

Abstract
Background
Autosomal recessive congenital ichthyosis (ARCI) is a genetically and phenotypically heterogeneous skin disease, associated with defects in the skin permeability barrier. Several but not all genes with underlying mutations have been identified, but a clear correlation between genetic causes and clinical picture has not been described to date.
Methods
Our study included 19 families from Saudi Arabia, Yemen, and Pakistan. All patients were born to consanguineous parents and diagnosed with ARCI. Mutations were analyzed by homozygosity mapping and direct sequencing.
Results
We have detected mutations in all families in five different genes: TGM1, ABCA12, CYP4F22, NIPAL4, and ALOXE3. Five likely pathogenic variants were unknown so far, a splice site and a missense variant in TGM1, a splice site variant in NIPAL4, and missense variants in ABCA12 and CYP4F22. We attributed TGM1 and ABCA12 mutations to the most severe forms of lamellar and erythematous ichthyoses, respectively, regardless of treatment. Other mutations highlighted the presence of a phenotypic spectrum in ARCI.
Conclusion
Our results contribute to expanding the mutational spectrum of ARCI and revealed new insights into genotype/phenotype correlations. The findings are instrumental for a faster and more precise diagnosis, a better understanding of the pathophysiology, and the definition of targets for more specific therapies for ARCI.
Keywords: congenital ichthyosis, erythema, genotype/phenotype correlation, homozygosity mapping, Skin permeability barrier, skin scaling
1. INTRODUCTION
The skin barrier is imperative for protecting the organism from internal water loss as well as from outside pathogens and toxic compounds. Defects in the skin barrier function can lead to several skin disorders including autosomal recessive congenital ichthyosis (ARCI) (Feingold & Elias, 2014; Schmuth et al., 2013; Traupe, Fischer, & Oji, 2014).
Autosomal recessive congenital ichthyosis (MIM 242300, 242100, 606545, 601277, 242500, 604777, 612281, 615022, 613943, 615023, 602400, 615024, 617320, 617574, 617571) refers to a group of nonsyndromic congenital ichthyoses that include Harlequin ichthyosis (HI), lamellar ichthyosis (LI), and congenital ichthyosiform erythroderma (CIE) (Oji et al., 2010). Even though ARCI is a very heterogeneous disorder, common features include a generalized scaling of the skin, often with underlying erythema. Newborns usually present a collodion membrane that is lost during the first weeks of life. To date, 12 genes have been associated with ARCI: TGM1 (MIM 190195), ALOX12B (MIM 603741), ALOXE3 (MIM 607206), ABCA12 (MIM 607800), CYP4F22 (MIM 611495), NIPAL4 (MIM 609383), LIPN (MIM 613924), CERS3 (MIM 615276), PNPLA1 (MIM 612121), CASP14 (MIM 605848), SDR9C7 (MIM 609769), and SULT2B1 (MIM 604125), and at least 10%–15% of affected individuals do not have mutations in any of the known genes (Hellström Pigg et al., 2016; Vahlquist, Fischer, & Törmä, 2018). These genes have been linked to the maintenance of skin barrier function as their products are involved in the formation of the cornified lipid envelope in the stratum corneum and ceramide formation and processing in the epidermis (Eckl et al., 2013; Li, Loriè, Fischer, Vahlquist, & Törmä, 2012; Ohno et al., 2015).
HI is the most severe and often fatal form of the disease. Truncating mutations in ABCA12 have been detected as the main cause for HI (Akiyama, 2014; Akiyama et al., 2005). LI and CIE, in contrast, have not yet been associated with a clear genotype/phenotype correlation. Patients with LI exhibit large, thick scales over the entire body without a severe background erythroderma. Patients with CIE normally present fine, whitish scales as well as generalized erythroderma. Both clinical forms can show partially overlapping phenotypes, ranging from coarse to fine scaling and mild to severe erythema (Rodriguez‐Pazos, Ginarte, Vega, & Toribio, 2013). In more than 50% of the patients, LI is caused by mutations in TGM1, however, TGM1 mutations have also been reported to cause CIE (Becker et al., 2003; Herman et al., 2009), as well as other subtypes of ARCI, such as bathing suit ichthyosis (BSI) and self‐improving ichthyosis (SII) (Hackett, Fitzgerald, Watson, Hol, & Irvine, 2010; Raghunath et al., 2003). SII refers to a patient who is born with a collodion membrane but presents only a particularly mild phenotype after the first weeks of life.
Mutations in ALOXE3 and ALOX12B, that code for lipoxygenases eLOX‐3 and 12R‐LOX, and NIPAL4 have been associated to LI, CIE and also SII (Eckl et al., 2009; Jobard et al., 2002; Vahlquist et al., 2010). Other ARCI genes include cytochrome P450 member CYP4F22, which encodes a fatty acid hydroxylase (Ohno et al., 2015), and defects of this gene have been linked to LI (Lefevre et al., 2006). Mutations in CERS3, PNPLA1, SDR9C7, and SULT2B1, have been identified in few families with ARCI presenting mainly LI phenotypes (Eckl et al., 2013; Grall et al., 2012; Heinz et al., 2017; Israeli et al., 2011; Shigehara et al., 2016).
We present the findings of a study that involved 19 consanguineous families from Saudi Arabia, Yemen, and Pakistan with affected members diagnosed with different forms of ARCI. For all of them, we have identified the underlying mutations causing the disease, using homozygosity mapping combined with Sanger sequencing. We have detected eleven different mutations in various ARCI‐associated genes, five of which are being reported for the first time.
2. MATERIALS AND METHODS
2.1. Ethical compliance
All procedures involving human participants were approved by institutional Research Ethics Committees and in accordance with the 1964 Helsinki declaration and its later amendments. Informed consent was obtained from all individual participants included in the study.
2.2. Patients and phenotypic features
Samples from a total of 37 individuals diagnosed with different forms of ARCI and 72 unaffected members were collected. We have studied 19 consanguineous families, 13 from Saudi Arabia, 1 from Yemen, and 5 from Pakistan. At least one affected individual (index case) from each pedigree was tested by homozygosity mapping. The detailed clinical features of all index cases are shown in Table 1 and Figure 1.
Table 1.
Patient information and clinical diagnosis
| Patient | Gender | Birth year | Affected in family | Origin | Clinical diagnosis | Phenotypic features | Treatment | |
|---|---|---|---|---|---|---|---|---|
| Scaling | Erythema | |||||||
| SA‐01 | M | 1997 | 1 | Saudi Arabia | LI | Severe, generalized; coarse brown scales; palmoplantar hyperkeratosis | No | Acitretin (3–4 years, 11–20 mg) |
| SA‐02 | M | 1994 | 2 | Saudi Arabia | CIE | Severe, generalized; fine white scales; palmoplantar hyperkeratosis | Severe | ‐ |
| SA‐04 | F | 1995 | 1 | Saudi Arabia | LI | Severe generalized; coarse brown scales; palmoplantar hyperkeratosis | No | Acitretin (11–15 years, 21–30 mg) |
| SA‐05 | F | 2007 | 1 | Saudi Arabia | LI | Severe generalized; coarse brown scales; palmoplantar hyperkeratosis | No | Acitretin (1–2 years, 5–10 mg) |
| SA‐06 | F | 1999 | 2 | Saudi Arabia | LI | Severe generalized; coarse brown scales; palmoplantar hyperkeratosis | No | Acitretin (1–4 weeks, 5–10 mg) |
| SA‐08 | F | 1986 | 1 | Saudi Arabia | LI | Mild generalized; fine light brown scales; plantar hyperkeratosis | Mild | Acitretin (5–6 years, 5–10 mg) |
| SA‐09 | M | 2003 | 2 | Saudi Arabia | LI | Moderate generalized; fine light brown scales; plantar hyperkeratosis | No | NA |
| SA‐10 | M | 2003 | 2 | Saudi Arabia | LI | Moderate generalized; coarse, large light brown scales; plantar hyperkeratosis | No | ‐ |
| SA‐11 | F | 2005 | 1 | Saudi Arabia | LI | Severe generalized; coarse, large light brown scales; plantar hyperkeratosis | No | ‐ |
| SA‐12 | M | 2010 | 1 | Saudi Arabia | LI | Severe generalized; coarse, large light brown, plate‐like scales; palmoplantar hyperkeratosis | No | ‐ |
| SA‐13 | F | 2006 | 2 | Saudi Arabia | SII | No | No | ‐ |
| SA‐14 | M | 2006 | 1 | Saudi Arabia | LI | Severe generalized scaling; coarse and large brown scales; palmoplantar hyperkeratosis | No | Acitretin (3–4 years, 5–10 mg) |
| SA‐15 | M | 2007 | 1 | Saudi Arabia | LI | Moderate, more present in upper body; coarse, large light brown scales; plantar hyperkeratosis | No | ‐ |
| YE‐01 | F | 2006 | 1 | Yemen | LI | Severe, generalized; coarse brown scales; palmoplantar hyperkeratosis | No | ‐ |
| PK01‐01 | F | NA | 4 | Pakistan | CI | Generalized scaling, more visible in hands, neck and face; fine light‐colored scales | Moderate | NA |
| PK01‐02 | M | NA | NA | |||||
| PK02‐01 | F | NA | 2 | Pakistan | CI | Scaling more severe in face, neck and hands; coarse brownish scales | No | NA |
| PK02‐02 | M | NA | NA | |||||
| PK03‐01 | M | NA | 2 | Pakistan | CI | More visible in hands, neck and face; fine lighter‐colored, yellowish scales | Moderate | NA |
| PK03‐04 | M | NA | 4 | PLSa | Severe localized in hands, feet, knees; nail and teeth malformation; transgredient hyperkeratosis | No | NA | |
| PK04‐01 | M | NA | 1 | Pakistan | CI | Scaling localized visible in hands, neck and face; lighter‐colored and less compact scales | Moderate | NA |
| PK05‐01 | M | NA | 5 | Pakistan | CI | Scaling more severe in face, neck and hands; coarse brownish scales | No | NA |
| PK05‐03 | F | NA | NA | |||||
| PK05‐04 | M | NA | NA | |||||
CI: Congenital ichthyosis; CIE: Congenital ichthyosiform erythroderma; LI: Lamellar ichthyosis; NA: information not available; PLS: Papillon–Lefèvre syndrome; SII: Self‐improving ichthyosis.
On reinvestigation following molecular analysis.
Figure 1.
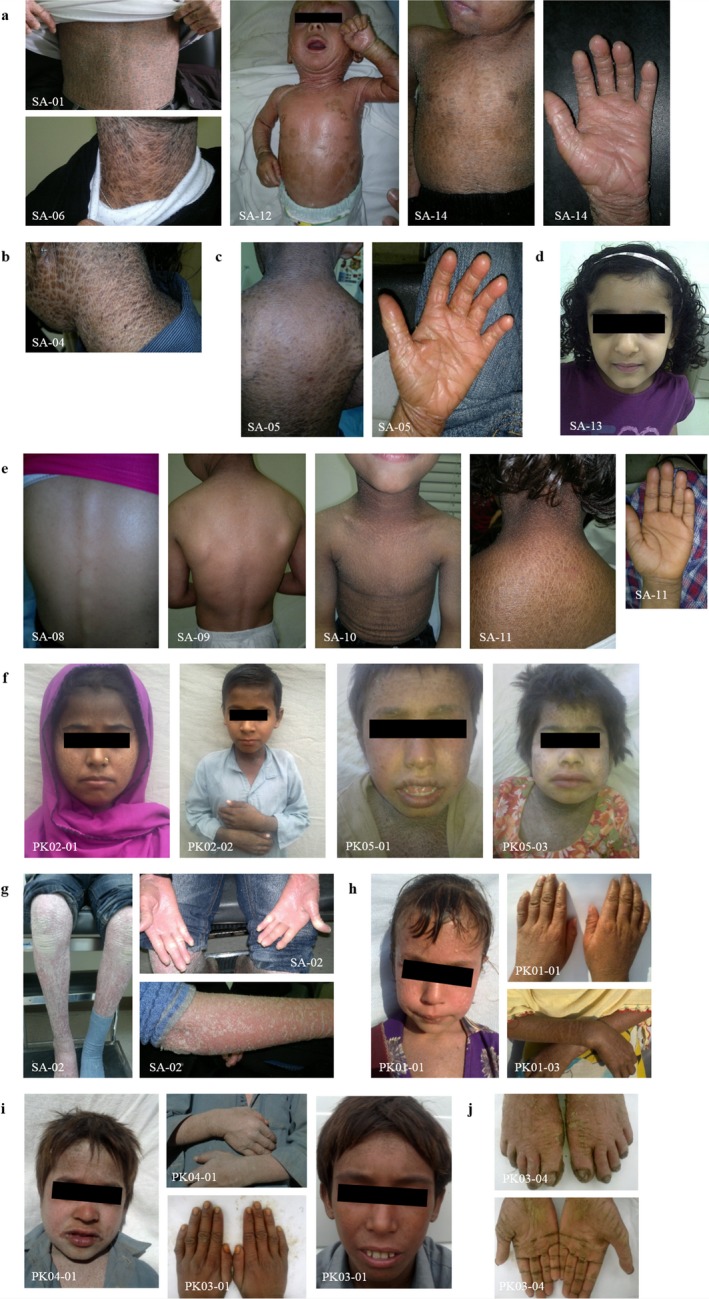
Clinical features of patients with ARCI, categorized by mutant genes. (a–c) TGM1 affected patients showed generalized severe, dark colored scales, and palmoplantar hyperkeratosis, apparently independent of type of mutation or treatment. (d) Patient diagnosed with SII, with no visible skin alterations at 6 years of age. (e–f) Patients with NIPAL4 splice site (e) and missense (f) mutations presented a variable range of symptoms like milder to moderate scaling more prominent in the upper body half but no palmar hyperkeratosis. (g–h) Patients with ABCA12 mutations with fine to medium‐sized whitish scaling and erythema (SA‐02), and with slightly milder phenotype and white scales (PK01). (i) Patients from families PK04 and PK03 with generalized whitish scaling, quite visible on the hands, diagnosed with ALOXE3 nonsense mutations. (j) Affected individuals from another branch of family PK03, initially also diagnosed with ARCI. Upon reinvestigation, they showed more localized severe scaling on the hands and feet as well as nail and teeth (not shown) anomalies consistent with a diagnosis of Papillon–Lefèvre syndrome caused by CTSC mutation
Affected individuals were clinically diagnosed with different forms of ARCI by experienced dermatologists and presented heterogeneous phenotypes. The majority of Saudi Arabian (SA) patients and the Yemenite (YE) patient showed a phenotype of LI, with generalized scaling and mild to no erythema. SA‐02 was the only patient with severe erythema and was hence diagnosed with CIE. SA‐13 was diagnosed with SII, as she presented no scaling or erythema but was reported to have had a collodion membrane at birth. Some patients were being treated with the retinoid acitretin, which modulates keratinocyte differentiation, at the time of the sample collections, but with different durations, ranging from few weeks to nearly 15 years of treatment.
Pakistani affected individuals (PK) showed more apparent generalized and severe localized scaling. The families had limited access to medical care and no pharmacological treatment options. Patients from families PK01 and PK04 presented moderate erythema. Family PK03 (Supporting information Figure S1) showed more dissimilar phenotypes among the patients: patient PK03‐01 had a more generalized but less severe scaling phenotype but patient PK03‐04 presented with more localized and severe scaling on hands and feet associated with visible tooth and nail malformations(Figure 1h,i).
2.3. Homozygosity mapping
Genomic DNA was extracted from peripheral blood or saliva samples with standard methods. Homozygosity mapping was performed in samples of index patients using HumanCytoSNP‐12 (Illumina, San Diego, CA, USA), following the manufacturer's instructions. Generated data were analyzed with Nexus Copy Number™ software (BioDiscovery, Hawthorne CA, USA). The longest regions of homozygosity were extracted for each index case and compared to the locations of known ARCI genes.
2.4. Sanger sequencing
Target exons were amplified by PCR using standard cycle conditions and in‐house designed primers (primers lists available upon request). Products were sequenced using BigDye Terminator v1.1 kit (Applied Biosystems, Foster City, CA, USA) and run on a Sequence Analyzer 3130XL (Applied Biosystems).
2.5. Next‐generation sequencing
Two index patients with a homozygous region matching ABCA12 location were further analyzed with a custom‐made dermatogenetics gene panel comprising 56 genes including all known ARCI genes using an Illumina HiSeq sequencing system (Illumina).
Because no homozygous mutation was found in any known ARCI gene in PK03‐04, in contrast to his nephew PK03‐01, exome sequencing was performed using enrichment with SureSelect Human All Exon V6 (Agilent, Santa Clara, CA) and a HiSeq sequencing system (Illumina). Data analysis of filter‐passed reads was done with BWA‐short in combination with GATK and SAMTOOLS as implemented in the in‐house analysis tool Varbank (Cologne Center for Genomics).
2.6. In silico data analysis
Sequences were analyzed with SeqmanPro (DNASTAR Inc., Madison, WI, USA). Unknown missense variants were regarded as likely pathogenic if they were predicted to be damaging by at least two of the algorithms MutTaster (Schwarz, Cooper, Schuelke, & Seelow, 2014), SIFT (Kumar, Henikoff, & Ng, 2009) and PolyPhen‐2 (Adzhubei et al., 2010), affected highly conserved amino acids, and were not found as homozygous variants in control DNA sequences as analyzed by the Exome Aggregation Consortium (exac.broadinstitute.org) and the 1,000 Genomes Project (www.internationalgenome.org). Splice site variants were analyzed with Human Splicing Finder 3.0 (Desmet et al., 2009). Protein domains were determined using PFAM (Finn, Coggill, Eberhardt, & Eddy, 2016).
2.7. GenBank Accession numbers
TGM1: NM_00359.2/NP_000350.1
NIPAL4: NM_001099287.1/NP_001092757.1
ABCA12: NM_173076.2/NP_775099.2.
CYP4F22: NM_173483.3/NP_775754.2.
ALOXE3: NM_001165960.1/NP_001159432.1
CTSC: NM_001814.4/NP_001805.3
3. RESULTS
DNA samples from 13 unrelated ARCI families from Saudi Arabia, one from Yemen as well as five extended families from Pakistan were used in this study to investigate the genetic causes of ARCI. After aligning the location of known ARCI genes with regions of homozygosity obtained from genome‐wide homozygosity mapping, one to three candidate intervals containing one or two of these genes each were found for all index cases (Supporting information Table S1). Mutations were identified with conventional direct sequencing of candidate genes, except for ABCA12, which comprises 53 exons and was therefore analyzed with gene panel sequencing. Homozygous causal mutations were found for each index case, and co‐segregation of mutations with phenotypes was confirmed in all available family members. Previously unknown variants in ARCI genes were assessed in silico and classified as defined in Methods. A summary of the mutations is shown in Table 2.
Table 2.
Mutations identified in Saudi Arabian and Pakistani families
| Patient | Genea | Mutation cDNAb | Mutation protein | Prediction (SpliceFinder, Polyphen, SIFT, MutTaster) | MAFc | References |
|---|---|---|---|---|---|---|
| SA‐01 | TGM1 | c.398_407dupAGTATGAGTA | p.(Tyr136*) | Premature termination | 0.000004d | Wakil et al. (2016) |
| SA‐02 | ABCA12 | c.4541G>T | p.(Arg1514Leu) | Damaging | ‒ | This study |
| SA‐04 | TGM1 | c.1340A>C | p.(Asp447Ala) | Damaging | ‒ | This study |
| SA‐05 | TGM1 | c.758‐1G>C | ‐ | Aberrant splicing 100% | ‒ | This study |
| SA‐06 | TGM1 | c.398_407dupAGTATGAGTA | p.(Tyr136*) | Premature termination | 0.000004d | Wakil et al. (2016) |
| SA‐08 | NIPAL4 | c.223+5_223+22delGTACGGCAGGGCTGGGGA | ‐ | Aberrant splicing 61.4% | ‒ | This study |
| SA‐09 | NIPAL4 | c.223+5_223+22delGTACGGCAGGGCTGGGGA | ‐ | Aberrant splicing 61.4% | ‒ | This study |
| SA‐10 | NIPAL4 | c.223+5_223+22delGTACGGCAGGGCTGGGGA | ‐ | Aberrant splicing 61.4% | ‒ | This study |
| SA‐11 | NIPAL4 | c.223+5_223+22delGTACGGCAGGGCTGGGGA | ‐ | Aberrant splicing 61.4% | ‒ | This study |
| SA‐12 | TGM1 | c.398_407dupAGTATGAGTA | p.(Tyr136*) | Premature termination | 0.000004d | Wakil et al. (2016) |
| SA‐13 | TGM1 | c.871G>A | p.(Gly291Ser) | Damaging | 0.00001 | Sakai et al. (2016) |
| SA‐14 | TGM1 | c.398_407dupAGTATGAGTA | p.(Tyr136*) | Premature termination | 0.000004d | Wakil et al. (2016) |
| SA‐15 | CYP4F22 | c.982G>A | p.(Glu328Lys) | Damaging | 0.000004 | This study |
| YE‐01 | TGM1 | c.398_407dupAGTATGAGTA | p.(Tyr136*) | Premature termination | 0.000004d | Wakil et al. (2016) |
|
PK01‐01 PK01‐02 PK01‐04 |
ABCA12 | c.4676G>T | p.(Gly1559Val) | Damaging | 0.000004 | Nawaz et al. (2012) |
|
PK02‐01 PK02‐02 |
NIPAL4 | c.527C>A | p.(Ala176Asp) | Damaging | 0.0007 | Lefevre et al. (2004) |
|
PK03‐01 PK03‐04 |
ALOXE3
CTSC |
c.814C>T c.901G>A | p.(Arg272*) p.(Gly301Ser) | Premature termination |
0.00001 0.00003 |
Ullah et al. (2016) Toomes et al. (1999) |
| PK04‐01 | ALOXE3 | c.814C>T | p.(Arg272*) | Premature termination | 0.00001 | Ullah et al. (2016) |
|
PK05‐01 PK05‐03 PK05‐04 |
NIPAL4 | c.527C>A | p.(Ala176Asp) | Damaging | 0.0007 | Lefevre et al. (2004) |
TGM1: NM_00359.2; ABCA12: NM_173076.2; NIPAL4: NM_001099287.1; CYP4F22: NM_173483.3 ALOXE3: NM_001165960.1; CTSC: NM_001814.4.
Mutations first identified in this study are shown in bold.
Minor allele frequency (MAF) according to the Genome Aggregation Database (gnomad.broadinstitute.org).
Same codon change resulting from a different variant.
3.1. Families from Saudi Arabia and Yemen
Mutations in TGM1 were the most common in this study, found in seven of the thirteen index patients from Saudi Arabia. Aside from SA‐13 diagnosed with SII, all patients with TGM1 mutations had been diagnosed with LI and presented with similar clinical features (Figure 1a‐d). SA‐01, SA‐06, SA‐12, SA‐14, and YE‐01 had the same homozygous duplication c.398_407dupAGTATGAGTA in exon 3, which leads to a premature stop codon (p.Tyr136*). This mutation has been linked to two ARCI families from Saudi Arabia (Wakil et al., 2016), pointing to a potential founder mutation on the Arabian Peninsula. SA‐04, also diagnosed with LI, presented a missense change in exon 9, c.1340A>C (p.Asp447Ala). This variant is located within the catalytic core of transglutaminase 1 (Figure 2b). A splice acceptor site change c.758‐1G>C in intron 4 of TGM1 was found in SA‐05 (Figure 2a). Patient SA‐13, diagnosed with SII based on the lack of signs for congenital ichthyosis aside from the presence of a collodion membrane at birth, presented a homozygous missense variant in exon 5 of TGM1, c.871G>A (p.Gly291Ser). This variant was previously reported in a Japanese LI patient with compound heterozygosity, which might explain the different forms of the disease (Sakai et al., 2009). This finding is in accordance with Hackett et al, who reviewed the phenotypes associated with more than 40 mutations reported in TGM1 and noted that mutations associated with SII seemed to cluster in exons 5, 6, and 7 of TGM1 (Hackett et al., 2010).
Figure 2.
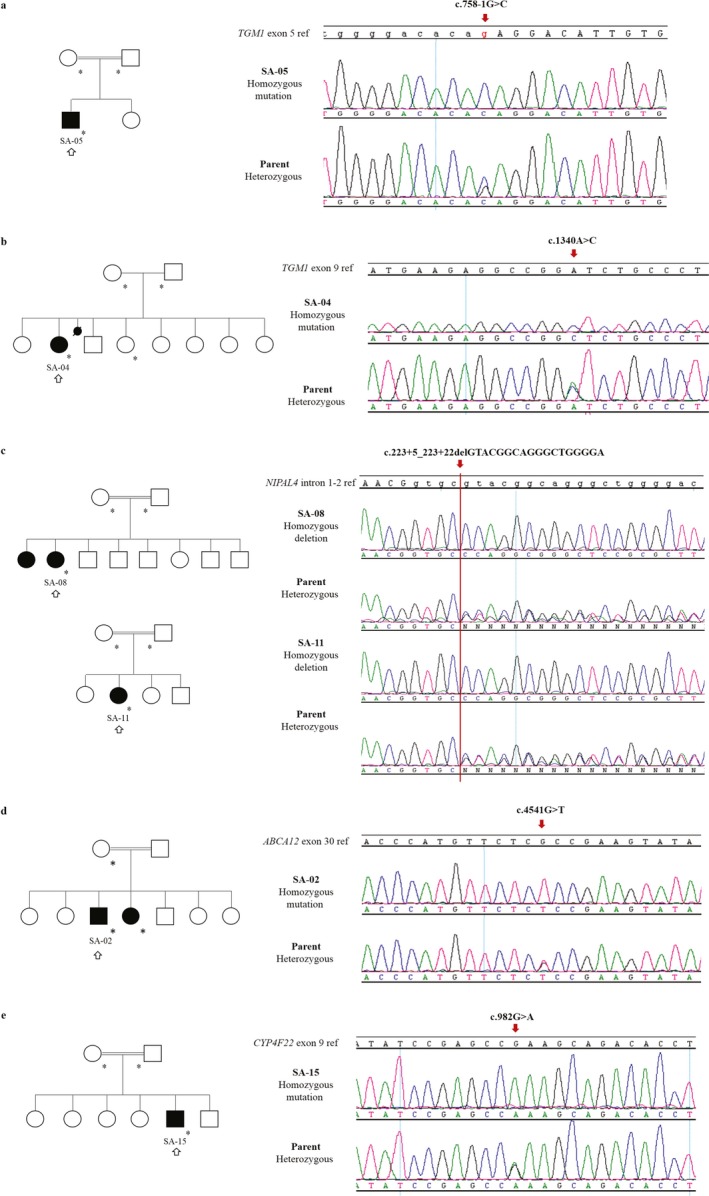
Family pedigrees and sequences of unknown mutations identified in this study. Pedigrees show index patients used for homozygosity mapping marked with a black outlined arrow (↑) and all samples confirmed by Sanger sequencing marked with a star (*). All parents of patients have a consanguinity relation. At least one parent is included in the displayed sequences to demonstrate co‐segregation. Red arrows indicate the positions of nucleotide substitutions or deletions. In the reference sequences, uppercase letters indicate exon nucleotides and lowercase letters intronic bases. (a) Examples of two Saudi Arabian pedigrees with patients with a homozygous intronic deletion in intron 1 of NIPAL4. (b) A homozygous missense mutation c.1340A>C was detected in exon 9 of TGM1 in SA‐04. (c) Patient SA‐05 was identified with a homozygous mutation in the acceptor splice site of TGM1 intron 4. (d) A previously unknown missense mutation in exon 9 of CYP4F22 was found in SA‐15
Four patients diagnosed with LI (SA‐08, SA‐09, SA‐10, and SA‐11) presented a common, previously unreported homozygous intronic deletion of 18 nucleotides in NIPAL4, c.223+5_223+22delGTACGGCAGGGCTGGGGA, which is predicted to affect the donor splice site of intron 1 (Figure 2c).
SA‐02, a patient diagnosed with CIE, was found to have a homozygous missense variant, c.4541G>T (p.Arg1514Leu), in exon 30 of ABCA12 (Figure 2d). SA‐15, diagnosed with LI, has a missense variant c.982G>A (p.Glu328Lys) in exon 9 of CYP4F22, which has not been described before (Figure 2e). This variant was predicted as deleterious and is located inside the predicted cytochrome P450 domain.
3.2. Families from Pakistan
We found a homozygous missense variant in exon 31 of ABCA12, c.4676G>T (p.Gly1559Val), in family PK01. Nawaz et al described this variant before in a consanguineous family from Pakistan with non‐bullous CIE (Nawaz et al., 2012).
Patients from families PK02 and PK05 were identified with the same homozygous missense variant c.527C>A (p.Ala176Asp) in exon 4 of NIPAL4. This variant was reported before (Lefevre et al., 2004) and is located within the suggested transporter domain of the NIPAL4 protein.
A homozygous nonsense mutation, c.814C>T (p.Arg272*), in exon 4 of ALOXE3 was found in patients of family PK04. The mutation was reported in a Pakistani family diagnosed with CIE (Ullah et al., 2016).
Homozygosity mapping in index cases of family PK03 (Supporting information Figure S1) showed a homozygous region matching the ALOXE3/ALOX12B genes location, and Sanger sequencing revealed the same homozygous mutation as seen in PK04, c.814C>T (p.Arg272*) in ALOXE3, in one branch of the family. However, affected individuals from another branch were either heterozygous for the mutation or even homozygous for the reference allele. Reinvestigation demonstrated that these patients were also affected with nail malformation and early loss of teeth and the skin lesions were more limited to the extremities, in contrast to PK03‐01 and his affected sister. PK03‐04 was then analyzed by exome sequencing and the likely pathogenic variant c.901G>A (p.Gly301Ser) in CSTC, the gene encoding cathepsin C, was found.
4. DISCUSSION
Using a combination of homozygosity mapping and candidate gene sequencing, we identified eleven different mutations in five genes in a total of 19 consanguineous families with ARCI from Saudi Arabia, Yemen, and Pakistan. All variants were classified as pathogenic or likely pathogenic and were homozygous as expected, and thus represent a particularly useful resource to search for genotype/phenotype correlations, although the clinical appearance was different between Saudi Arabian and Pakistani patients based on the level of medical care available. All patients with TGM1 mutations, except SII‐diagnosed SA‐13, presented the most severe phenotypes, with generalized coarse brown scaling (Figure 1a–d). Interestingly, this phenotype seems independent of the type of mutation and even prominent after oral treatment with acitretin. Two unknown disease‐causing mutations were found in TGM1: p.Asp447Ala (c.1340A>C) in exon 9 and c.758‐1G>C at the splice acceptor site of intron 4. Even after treatment with acitretin for approximately 15 years, the patient with mutation p.Asp447Ala presented a severe phenotype, similar to non‐treated patients with TGM1 mutations. The second mutation was found in a patient with LI and a similar phenotype with severe scaling and rough and brownish scales.
In contrast, the patients from the Saudi Arabian cohort carrying the same NIPAL4 intronic deletion, c.223+5_223+22delGTACGGCAGGGCTGGGGA, showed variable phenotypes, ranging from mild to severe generalized scaling, with white to light brown‐colored scales, fine or coarse. SA‐08 had the mildest symptoms, with mild scaling and skin dryness, likely improved by the treatment with acitretin. On the other hand, SA‐11 was never treated with acitretin and presented the most severe phenotype in this group, with coarse light brown scales and severe scaling and dryness (Figure 1e). Patient SA‐15 with a CYP4F22 mutation, also diagnosed with LI, presented moderate skin dryness and more localized scaling in the face, neck, abdomen, back, and arms (photos not available).
SA‐02, diagnosed with CIE and with the unreported ABCA12 missense mutation p.Arg1514Leu (c.4541G>T), presented a severe skin phenotype, including generalized severe scaling with fine white scales, pronounced erythema and palmoplantar keratoderma (Figure 1g). A homozygous G to A change at the same position leading to p.Arg1514His was described in ARCI before (Lefevre et al., 2003).
Notably, we identified two autosomal recessive skin disorders in the same consanguineous family, PK03, initially diagnosed only with ARCI. A homozygous nonsense mutation in exon 4 of ALOXE3 was found in index patient PK03‐01 and segregation was confirmed by analysis of the parents and siblings. The phenotype of PK03‐01 also matched the phenotype of PK04‐01 who showed the same mutation (Figure 1h). However, the index patient PK03‐04 of the second branch of the family and his affected siblings, initially diagnosed with ARCI, showed localized scaling mostly on hands and feet associated with nail malformations and teeth loss at an early age on reinvestigation (Figure 1i). They did not carry the homozygous ALOXE3 mutation, and exome sequencing revealed a homozygous mutation in CTSC, which was previously reported in patients with Papillon–Lefèvre syndrome (PLS) (Nagy et al., 2014; Noack et al., 2008; Toomes et al., 1999). PLS (MIM #245000) is a rare autosomal recessive condition presenting diffuse keratoderma together with rapidly progressive periodontitis (Dhanrajani, 2009). This finding confirmed two different diagnoses of ARCI and PLS, respectively, in two branches of the family (Supporting information Figure S1).
The identification of various homozygous mutations in different genes facilitated an important contribution to defining a genotype/phenotype correlation for ARCI (Figure 3). Mutations in TGM1 and ABCA12 cause the most severe phenotypes, compared to NIPAL4, CYP4F22, and ALOXE3 mutations. We could also establish parameters regarding the color and appearance of the scales, where patients with defects in TGM1 seem to present the darkest and coarsest scales, followed by CYP4F22, NIPAL4 and lastly ALOXE3, which ranged from light brown to white. ABCA12 affected patients presented the lightest colored scales, all of them white and fine. Furthermore, all Saudi Arabian patients showed palmar hyperlinearity but only TGM1 and ABCA12 affected individuals presented palmoplantar hyperkeratosis, while NIPAL4 and CYP4F22 presented only plantar keratosis.
Figure 3.
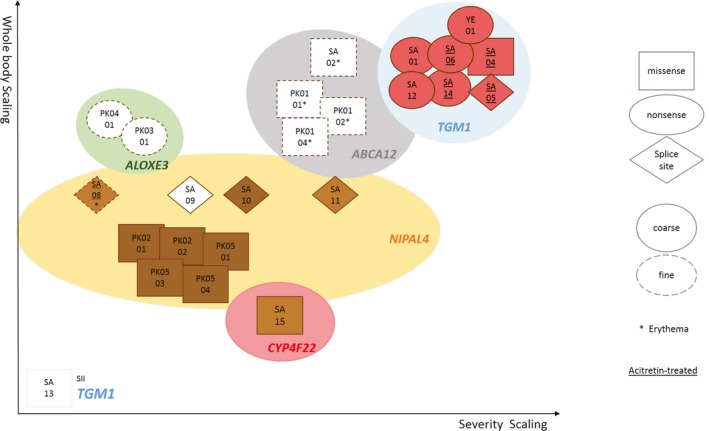
Schematic representation of genotype/phenotype correlations of ARCI found in this study. All phenotypes and genotypes were grouped according to our findings. Each index case is named and boxed according to the severity of the scaling (X axis) and its extension (Y axis). Types of mutations are indicated by the shape of the boxes, that is, squares for missense, oval for nonsense, and diamonds for splice site mutations. Scaling type was also included, with coarse scales represented by a full‐outline box and fine scales by dashed outlines. Patients with an erythematous phenotype are represented by an asterisk (*) and those treated with acitretin are underlined
Our study demonstrated that homozygosity mapping coupled with Sanger sequencing is still a valid and cost‐efficient tool to identify rare disease‐causing mutations in consanguineous families. While establishing a genotype/phenotype correlation for ARCI patients is a difficult task, our findings have added strong and new insights to this matter, hopefully leading to a better clinical and molecular understanding of this heterogeneous disease and to novel approaches for pathophysiology‐based therapies.
CONFLICT OF INTEREST
The authors have no potential conflict of interest to declare.
Supporting information
ACKNOWLEDGMENTS
We are grateful to all patients and family members who participated in the study. We wish to thank Margit Rauch for excellent technical assistance. The project was supported in part by grants from the Imam Abdulrahman Bin Faisal University (formerly University of Dammam), the Austrian National Bank (OeNB 15620), the German Federal Ministry for Education and Research (E‐Rare‐2 01GM1201), the Austrian Science Fund (I2259‐B26), and the Köln Fortune Program of the Faculty of Medicine, University of Cologne.
Lima Cunha D, Alakloby OM, Gruber R, et al. Unknown mutations and genotype/phenotype correlations of autosomal recessive congenital ichthyosis in patients from Saudi Arabia and Pakistan. Mol Genet Genomic Med. 2019;7:e539 10.1002/mgg3.539
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , … Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama, M. (2014). The roles of ABCA12 in epidermal lipid barrier formation and keratinocyte differentiation. Biochimica Et Biophysica Acta, 1841(3), 435–440. 10.1016/j.bbalip.2013.08.009. [DOI] [PubMed] [Google Scholar]
- Akiyama, M. , Sugiyama‐Nakagiri, Y. , Sakai, K. , McMillan, J. R. , Goto, M. , Arita, K. , … Shimizu, H. (2005). Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. The Journal of Clinical Investigation, 115(7), 1777–1784. 10.1172/jci24834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, K. , Csikos, M. , Sardy, M. , Szalai, Z. S. , Horvath, A. , & Karpati, S. (2003). Identification of two novel nonsense mutations in the transglutaminase 1 gene in a Hungarian patient with congenital ichthyosiform erythroderma. Experimental Dermatology, 12(3), 324–329. 10.1034/j.1600-0625.2003.120313.x [DOI] [PubMed] [Google Scholar]
- Desmet, F. O. , Hamroun, D. , Lalande, M. , Collod‐Beroud, G. , Claustres, M. , & Beroud, C. (2009). Human splicing finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Research, 37(9), e67 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanrajani, P. J. (2009). Papillon‐Lefevre syndrome: Clinical presentation and a brief review. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics, 108(1), e1–7. 10.1016/j.tripleo.2009.03.016. [DOI] [PubMed] [Google Scholar]
- Eckl, K. M. , de Juanes, S. , Kurtenbach, J. , Natebus, M. , Lugassy, J. , Oji, V. , … Hennies, H. C. (2009). Molecular analysis of 250 patients with autosomal recessive congenital ichthyosis: Evidence for mutation hotspots in ALOXE3 and allelic heterogeneity in ALOX12B. Journal of Investigative Dermatology, 129(6), 1421–1428. 10.1038/jid.2008.409. [DOI] [PubMed] [Google Scholar]
- Eckl, K. M. , Tidhar, R. , Thiele, H. , Oji, V. , Hausser, I. , Brodesser, S. , … Hennies, H. C. (2013). Impaired epidermal ceramide synthesis causes autosomal recessive congenital ichthyosis and reveals the importance of ceramide acyl chain length. Journal of Investigative Dermatology, 133(9), 2202–2211. 10.1038/jid.2013.153. [DOI] [PubMed] [Google Scholar]
- Feingold, K. R. , & Elias, P. M. (2014). Role of lipids in the formation and maintenance of the cutaneous permeability barrier. Biochimica Et Biophysica Acta, 1841(3), 280–294. 10.1016/j.bbalip.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Finn, R. D. , Coggill, P. , Eberhardt, R. Y. , & Eddy, S. R. (2016). The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Research, 44(D1), D279–285. 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grall, A. , Guaguere, E. , Planchais, S. , Grond, S. , Bourrat, E. , Hausser, I. , … Fischer, J. (2012). PNPLA1 mutations cause autosomal recessive congenital ichthyosis in golden retriever dogs and humans. Nature Genetics, 44(2), 140–147. 10.1038/ng.1056. [DOI] [PubMed] [Google Scholar]
- Hackett, B. C. , Fitzgerald, D. , Watson, R. M. , Hol, F. A. , & Irvine, A. D. (2010). Genotype‐phenotype correlations with TGM1: Clustering of mutations in the bathing suit ichthyosis and self‐healing collodion baby variants of lamellar ichthyosis. British Journal of Dermatology, 162(2), 448–451. 10.1111/j.1365-2133.2009.09537.x. [DOI] [PubMed] [Google Scholar]
- Heinz, L. , Kim, G. J. , Marrakchi, S. , Christiansen, J. , Turki, H. , Rauschendorf, M. A. , … Fischer, J. (2017). Mutations in SULT2B1 cause autosomal‐recessive congenital ichthyosis in humans. American Journal of Human Genetics, 100(6), 926–939. 10.1016/j.ajhg.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellström Pigg, M. H. , Bygum, A. , Ganemo, A. , Virtanen, M. , Brandrup, F. , Zimmer, A. D. , … Fischer, J. (2016). Spectrum of autosomal recessive congenital Ichthyosis in scandinavia: Clinical characteristics and novel and recurrent mutations in 132 patients. Acta Dermato‐Venereologica, 96(7), 932–937. 10.2340/00015555-2418. [DOI] [PubMed] [Google Scholar]
- Herman, M. L. , Farasat, S. , Steinbach, P. J. , Wei, M. H. , Toure, O. , Fleckman, P. , … Toro, J. R. (2009). Transglutaminase‐1 gene mutations in autosomal recessive congenital ichthyosis: Summary of mutations (including 23 novel) and modeling of TGase‐1. Human Mutation, 30(4), 537–547. 10.1002/humu.20952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israeli, S. , Khamaysi, Z. , Fuchs‐Telem, D. , Nousbeck, J. , Bergman, R. , Sarig, O. , & Sprecher, E. (2011). A mutation in LIPN, encoding epidermal lipase N, causes a late‐onset form of autosomal‐recessive congenital ichthyosis. American Journal of Human Genetics, 88(4), 482–487. 10.1016/j.ajhg.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobard, F. , Lefevre, C. , Karaduman, A. , Blanchet‐Bardon, C. , Emre, S. , Weissenbach, J. , … Fischer, J. (2002). Lipoxygenase‐3 (ALOXE3) and 12(R)‐lipoxygenase (ALOX12B) are mutated in non‐bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Human Molecular Genetics, 11(1), 107–113. 10.1093/hmg/11.1.107 [DOI] [PubMed] [Google Scholar]
- Kumar, P. , Henikoff, S. , & Ng, P. C. (2009). Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4(7), 1073–1081. 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Lefevre, C. , Audebert, S. , Jobard, F. , Bouadjar, B. , Lakhdar, H. , Boughdene‐Stambouli, O. , … Fischer, J. (2003). Mutations in the transporter ABCA12 are associated with lamellar ichthyosis type 2. Human Molecular Genetics, 12(18), 2369–2378. 10.1093/hmg/ddg235. [DOI] [PubMed] [Google Scholar]
- Lefevre, C. , Bouadjar, B. , Ferrand, V. , Tadini, G. , Megarbane, A. , Lathrop, M. , … Fischer, J. (2006). Mutations in a new cytochrome P450 gene in lamellar ichthyosis type 3. Human Molecular Genetics, 15(5), 767–776. 10.1093/hmg/ddi491. [DOI] [PubMed] [Google Scholar]
- Lefevre, C. , Bouadjar, B. , Karaduman, A. , Jobard, F. , Saker, S. , Ozguc, M. , … Fischer, J. (2004). Mutations in ichthyin a new gene on chromosome 5q33 in a new form of autosomal recessive congenital ichthyosis. Human Molecular Genetics, 13(20), 2473–2482. 10.1093/hmg/ddh263. [DOI] [PubMed] [Google Scholar]
- Li, H. , Loriè, E. P. , Fischer, J. , Vahlquist, A. , & Törmä, H. (2012). The expression of epidermal lipoxygenases and transglutaminase‐1 is perturbed by NIPAL4 mutations: Indications of a common metabolic pathway essential for skin barrier homeostasis. Journal of Investigative Dermatology, 132(10), 2368–2375. 10.1038/jid.2012.160. [DOI] [PubMed] [Google Scholar]
- Nagy, N. , Valyi, P. , Csoma, Z. , Sulak, A. , Tripolszki, K. , Farkas, K. , … Szell, M. (2014). CTSC and Papillon‐Lefevre syndrome: Detection of recurrent mutations in Hungarian patients, a review of published variants and database update. Molecular Genetics & Genomic Medicine, 2(3), 217–228. 10.1002/mgg3.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawaz, S. , Tariq, M. , Ahmad, I. , Malik, N. A. , Baig, S. M. , Dahl, N. , & Klar, J. (2012). Non‐bullous congentital ichthyosiform erythroderma associated with homozygosity for a novel missense mutation in an ATP binding domain of ABCA12. European Journal of Dermatology, 22(2), 178–181. 10.1684/ejd.2011.1638. [DOI] [PubMed] [Google Scholar]
- Noack, B. , Gorgens, H. , Schacher, B. , Puklo, M. , Eickholz, P. , Hoffmann, T. , & Schackert, H. K. (2008). Functional Cathepsin C mutations cause different Papillon‐Lefevre syndrome phenotypes. Journal of Clinical Periodontology, 35(4), 311–316. 10.1111/j.1600-051X.2008.01201.x. [DOI] [PubMed] [Google Scholar]
- Ohno, Y. , Nakamichi, S. , Ohkuni, A. , Kamiyama, N. , Naoe, A. , Tsujimura, H. , … Kihara, A. (2015). Essential role of the cytochrome P450 CYP4F22 in the production of acylceramide, the key lipid for skin permeability barrier formation. Proceedings of the National Academy of Sciences of the United States of America, 112(25), 7707–7712. 10.1073/pnas.1503491112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oji, V. , Tadini, G. , Akiyama, M. , Blanchet Bardon, C. , Bodemer, C. , Bourrat, E. , … Traupe, H. (2010). Revised nomenclature and classification of inherited ichthyoses: Results of the First Ichthyosis Consensus Conference in Soreze 2009. Journal of the American Academy of Dermatology, 63(4), 607–641. 10.1016/j.jaad.2009.11.020. [DOI] [PubMed] [Google Scholar]
- Raghunath, M. , Hennies, H. C. , Ahvazi, B. , Vogel, M. , Reis, A. , Steinert, P. M. , & Traupe, H. (2003). Self‐healing collodion baby: A dynamic phenotype explained by a particular transglutaminase‐1 mutation. Journal of Investigative Dermatology, 120(2), 224–228. 10.1046/j.1523-1747.2003.12032.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Pazos, L. , Ginarte, M. , Vega, A. , & Toribio, J. (2013). Autosomal recessive congenital ichthyosis. Actas Dermo‐Sifiliográficas, 104(4), 270–284. 10.1016/j.adengl.2011.11.021. [DOI] [PubMed] [Google Scholar]
- Sakai, K. , Akiyama, M. , Yanagi, T. , McMillan, J. R. , Suzuki, T. , Tsukamoto, K. , … Shimizu, H. (2009). ABCA12 is a major causative gene for non‐bullous congenital ichthyosiform erythroderma. Journal of Investigative Dermatology, 129(9), 2306–2309. 10.1038/jid.2009.23. [DOI] [PubMed] [Google Scholar]
- Schmuth, M. , Martinz, V. , Janecke, A. R. , Fauth, C. , Schossig, A. , Zschocke, J. , & Gruber, R. (2013). Inherited ichthyoses/generalized Mendelian disorders of cornification. European Journal of Human Genetics, 21(2), 123–133. 10.1038/ejhg.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, J. M. , Cooper, D. N. , Schuelke, M. , & Seelow, D. (2014). MutationTaster2: Mutation prediction for the deep‐sequencing age. Nature Methods, 11(4), 361–362. 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- Shigehara, Y. , Okuda, S. , Nemer, G. , Chedraoui, A. , Hayashi, R. , Bitar, F. , … Shimomura, Y. (2016). Mutations in SDR9C7 gene encoding an enzyme for vitamin A metabolism underlie autosomal recessive congenital ichthyosis. Human Molecular Genetics, 25(20), 4484–4493. 10.1093/hmg/ddw277. [DOI] [PubMed] [Google Scholar]
- Toomes, C. , James, J. , Wood, A. J. , Wu, C. L. , McCormick, D. , Lench, N. , … Thakker, N. S. (1999). Loss‐of‐function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nature Genetics, 23(4), 421–424. 10.1038/70525. [DOI] [PubMed] [Google Scholar]
- Traupe, H. , Fischer, J. , & Oji, V. (2014). Nonsyndromic types of ichthyoses ‐ an update. Journal Der Deutschen Dermatologischen Gesellschaft, 12(2), 109–121. 10.1111/ddg.12229. [DOI] [PubMed] [Google Scholar]
- Ullah, R. , Ansar, M. , Durrani, Z. U. , Lee, K. , Santos‐Cortez, R. L. , Muhammad, D. , … Ahmad, W. (2016). Novel mutations in the genes TGM1 and ALOXE3 underlying autosomal recessive congenital ichthyosis. International Journal of Dermatology, 55(5), 524–530. 10.1111/ijd.12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahlquist, A. , Fischer, J. , & Törmä, H. (2018). Inherited nonsyndromic Ichthyoses: An update on pathophysiology, diagnosis and treatment. American Journal of Clinical Dermatology, 19(1), 51–66. 10.1007/s40257-017-0313-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahlquist, A. , Bygum, A. , Ganemo, A. , Virtanen, M. , Hellstrom‐Pigg, M. , Strauss, G. , … Fischer, J. (2010). Genotypic and clinical spectrum of self‐improving collodion ichthyosis: ALOX12B, ALOXE3, and TGM1 mutations in Scandinavian patients. Journal of Investigative Dermatology, 130(2), 438–443. 10.1038/jid.2009.346. [DOI] [PubMed] [Google Scholar]
- Wakil, S. M. , Binamer, Y. , Al‐Dossari, H. , Al‐Humaidy, R. , Thuraya, R. A. , Khalifa, O. , … Al Owain, M. (2016). Novel mutations in TGM1 and ABCA12 cause autosomal recessive congenital ichthyosis in five Saudi families. International Journal of Dermatology, 55(6), 673–679. 10.1111/ijd.13279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials