

Summary
The immune system is essential for maintaining a delicate balance between eliminating pathogens and maintaining tolerance to self‐tissues to avoid autoimmunity. An enormous and complex community of gut microbiota provides essential health benefits to the host, particularly by regulating immune homeostasis. Many of the metabolites derived from commensals can impact host health by directly regulating the immune system. Many autoimmune diseases arise from an imbalance between pathogenic effector T cells and regulatory T (Treg) cells. Recent interest has emerged in understanding how cross‐talk between gut microbiota and the host immune system promotes autoimmune development by controlling the differentiation and plasticity of T helper and Treg cells. At the molecular level, our recent study, along with others, demonstrates that asymptomatic colonization by commensal bacteria in the gut is capable of triggering autoimmune disease by molecular mimicking self‐antigen and skewing the expression of dual T‐cell receptors on T cells. Dysbiosis, an imbalance of the gut microbiota, is involved in autoimmune development in both mice and humans. Although it is well known that dysbiosis can impact diseases occurring within the gut, growing literature suggests that dysbiosis also causes the development of gut‐distal/non‐gut autoimmunity. In this review, we discuss recent advances in understanding the potential molecular mechanisms whereby gut microbiota induces autoimmunity, and the evidence that the gut microbiota triggers gut‐distal autoimmune diseases.
Keywords: autoimmunity, microbiota, mucosal immunology, T cell
Introduction
The mammalian gastrointestinal tract is home to an enormous and complex community of commensal bacteria.1, 2, 3 This gut microbial community (microbiota) has co‐evolved with its host over millennia, suggested by the fact that out of 52 bacterial phyla, two dominant phyla, Bacteroidetes and Firmicutes, represent 98% of the bacteria in the gut.4, 5 These gut microbes provide benefits to their host in many ways, including, but not limited to, digestion, production of nutrients, detoxification, protection against pathogens and regulation of the immune system.1, 2, 3, 6, 7 As a result of its collective metabolic activity, and necessity for human health, the gut microbiota is often referred to as the ‘forgotten’ organ.8 Interestingly, many of the metabolites derived from commensal bacteria have recently been found to directly impact the immune system. Commensal‐mediated immunomodulation includes the promotion of T helper cell subset differentiation and T helper cell plasticity, the ability of a differentiated CD4+ T cell to take on characteristics of other T‐cell subsets simultaneously or at different times.9 Plasticity is an especially important subject for mucosal immunity, as mucosal tissues are major sites where T‐cell plasticity has been observed.10, 11, 12, 13, 14 Therefore, the mucosa harbors the frontline immune response to commensal bacteria, and their interactions may subsequently control health versus disease status through T‐cell differentiation and plasticity.
An autoimmune condition develops when the immune system mistakenly attacks our own self‐tissues. A long‐standing question in the field of microbe–host communication is how interaction of microbes with T cells promotes autoimmune development. Molecular mimicry is one prominent hypothesis, theorizing that microbes trigger autoimmunity by a shared immunogenic epitope between microbes and a self‐peptide, which leads to the activation of autoreactive T cells.15, 16 Another less well‐known theory is that expression of dual T‐cell receptors (TCRs) on T cells promotes autoimmunity by allowing autoreactive T cells to escape thymic clonal deletion.17, 18, 19 Alterations in the composition of the gut microbiome, known as dysbiosis, have been implicated in many diseases and disease models, including those that show clear association with the gut such as inflammatory bowel disease, colorectal cancer, enteric infections, and obesity.20, 21, 22, 23, 24, 25, 26, 27 Dysbiosis has also been implicated in immune disorders that occur outside the gut, such as asthma, eczema, allergies, and gut‐distal autoimmune diseases (autoimmune arthritis, type 1 diabetes, experimental autoimmune encephalomyelitis).20, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37 Although it is easy to see how the gut microbiota can influence immune responses within the gut, over the last decade an appreciation for the impact of the gut microbiome on gut‐distal autoimmunity disease has developed. For example, a strong interest has emerged to characterize the impact of gut microbiota on health and disease status in the lung and brain, namely ‘gut–lung axis’ and ‘gut–brain axis’, respectively.38, 39
This article aims to review the influence of microbiota on the immune system by producing key commensal‐derived metabolites as well as controlling T‐cell subsets and plasticity. Additionally, we discuss the recent findings on how gut commensals affect autoimmunity by molecular mimicry and skewing the expression of dual TCRs. Finally, we discuss the role of dysbiosis in the pathogenesis of common autoimmune diseases. As dysbiosis in inflammatory bowel disease has been extensively reviewed elsewhere (see ref. 40, 41, 42), this review will focus on the impact of gut microbiota dysbiosis in the development of gut‐distal autoimmune diseases. Understanding the mechanisms of how gut commensals affect the immune system will be crucial for us to elucidate autoimmune pathogenesis and to generate novel therapies. This is an urgent subject, as dysbiosis‐related diseases have emerged as new epidemics in the industrialized world.43, 44, 45
Commensal bacteria‐mediated metabolites and immunity
Gut commensals are well‐known for their function in digestion, a process involving extraction and synthesis of many metabolites, some of which are produced only by commensal bacteria and are crucial for host health.46 Recently, several advancements have been made determining how the metabolites generated by commensal bacteria can directly influence the development and function of the immune system, directly impacting health and diseases (Table 1). Here, we review the most recent reports in this field.
Table 1.
The impact of commensal‐derived metabolites on immunomodulation and disease development
| Commensal | Metabolite | Disease phenotype | Immunomodulation | References |
|---|---|---|---|---|
| Clusters IV and XIVa of Clostridium | ↑ IDO, enzyme involved in tryptophan catabolism, on IEC | ↓ Colitis |
↑ Colon Treg ↓ Th1 |
63, 66 |
| Lactobacillus reuteri | ↑ Indole derivatives by metabolizing tryptophan | ↑ Differentiation of DP IEL | 69 | |
| Clostridium sporogenes | ↑ IPA derivatives by metabolizing tryptophan, phenylalanine, and tyrosine | ↓ Permeable intestines | ↓ Neutrophils, monocytes, and memory T cells | 72 |
| General microbiota signal through MyD88 | ↑ RALDH, enzyme able to catalyze the synthesis of RA from Vitamin A |
↓ EAE ↓ Autoimmune arthritis |
↑ Treg ↓ Th17 ↑ Gut‐homing integrin α 4 β 7 ↑ Maintenance of peripheral lymph nodes |
78, 79, 92 |
| Ruminococcaceae eubacterium | ↑ SCFAs signal through GPR43 |
↓ EAE ↓ Type 1 diabetes |
↑ Lamina propria Treg ↑ Antimicrobial peptides |
53, 55, 57 |
Abbreviations: DP IEL, double‐positive intraepithelial T lymphocytes; EAE, experimental autoimmune encephalomyelitis; IDO, indoleamine 2,3‐dioxygenase; IEC, intestinal epithelial cells; IPA, indolepropionic acid; RA, rheumatoid arthritis; RALDH, retinal dehydrogenase; SCFA, short‐chain fatty acids; Th1, T helper type 1; Treg, regulatory T.
Short‐chain fatty acids
Short‐chain fatty acids (SCFAs) including butyric acid, propionic acid, and acetic acid are the main metabolic products of undigested carbohydrates by gut commensal bacteria and have broad effects on the host immune system.47 Among the fatty acid receptors, two orphan G protein‐coupled receptors, GPR41 and GPR43, are activated by SCFAs.48, 49 SCFAs are significantly reduced in the colon of germ‐free mice indicating that the gut microbiota is essential for their production.50 Recently, one group reported that long‐chain fatty acids enhanced the differentiation and proliferation of T helper type 1 (Th1) and/or Th17 cells, whereas SCFAs expanded gut regulatory T (Treg) cells.51, 52 Using experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis, Haghikia et al. showed that long‐chain fatty acids decreased SCFAs in the gut, leading to exacerbated EAE by expanding pathogenic Th1 and/or Th17 cells in the small intestine.51 Treatment with SCFAs ameliorated EAE by inducing lamina‐propria‐derived Treg cells. Consistent with the anti‐inflammatory properties of SCFAs, treatment of germ‐free mice with acetate, a type of SCFA, is sufficient to reduce intestinal inflammation in the dextran sulfate sodium model of colitis.53 The protective effect of acetate was lost in GPR43−/− mice, indicating that signaling of acetate through GPR43 is necessary for the anti‐inflammatory effect of SCFAs. Remarkably, administration of the SCFA butyrate ameliorates inflammation in patients with ulcerative colitis.54 SCFAs also indirectly control type 1 diabetes through the production of antimicrobial peptides. Mechanistically, SCFAs produced by the gut microbiota have been shown to stimulate the production of antimicrobial peptides by pancreatic beta cells.55 Furthermore, systemic administration of these antimicrobial peptides induce Treg cells in the pancreatic islets of pre‐diabetic mice, reducing the incidence of autoimmune diabetes. Additionally, it has been recently demonstrated that administration of the SCFA propionate significantly attenuated HLA‐B27‐associated intestinal inflammation in Fischer 344 HLA‐B27/β 2m transgenic rats.56 Interestingly, this propionate‐mediated inhibition of inflammation is independent of its role in regulating the Treg cell response.
Amino acids
An early study gives an indication that gut bacteria may play an important role in host amino acid homeostasis and health by showing that germ‐free mice had an altered distribution of free amino acids along the gastrointestinal tract compared with conventionalized mice.58 More recent reports suggest that the most abundant amino‐acid‐fermenting bacteria in the human small intestine are bacteria belonging to the Clostridia, the Bacillus–Lactobacillus–Streptococcus groups, Proteobacteria, and Peptostreptococcus.59, 60, 61 These bacteria are therefore likely to be important for amino acid absorption in the gastrointestinal tract. Among the 20 amino acids, tryptophan belongs to one of the nine ‘essential’ amino acids that humans and higher vertebrates cannot synthesize, and that must be supplied in their diet.62 Numerous studies suggest that tryptophan metabolites generated by gut commensal bacteria serve as important signaling molecules in host–microbe cross‐talk. For example, the colonic intestinal epithelial cells from mice colonized with clusters IV, XIVa, and XVIII of Clostridium express high levels of indoleamine 2,3‐dioxygenase,63 an enzyme involved in the initial and rate‐limiting step of tryptophan catabolism, which has been implicated in Treg cell induction.64 Specifically, it has been demonstrated that kynurenine, the first product in the indoleamine 2,3‐dioxygenase‐dependent tryptophan degradation pathway, activates the aryl hydrocarbon receptor, leading to aryl hydrocarbon receptor‐dependent Treg cell generation.65 Indeed, an induction of colonic Treg cells was observed in mice colonized with Clostridium species.63
The small intestinal epithelium contains a unique population of CD4+ CD8αα + double‐positive intraepithelial T lymphocytes (DP IELs) which exhibit anti‐inflammatory properties.54, 67 It has been shown that upon migration to the epithelium, Treg cells lose Foxp3 and convert to DP IELs in a microbiota‐dependent manner, as microbiota depletion by treating mice with broad‐spectrum antibiotics prevents Foxp3 loss.68 Whereas all Treg cells express the CD4‐lineage transcription factor T helper‐inducing POZ/Krüppel‐like factor (ThPOK), CD8αα + CD4+ and > 50% of Foxp3– CD8α – CD4+ cells in the small intestinal epithelium lack ThPOK expression. Later, it was found that loss of ThPOK corresponds to an IEL‐like behavior in CD4+ T cells. Interestingly, supplement of TCR stimulation can overcome the microbiota requirement for the DP IEL differentiation, as demonstrated by the relatively normal DP IEL differentiation in antibiotics‐treated Rag1 −/− OT‐II (ova‐specific) mice exposed to oral ovalbumin. By comparing the microbiota profile between mice housed at two vivaria, with mice at one vivarium displaying significant numbers and another displaying negligible or absent DP IELs, Cervantes‐Barragan et al. discovered that the commensal Lactobacillus reuteri was responsible for the differentiation of DP IELs.69 Lactobacillus reuteri generated indole derivatives by metabolizing tryptophan, which activated the aryl‐hydrocarbon receptor in CD4+ T cells, leading to their ThPOK down‐regulation and differentiation into DP IELs. Hence, L. reuteri together with a tryptophan‐rich diet can promote gut immune homeostasis by allowing the differentiation of intraepithelial CD4+ T cells into anti‐inflammatory DP IELs.
Only a few species of gut commensals, including Clostridium sporogenes, can break down tryptophan and produce the metabolite indolepropionic acid (IPA), a deamination product of tryptophan.70 Bacterial tryptophanase catalyzes the conversion of dietary tryptophan to indole and subsequently to IPA.71 A recent study identified a total of 12 compounds that can be produced in this process, nine of which can accumulate in the blood and three of which are produced exclusively by bacteria.72 Specifically, the C. sporogenes‐expressed gene fldC is necessary for the production of IPA. In fact, fldC is essential for the reductive metabolism of all three aromatic amino acids (tryptophan, phenylalanine, and tyrosine). Germ‐free mice receiving wild‐type C. sporogenes exhibit high levels (~80 μm) of serum IPA whereas germ‐free mice receiving the mutated version of C. sporogenes lacking fldC, had undetectable serum IPA. Importantly, mice with undetectable IPA had higher levels of immune cells, including neutrophils, classical monocytes, and memory T cells. In addition, the mice with the engineered version of C. sporogenes had increased intestinal permeability, a defect that is often seen in inflammatory bowel disease.
In addition to having a direct impact on the immune system, undigested amino acids have the potential to become supplemental precursors for SCFA generation by the gut microbiota, in addition to indigestible carbohydrates.73 Therefore, amino acids could have indirect impact on the immune system through SCFAs as we discussed earlier. Numerous amino acids including glycine, threonine, glutamate, and ornithine can be metabolized by anaerobic bacteria to generate acetate, whereas threonine, lysine, and glutamate can be used for butyrate synthesis.74 Moreover, at the molecular level, it has been shown that intracellular leucine concentrations can be sensed by the multiprotein complex leucyl‐tRNA synthetase,75, 76 which activates the mechanistic target of rapamycin kinase, proving to be a vital link between immune function and metabolism.77
Retinoic acid
Retinoic acid, a metabolite of vitamin A, is one of the most active physiological retinoid metabolites and has a wide range of biological activity including regulating immune responses.78 A major part of retinoic acid's anti‐inflammatory effects depends on the inhibition of Th17 cells and promotion of Foxp3+ Treg cell responses.78, 79 In addition, retinoic acid has been shown to be important for the expression of the gut homing receptor integrin α 4 β 7 on T cells.80, 81, 82 The α 4 β 7 integrin receptors are imprinted on lymphocytes by dendritic cells (DCs) from Peyer's patches (PPs), and mesenteric lymph nodes.82, 83 In the absence of microbial toll‐like receptor signaling in Myd88−/− mice, gut DCs express low levels of retinal dehydrogenase (RALDH) required for retinoic acid biosynthesis, so these Myd88−/− mice are unable to generate gut‐homing lymphocytes.84 AM80 is a synthetic retinoic acid that is characterized by higher stability and fewer potential adverse effects compared with retinoic acid.85, 86 It has been reported that both retinoic acid and AM80 ameliorate many autoimmune responses, including experimental autoimmune myositis, experimental autoimmune encephalitis, and collagen‐induced arthritis.87, 88, 89, 90 We recently showed that oral administration of AM80 inhibits autoimmune disease in the joints as well as in the lung.91 We elucidated a novel mechanism whereby AM80 suppresses the autoimmune pathology in both the lung and joints by inhibiting T follicular helper (Tfh) cells in addition to inhibiting Th17 responses. Specifically, AM80 increased the expression of the gut‐homing integrin α 4 β 7 on Tfh cells, which diverted Tfh cells from systemic (non‐gut) inflamed sites such as the lung draining lymph nodes into the gut (the non‐immunopathological site) and so reduced systemic autoantibody production.
Moreover, the impact of retinoic acid can go beyond the cellular level and impact the development of whole lymphoid tissues.92 It has been reported that cellular expansion in peripheral lymphoid tissues is controlled by gut microbiota in a retinoic acid‐dependent manner.93 The mucosal addressin MAdCAM‐1 is the receptor for the gut‐homing integrin α 4 β 7, and peripheral node addressin PNAd is the receptor for CD62L.94 In neonatal mice, MAdCAM‐1 expression in lymph nodes is elevated shortly after birth,95 followed by a switch to a PNAd over a course of 2–3 weeks.93 Zhang et al. demonstrated that commensal fungi drive a wave of RALDH+ DCs to migrate to the peripheral lymph nodes after birth.92 The arrival of these cells increases the amounts of retinoic acid in situ, mediates the neonatal MAdCAM‐1 to adult PNAd addressin switch on endothelial cells, and directs the homing of lymphocytes to gut‐associated lymphoid tissues. Finally, the authors found that a diet deficient in vitamin A causes reduced homing of RALDH+ DCs into peripheral lymph nodes and a lack of maintenance of peripheral lymph node structures, suggesting a dependence on retinoic acid signaling for structural and functional maintenance of peripheral immune tissues.
Microbiota and T‐cell subset determination
Understanding the role of microbiota in T‐cell subset commitment and plasticity holds the key for unveiling the pathogenesis and therapeutic options for autoimmune diseases, as well as diseases associated with unbalanced immune responses such as cancers and infections. Not surprisingly, addressing this question has become a focused area of research in recent years. One study using transgenic mice expressing a limited but diverse TCR repertoire, through fixed TCR‐β expression, showed that the TCR repertoire of colonic Treg cells is unique compared with thymically derived Treg cells.96 Furthermore, these colonic Treg cells were shown to react with bacterial isolates, suggesting that encountering gut microbes in the intestines leads to peripheral Treg cell induction, and hence to tolerance to the gut microbiota.96 A more recent study, also using transgenic mice expressing a constrained TCR repertoire, through fixed TCR‐β expression and limited TCR‐α rearrangement (TCRmini mice97) showed that thymic and intestinal Treg cells expressed overlapping TCR repertoires, many of which recognize microbial antigens.98 Alteration of the gut microbiota through treatment of TCRmini mice with a cocktail of antibiotics concurrently altered the colonic and thymic Treg TCR repertoire.98 This suggests that the repertoire of thymically derived Treg cells is heavily influenced by the microbiota. Although these studies have differing results regarding the initial site of gut microbiota influence, in either case it is clear that direct recognition of microbial antigens by T cells can promote Treg cell lineage commitment. Beyond Treg cells, the gut microbiota can also influence T helper subsets. In the presence of a normal microbiota, CX3CR1+ intestinal antigen‐presenting cells act to limit Th1 cell development and expansion.99 However, antibiotic‐induced dysbiosis leads to a shift in the function of CX3CR1+ antigen‐presenting cells, allowing them to promote pathogenic Th1 cell development.99
Our laboratory and others have also demonstrated that a single type of commensal bacteria, segmented filamentous bacteria (SFB), can promote Th17 cell responses. In C57BL/6 mice, SFB specifically induce intestinal Th17 cells.25 More recent studies, however, have demonstrated that SFB can also influence T helper subsets outside the gut. In 2011, Lee et al. discovered that SFB alone was able to promote Th17 cells specifically in the spinal cord of EAE mice.33 Similarly, we have shown that in the K/BxN mouse model of autoimmune arthritis, SFB specifically promotes Th17 cell responses in the lung.100 Additionally, SFB can also drive the differentiation of Tfh cells and promote their migration to systemic sites, leading to exacerbated autoimmune arthritis in K/BxN mice.101 However, the exact molecule(s) employed by SFB to promote the differentiation of T helper cell subsets remain elusive. Although many microbiome studies focus on the microbes residing in the intestines, the entire digestive tract is colonized with bacteria. Hence, the oral microbiome has recently become an area of interest with regards to mucosal immunity. Data from Dutzan et al. showed that oral homeostatic Th17 responses, unlike gut Th17 responses, were unaffected by shifts in the oral or gut microbiomes.25, 102 This suggests that not all mucosal T cells are influenced by the microbiome, and that other environmental cues such as chewing activity in the oral cavity and chewing‐associated damage can maintain the Th17 cell population. Interestingly, a more recent study from this same group found that inflammatory Th17 cells isolated directly from periodontal lesions were in fact heavily influenced by oral microbiome dysbiosis.103 By focusing on Th17 isolated from periodontal lesions of both mice and humans, they found that increased abundance of Th17 cells and enhanced production of inflammatory cytokine IL‐17 was associated with shifts in the oral microbiome. Specific outgrowth of Enterococcus and Actinobacteria along with loss of Streptococcus was associated with periodontitis and enhanced Th17 cell responses in the ligature‐induced periodontitis mouse model. Together the studies suggest that only pathogenic but not homeostatic Th17 cell responses are influenced by dysbiosis. We recently addressed how both age and gut microbiota affect T‐cell subsets in the context of autoimmunity.104 Our results show an augmented autoimmune disease phenotype in both the joints and the lung of middle‐aged compared with young mice. Mechanistically, we saw a soaring accumulation of Tfh, but surprisingly not Th17 cells with age. Our data suggest exposure to immunomodulatory commensals such as SFB may allow the young host to develop an overactive immune system similar to that found in middle‐aged hosts. Our study also shows that age can independently increase the Tfh cell response without the help of immunomodulatory commensals.
CD4 T helper cell plasticity and the microbiota
The development of specific transcription factor reporter mice has proven to be an invaluable tool in dissecting the plasticity of T‐cell subsets. An early study using adoptive transfer of cells from Foxp3eGFP reporter mice showed that 80% of Treg cells lost GFP expression in the PPs compared with 50% in spleen.12 Within the GFP‐negative PP population, > 60% of the cells expressed CXCR5, consistent with a Tfh phenotype,12 suggesting that PP, but not spleen, is the preferred site for a Treg to Tfh reprogramming. Two other studies found that retinoic‐acid‐receptor‐related orphan receptor γ t (Rorγt), the hallmark Th17 transcription factor, was preferentially expressed by colonic Treg cells, and furthermore, that in the absence of gut bacteria, the Rorγt+ Treg cell population was substantially diminished.10, 14 Interestingly, several bacterial species were able to induce Rorγt+ Treg cells, which were indispensable for maintaining gut homeostasis as determined by enhanced IL‐17, interferon‐γ (IFN‐γ), and colitis in their absence.10 Another study showed that the pathogenic bacterium Helicobacter hepaticus promoted expansion of Rorγt+ Treg cells in a c‐MAF‐dependent manner, which selectively suppressed pro‐inflammatory Th17 cells in the intestines.105 However, in the absence of Treg cells (in Il10 −/− mice), H. hepaticus predominantly induced Th17 cells, suggesting that pathobionts can have an impact on T‐cell plasticity by expanding potent suppressive T cells, which in turn, inhibit the inflammatory T helper cells necessary for pathogen elimination. Plasticity among T helper subsets has also been observed. Using cell fate reporter mice in which Th17 cells are permanently marked by YFP (Il17 cre R26 eYFP), Hirota et al. found that selectively in the PP, but not spleen, Th17 cells were reprogramed to a Tfh (PD‐1hi CXCR5hi) phenotype.13 Furthermore, these ex‐Th17‐Tfh cells localized in germinal centers where they assisted in T‐cell‐dependent IgA production. In a Th17 adoptive transfer model of colitis, it was demonstrated that Th17 cell conversion to a Th1 phenotype was necessary for colitis development as Th17 cells unable to produce IFN‐γ (Ifng −/−) were incapable of inducing colitis.106 Interestingly, not only did Th17 cells convert to Th1 and contribute to colitis development directly, but also Th17 were capable of providing help to naive T cells for Th1 lineage commitment. Together these studies suggest that gut microbiota is able to influence T‐cell plasticity within the gut microenvironment and influence the development of autoimmunity and other inflammatory diseases.
Gut microbiota induces autoimmunity by TCR‐mediated mechanisms
Central to T‐cell function is the TCR, through which T cells recognize their cognate antigen and become activated. An important feature of the TCR is its ability to recognize multiple antigens, known as TCR cross‐reactivity. Sequencing‐based methods estimate that humans and mice express 107 and 106 productive unique αβ TCRs, respectively, yet this is insufficient to cover the possible foreign antigen repertoire.107, 108 Therefore, TCRs possess the ability to recognize multiple antigens in order to recognize the plethora of possible foreign antigens (see ref. 109). One potential mechanism through which pathogenic autoreactive T cells may become activated is through weak recognition of a microbial antigen for which their TCR is cross‐reactive. This is also known as molecular mimicry, a phenomenon in which a foreign antigen shares significant structural or sequence similarities with a self‐antigen.15, 16 Later, this now activated autoreactive T cell can home to the tissue where its cognate self‐antigen is expressed and elicit autoimmune pathology. In fact, cross‐reactive TCR recognition of microbial peptides by autoreactive T cells has been seen in several autoimmune settings (Fig. 1). In 2015, Horai et al. showed that T cells restricted to expressing only the retina‐specific TCR (R161H), became activated in the intestines in response to an unidentified microbial antigen.110 T‐cell activation was in response to TCR‐signaling to a non‐cognate microbial antigen, as T cells in the small intestines of R161H‐Tcrα −/− mice had clear co‐localization of phosphorylated Zap‐70 with CD4, known to be downstream of TCR signaling.110, 111, 112 Later, it was shown that Islet‐specific glucose‐6‐phosphate catalytic subunit‐related protein (IGRP)‐specific CD8+ T cells in non‐obese diabetic (NOD) mice were activated through recognition of a peptide from Fusobacteria and elicited autoimmune diabetes.113 Interestingly, the Fusobacteria peptide shared significant homology with the IGRP peptide targeted by the IGRP‐specific transgenic TCR,113 suggesting that molecular mimicry in microbial peptides may pose a threat because they can activate pathogenic autoreactive T cells. Most recently, it was shown that gut commensal bacteria expressing an ortholog to human Ro60, a nuclear RNA‐binding protein and primary targeted self‐antigen in lupus, were able to activate human Ro60‐specific CD4+ T cells through cross‐reactive TCR recognition.114 Furthermore, germ‐free mice monocolonized with Ro60‐ortholog‐expressing commensal bacteria spontaneously developed lupus‐like disease, as indicated by glomerular immune complex deposits. Together, these studies suggest that molecular mimicry is a mechanism through which the intestinal microbiota is able to propagate autoimmune responses leading to disease.
Figure 1.
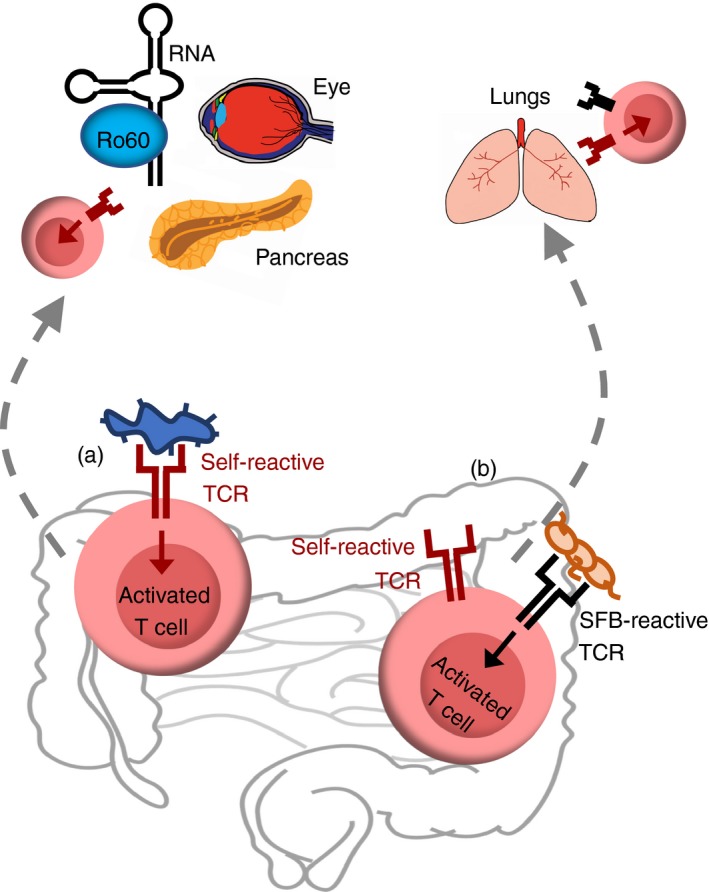
Mechanism leads to autoimmunity: recognition of commensal bacteria by T‐cell receptor (TCR). (a) Molecular mimicry. Several studies have demonstrated, as a result of the cross‐reactive nature of TCRs, that autoreactive T cells can recognize both a self‐antigen and a gut commensal‐antigen. Recognition of a microbial antigen is able to activate autoreactive T cells, which in turn migrate to the tissue where their cognate self‐antigen is expressed and elicit autoimmune diabetes,113 autoimmune uveitis,110 and lupus.114 (b) Dual TCRs. Another mechanism involves autoreactive T cells that are able to recognize gut commensal‐antigens through expression of a secondary TCR in addition to self‐antigen recognizing TCR and differentiate into T helper type 17 (Th17) cells.100 These T cells differentiate into Th17 cells through TCR recognition of segmented filamentous bacteria (SFB), then traffic to the lung where they mediate lung pathology.
In addition to molecular mimicry, it has long been thought that pathogenic autoreactive T cells may arise from a subset of thymocytes expressing two unique TCRs, one of which allows them to evade negative selection along with one that has a high affinity for a self‐antigen.115, 116, 117 Approaching from a different angle, outside the central immune system, we found that in peripheral tissues, SFB selectively expand dual TCR‐expressing T cells, leading to the augmentation of lung autoimmune pathology (Fig. 1).100 This is the first study to show that dual TCR T cells can be activated through the recognition of the cognate commensal‐derived antigen and migrate to elicit autoimmune pathology in the lung, a gut‐distal site. At the molecular level, we demonstrate that SFB selectively expand autoimmune T cells co‐expressing SFB‐specific TCRs in addition to their self‐reactive TCRs. This additional SFB‐specific TCR provides a proliferative advantage for autoreactive Th17 cells in SFB(+) hosts. Our data suggest that the induction of robust SFB‐specific Th17 response by transferring SFB(+) recipients with SFB‐specific 7B8 T cells could not rescue the autoimmune activity of Rag−/−.KRN T cells through bystander activation. Instead, our data suggest that the same T cells need to specifically recognize self‐ as well as SFB‐antigen to induce IL‐17‐expressing pathogenic T cells to cause autoimmunity. This is supported by previous data showing that in healthy mice, cognate TCR recognition of SFB‐antigen is crucial for SFB‐mediated Th17 induction.118, 119 Taken together, our study suggests that the same T cell needs to recognize both self‐antigen and be activated by SFB to undergo preferential Th17 cell expansion and so enhance autoimmunity in an SFB‐dependent manner. Beyond molecular mimicry, our study suggests an alternative, dual TCR‐based mechanism for commensal‐mediated autoimmunity.
Role of gut microbiota in human and murine gut‐distal autoimmunity
Autoimmune disorders have been sharply increasing worldwide in recent decades.26 The dramatic changes in the disease onset rate cannot be explained by genetic basis as it occurs in such a short period of time. On the contrary, these data suggest that environmental factors play a key role in the recent surge of autoimmune diseases. In recent years, we have begun to appreciate that the gut microbiota provides environmental cues controlling human health and disease, including its potential impact on autoimmune responses. Under healthy conditions, the human intestinal microbiota is composed primarily of bacteria of the phylum Firmicutes, with Bacteroidetes and Actinobacteria also represented.120 On the family and species level, the gut microbiota varies widely among individuals, although it remains largely stable within an individual over the course of several years.121 Studies using mouse models or samples from human subjects transplanted into mice have shown that age, gender, diet, smoking, autoimmunity, and other factors can alter the microbiota, and that the altered microbiota can be transferred and exert influence over the metabolism and immune system of the recipient.122 These studies demonstrate that the host–microbiota relationship is not a one‐way street, but rather a dynamic conversation that can have long‐lasting impacts.
Many studies have investigated a correlation between gut microbiome composition and autoimmune disease in humans, but these studies are limited in their ability to determine causality. Exploring this field using animal models allows for perturbation of the gut microbiome to better distinguish cause from effect, permit mechanistic dissection and allow pre‐clinical evaluation of suggested therapeutic strategies. Throughout this section, we will highlight the important observations made in both human patients and mouse models.
Multiple sclerosis (MS) is an autoimmune disease in which pathogenic CD4+ T cells penetrate the blood–brain barrier and cause damage to the central nervous system.123 Numerous studies have shown that patients with MS display intestinal microbiome dysbiosis.124, 125, 126 Two independent studies comparing patients with relapsing–remitting MS and healthy controls found significant alterations in the composition of the microbiome from patients with MS.124, 127 Perhaps even more intriguing, when compared with treatment‐naive patients, those receiving treatment displayed some restoration in their microbiome composition.127 Untreated MS patients displayed increased relative abundance of Methanobrevibacter and Akkermansia, which were lower in healthy controls and treated MS patients. MS patients receiving treatment had increased Sutterella, which has been previously observed to be increased in healthy controls compared with treatment‐naive patients with inflammatory bowel disease (Fig. 2).128 This suggests that although dysbiosis may contribute to a predisposition for developing MS, perhaps treatment may act to normalize pro‐inflammatory microbiota. A recent study by Cekanaviciute et al. showed that intestinal microbiome samples from MS patients displayed specific outgrowth of Acinetobacter and Akkermansia.129 Importantly, they further demonstrated that in the presence of Acinetobacter calcoaceticus, human peripheral blood mononuclear cells preferentially shifted towards an inflammatory phenotype (IFN‐γ) and away from a regulatory phenotype (Foxp3+ CD25+). This suggests that the specific dysbiosis observed in patients with MS favors an inflammatory T‐cell response. More definitive studies using the EAE mouse model of MS have shown that in germ‐free mice lacking gut microflora, EAE disease severity is reduced to an almost undetectable level, due to a more than twofold reduction in IFN‐γ and IL‐17 production from CD4+ T cells.33 Importantly, if CD4+ T cells from germ‐free mice were transferred to specific pathogen‐free mice harboring a diverse intestinal microflora, these cells gained the ability to mediate EAE disease development, suggesting that the gut microbiota is necessary to elicit the pathogenic Th1 and Th17 responses seen in EAE and perhaps MS.33 Importantly, it was recently demonstrated that colonization of germ‐free Myelin oliodendrocyte glycoprotein (MOG)‐specific TCR transgenic mice (RR mice) with gut microbiota from patients with MS resulted in increased incidence of spontaneous EAE development.130 This suggests that the microbiota of patients is sufficient to precipitate autoimmune disease. Together these studies suggest a gut–brain axis of communication that allows gut microbes to elicit autoimmune responses in the brain.
Figure 2.
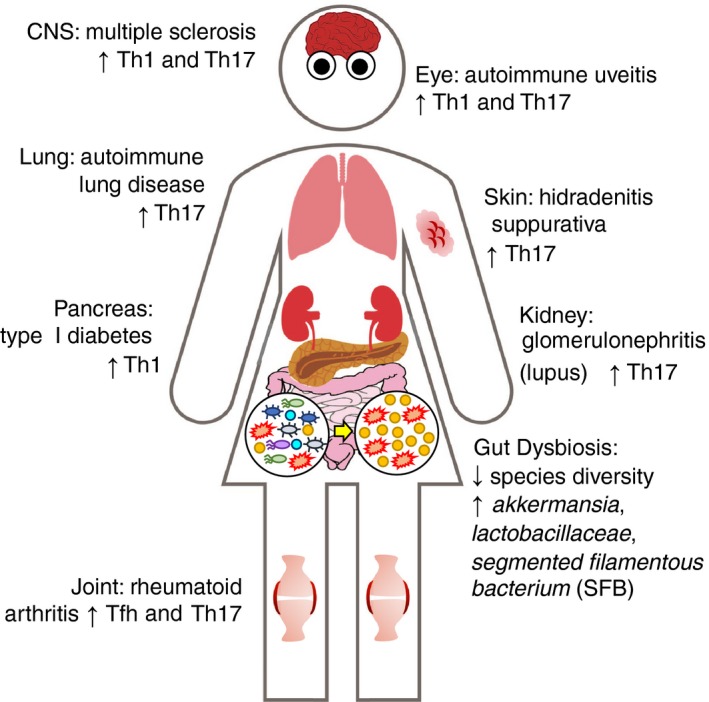
Dysbiosis leads to gut‐distal autoimmunity. Several studies have demonstrated a link between alterations in the composition of the gut microbiota and the development of autoimmune disease. Multiple sclerosis (MS) and the C57BL/6 proteolipid protein‐induced experimental autoimmune encephalomyelitis mouse model, which have autoimmune manifestations mediated by T helper type 1 (Th1) and Th17 cells in the central nervous system (brain and spinal cord).33, 123, 124, 127, 128, 129, 130 Autoimmune uveitis and the R161H‐TCR transgenic mouse model display autoimmune manifestations mediated by Th1 and Th17 cells in the retina of the eye.110 Rheumatoid arthritis (RA) and the RA‐associated lung pathology in the K/BxN mouse model have autoimmune manifestations mediated by follicular helper T (Tfh) and Th17 cells in the joints and by Th17 cells in the lung.131, 145, 146, 147, 148, 149 Hidradenitis suppurativa (HS) is an autoimmune skin condition associated with an increase of Th17 cells.157 Currently, there is no good mouse model for HS, so the role of gut microbiota in this disease remains elusive. Systemic lupus erythematosus (SLE) and the NZB/W F1 mouse model develop autoimmune manifestations mediated by Th17 cells and immune complex deposition specifically in the kidneys.152, 155, 156 Type 1 diabetes and the various mouse models (C57BL/6 streptozotocin‐induced model and IGRP‐specific TCR transgenic model) develop autoreactive Th1 responses in the pancreatic islets.113, 160, 161, 163
Rheumatoid arthritis (RA) is an autoimmune disease that primarily affects joints but may also affect other parts of the body including the lungs and heart.131, 132, 133 Several studies have suggested that many patients with RA display antigen–antibody complexes with complement fixation in joints that can cause tissue damage and tumor necrosis factor‐α production.134, 135, 136, 137, 138, 139 Recent studies also suggest that anti‐citrullinated protein antibodies are involved in RA pathogenesis.140, 141 Tfh cells are a T effector cell type specialized in helping B cells.142, 143, 144 Over‐reactive Tfh responses pose a threat of triggering excessive autoantibody production and autoimmune disease.145, 146, 147, 148, 149 In that regard, the presence of circulating CXCR5+ Tfh (cTfh) cells in the blood has been observed in patients with different autoimmune diseases, including RA.20 In a recent study assessing 77 treatment‐naive patients with RA, 21 treated patients with RA, and 80 healthy controls, the gut microbiome was found to be altered in patients with RA.150 In particular, Haemophilus spp. were depleted in individuals with RA and negatively correlated with serum autoantibodies titers, whereas Lactobacillus salivarius was over‐represented in individuals with RA and abundance correlated with disease severity (Fig. 2). Interestingly, similar to what has been observed in patients with MS, patients with RA undergoing treatment displayed partial normalization of their intestinal microflora compared with treatment‐naive patients.
By using a photo‐labeling mouse model to track cell migration, our laboratory has demonstrated that a commensal bacterium, SFB, is able to promote the differentiation and migration Tfh cells from the PPs in the small intestine to systemic sites where they elicit autoimmune arthritis development.101 At the molecular level, SFB induce PP Tfh cell differentiation by limiting the access of IL‐2 to PP CD4+ T cells and DCs are required for SFB‐mediated IL‐2Rα suppression and up‐regulation of Bcl‐6, a master regulator of Tfh cells, in PPs. Furthermore, we have recently shown that SFB can also promote RA‐associated autoimmune lung disease through propagation and mobilization of gut‐derived Th17 cells.100 Importantly, this highlights a gut‐lung axis of communication through which specific gut commensal bacteria are able to promote pathogenic T‐cell responses leading to gut‐distal autoimmune pathology. In the collagen‐induced arthritis model of RA, dysbiosis characterized by outgrowth of Lachnospiraceae and Lactobacillaceae was detected before arthritis development.151 Furthermore, antibiotic‐induced depletion of the microbiota before induction of collagen‐induced arthritis reduced disease severity by 40% and was accompanied by reduction in the inflammatory cytokines IL‐17, IL‐22, and IL‐23 in the intestines along with reduced anti‐collagen antibodies. This suggests that the gut microbiota is necessary for autoimmune disease development and that alterations in the composition of the gut microbiota precipitate autoimmune disease.
Systemic lupus erythematosus (SLE) is an autoimmune disease affecting almost all organs of the body, in which both autoreactive T cells and autoantibody‐producing B cells contribute to the pathogenesis of SLE.152 Like many gut‐distal autoimmune diseases, recent studies have linked gut microbiota dysbiosis with SLE.153 In a 2016 study from Lopez et al., it was shown that intestinal microbiota isolates from patients with SLE promoted naive T‐cell differentiation into Th17 cells.154 Furthermore, peripheral blood mononuclear cells from patients with SLE were enriched for both Th17 and Foxp3+ IL‐17+ cells compared with healthy controls. As described earlier, due to T cell plasticity, Treg cells are able to shift between regulatory and potentially pathogenic phenotypes. Hence, the presence of Foxp3+ IL‐17+ cells in the peripheral blood of patients with SLE suggests that the shift observed in the gut microbiome of these patients may promote inflammatory T‐cell responses and may negatively impact Treg cell stability. More recently, in a study by Luo et al. which assessed the intestinal microbiota dynamics of both SLE patients and the NZB/W F1 mouse model of SLE, it was shown that alteration in the gut microbiota resulted in decreased diversity with increased representation of Gram‐negative bacteria (Fig. 2).155 Interestingly, as NZB/W F1 mice progressed to overt disease, the relative abundance of Lactobacillaceae increased from 0·1% to 10%. Furthermore, there was a positive correlation between Lactobacillaceae abundance and disease severity in female NZB/W F1 mice. This suggests that reduction in α diversity, a measure of the number of different species present in a given site, and outgrowth of specific commensal microbes may promote autoreactive immune responses in susceptible individuals. Another report found a high frequency of Th17 cells in the kidneys of patients with glomerulonephritis, a form of SLE.156 By using Kaede mice to track intestinal T‐cell migration during glomerulonephritis induction, they observed that Th17 cells egress from the intestine and subsequently migrate to the kidney through CCR6 recognition of CCL20. Furthermore, depletion of gut Th17 cells in germ‐free and antibiotics‐treated mice ameliorated the autoimmune glomerulonephritis, suggesting that targeting the intestinal Th17 cells may offer a therapeutic strategy for autoimmune diseases.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition of the hair follicles thought to be mediated in large part by Th17 cells (Fig. 2).157 With respect to perturbations in the microbiota, most studies assess changes in the cutaneous microbiota as this is the site of the disease manifestation. Recently, HS has been identified to have a strong concurrence rate with inflammatory bowel disease, with over 17% of Crohn's patients also having HS.158 Hence, while there are definite changes in the cutaneous microbiome of patients with HS,159 taking a closer look at the gut microbiome may prove fruitful in the quest for identifying a causal mechanism.
Type 1 diabetes (T1D) is a T‐cell‐mediated autoimmune disease characterized by the selective destruction of insulin secreting β‐cells in the pancreatic islets.160 Many patients with T1D present with increased intestinal permeability or ‘leaky gut’, which has been shown to precede the onset of clinical disease.161 In addition, patients with T1D also display an altered intestinal microbiome characterized by decreased α diversity and increased abundance of inflammatory species compared with healthy controls (Fig. 2).162 Hence, it is tempting to assume that in combination with increased intestinal permeability, dysbiosis could promote an inflammatory environment in the gut–proximal pancreatic tissue leading to activation of an autoimmune response. In a 2016 study from Costa et al., it was determined that autoimmune diabetes development in the streptozotocin‐induced model of T1D is dependent on translocation of gut microbiota to the pancreatic lymph node.163 Interestingly, islet‐infiltrating T cells seem to be of gut origin as they often express α4β7 integrin.83, 164, 165
We now know that the effect of commensal bacteria on autoimmune disease can be model dependent. For example, SFB is pathogenic in animal models of arthritis and multiple sclerosis32, 33 but protective in the NOD mouse model of type1 diabetes.166, 167 The beneficial and detrimental effects of SFB could depend on whether commensal‐mediated immunomodulation is enhancing or inhibiting the pathogenesis of each disease. For example, autoantibodies are a key pathogenic factor and diseases can be induced by passive transfer of autoantibodies in the K/BxN and EAE models.168, 169, 170 In contrast, type 1 diabetes in NOD mice is a T‐cell‐mediated autoimmune disease, and although B cells of NOD mice produce autoantibodies, these are not thought to play a diabetogenic role.171, 172 Hence, SFB, with their Tfh and autoantibody boosting effect, are more likely to play a pathogenic role in the K/BxN model and other antibody‐mediated autoimmune diseases than in T‐cell‐mediated autoimmune diseases such as in NOD mice. One feature that many of the autoimmune diseases have in common is evidence of gut‐derived T cells participating in the autoimmune response.83, 101, 156, 164, 165 This would suggest that perhaps one mechanism through which the gut microbiota modulates autoimmune responses is through mobilization of T cells from the tolerogenic environment of the gut to systemic sites where alleviated tolerogenic pressure allows these cells to become active and initiate autoimmune disease development.173, 174, 175 However, more studies are required to determine whether the altered microbiome drives changes in immunity or conversely the onset of immunological disease induces changes in the gut microflora in human patients. It is likely a combination, as changes in the gut microbiota precede onset of clinical autoimmune manifestations,162 yet some evidence suggests that perhaps T cells, Foxp3+ Treg cells in particular, regulate gut microbiota diversity.176
Conclusions and future directions
There is a surge of recent studies investigating the molecular mechanisms of dysbiosis‐related autoimmune diseases. In both molecular mimicry and dual TCR theories, until recently, only infectious pathogens including viruses and bacteria have been implicated as the primary culprits,15, 16, 17 and little is known about the molecular mechanism by which asymptomatic commensal colonization could trigger autoimmunity. Our recent study along with others demonstrates a previously unknown condition that asymptomatic colonization by commensal bacteria in the gut is capable of triggering systemic autoimmune disease by molecular mimicking self‐antigen and skewing the dual TCR expression in the host. Notably, dual TCR expression is not limited to transgenic mouse models. In wild‐type mice and in humans, up to 15% and 33%, respectively, of peripheral T cells have been reported to express dual TCRs, due to incomplete allelic exclusion at the Tcra locus.177, 178 Future investigation is required to determine whether dual TCR expression is involved in dysbiosis‐related immune disorders in human patients. Another important, but poorly understood, aspect of microbiota–T‐cell interaction is how different microbial signals impact T‐cell stability and plasticity at the mucosal frontline. Understanding the plasticity and stability of T‐cell subsets in mucosal tissues is highly relevant for future therapeutic strategies as we will not be able to manipulate T cells for therapeutic purposes without understanding the mechanisms by which T cells shift between alternative programs because of exposure to gut microbes.
As illustrated in this review, the use of animal models provides a powerful tool for mechanistic studies as it allows us to establish the causative effect of microbiota in disease development. By using germ‐free and specific pathogen‐free mouse models, numerous studies unravel the crucial roles of gut commensals in immune regulation in the context of health and disease. Although these studies provide promising clinical implications, various challenges need to be overcome by the field to harness these findings for future diagnostic and therapeutic approaches. One of the challenges is that the reductionist experiments in mice often do not reflect important features of the immune system in the adult human. In this regard, Beura et al. demonstrated that laboratory mice – like neonates, but not adult humans – lack effector‐differentiated and mucosal memory T cells.179 These cell populations are present in feral mice and pet store mice with constant and diverse microbial exposure. Furthermore, laboratory mice co‐housed with pet store mice display profound changes in immune systems, resulting in an immune signature that is more closely reflected by adult humans than by neonates. Hence, the ‘dirty’ mouse model may provide a unique advantage over the current reductionist mouse models and should be considered for studies with translational purposes in human disease.
One of the biggest dilemmas facing the field of gut microbiota research is how to apply laboratory findings to clinical therapies. Currently, microbiota‐based therapy relies on three major strategies: complete fecal microbiota transplants (FMTs), administering one or more species of bacteria orally (probiotics), or administering substrates to favor the expansion of certain kinds of bacteria or a shift in metabolite production, a strategy known as prebiotics.180 Each of these approaches has limitations; for example, probiotics often colonize only transiently because of ineffective competition with the existing microbiota.181 Additionally, despite the focus on taxonomic composition analyzed by 16S rRNA sequencing of microbial communities in many studies, the functional relevance of microbial communities is more likely to be revealed by their metagenomic gene expression and metabolomics profiles. This is further proved by the fact that bacterial isolates of the same strain can display very different immunoregulatory capabilities.182 FMTs depend on severe depletion of the constituent microbiota through antibiotics and bowel flushing before administration of the transfer.183, 184 Other researchers are working to find alternative methods to circumvent the difficulties of FMTs, such as filtering donor material to the point of removing intact cells, leaving only bacterial byproducts such as metabolites.185 An additional approach is to identify bacterial metabolites able to modulate human immune responses and artificially administer the identified metabolites as novel therapeutics.186 This approach would be appealing from a pharmaceutical perspective, because it would allow the development of drugs that would need to be taken regularly. It would also minimize the factors that need to be controlled, since administering metabolites would not depend on the engraftment of a foreign bacterial species. However, a careful determination on how to achieve a comparable delivery of the metabolite that mimics the one produced by the intestinal microbiota will be crucial for the effectiveness of the treatment. In conclusion, the gut microbiota is a complex community that engages in significant cross‐talk with the host. A better understanding of host–microbe interaction and the underlying microbiota‐derived molecules that modulate the immune system and disease development may help to pave the way for better patient‐tailored interventions and microbial molecule‐based therapies for immune disorders.
Disclosures
The authors have no competing interests to declare.
References
- 1. Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006; 124:837–48. [DOI] [PubMed] [Google Scholar]
- 2. Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol 2008; 6:776–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host–bacterial mutualism in the human intestine. Science 2005; 307:1915–20. [DOI] [PubMed] [Google Scholar]
- 4. Rappe MS, Giovannoni SJ. The uncultured microbial majority. Annu Rev Microbiol 2003; 57:369–94. [DOI] [PubMed] [Google Scholar]
- 5. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Human gut microbes associated with obesity. Nature 2006; 444:1022–23. [DOI] [PubMed] [Google Scholar]
- 6. Finke D. Induction of intestinal lymphoid tissue formation by intrinsic and extrinsic signals. Semin Immunopathol 2009; 31:151–69. [DOI] [PubMed] [Google Scholar]
- 7. Hill DA, Artis D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol 2010; 28:623–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. O'Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep 2006; 7:688–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DuPage M, Bluestone JA. Harnessing the plasticity of CD4+ T cells to treat immune‐mediated disease. Nat Rev Immunol 2016; 16:149–63. [DOI] [PubMed] [Google Scholar]
- 10. Sefik E, Geva‐Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM et al Mucosal immunology. Individual intestinal symbionts induce a distinct population of RORγ + regulatory T cells. Science 2015; 349:993–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brown CC, Esterhazy D, Sarde A, London M, Pullabhatla V, Osma‐Gracia I et al Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 2015; 42:499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T et al Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer's patches. Science 2009; 323:1488–92. [DOI] [PubMed] [Google Scholar]
- 13. Hirota K, Turner JE, Vila M, Duarte JH, Demengeot J, Steinmetz OM et al Plasticity of Th17 cells in Peyer's patches is responsible for the induction of T cell‐dependent IgA responses. Nat Immunol 2013; 14:372–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y et al Mucosal immunology. The microbiota regulates type 2 immunity through RORγt+ T cells. Science 2015; 349:989–93. [DOI] [PubMed] [Google Scholar]
- 15. Albert LJ, Inman RD. Molecular mimicry and autoimmunity. N Engl J Med 1999; 341:2068–74. [DOI] [PubMed] [Google Scholar]
- 16. Munz C, Lunemann JD, Getts MT, Miller SD. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol 2009; 9:246–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ji Q, Perchellet A, Goverman JM. Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat Immunol 2010; 11:628–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Elliott JI, Altmann DM. Dual T cell receptor alpha chain T cells in autoimmunity. J Exp Med 1995; 182:953–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Padovan E, Casorati G, Dellabona P, Giachino C, Lanzavecchia A. Dual receptor T‐cells. Implications for alloreactivity and autoimmunity. Ann N Y Acad Sci 1995; 756: 66–70. [DOI] [PubMed] [Google Scholar]
- 20. Wu HJ, Wu E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012; 3:4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell 2012; 148:1258–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science 2012; 336:1268–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W et al . Host‐gut microbiota metabolic interactions. Science 2012; 336:1262–7. [DOI] [PubMed] [Google Scholar]
- 24. Musso G, Gambino R, Cassader M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu Rev Med 2011; 62:361–80. [DOI] [PubMed] [Google Scholar]
- 25. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U et al Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009; 139:485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med 2002; 347:911–20. [DOI] [PubMed] [Google Scholar]
- 27. Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR et al A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 2009; 15:1016–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C et al Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 2011; 479:538–41. [DOI] [PubMed] [Google Scholar]
- 29. Toivanen P. Normal intestinal microbiota in the aetiopathogenesis of rheumatoid arthritis. Ann Rheum Dis 2003; 62:807–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scher JU, Abramson SB. The microbiome and rheumatoid arthritis. Nat Rev Rheumatol 2011; 7:569–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mathis D, Benoist C. Microbiota and autoimmune disease: the hosted self. Cell Host Microbe 2011; 10:297–301. [DOI] [PubMed] [Google Scholar]
- 32. Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y et al Gut‐residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010; 32:815–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T‐cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 2011; 108:4615–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Helm RM, Burks AW. Animal models of food allergy. Curr Opin Allergy Clin Immunol 2002; 2:541–6. [DOI] [PubMed] [Google Scholar]
- 35. Fujimura KE, Rauch M, Matsui E, Iwai S, Calatroni A, Lynn H et al Development of a standardized approach for environmental microbiota investigations related to asthma development in children. J Microbiol Methods 2012; 91:231–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abrahamsson TR, Jakobsson HE, Andersson AF, Björkstén B, Engstrand L, Jenmalm MC. Low diversity of the gut microbiota in infants with atopic eczema. J Allergy Clin Immunol 129, 434–440, 440 e431‐432 (2012). [DOI] [PubMed] [Google Scholar]
- 37. Ismail IH, Oppedisano F, Joseph SJ, Boyle RJ, Licciardi PV, Robins‐Browne RM et al Reduced gut microbial diversity in early life is associated with later development of eczema but not atopy in high‐risk infants. Pediatr Allergy Immunol 2012; 23:674–81. [DOI] [PubMed] [Google Scholar]
- 38. Colpitts SL, Kasper LH. Influence of the gut microbiome on autoimmunity in the central nervous system. J Immunol 2017; 198:596–604. [DOI] [PubMed] [Google Scholar]
- 39. Budden KF, Gellatly SL, Wood DL, Cooper MA, Morrison M, Hugenholtz P et al Emerging pathogenic links between microbiota and the gut–lung axis. Nat Rev Microbiol 2017; 15:55–63. [DOI] [PubMed] [Google Scholar]
- 40. Dalal SR, Chang EB. The microbial basis of inflammatory bowel diseases. J Clin Invest 2014; 124:4190–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang M, Sun K, Wu Y, Yang Y, Tso P, Wu Z. Interactions between intestinal microbiota and host immune response in inflammatory bowel disease. Front Immunol 2017; 8:942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol 2018; 11:1–10. [DOI] [PubMed] [Google Scholar]
- 43. Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature 2016; 535:75–84. [DOI] [PubMed] [Google Scholar]
- 44. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol 2017; 17:219–232. [DOI] [PubMed] [Google Scholar]
- 45. Yurkovetskiy LA, Pickard JM, Chervonsky AV. Microbiota and autoimmunity: exploring new avenues. Cell Host Microbe 2015; 17:548–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Resta SC. Effects of probiotics and commensals on intestinal epithelial physiology: implications for nutrient handling. J Physiol 2009; 587:4169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol 2016; 16:341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D et al The Orphan G protein‐coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem 2003; 278:11312–19. [DOI] [PubMed] [Google Scholar]
- 49. Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME et al Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem 2003; 278:25481–9. [DOI] [PubMed] [Google Scholar]
- 50. Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK et al The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab 2011; 13:517–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Haghikia A, Jorg S, Duscha A, Berg J, Manzel A, Waschbisch A et al Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity 2015; 43:817–29. [DOI] [PubMed] [Google Scholar]
- 52. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D et al Commensal microbe‐derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013; 504:446–50. [DOI] [PubMed] [Google Scholar]
- 53. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D et al Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009; 461:1282–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Scheppach W, Sommer H, Kirchner T, Paganelli GM, Bartram P, Christl S et al Effect of butyrate enemas on the colonic mucosa in distal ulcerative colitis. Gastroenterology 1992; 103:51–6. [DOI] [PubMed] [Google Scholar]
- 55. Sun J, Furio L, Mecheri R, van der Does AM, Lundeberg E, Saveanu L et al Pancreatic β‐cells limit autoimmune diabetes via an immunoregulatory antimicrobial peptide expressed under the influence of the gut microbiota. Immunity 2015; 43:304–17. [DOI] [PubMed] [Google Scholar]
- 56. Asquith M, Davis S, Stauffer P, Michell C, Janowitz C, Lin P et al Intestinal metabolites are profoundly altered in the context of HLA‐B27 expression and functionally modulate disease in a rat model of spondyloarthritis. Arthritis Rheumatol 2017; 69:1984–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Koh A, De Vadder F, Kovatcheva‐Datchary P, Backhed F. From dietary fiber to host physiology: short‐chain fatty acids as key bacterial metabolites. Cell 2016; 165:1332–45. [DOI] [PubMed] [Google Scholar]
- 58. Whitt DD, Demoss RD. Effect of microflora on the free amino acid distribution in various regions of the mouse gastrointestinal tract. Appl Microbiol 1975; 30:609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dai Z, Wu G, Zhu W. Amino acid metabolism in intestinal bacteria: links between gut ecology and host health. Front Biosci 2011; 16:1768–86. [DOI] [PubMed] [Google Scholar]
- 60. Davila AM, Blachier F, Gotteland M, Andriamihaja M, Benetti PH, Sanz Y et al Intestinal luminal nitrogen metabolism: role of the gut microbiota and consequences for the host. Pharmacol Res 2013; 68:95–107. [DOI] [PubMed] [Google Scholar]
- 61. Allison C, Macfarlane GT. Influence of pH, nutrient availability, and growth rate on amine production by Bacteroides fragilis and Clostridium perfringens . Appl Environ Microbiol 1989; 55:2894–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McGaha TL, Huang L, Lemos H, Metz R, Mautino M, Prendergast GC et al Amino acid catabolism: a pivotal regulator of innate and adaptive immunity. Immunol Rev 2012; 249:135–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y et al Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011; 331:337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Matteoli G, Mazzini E, Iliev ID, Mileti E, Fallarino F, Puccetti P et al Gut CD103+ dendritic cells express indoleamine 2,3‐dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut 2010; 59:595–604. [DOI] [PubMed] [Google Scholar]
- 65. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol 2010; 185:3190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yan Y, Zhang GX, Gran B, Fallarino F, Yu S, Li H et al IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J Immunol 2010; 185:5953–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mucida D, Kutchukhidze N, Erazo A, Russo M, Lafaille JJ, Curotto de Lafaille MA. Oral tolerance in the absence of naturally occurring Tregs. J Clin Invest 2005; 115:1923–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sujino T, London M, Hoytema van Konijnenburg DP, Rendon T, Buch T, Silva HM et al Tissue adaptation of regulatory and intraepithelial CD4⁺ T cells controls gut inflammation. Science 2016; 352:1581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cervantes‐Barragan L, Chai JN, Tianero MD, Di Luccia B, Ahern PP, Merriman J et al Lactobacillus reuteri induces gut intraepithelial CD4+ CD8αα + T cells. Science 2017; 357:806–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Donia MS, Fischbach MA. Human microbiota. Small molecules from the human microbiota. Science 2015; 349:1254766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wikoff WR, Anfora AT, Liu JL, Schultz PG, Lesley SA, Peters EC et al Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA 2009; 106:3698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dodd D, Spitzer MH, Van Treuren W, Merrill BD, Hryckowian AJ, Higginbottom SK et al A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017; 551:648–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ciarlo E, Heinonen T, Herderschee J, Fenwick C, Mombelli M, Le Roy D et al Impact of the microbial derived short chain fatty acid propionate on host susceptibility to bacterial and fungal infections in vivo. Sci Rep 2016; 6:37944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Neis EP, Dejong CH, Rensen SS. The role of microbial amino acid metabolism in host metabolism. Nutrients 2015; 7:2930–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK et al Leucyl‐tRNA synthetase is an intracellular leucine sensor for the mTORC1‐signaling pathway. Cell 2012; 149:410–24. [DOI] [PubMed] [Google Scholar]
- 76. Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR et al Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016; 351:43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol 2012; 30:39–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Larange A, Cheroutre H. Retinoic acid and retinoic acid receptors as pleiotropic modulators of the immune system. Annu Rev Immunol 2016; 34:369–94. [DOI] [PubMed] [Google Scholar]
- 79. Pino‐Lagos K, Benson MJ, Noelle RJ. Retinoic acid in the immune system. Ann N Y Acad Sci 2008; 1143:170–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Benson MJ, Pino‐Lagos K, Rosemblatt M, Noelle RJ. All‐trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co‐stimulation. J Exp Med 2007; 204:1765–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang C, Kang SG, HogenEsch H, Love PE, Kim CH. Retinoic acid determines the precise tissue tropism of inflammatory Th17 cells in the intestine. J Immunol 2010; 184:5519–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut‐homing specificity on T cells. Immunity 2004; 21:527–38. [DOI] [PubMed] [Google Scholar]
- 83. Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M et al Selective imprinting of gut‐homing T cells by Peyer's patch dendritic cells. Nature 2003; 424:88–93. [DOI] [PubMed] [Google Scholar]
- 84. Wang S, Villablanca EJ, De Calisto J, Gomes DC, Nguyen DD, Mizoguchi E et al MyD88‐dependent TLR1/2 signals educate dendritic cells with gut‐specific imprinting properties. J Immunol 2011; 187:141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kagechika H. Novel synthetic retinoids and separation of the pleiotropic retinoidal activities. Curr Med Chem 2002; 9:591–608. [DOI] [PubMed] [Google Scholar]
- 86. Mic FA, Molotkov A, Benbrook DM, Duester G. Retinoid activation of retinoic acid receptor but not retinoid X receptor is sufficient to rescue lethal defect in retinoic acid synthesis. Proc Natl Acad Sci USA 2003; 100:7135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ohyanagi N, Ishido M, Suzuki F, Kaneko K, Kubota T, Miyasaka N et al Retinoid ameliorates experimental autoimmune myositis, with modulation of Th cell differentiation and antibody production in vivo . Arthritis Rheum 2009; 60:3118–27. [DOI] [PubMed] [Google Scholar]
- 88. Klemann C, Raveney BJ, Klemann AK, Ozawa T, von Horsten S, Shudo K et al Synthetic retinoid AM80 inhibits Th17 cells and ameliorates experimental autoimmune encephalomyelitis. Am J Pathol 2009; 174:2234–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kwok SK, Park MK, Cho ML, Oh HJ, Park EM, Lee DG et al Retinoic acid attenuates rheumatoid inflammation in mice. J Immunol 2012; 189:1062–71. [DOI] [PubMed] [Google Scholar]
- 90. Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B et al Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF‐β‐driven Smad3 signaling and inhibiting IL‐6 and IL‐23 receptor expression. J Immunol 2008; 181:2277–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Naskar D, Teng F, Felix KM, Bradley CP, Wu HJ. Synthetic retinoid AM80 ameliorates lung and arthritic autoimmune responses by inhibiting T follicular helper and Th17 cell responses. J Immunol 2017; 198:1855–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhang Z, Li J, Zheng W, Zhao G, Zhang H, Wang X et al Peripheral lymphoid volume expansion and maintenance are controlled by gut microbiota via RALDH+ dendritic cells. Immunity 2016; 44:330–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Panoutsakopoulou V, Sanchirico ME, Huster KM, Jansson M, Granucci F, Shim DJ et al Analysis of the relationship between viral infection and autoimmune disease. Immunity 2001; 15:137–47. [DOI] [PubMed] [Google Scholar]
- 94. Rosen SD. Ligands for l‐selectin: homing, inflammation, and beyond. Annu Rev Immunol 2004; 22:129–56. [DOI] [PubMed] [Google Scholar]
- 95. Mebius RE, Streeter PR, Michie S, Butcher EC, Weissman IL. A developmental switch in lymphocyte homing receptor and endothelial vascular addressin expression regulates lymphocyte homing and permits CD4+ CD3– cells to colonize lymph nodes. Proc Natl Acad Sci USA 1996; 93:11019–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N et al Peripheral education of the immune system by colonic commensal microbiota. Nature 2011; 478:250–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L. Origin and T cell receptor diversity of Foxp3+ CD4+ CD25+ T cells. Immunity 2006; 25:249–59. [DOI] [PubMed] [Google Scholar]
- 98. Cebula A, Seweryn M, Rempala GA, Pabla SS, McIndoe RA, Denning TL et al Thymus‐derived regulatory T cells contribute to tolerance to commensal microbiota. Nature 2013; 497:258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kim M, Galan C, Hill AA, Wu WJ, Fehlner‐Peach H, Song HW et al Critical role for the microbiota in CX3CR1+ intestinal mononuclear phagocyte regulation of intestinal T cell responses. Immunity 2018; 49:151–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Bradley CP, Teng F, Felix KM, Sano T, Naskar D, Block KE et al Segmented filamentous bacteria provoke lung autoimmunity by inducing gut‐lung axis Th17 cells expressing dual TCRs. Cell Host Microbe 2017; 22:697–704 e694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Teng F, Klinger CN, Felix KM, Bradley CP, Wu E, Tran NL et al Gut microbiota drive autoimmune arthritis by promoting differentiation and migration of Peyer's patch T follicular helper cells. Immunity 2016; 44:875–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Dutzan N, Abusleme L, Bridgeman H, Greenwell‐Wild T, Zangerle‐Murray T, Fife ME et al On‐going mechanical damage from mastication drives homeostatic Th17 cell responses at the oral barrier. Immunity 2017; 46:133–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Dutzan N, Kajikawa T, Abusleme L, Greenwell‐Wild T, Zuazo CE, Ikeuchi T et al A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med 2018; 10:eaat0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Teng F, Felix KM, Bradley CP, Naskar D, Ma H, Raslan WA et al The impact of age and gut microbiota on Th17 and Tfh cells in K/BxN autoimmune arthritis. Arthritis Res Ther 2017; 19:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Xu M, Porkrovskii M, Ding Y, Yi R, Au C, Harrison OJ et al c‐MAF‐dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 2018; 554:373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci USA 2015; 112:7061–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human αβ T cell receptor diversity. Science 1999; 286:958–61. [DOI] [PubMed] [Google Scholar]
- 108. Casrouge A, Beaudoing E, Dalle S, Pannetier C, Kanellopoulos J, Kourilsky P. Size estimate of the TCR repertoire of naive mouse splenocytes. J Immunol 2000; 164:5782–7. [DOI] [PubMed] [Google Scholar]
- 109. Petrova G, Ferrante A, Gorski J. Cross‐reactivity of T cells and its role in the immune system. Crit Rev Immunol 2013; 32:349–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Horai R, Zarate‐Blades CR, Dillenburg‐Pilla P, Chen J, Kielczewski JL, Silver PB et al Microbiota‐dependent activation of an autoreactive T cell receptor provokes autoimmunity in an immunologically privileged site. Immunity 2015; 43:343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wang H, Kadlecek TA, Au‐Yeung BB, Goodfellow HE, Hsu LY, Freedman TS et al ZAP‐70: an essential kinase in T‐cell signaling. Cold Spring Harb Perspect Biol 2010; 2:a002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Courtney AH, Lo WL, Weiss A. TCR signaling: mechanisms of initiation and propagation. Trends Biochem Sci 2018; 43:108–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Tai N, Peng J, Liu F, Gulden E, Hu Y, Zhang X et al Microbial antigen mimics activate diabetogenic CD8 T cells in NOD mice. J Exp Med 2016; 213:2129–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Greiling TM, Dehner C, Chen X, Hughes K, Iñiguez AJ, Boccitto M et al Commensal orthologs of the human autoantigen Ro60 as triggers of autoimmunity in lupus. Sci Transl Med 2018; 10:eaan2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Paterson RK, Bluethmann H, Tseng P, Dunlap A, Finkel TH. Development and function of autospecific dual TCR T lymphocytes. Int Immunol 1999; 11:113–9. [DOI] [PubMed] [Google Scholar]
- 116. Auger JL, Haasken S, Steinert EM, Binstadt BA. Incomplete TCR‐β allelic exclusion accelerates spontaneous autoimmune arthritis in K/BxN TCR transgenic mice. Eur J Immunol 2012; 42:2354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Balakrishnan A, Morris GP. The highly alloreactive nature of dual TCR T cells. Curr Opin Organ Transplant 2016; 21:22–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG et al Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 2014; 40:594–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL et al Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature 2014; 510:152–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Dominianni C, Sinha R, Goedert JJ, Pei Z, Yang L, Hayes RB et al Sex, body mass index, and dietary fiber intake influence the human gut microbiome. PLoS ONE 2015; 10:e0124599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL et al The long‐term stability of the human gut microbiota. Science 2013; 341:1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL et al Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013; 341:1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol 2009; 9:393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Chen J, Chia N, Kalari KR, Yao JZ, Novotna M, Paz Soldan MM et al Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci Rep 2016; 6:28484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Pröbstel A‐K, Baranzini SE. The role of the gut microbiome in multiple sclerosis risk and progression: towards characterization of the “MS Microbiome”. Neurotherapeutics 2017; 15:126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Shahi SK, Freedman SN, Mangalam AK. Gut microbiome in multiple sclerosis: the players involved and the roles they play. Gut Microbes 2017; 8:607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Jangi S, Gandhi R, Cox LM, Li N, von Glehn F, Yan R et al Alterations of the human gut microbiome in multiple sclerosis. Nat Commun 2016; 7:12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Gevers D, Kugathasan S, Denson LA, Vazquez‐Baeza Y, Van Treuren W, Ren B et al The treatment‐naive microbiome in new‐onset Crohn's disease. Cell Host Microbe 2014; 15:382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA et al Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci USA 2017; 114:10713–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Berer K, Gerdes LA, Cekanaviciute E, Jia X, Xiao L, Xia Z et al Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci USA 2017; 114:10719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Shaw M, Collins BF, Ho LA, Raghu G. Rheumatoid arthritis‐associated lung disease. Eur Respir Rev 2015; 24:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kim EJ, Collard HR, King TE Jr. Rheumatoid arthritis‐associated interstitial lung disease: the relevance of histopathologic and radiographic pattern. Chest 2009; 136:1397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Crowson CS, Liao KP, Davis JM, Solomon DH, Matteson EL, Knutson KL et al Rheumatoid arthritis and cardiovascular disease. Am Heart J 2013;166:622–628 e621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Pekin TJ Jr, Zvaifler NJ. Hemolytic complement in synovial fluid. J Clin Investig 1964; 43:1372–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Ruddy S, Austen KF. The complement system in rheumatoid synovitis. I. An analysis of complement component activities in rheumatoid synovial fluids. Arthritis Rheum 1970; 13:713–23. [DOI] [PubMed] [Google Scholar]
- 136. Britton MC, Schur PH. The complement system in rheumatoid synovitis II. Intracytoplasmic inclusions of immunoglobulins and complement. Arthritis Rheum 1971; 14:87–95. [DOI] [PubMed] [Google Scholar]
- 137. Olsen NJ, Ho E, Barats L. Clinical correlations with serum C1q levels in patients with rheumatoid arthritis. Arthritis Rheum 1991; 34:187–91. [DOI] [PubMed] [Google Scholar]
- 138. Mathsson L, Lampa J, Mullazehi M, Ronnelid J. Immune complexes from rheumatoid arthritis synovial fluid induce FcγRIIa dependent and rheumatoid factor correlated production of tumour necrosis factor‐α by peripheral blood mononuclear cells. Arthritis Res Ther 2006; 8:R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Halla JT, Volanakis JE, Schrohenloher RE. Immune complexes in rheumatoid arthritis sera and synovial fluids: a comparison of three methods. Arthritis Rheum 1979; 22:440–8. [DOI] [PubMed] [Google Scholar]
- 140. Ge C, Tong D, Liang B, Lönnblom E, Schneider N, Hagert C et al Anti‐citrullinated protein antibodies cause arthritis by cross‐reactivity to joint cartilage. JCI Insight 2017; 2:93688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Harre U, Georgess D, Bang H, Bozec A, Axmann R, Ossipova E et al Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Investig 2012; 122:1791–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol 2011; 29:621–63. [DOI] [PubMed] [Google Scholar]
- 143. Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med 2012; 209:1241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. King C, Tangye SG, Mackay CR. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu Rev Immunol 2008; 26:741–66. [DOI] [PubMed] [Google Scholar]
- 145. Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, Cannons JL et al Follicular helper T cells are required for systemic autoimmunity. J Exp Med 2009; 206:561–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol 2012; 8:337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Nguyen V, Luzina I, Rus H, Tegla C, Chen C, Rus V. IL‐21 promotes lupus‐like disease in chronic graft‐versus‐host disease through both CD4 T cell‐ and B cell‐intrinsic mechanisms. J Immunol 2012; 189:1081–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. DiPlacido LD, Craft J. Emerging from the shadows: follicular helper T cells in autoimmunity. Arthritis Rheum 2010; 62:6–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol 2005; 5:853–65. [DOI] [PubMed] [Google Scholar]
- 150. Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D et al The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med 2015; 21:895–905. [DOI] [PubMed] [Google Scholar]
- 151. Jubair WK, Hendrickson JD, Severs EL, Schulz HM, Adhikari S, Ir D et al Modulation of inflammatory arthritis in mice by gut microbiota through mucosal inflammation and autoantibody generation. Arthritis Rheumatol 2018; 70:1220–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Mak A, Kow NY. The pathology of T cells in systemic lupus erythematosus. J Immunol Res 2014; 2014:419029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Hevia A, Milani C, Lopez P, Cuervo A, Arboleya S, Duranti S et al Intestinal dysbiosis associated with systemic lupus erythematosus. MBio 2014; 5:e01548–01514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Lopez P, de Paz B, Rodriguez‐Carrio J, Hevia A, Sanchez B, Margolles A et al Th17 responses and natural IgM antibodies are related to gut microbiota composition in systemic lupus erythematosus patients. Sci Rep 2016; 6:24072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Luo XM, Edwards MR, Mu Q, Yu Y, Vieson MD, Reilly CM et al Gut microbiota in human systemic lupus erythematosus and a mouse model of lupus. Appl Environ Microbiol 2018; 84:e02288–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Krebs CF, Paust HJ, Krohn S, Koyro T, Brix SR, Riedel JH et al Autoimmune renal disease is exacerbated by S1P‐receptor‐1‐dependent intestinal Th17 cell migration to the kidney. Immunity 2016; 45:1078–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Moran B, Sweeney CM, Hughes R, Malara A, Kirthi S, Tobin AM et al Hidradenitis suppurativa is characterized by dysregulation of the Th17: Treg cell axis, which is corrected by Anti‐TNF therapy. J Invest Dermatol 2017; 137:2389–95. [DOI] [PubMed] [Google Scholar]
- 158. Principi M, Cassano N, Contaldo A, Iannone A, Losurdo G, Barone M et al Hydradenitis suppurativa and inflammatory bowel disease: an unusual, but existing association. World J Gastroenterol 2016; 22:4802–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ring HC, Thorsen J, Saunte DM, Lilje B, Bay L, Riis PT et al The follicular skin microbiome in patients with hidradenitis suppurativa and healthy controls. JAMA Dermatol 2017; 153:897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010; 464:1293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Bosi E, Molteni L, Radaelli MG, Folini L, Fermo I, Bazzigaluppi E et al Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia 2006; 49:2824–7. [DOI] [PubMed] [Google Scholar]
- 162. Kostic AD, Gevers D, Siljander H, Vatanen T, Hyotylainen T, Hamalainen AM et al The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015; 17:260–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Costa FR, Francozo MC, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS et al Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med 2016; 213:1223–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Hanninen A, Nurmela R, Maksimow M, Heino J, Jalkanen S, Kurts C. Islet β‐cell‐specific T cells can use different homing mechanisms to infiltrate and destroy pancreatic islets. Am J Pathol 2007; 170:240–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Ryan GA, Wang CJ, Chamberlain JL, Attridge K, Schmidt EM, Kenefeck R et al B1 cells promote pancreas infiltration by autoreactive T cells. J Immunol 2010; 185:2800–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, Mathis D. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci USA 2011; 108:11548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Yurkovetskiy L, Burrows M, Khan AA, Graham L, Volchkov P, Becker L et al Gender bias in autoimmunity is influenced by microbiota. Immunity 2013; 39:400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Ditzel HJ. The K/BxN mouse: a model of human inflammatory arthritis. Trends Mol Med 2004; 10:40–5. [DOI] [PubMed] [Google Scholar]
- 169. Urich E, Gutcher I, Prinz M, Becher B. Autoantibody‐mediated demyelination depends on complement activation but not activatory Fc‐receptors. Proc Natl Acad Sci USA 2006; 103:18697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Lyons JA, Ramsbottom MJ, Cross AH. Critical role of antigen‐specific antibody in experimental autoimmune encephalomyelitis induced by recombinant myelin oligodendrocyte glycoprotein. Eur J Immunol 2002; 32:1905–13. [DOI] [PubMed] [Google Scholar]
- 171. Wong FS, Wen L, Tang M, Ramanathan M, Visintin I, Daugherty J et al Investigation of the role of B‐cells in type 1 diabetes in the NOD mouse. Diabetes 2004; 53:2581–7. [DOI] [PubMed] [Google Scholar]
- 172. Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen‐presenting cells for the initiation of T cell‐mediated autoimmune diabetes in nonobese diabetic mice. J Immunol 1998; 161:3912–8. [PubMed] [Google Scholar]
- 173. Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY et al Control of TH17 cells occurs in the small intestine. Nature 2011; 475:514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A et al Induction and molecular signature of pathogenic TH17 cells. Nat Immunol 2012; 13:991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE et al Generation of pathogenic TH17 cells in the absence of TGF‐β signalling. Nature 2010; 467:967–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y et al Foxp3+ T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 2014; 41:152–65. [DOI] [PubMed] [Google Scholar]
- 177. Padovan E, Casorati G, Dellabona P, Meyer S, Brockhaus M, Lanzavecchia A. Expression of two T cell receptor α chains: dual receptor T cells. Science 1993; 262:422–4. [DOI] [PubMed] [Google Scholar]
- 178. Heath WR, Carbone FR, Bertolino P, Kelly J, Cose S, Miller JF. Expression of two T cell receptor α chains on the surface of normal murine T cells. Eur J Immunol 1995; 25:1617–23. [DOI] [PubMed] [Google Scholar]
- 179. Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA et al Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 2016; 532:512–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Bindels LB, Delzenne NM, Cani PD, Walter J. Towards a more comprehensive concept for prebiotics. Nat Rev Gastroenterol Hepatol 2015; 12:303–10. [DOI] [PubMed] [Google Scholar]
- 181. Maldonado‐Gomez MX, Martínez I, Bottacini F, O'Callaghan A, Ventura M, van Sinderen D et al Stable engraftment of Bifidobacterium longum AH1206 in the human gut depends on individualized features of the resident microbiome. Cell Host Microbe 2016; 20:515–26. [DOI] [PubMed] [Google Scholar]
- 182. Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG et al IgA‐coated E. coli enriched in Crohn's disease spondyloarthritis promote TH17‐dependent inflammation. Sci Transl Med 2017; 9:eaaf9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Hamilton MJ, Weingarden AR, Sadowsky MJ, Khoruts A. Standardized frozen preparation for transplantation of fecal microbiota for recurrent Clostridium difficile infection. Am J Gastroenterol 2012; 107:761–7. [DOI] [PubMed] [Google Scholar]
- 184. Kang DW, Adams JB, Gregory AC, Borody T, Chittick L, Fasano A et al Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: an open‐label study. Microbiome 2017; 5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Ott SJ, Waetzig GH, Rehman A, Moltzau‐Anderson J, Bharti R, Grasis JA et al Efficacy of sterile fecal filtrate transfer for treating patients with Clostridium difficile infection. Gastroenterology 2017; 152:799–811 e797. [DOI] [PubMed] [Google Scholar]
- 186. den Besten G, Bleeker A, Gerding A, van Eunen K, Havinga R, van Dijk TH et al Short‐chain fatty acids protect against high‐fat diet‐induced obesity via a PPARγ‐dependent switch from lipogenesis to fat oxidation. Diabetes 2015; 64:2398–408. [DOI] [PubMed] [Google Scholar]