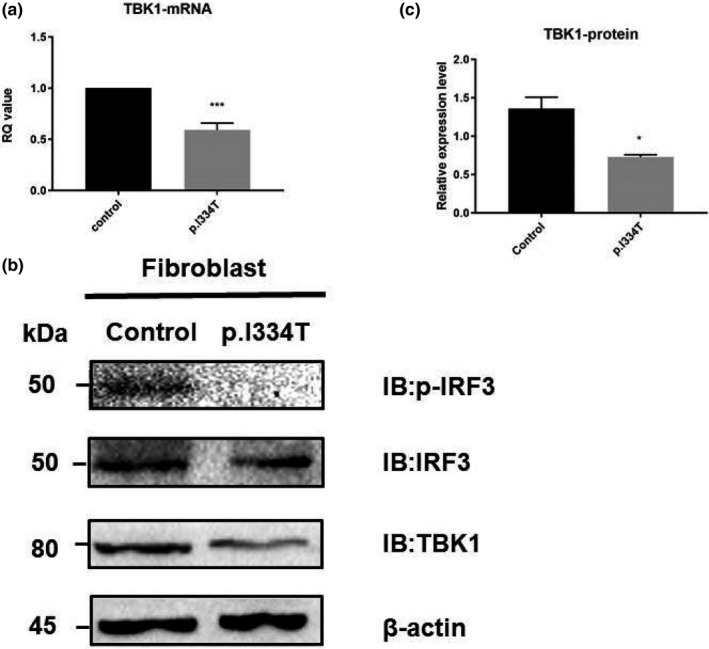
Quantification of mRNA and protein expression level of TBK1 in patient‐derived fibroblasts. (a) Quantitative RT‐PCR analysis of TBK1 mRNA level in fibroblasts from healthy control and the patient with p.Ile334Thr mutation. Values are expressed as mean ± SEM. ***p < 0.0001; unpaired t test. (b) Western blot analysis of TBK1 and phosphorylated IRF3 protein expression level in patient‐derived fibroblasts. β‐actin was used as a loading control. (c) Quantification of TBK1 level. Values are expressed as mean ± SEM. *p < 0.05; unpaired t test
TBK1 is known to phosphorylate IRF3 and regulate its dimerization and nucleus translocation, which participate significantly in the activation of IFN pathway in inflammation. Thus, the protein level of phosphorylated IRF3 in the fibroblasts of p.Ile334Thr variant was also evaluated and found decreased, suggesting a possible disruption of the downstream IFN pathway, which was then measured by the activation of IFN‐β promoter activity using luciferase reporter assay. Compared with the wild‐type TBK1, the p.Ile334Thr variant overexpressed in the HEK293T cells showed approximately 25% reduced IFN‐β signaling (Figure 4a), suggesting a potential disruption of TBK1 anti‐inflammation function.
Figure 4.
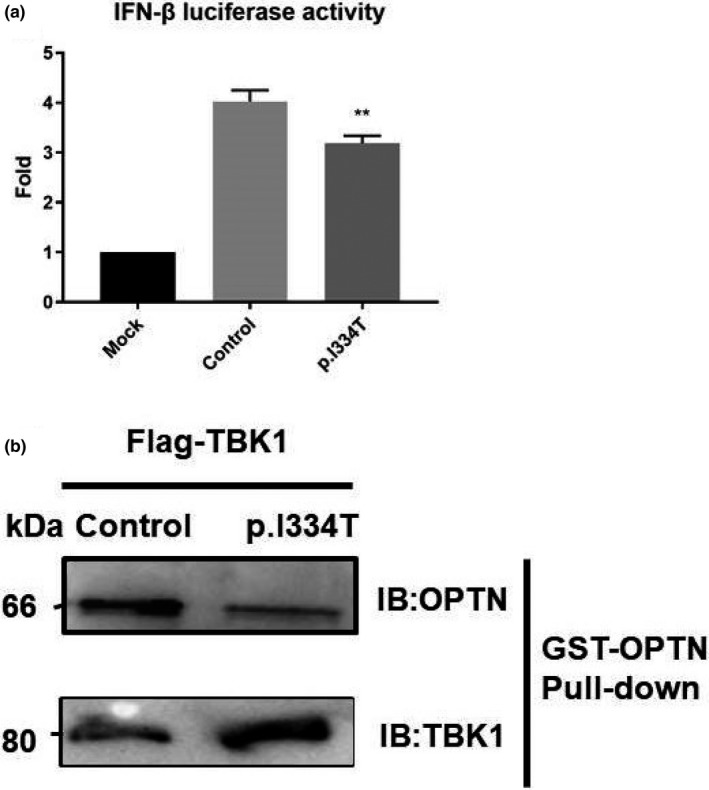
In vitro functional analysis of TBK1 mutation. (a) Luciferase activity in HEK293T cells cotransfected with an IFN‐β reporter plasmid and a GFP‐tagged wild‐type plasmid or a p.Ile334Thr mutation TBK1 plasmid for 24 hr. Firefly luciferase activity served as an internal control. Values are expressed as mean ± SEM, **p < 0.05. (b) Interaction between TBK1 and its downstream autophagy receptor optineurin. Lysates of a GFP‐tagged wild‐type TBK1 or p.Ile334Thr from HEK293T cells were incubated with GST‐OPTN. Both cell lysates and bound proteins were detected by western blotting
CCD2 domain of TBK1 phosphorylated its downstream binding protein optineurin. Phosphorylated optineurin then took part in the autophagy. It would be of interest to further test whether autophagy defects are observed in p.Ile334Thr mutant. Here, we examined the direct interactions between TBK1 and optineurin through GST pull‐down assay. We transfected HEK293T cells with GFP‐tagged wild‐type or p.Ile334Thr form of TBK1. The whole cell lysates of HEK293T cell were subjected to GST pull‐down assay with GST‐OPTN. Immunoblotting showed that both wild‐type and p.Ile334Thr TBK1 could bind with optineurin (Figure 4b), indicating normal function of TBK1 CCD2 domain.
According to ACMG guideline, the variant was classified as likely pathogenic (Richards et al., 2015).
4. DISCUSSION
In the present study, we described an early‐adult onset patient of FTD, carrying a TBK1 mutation of p.Ile334Thr. The patient displayed severe behavior changes and cognitive impairment especially in executive functions but no evidence of ALS. Further experiments showed reduced transcription and expression level of TBK1 as well as decreased IFN‐β signaling but normal binding ability with downstream autophagy receptor optineurin, indicating dysfunction of inflammatory pathway with this mutation.
The FTD‐ALS causative gene TBK1 contributed to about 1.3% ALS, 3%–4% ALS‐FTD, and <1% FTD with TDP‐43 pathology (Nguyen, Van Broeckhoven, & van der Zee, 2018), of whom great clinical heterogeneity was presented. Considering the TBK1 variants, clinical manifestations of reported mutations in FTD‐ALS spectrum were listed (Table 2). Among the patients with TBK1 mutations, over 50% patients were diagnosed as ALS, as well as 18% FTD, 14% FTD‐ALS, and 1.3% AD. The age at onset varied from 26 to 86, with a disease course ranging from <1 year to more than 10 years, and approximately 18% patients died within 2 years. The most common initial symptoms included limb weakness, cognitive deficits, bulbar signs such as dysarthria and dysphagia. Interestingly, about half of the patients with TBK1 mutations were identified with cognitive impairment and one‐thirds with bulbar symptoms. Intrafamilial and interfamilial heterogeneity were also seen in patients carrying the same mutants (Freischmidt et al., 2015). The TBK1 p.Ile334Thr mutation was first reported in a sporadic ALS (sALS) patient without cognitive impairment (Shu et al., 2016). However, in our case, the patient displayed bvFTD, suggesting this mutation may be obligated to FTD‐ALS spectrum.
Table 2.
The clinical characteristic of patients harboring TBK1 mutations
| Function | Population | Mutation | Diagnosis | Age at onset (y) | Disease duration | Family history | Initial symptoms | Cognitive impairment | MNp | Bulbar sign | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| LoF | Japanese | c.1644‐1G>A | ALS | 58 | >7 | Y | Bulbar, lower limbs | N | UMN+LMN | Y | Naruse et al. (2018) |
| LoF | Italian | c.358+5G>A | ALS | 62 | >1.4 | N | Spinal | N | LMN | NA | Pozzi et al. (2017) |
| NA | Italian | c.1644–5_1644‐2delAATA | ALS | 43 | >2.2 | N | Spinal | N | UMN+LMN | NA | Pozzi et al. (2017) |
| LoF | French | IVS18‐2A>G | FTD‐ALS | NA | NA | NA | NA | Y | NA | NA | Le Ber et al. (2015) |
| LoF | Spanish | p.Gln*2 | FTD | 56 | >4.41 | NA | NA | Y | NA | NA | van der Zee et al. (2017) |
| NA | French | p.Thr4Ala | FTD | NA | NA | NA | NA | Y | NA | NA | Le Ber et al. (2015) |
| NA | French | p.Gly26Glu | ALS | NA | NA | NA | NA | NA | NA | NA | Le Ber et al. (2015) |
| LoF | German | p.Lys29Argfs*15 | FTD | 73 | 4 | NA | NA | Y | NA | NA | van der Zee et al. (2017) |
| LoF | NA | p.Lys30 Glu76del | ALS | NA | NA | NA | NA | NA | NA | NA | van der Zee et al. (2017) |
| FMV | German | p.Arg47Hisa | ALS | NA | NA | NA | NA | NA | NA | NA | Freischmidt et al. (2015) |
| ALS | NA | NA | NA | NA | NA | NA | NA | ||||
| ALS | NA | NA | NA | Spinal | NA | NA | NA | ||||
| LoF | Italian | p.Leu59Phefs*16 | ALS | 36 | 4.3 | N | Spinal | N | NA | NA | Pozzi et al. (2017) |
| NA | Chinese | p.Leu62Pro | ALS | 47 | 2.2 | N | Limb | N | NA | NA | Shu et al. (2016) |
| LoF | German | p.Thr77Trpfs*4a | ALS | 35 | >5 | Y | Spinal | NA | NA | NA | Freischmidt et al. (2015) |
| ALS | 58 | 3 | Y | NA | NA | NA | NA | ||||
| LoF | Spanish | p.Thr79del | ALS‐FTD | 56 | 1.5 | NA | NA | Y | NA | Y | van der Zee et al. (2017) |
| FMV | Bulgarian | p.Leu94Ser | ALS | 44 | >10 | Y | Spinal | NA | NA | NA | van der Zee et al. (2017) |
| NA | Chinese | p.Leu94Ser | ALS | NA | 3.2 | NA | NA | NA | NA | NA | Pang et al. (2017) |
| LoF | Swedish | p.Val97Phefs*2 | ALS | 62 | <1 | Y | NA | NA | NA | NA | van der Zee et al. (2017) |
| FMV | Swedish | p.Tyr105Cys | ALS | NA | NA | N | NA | NA | NA | NA | Freischmidt et al. (2015) |
| LoF | Italian | p.Arg117* | FTD | 67 | 7.1 | Y | NA | Y | NA | NA | van der Zee et al. (2017) |
| American | p.Arg117*b | FTD | 68 | 4 | N | NA | Y | NA | Y | Pottier et al. (2015) | |
| FMV | Spanish | p.Gly121Asp | ALS | 34 | >5 | NA | Spinal | NA | NA | NA | van der Zee et al. (2017) |
| LoF | German | p.Arg127* | ALS | 70 | NA | NA | NA | NA | NA | Y | van der Zee et al. (2017) |
| FMV | German | p.Arg143Cys | FTD | 45 | NA | Y | NA | Y | NA | NA | van der Zee et al. (2017) |
| NA | French | p.Arg143Cys | ALS | NA | NA | NA | NA | NA | NA | NA | Le Ber et al. (2015) |
| LoF | French | p.Thr156Argfs*6 | FTD‐ALS | NA | NA | NA | NA | Y | NA | NA | Le Ber et al. (2015) |
| LoF | Belgian | p.Asp167del | ALS | 60 | <1 | NA | NA | NA | NA | NA | van der Zee et al. (2017) |
| LoF | German | p.Tyr185*a | ALS | 47 | >6 | Y | Spinal | N | UMN+LMN | N | Freischmidt et al. (2015) |
| ALS | 37 | 3 | Y | NA | NA | NA | NA | ||||
| ALS | 41 | 6 | Y | NA | NA | NA | NA | ||||
| ALS | 40 | 3 | Y | NA | NA | NA | NA | ||||
| LoF | Italian | p.Asp118Asn | ALS | 81 | >2.9 | N | Spinal | Y | LMN | NA | Pozzi et al. (2017) |
| FMV | German | p.Arg229Ser | ALS | 47 | NA | NA | NA | NA | NA | NA | van der Zee et al. (2017) |
| FMV | Portuguese | p.Gly244Valc | FTD‐ALS | 41 | <2 | Y | Bulbar | Y | NA | Y | van der Zee et al. (2017) |
| FMV | German | p.Ile246Thr | ALS | 57 | <2 | NA | Bulbar | NA | NA | Y | van der Zee et al. (2017) |
| NA | Belgian | p.Arg271Leu | FTD | 80 | 7 | Y | NA | Y | NA | NA | Gijselinck et al. (2015) |
| LoF | Belgian | p.Gly272_Thr331del | FTD | 48 | 2.4 | Y | NA | Y | NA | NA | van der Zee et al. (2017) |
| NA | Scottish | p.Leu277Val | MND | 26 | 11.8 | N | limb | NA | NA | NA | Black et al. (2017) |
| FMV | Italian | p.Lys291Glu | ALS | 74 | >2.75 | N | Spinal | Y | LMN | NA | Pozzi et al. (2017) |
| FMV | Belgian | p.Lys291Glu | FTD | 52 | <9 | Y | NA | Y | NA | NA | van der Zee et al. (2017) |
| Belgian | p.Lys291Glu | FTD | 60 | 4 | Y | NA | Y | NA | NA | Gijselinck et al. (2015) | |
| FMV | Swedish | p.Ile305Thr | ALS | NA | NA | N | NA | NA | NA | NA | Freischmidt et al. (2015) |
| FMV | American | p.Leu306Ile | FTD‐ALS | 70 | 2 | N | Y | Y | NA | NA | Pottier et al. (2015) |
| FMV | Swedish | p.Arg308Gln | ALS | 37.9 | 3.5 | N | Spinal | N | UMN+LMN | Y | Freischmidt et al. (2015) |
| NA | French | p.Thr320Ile | ALS | NA | NA | NA | NA | NA | NA | NA | Le Ber et al. (2015) |
| LoF | Swedish | p.Thr320Glnfs*40a | ALS | 60.1 | 1.9 | Y | Spinal | NA | UMN+LMN | Y | Freischmidt et al. (2015) |
| ALS | NA | NA | Y | NA | NA | NA | NA | ||||
| NA | Belgian | p.His322Tyr | ALS | 64 | 2 | N | NA | NA | NA | NA | Gijselinck et al. (2015) |
| NA | Chinese | p.Ile334Thr | ALS | 51 | 4 | N | Limb | N | NA | NA | Shu et al. (2016) |
| NA | Chinese | p.His336Arg | ALS | NA | 3.5 | NA | NA | NA | NA | NA | Pang et al. (2017) |
| FMV | Italian | p.Arg357Gln | ALS | 36 | >1.7 | N | Spinal | N | NA | NA | Pozzi et al. (2017) |
| Swedish | p.Arg357Gln | ALS | 61.1 | 3 | Y | Bulbar | N | UMN+LMN | Y | Freischmidt et al. (2015) | |
| NA | Sardinian | p.Arg384Thr | ALS | NA | 1.6 | Y | Limb | N | UMN+LMN | Y | Borghero et al. (2016) |
| NA | Italian | p.Ile397Thr | ALS | 65 | >5 | N | Spinal | Y | UMN+LMN | NA | Pozzi et al. (2017) |
| LoF | Belgian | p.Ser398Profs*11 | ALS | 59 | >6.25 | Y | NA | NA | NA | Y | van der Zee et al. (2017) |
| NA | Chinese | p.Leu399fsa | ALS | 60 | 5.7 | Y | Right arm | Y | LMN | N | Williams et al. (2015) |
| ALS | 73 | 8 | Y | Right leg | NA | UMN | NA | ||||
| NA | American | p.Lys401Glu | AD | 80 | 10 | NA | Y | Y | NA | NA | Pottier et al. (2015) |
| LoF | Swedish | p.Ala417* | FTD | 68 | 2.25 | Y | NA | Y | NA | NA | van der Zee et al. (2017) |
| Swedish | p.Ala417*a | ALS | 65 | >7.3 | Y | Spinal | Y | UMN+LMN | Y | Freischmidt et al. (2015) | |
| FTD | NA | NA | Y | Spinal | Y | NA | Y | ||||
| ALS | 56 | 2 | Y | Spinal | N | UMN+LMN | Y | ||||
| Swedish | p.Ala417* | ALS | 62.2 | 1.7 | N | Bulbar | N | UMN+LMN | Y | Freischmidt et al. (2015) | |
| Swedish | p.Ala417* | FTD‐ALS | 61.9 | 6.3 | N | Spinal | Y | UMN+LMN | Y | Freischmidt et al. (2015) | |
| Swedish | p.Ala417* | ALS | 64.7 | 2.7 | N | Bulbar | N | UMN+LMN | Y | Freischmidt et al. (2015) | |
| FMV | Portuguese | p.Ile418Val | FTD | 53 | <1 | Y | NA | Y | NA | NA | van der Zee et al. (2017) |
| NA | Italian | p.Tyr424Asp | ALS | 68 | >0.75 | NA | Spinal | Y | UMN+LMN | Y | Piaceri et al. (2018) |
| LoF | French | p.Arg440X | ALS | NA | NA | Y | NA | NA | NA | NA | Le Ber et al. (2015) |
| French | p.Arg440Xa | ALS | 47 | 13.0 | Y | Spinal | N | UMN+LMN | Y | Freischmidt et al. (2015) | |
| FTD‐ALS | 58 | 3.0 | Y | Spinal | Y | UMN | NA | ||||
| ALS | 73 | 4.0 | Y | Spinal | NA | UMN | Y | ||||
| ALS | Y | NA | NA | NA | NA | ||||||
| FTD | 60 | 16.0 | Y | NA | Y | NA | NA | ||||
| NA | Sardinian | p.Arg444Gln | FTD‐ALS | 72 | >0.8 | N | Cognition impairment | Y | UMN+LMN | Y | Borghero et al. (2016) |
| LoF | Chinese | p.Arg444X | FTD‐ALS | 55 | 4 | N | Cognition impairment+Limb | Y | NA | Y | Tsai et al. (2016) |
| LoF | Spanish | p.Trp445* | FTD+CBS | 78 | >6 | NA | NA | Y | NA | NA | van der Zee et al. (2017) |
| LoF | Danish | p.Ile450Lysfs*15a | ALS | 56.9 | 1.9 | Y | Spinal | N | UMN+LMN | Y | Freischmidt et al. (2015) |
| ALS+D | 71 | 3 | Y | Spina | Y | NA | Y | ||||
| ALS | 77 | >0.7 | Y | Bulbar | N | UMN+LMN | Y | ||||
| ALS | 71 | 2 | Y | Spinal | N | NA | Y | ||||
| ALS | 51 | 2.2 | Y | Spinal | N | UMN+LMN | N | ||||
| Swedish | p.Ile450Lysfs*15a | ALS | 54.9 | >3.2 | Y | Spinal | N | UMN+LMN | Y | Freischmidt et al. (2015) | |
| ALS | 54 | 3 | Y | Spinal | N | UMN+LMN | Y | ||||
| LoF | German | p.Thr462Lysfs*3 | ALS+D | 74 | 0.9 | Y | NA | Y | LMN | NA | van der Zee et al. (2017) |
| NA | German | p.Thr462Lysfs† | FTD‐ALS | 75 | <1 | Y | NA | Y | NA | Y | Schonecker et al. (2016) |
| FTD‐ALS | 77 | <1 | Y | NA | Y | NA | Y | ||||
| LoF | Korean | p.Ile472Serfs*8 | ALS | 53 | >3.8 | N | Bulbar | N | UMN | Y | Kim et al. (2017) |
| LoF | Scottish | p.Glu476fsc | MND | 44 | 2.83 | Y | Limb | NA | NA | NA | Black et al. (2017) |
| LoF | Swedish | p.Val479Glufs*4a | FTD‐ALS | 64.7 | 1.2 | Y | Spinal | Y | UMN+LMN | Y | Freischmidt et al. (2015) |
| FTD‐ALS | 58 | 3.0 | Y | Spinal | Y | NA | NA | ||||
| LoF | French | p.Tyr482X | FTD‐ALS | NA | NA | NA | NA | Y | NA | NA | Le Ber et al. (2015) |
| NA | Belgian | p.Ile515Thr | ALS | 59 | >10 | NA | NA | NA | NA | NA | Gijselinck et al. (2015) |
| LoF | Belgian | p.Ser518Leufs*32 | ALS | 64 | 0.5 | Y | NA | NA | NA | Y | van der Zee et al. (2017) |
| NA | Belgian | p.Ala535Thr | FTD | 52 | 10 | Y | NA | Y | NA | NA | Gijselinck et al. (2015) |
| FMV | Portuguese | p.Met559Arg | ALS | 60 | 6 | Y | Spinal | N | UMN+LMN | NA | Freischmidt et al. (2015) |
| FMV | Swedish | p.Ala571Val | ALS | 66.6 | 1.9 | N | Bulbar | N | UMN+LMN | Y | Freischmidt et al. (2015) |
| NA | Spanish | p.Arg573Glya | FTD | 65 | 13 | NA | NA | NA | NA | NA | Gomez‐Tortosa et al. (2017) |
| FTD | 61 | 6 | Y | NA | NA | NA | NA | ||||
| Dysarthria | 60 | 17 | Y | Memory deficit | Y | N | Y | ||||
| D | 65 | 4 | Y | Memory deficit | Y | N | Y | ||||
| PLS | 66 | 9 | Y | Bulbar | NA | UMN | Y | ||||
| PLS | 61 | 6 | Y | Bulbar | NA | N | Y | ||||
| FMV | Swedish | p.Met598Val | ALS | 61.8 | >2.5 | N | Bulbar | N | UMN+LMN | Y | Freischmidt et al. (2015) |
| LoF | Belgian | p.Glu643dela | FTD‐ALS | 62 | 11.3 | Y | NA | Y | NA | NA | Gijselinck et al. (2015) and van der Zee et al. (2017) |
| D | 71 | 10 | Y | NA | Y | NA | NA | ||||
| D | 86 | 4 | Y | Spinal | Y | NA | NA | ||||
| ALS | 69 | 3 | Y | NA | NA | NA | NA | ||||
| FTD | 69 | 6 | Y | NA | Y | NA | NA | ||||
| D | 70 | 3 | Y | NA | Y | NA | NA | ||||
| FTD | 63 | >3 | Y | NA | Y | NA | NA | ||||
| FTD | 66 | 6 | Y | NA | Y | NA | NA | ||||
| FTD | 61 | 13 | Y | NA | Y | NA | NA | ||||
| D | 63 | 6 | Y | NA | Y | NA | NA | ||||
| D | 73 | 11 | NA | NA | Y | NA | NA | ||||
| D | 82 | 4 | NA | NA | Y | NA | NA | ||||
| ALS | 63 | 1 | NA | NA | NA | NA | NA | ||||
| German | p.Glu643del | ALS | NA | NA | N | NA | NA | NA | NA | Freischmidt et al. (2015) | |
| German | p.Glu643del | ALS | NA | NA | N | Spinal | NA | UMN | Y | Freischmidt et al. (2015) | |
| Belgian | p.Glu643del | FTD | 64 | >9.1 | Y | NA | Y | NA | NA | van der Zee et al. (2017) | |
| Belgian | p.Glu643del | ALS | 51 | 1.7 | Y | NA | NA | NA | Y | van der Zee et al. (2017) | |
| Belgian | p.Glu643del | ALS | 63 | 3 | NA | NA | NA | NA | NA | van der Zee et al. (2017) | |
| Belgian | p.Glu643del | FTD | 70 | 3.5 | Y | NA | Y | NA | NA | van der Zee et al. (2017) | |
| Belgian | p.Glu643del | FTD | 69 | >8.3 | NA | NA | Y | NA | NA | van der Zee et al. (2017) | |
| Belgian | p.Glu643del | FTD | 70 | > 6 | N | NA | Y | NA | NA | Gijselinck et al. (2015) | |
| Belgian | p.Glu643del | FTD | 69 | > 7 | N | NA | Y | NA | NA | Gijselinck et al. (2015) | |
| Belgian | p.Glu643del | ALS | 41 | <1 | N | Bulbar | NA | NA | Y | Gijselinck et al. (2015) | |
| LoF | French | p.Gln655X | FTD‐ALS | NA | NA | NA | NA | Y | NA | NA | Le Ber et al. (2015) |
| NA | French | p.Met662Thr | FTD‐ALS | NA | NA | NA | NA | Y | NA | NA | Le Ber et al. (2015) |
| NA | Sardinian | p.Met690fsd | ALS | 66 | >8.6 | N | Limb | N | LMN | Y | Borghero et al. (2016) |
| LoF | Danish | p.690‐713dela | ALS | 62.7 | 3.8 | Y | Spinal | N | UMN+LMN | Y | Freischmidt et al. (2015) |
| ALS | 52 | 2.0 | Y | Spinal | Y | NA | Y | ||||
| ALS‐FTD | 74 | 1.2 | Y | Spinal | Y | UMN+LMN | Y | ||||
| mono paresis | 52 | >0.5 | Y | Spinal | N | LMN | N | ||||
| possible FTD | NA | NA | Y | Spinal | Y | NA | NA | ||||
| Swedish | p.690‐713dela | ALS | 64 | 2.2 | Y | Bulbar | N | UMN+LMN | Y | Freischmidt et al. (2015) | |
| FTD‐ALS | 65 | 5.0 | Y | Spinal | Y | UMN+LMN | Y | ||||
| possible FTD | 78 | 3.0 | Y | NA | Y | NA | NA | ||||
| D | 65 | 2.0 | Y | NA | Y | NA | NA | ||||
| FMV | American | p.Glu696Lys | AD | 78 | 6.0 | NA | NA | Y | NA | Y | Pottier et al. (2015) |
| Swedish | p.Glu696Lys | ALS‐FTD | 61 | 1.3 | N | Bulbar | Y | UMN+LMN | Y | Freischmidt et al. (2015) | |
| Swedish | p.Glu696Lysa | ALS | 65.5 | 1.7 | Y | Bulbar | N | UMN+LMN | Y | Freischmidt et al. (2015) | |
| ALS | NA | Y | Bulbar | NA | NA | Y | |||||
| LoF | Scottish | p.Ala705fse | MND | 46 | 1.91 | N | Bulbar | NA | NA | Y | Black et al. (2017) |
NCBI Reference Sequence of TBK1 protein: NP_037386.1.
AD: Alzheimer's disease; ALS: amyotrophic lateral sclerosis; CBS: corticobasal syndrome; D:dementia; FMV: functional missense variant; FTD: frontotemporal dementia; LoF: loss of function; MND: motor neuron disease; N: no; NA: not assessed; PLS: primary lateral sclerosis; Y: yes.
aMembers of a family carrying the same TBK1 variant. bCo‐exist with OPTN mutation p.Gly538Glufs*27. cCo‐exist with C9orf72 repeat expansion. dCo‐exist with SQSTM1 mutation p.Arg33Val. eCo‐exist with OPTN mutation p.Gln314Leu.
Although the Caucasian accounts for most of TBK1 mutants, mutations in TBK1 also accounted for sALS patients of Asian population. The frequency of pathogenic or potentially toxic variants of TBK1 in Korean and Japanese sALS patients was estimated to be 0.8% and 1.26% separately (Kim et al., 2017; Tohnai et al., 2018). Besides, five missense variants of TBK1 have been reported in ALS patients of Chinese population so far, among which only the patient with p.Arg444X displayed FTD‐ALS, while the others reported no cognitive dysfunction (Tsai et al., 2016). In addition, one frameshift mutation (p.Leu339fs) of Chinese origin was also found in an Australian cohort study of ALS (Williams et al., 2015). Taken together, although infrequent, TBK1 mutants can still be considered responsible for ALS‐FTD spectrum in Chinese patients.
TBK1 encodes a multimeric kinase regulating multiple cellular pathways, especially inflammation and autophagy. It consists of 4 domains: a kinase domain (KD), an ubiquitin‐like domain (ULD), a helical scaffold dimerization domain (SDD), and a C‐terminal domain (Larabi et al., 2013). TBK1 phosphorylates transcription factor IRF3 in the C‐terminal regulatory domain, leading to its dimeration. Phosphorylated IRF3 then translocates to the nucleus, initiating the transcription of type I interferon genes. The ULD is a regulatory component of TBK1 which controls homodimerization, kinase activation, and interactions with other molecules in the IFN pathway (Li et al., 2012). The p.Ile334Thr mutation locates in the ULD region. Via in vitro functional study, we found reduced expression level of TBK1 mRNA and protein, as well as disruption in the luciferase activity of IFN‐β, the same as p.Arg357Gln variant which is also located in the ULD region (Freischmidt et al., 2015). Our results implied that TBK1 p.Ile334Thr mutation disturbed TBK1 homodimerization and kinase activation, resulting in impairment of downstream neuroinflammatory pathways.
CCD2 mutations abrogated optineurin binding, which is necessary for selective autophagy, but not phosphorylation and activation of IRF3. TBK1 interacts with autophagy receptor proteins which can bind with ubiquitinated cargos and transport them into autophagosomes. In addition, TBK1 also participates in the process of transferring autophagosomes into hydrolytic autophagolysosome, leading to the final degradation of autophagy receptors and their target cargo (Freischmidt et al., 2015). In our work, p.Ile334Thr TBK1 seemed to have normal function in selective autophagy as its maintenance of the binding ability with optineurin through GST pull‐down assay. However, further analysis such as autophagy flux assays should be performed so as to unequivocally identify the full function of TBK1 p.Ile334Thr in autophagy.
Although aberrant autophagy is considered as a main cause of patients with TBK1 variants predisposed to FTD‐ALS spectrum, evidence of neuroinflammation in TBK1 mutations also displayed its significance. Impaired migration of T cells was observed in TBK1 knockout mice, suggesting a decreased T‐cell regulation which led to less stabilization of glial cell and proinflammatory cytokines (Komine & Yamanaka, 2015). On the other hand, multiform effects of type I IFNs and cytokines have been demonstrated in neuron survival, suggesting IFN impairment in TBK1 mutations may also lead to the pathogenesis of FTD‐ALS spectrum. In addition, possible relationship between IFN pathways and autophagy in disease has been considered (Watson et al., 2015), implying that optineurin–TBK1 complexes may regulate IRF3–IFN responses. Whether such IFN impairment might also contribute to FTD pathogenesis is a possibility that remains unexplored.
There exist some limitations in this study. First, limited by the number of patients, we only detected one patient with p.Ile334Thr of TBK1. Although we detect that the mutation is likely pathogenic and can partially disturb neuroinflammatory pathway without interfering with the autophagy function of TBK1, additional patients with this mutation need to be identified with functional work. On the other hand, the decreased level of IFN‐β signaling of this mutation indicated that besides the disturbance of neuroinflammatory pathway, there may still be some unknown pathogenic mechanism under this mutation that we have not yet discovered.
5. CONCLUSION
In this article, we reported a TBK1 mutation of p.Ile334Thr in a Chinese bvFTD patient and classified it as likely pathogenic. Clinical evaluation confirmed the diagnosis of bvFTD, and functional analysis of the mutation demonstrated partially dysfunction of protein expression and activity of TBK1. Whether dysfunction of neuroinflammatory pathways reveals a general feature of p.Ile334Thr mutation of TBK1 remains to be explored. Further study will focus on the pathogenesis of this mutation on FTD and its relationship with ALS.
CONFLICT OF INTEREST
The authors report no conflicts of interest.
ACKNOWLEDGMENT
The authors sincerely thank the participants for their help and willingness to participate in this study, and thank the anonymous reviewers for helping to improve this manuscript.
Yu H, Yu W, Luo S‐S, et al. Association of the TBK1 mutation p.Ile334Thr with frontotemporal dementia and literature review. Mol Genet Genomic Med. 2019;7:e547 . 10.1002/mgg3.547
Funding information
The authors also thank the National Nature Science Foundation of China (No. 81401048 (Yi‐min Sun), 81701250 (Feng‐Tao Liu)) and the China Postdoctoral Science Foundation (No 2017M610227, 2018T110350, Yu‐Jie Yang) for supporting this research.
Contributor Information
Yi‐min Sun, Email: ys2504@sina.com.
Jian‐jun Wu, Email: wujianjun@fudan.edu.cn.
REFERENCES
- Ahmad, L. , Zhang, S. Y. , Casanova, J. L. , & Sancho‐Shimizu, V. (2016). Human TBK1: A gatekeeper of neuroinflammation. Trends in Molecular Medicine, 22(6), 511–527. 10.1016/j.molmed.2016.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black, H. A. , Leighton, D. J. , Cleary, E. M. , Rose, E. , Stephenson, L. , Colville, S. , … Chandran, S. (2017). Genetic epidemiology of motor neuron disease‐associated variants in the Scottish population. Neurobiology of Aging, 51, 178.e11–178.e20. 10.1016/j.neurobiolaging.2016.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghero, G. , Pugliatti, M. , Marrosu, F. , Marrosu, M. G. , Murru, M. R. , Floris, G. , … Consortia, S. (2016). TBK1 is associated with ALS and ALS‐FTD in Sardinian patients. Neurobiology of Aging, 43(180), e181–185. 10.1016/j.neurobiolaging.2016.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, J. , Randall, A. Z. , Sweredoski, M. J. , & Baldi, P. (2005). SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Research, 33(Web Server), W72–W76. 10.1093/nar/gki396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli, E. T. , Lasseigne, B. N. , Petrovski, S. , Sapp, P. C. , Dion, P. A. , Leblond, C. S. , … Goldstein, D. B. (2015). Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science, 347(6229), 1436–1441. 10.1126/science.aaa3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossen, J. R. , & Wiens, A. N. (1994). Comparison of the Auditory‐Verbal Learning Test (AVLT) and California Verbal Learning Test (CVLT) in a sample of normal subjects. Journal of Clinical and Experimental Neuropsychology, 16(2), 190–194. 10.1080/01688639408402630 [DOI] [PubMed] [Google Scholar]
- Freischmidt, A. , Wieland, T. , Richter, B. , Ruf, W. , Schaeffer, V. , Muller, K. , … Weishaupt, J. H. (2015). Haploinsufficiency of TBK1 causes familial ALS and fronto‐temporal dementia. Nature Neuroscience, 18(5), 631–636. 10.1038/nn.4000 [DOI] [PubMed] [Google Scholar]
- Fujioka, S. , & Wszolek, Z. K. (2011). Clinical aspects of familial forms of frontotemporal dementia associated with parkinsonism. Journal of Molecular Neuroscience, 45(3), 359–365. 10.1007/s12031-011-9568-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijselinck, I. , VanMossevelde, S. , van derZee, J. , Sieben, A. , Philtjens, S. , Heeman, B. , … Consortium, B. (2015). Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology, 85(24), 2116–2125. 10.1212/WNL.0000000000002220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Tortosa, E. , Van derZee, J. , Ruggiero, M. , Gijselinck, I. , Esteban‐Perez, J. , Garcia‐Redondo, A. , … Consortium, E. E. (2017). Familial primary lateral sclerosis or dementia associated with Arg573Gly TBK1 mutation. Journal of Neurology, Neurosurgery and Psychiatry, 88(11), 996–997. 10.1136/jnnp-2016-315250 [DOI] [PubMed] [Google Scholar]
- Jefferson, A. L. , Wong, S. , Gracer, T. S. , Ozonoff, A. , Green, R. C. , & Stern, R. A. (2007). Geriatric performance on an abbreviated version of the Boston naming test. Applied Neuropsychology, 14(3), 215–223. 10.1080/09084280701509166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz, A. , Davidson, W. , & Fox, H. (1997). Frontal behavioral inventory: Diagnostic criteria for frontal lobe dementia. Canadian Journal of Neurological Sciences, 24(1), 29–36. 10.1017/S0317167100021053 [DOI] [PubMed] [Google Scholar]
- Kim, Y. E. , Oh, K. W. , Noh, M. Y. , Nahm, M. , Park, J. , Lim, S. M. , … Kim, S. H. (2017). Genetic and functional analysis of TBK1 variants in Korean patients with sporadic amyotrophic lateral sclerosis. Neurobiology of Aging, 50, 170.e1–170.e6. 10.1016/j.neurobiolaging.2016.11.003 [DOI] [PubMed] [Google Scholar]
- Komine, O. , & Yamanaka, K. (2015). Neuroinflammation in motor neuron disease. Nagoya Journal of Medical Science, 77(4), 537–549. [PMC free article] [PubMed] [Google Scholar]
- Lai, C. K. (2014). The merits and problems of Neuropsychiatric Inventory as an assessment tool in people with dementia and other neurological disorders. Clinical Interventions in Aging, 9, 1051–1061. 10.2147/CIA.S63504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larabi, A. , Devos, J. M. , Ng, S. L. , Nanao, M. H. , Round, A. , Maniatis, T. , & Panne, D. (2013). Crystal structure and mechanism of activation of TANK‐binding kinase 1. Cell Reports, 3(3), 734–746. 10.1016/j.celrep.2013.01.034 [DOI] [PubMed] [Google Scholar]
- LeBer, I. , DeSeptenville, A. , Millecamps, S. , Camuzat, A. , Caroppo, P. , Couratier, P. , … Genetic Research Network on, F. F.‐A. (2015). TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiology of Aging, 36(11), 3116.e5–3116.e8. 10.1016/j.neurobiolaging.2015.08.009 [DOI] [PubMed] [Google Scholar]
- Li, J. , Li, J. , Miyahira, A. , Sun, J. , Liu, Y. , Cheng, G. , & Liang, H. (2012). Crystal structure of the ubiquitin‐like domain of human TBK1. Protein Cell, 3(5), 383–391. 10.1007/s13238-012-2929-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomen‐Hoerth, C. , Anderson, T. , & Miller, B. (2002). The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology, 59(7), 1077–1079. 10.1212/WNL.59.7.1077 [DOI] [PubMed] [Google Scholar]
- Mackenzie, I. R. , & Neumann, M. (2016). Molecular neuropathology of frontotemporal dementia: Insights into disease mechanisms from postmortem studies. Journal of Neurochemistry, 138(Suppl 1), 54–70. 10.1111/jnc.13588 [DOI] [PubMed] [Google Scholar]
- Mercy, L. , Hodges, J. R. , Dawson, K. , Barker, R. A. , & Brayne, C. (2008). Incidence of early‐onset dementias in Cambridgeshire, United Kingdom. Neurology, 71(19), 1496–1499. 10.1212/01.wnl.0000334277.16896.fa [DOI] [PubMed] [Google Scholar]
- Naruse, H. , Ishiura, H. , Mitsui, J. , Date, H. , Takahashi, Y. , Matsukawa, T. , … Tsuji, S. (2018). Molecular epidemiological study of familial amyotrophic lateral sclerosis in Japanese population by whole‐exome sequencing and identification of novel HNRNPA1 mutation. Neurobiology of Aging, 61, 255.e9–255.e16. 10.1016/j.neurobiolaging.2017.08.030 [DOI] [PubMed] [Google Scholar]
- Nguyen, H. P. , Van Broeckhoven, C. , & van der Zee, J. (2018). ALS genes in the genomic era and their implications for FTD. Trends in Genetics, 34(6), 404–423. 10.1016/j.tig.2018.03.001 [DOI] [PubMed] [Google Scholar]
- O'Bryant, S. E. , Lacritz, L. H. , Hall, J. , Waring, S. C. , Chan, W. , Khodr, Z. G. , … Cullum, C. M. (2010). Validation of the new interpretive guidelines for the clinical dementia rating scale sum of boxes score in the national Alzheimer's coordinating center database. Archives of Neurology, 67(6), 746–749. 10.1001/archneurol.2010.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, S. Y. , Hsu, J. S. , Teo, K. C. , Li, Y. , Kung, M. H. W. , Cheah, K. S. E. , … Ho, S. L. (2017). Burden of rare variants in ALS genes influences survival in familial and sporadic ALS. Neurobiology of Aging, 58, 238.e9–238.e15. 10.1016/j.neurobiolaging.2017.06.007 [DOI] [PubMed] [Google Scholar]
- Pedrero‐Perez, E. J. , Ruiz‐Sanchez de Leon, J. M. , Lozoya‐Delgado, P. , Llanero‐Luque, M. , Rojo‐Mota, G. , & Puerta‐Garcia, C. (2011). Prefrontal symptoms assessment: Psychometric properties and normative data of the Dysexecutive Questionnaire (DEX) in a sample from the Spanish population. Revista De Neurologia, 52(7), 394–404. [PubMed] [Google Scholar]
- Perry, D. C. , & Miller, B. L. (2013). Frontotemporal dementia. Seminars in Neurology, 33(4), 336–341. 10.1055/s-0033-1359316 [DOI] [PubMed] [Google Scholar]
- Piaceri, I. , Bessi, V. , Mata, S. , Polito, C. , Tedde, A. , Berti, V. , … Nacmias, B. (2018). Association of the new variant Tyr424Asp at TBK1 gene with amyotrophic lateral sclerosis and cognitive decline. Journal of Alzheimer's Disease, 61(1), 41–46. 10.3233/JAD-170694 [DOI] [PubMed] [Google Scholar]
- Pottier, C. , Ravenscroft, T. A. , Sanchez‐Contreras, M. , & Rademakers, R. (2016). Genetics of FTLD: Overview and what else we can expect from genetic studies. Journal of Neurochemistry, 138(Suppl 1), 32–53. 10.1111/jnc.13622 [DOI] [PubMed] [Google Scholar]
- Pottier, C. , Bieniek, K. F. , Finch, N. , van de Vorst, M. , Baker, M. , Perkersen, R. , … Rademakers, R. (2015). Whole‐genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathologica, 130(1), 77–92. 10.1007/s00401-015-1436-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi, L. , Valenza, F. , Mosca, L. , Dal Mas, A. , Domi, T. , Romano, A. , … Riva, N. (2017). TBK1 mutations in Italian patients with amyotrophic lateral sclerosis: Genetic and functional characterisation. Journal of Neurology, Neurosurgery and Psychiatry, 88(10), 869–875. 10.1136/jnnp-2017-316174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovsky, K. , Hodges, J. R. , Knopman, D. , Mendez, M. F. , Kramer, J. H. , Neuhaus, J. , … Miller, B. L. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain, 134(Pt 9), 2456–2477. 10.1093/brain/awr179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton, A. E. , Majounie, E. , Waite, A. , Simon‐Sanchez, J. , Rollinson, S. , Gibbs, J. R. , … Traynor, B. J. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron, 72(2), 257–268. 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Committee, A. L. Q. A. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpina, F. , & Tagini, S. (2017). The stroop color and word test. Frontiers in Psychology, 8, 557 10.3389/fpsyg.2017.00557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonecker, S. , Brendel, M. , van der Zee, J. , van Broeckhoven, C. , Rominger, A. , Danek, A. , & Levin, J. (2016). A pair of siblings with frontotemporal dementia and amyotrophic lateral sclerosis and a novel Thr462Lysfs mutation in the TBK1 gene. Fortschritte Der Neurologie‐Psychiatrie, 84(8), 494–498. 10.1055/s-0042-110653 [DOI] [PubMed] [Google Scholar]
- Schwarz, J. M. , Cooper, D. N. , Schuelke, M. , & Seelow, D. (2014). MutationTaster2: Mutation prediction for the deep‐sequencing age. Nature Methods, 11(4), 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Shu, S. , Li, X. L. , Liu, Q. , Liu, F. , Cui, B. , Liu, M. S. , … Zhang, X. (2016). Screening of the TBK1 gene in familial and sporadic amyotrophic lateral sclerosis patients of Chinese origin. Amyotroph Lateral Scler Frontotemporal Degener, 17(7–8), 605–607. 10.1080/21678421.2016.1183681 [DOI] [PubMed] [Google Scholar]
- Strong, M. J. , Grace, G. M. , Freedman, M. , Lomen‐Hoerth, C. , Woolley, S. , Goldstein, L. H. , … Figlewicz, D. (2009). Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler, 10(3), 131–146. 10.1080/17482960802654364 [DOI] [PubMed] [Google Scholar]
- Tohnai, G. , Nakamura, R. , Sone, J. , Nakatochi, M. , Yokoi, D. , & Katsuno, M. , … Japanese Consortium for Amyotrophic Lateral Sclerosis, R. (2018). Frequency and characteristics of the TBK1 gene variants in Japanese patients with sporadic amyotrophic lateral sclerosis. Neurobiology of Aging, 64, 158.e15–158.e19. 10.1016/j.neurobiolaging.2017.12.005 [DOI] [PubMed] [Google Scholar]
- Tsai, P. C. , Liu, Y. C. , Lin, K. P. , Liu, Y. T. , Liao, Y. C. , Hsiao, C. T. , … Lee, Y. C. (2016). Mutational analysis of TBK1 in Taiwanese patients with amyotrophic lateral sclerosis. Neurobiology of Aging, 40, 191.e11–191.e16. 10.1016/j.neurobiolaging.2015.12.022 [DOI] [PubMed] [Google Scholar]
- van der Zee, J. , Gijselinck, I. , Van Mossevelde, S. , Perrone, F. , Dillen, L. , Heeman, B. , … Testi, S. (2017). TBK1 mutation spectrum in an extended European Patient Cohort with frontotemporal dementia and amyotrophic lateral sclerosis. Human Mutation, 38(3), 297–309. 10.1002/humu.23161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Mossevelde, S. , van der Zee, J. , Cruts, M. , & Van Broeckhoven, C. (2017). Relationship between C9orf72 repeat size and clinical phenotype. Current Opinion in Genetics & Development, 44, 117–124. 10.1016/j.gde.2017.02.008 [DOI] [PubMed] [Google Scholar]
- Watson, R. O. , Bell, S. L. , MacDuff, D. A. , Kimmey, J. M. , Diner, E. J. , Olivas, J. , … Cox, J. S. (2015). The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host & Microbe, 17(6), 811–819. 10.1016/j.chom.2015.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, K. L. , McCann, E. P. , Fifita, J. A. , Zhang, K. , Duncan, E. L. , Leo, P. J. , … Blair, I. P. (2015). Novel TBK1 truncating mutation in a familial amyotrophic lateral sclerosis patient of Chinese origin. Neurobiology of Aging, 36(12), 3334.e1–3334.e5. 10.1016/j.neurobiolaging.2015.08.013 [DOI] [PubMed] [Google Scholar]