

Abstract
Pathogenic bacteria must rapidly adapt to ever‐changing environmental signals resulting in metabolism remodeling. The carbon catabolite repression, mediated by the catabolite control protein A (CcpA), is used to express genes involved in utilization and metabolism of the preferred carbon source. Here, we have identified RsaI as a CcpA‐repressed small non‐coding RNA that is inhibited by high glucose concentrations. When glucose is consumed, RsaI represses translation initiation of mRNAs encoding a permease of glucose uptake and the FN3K enzyme that protects proteins against damage caused by high glucose concentrations. RsaI also binds to the 3′ untranslated region of icaR mRNA encoding the transcriptional repressor of exopolysaccharide production and to sRNAs induced by the uptake of glucose‐6 phosphate or nitric oxide. Furthermore, RsaI expression is accompanied by a decreased transcription of genes involved in carbon catabolism pathway and an activation of genes involved in energy production, fermentation, and nitric oxide detoxification. This multifaceted RNA can be considered as a metabolic signature when glucose becomes scarce and growth is arrested.
Keywords: carbon metabolism, catabolite control protein A, pathogenic bacteria, regulatory RNAs, sRNA, translational regulation
Subject Categories: Metabolism; Microbiology, Virology & Host Pathogen Interaction; RNA Biology
Introduction
All bacteria require a carbon source, providing energy for their growth and for the synthesis of all macromolecules. Besides, pathogenic bacteria during the host infectious process must cope with hostile conditions such as nutrient deficiency, temperature, and oxidative and osmotic shocks, and must overcome innate immune responses. For instance, Staphylococcus aureus uses carbohydrates to grow under high nitric oxide (NO) and anaerobiosis (Vitko et al, 2016). To survive in these complex environments and to counteract the host defense, S. aureus produces a plethora of virulence factors. The synthesis of these factors is fine‐tuned by intricate interactions between multiple regulators involving both proteins and RNAs (Novick, 2003). Additionally, biosynthetic intermediates, generated from the central metabolism of S. aureus, have strong impacts on the synthesis of virulence factors. Besides, several metabolite‐sensing regulatory proteins (CcpA, CodY, Rex, and RpiR) act as key regulatory factors to coordinate the synthesis of genes involved in metabolic pathways, in stress responses, and in pathogenesis (Somerville & Proctor, 2009; Richardson et al, 2015). Through the adaptation of the metabolism of the bacteria to specific host microenvironment, these proteins contribute to S. aureus pathogenesis (Richardson et al, 2015).
Among these proteins, the catabolite control protein A (CcpA) acts as the main global regulator of carbon catabolite repression, allowing the bacteria to use the preferred carbon source (i.e., glucose) in a hierarchical manner (Seidl et al, 2008a, 2009). Catabolite control protein A belongs to the LacI repressor family and binds to a specific DNA sequence, called the cre (catabolite‐responsive element) sequence, which is conserved in many Gram‐positive bacteria. Transcription of CcpA is constitutive, and the protein is activated through the binding of its co‐regulator histidine‐containing phosphocarrier protein HPr in the presence of glucose. Inactivation of ccpA gene decreases transiently the growth rate and yield as long as glucose is present in the medium (Seidl et al, 2006) and affects the expression of a large number of metabolic encoding genes in a glucose‐dependent and glucose‐independent manner (Seidl et al, 2008a). Additionally, CcpA has a strong impact on the expression of S. aureus virulon. It enhances the yield of the quorum‐sensing‐induced RNAIII, which represses protein A and various adhesion factors at the post‐transcriptional level, and conversely activates the synthesis of many exotoxins. However, CcpA also modulates the transcription of mRNAs encoding protein A, α‐hemolysin (hla), and toxic shock syndrome toxin (TSST; Seidl et al, 2006, 2008b), represses capsule formation, and activates biofilm formation in a glucose‐rich environment (Seidl et al, 2008a). Indeed, a S. aureus ccpA deletion mutant strain was less pathogenic than the wild‐type strain in a murine abscess model (Li et al, 2010) and ccpA inactivation increased the susceptibility of hyperglycemic animals to acute pneumonia infections (Bischoff et al, 2017). Nevertheless, the mechanism by which CcpA affected S. aureus pathogenesis cannot be simply resumed as a modulation of the RNAIII‐dependent regulatory networks. Therefore, it has been suggested that CcpA can also act indirectly on gene expression through the action of other regulatory proteins or small non‐coding RNAs (sRNAs; Somerville & Proctor, 2009; Richardson et al, 2015).
In Enterobacteriaceae, several sRNAs have been shown as key actors of the uptake and the metabolism of carbohydrates (reviewed in Bobrovskyy & Vanderpool, 2016). For instance, they participate in the regulation of the galactose operon and carbon catabolite repression, metabolism of amino acids, and contribute to bacterial survival during phospho‐sugar stress. The importance of sRNAs in regulatory networks is now well recognized to rapidly adjust cell growth to various stresses and changes in the environment. Thus, they are obvious candidates creating the links between virulence and metabolism. One example is RsaE, a sRNA conserved in S. aureus and Bacillus subtilis, that controls enzymes involved in the TCA cycle (Geissmann et al, 2009; Bohn et al, 2010) and in arginine degradation pathway (Rochat et al, 2018). In B. subtilis, the transcriptional repressor ResD represses RoxS (homologous to S. aureus RsaE) to readjust the pool of NAD+/NADH in responses to various stress and stimuli (Durand et al, 2017). Its promoter is also highly conserved among Staphylococceae and is recognized by the orthologous response regulator SrrA in S. aureus. Responding to reactive oxygen species through SrrAB, S. aureus RsaE may also intervene in the survival of cells against host immune reactions (Durand et al, 2015, 2017).
Here, we have identified a signaling pathway responding to glucose uptake, which involves a sRNA, called RsaI (or RsaOG). This 144 nucleotides‐long sRNA is highly conserved among Staphylococcacea and carries two conserved regions including two G‐track sequences and a long unpaired interhelical region rich in pyrimidines (Geissmann et al, 2009; Marchais et al, 2010). The expression of RsaI is observed exclusively at the stationary phase of growth in rich medium (Geissmann et al, 2009) and is enhanced after vancomycin exposure (Howden et al, 2013). In this study, we revealed that CcpA is the main repressor of RsaI expression in the presence of glucose and that this inhibition is alleviated after the utilization of glucose. The identification of the targetome of RsaI using the MS2‐affinity purification approach coupled with RNA sequencing (MAPS) unexpectedly showed two classes of RNA targets, including mRNAs involved in glucose uptake, sugar metabolism, and biofilm formation, as well as various sRNAs. Using site‐directed mutagenesis, we identified two distinct and functional regions of RsaI. All in all, our data showed the existence of two sRNAs involved in essential pathways responding to either glucose or glucose‐6 phosphate uptake, with RsaI acting as a signature for a metabolic switch when the preferred carbon source is metabolized.
We will discuss the importance of sRNA‐mediated regulation in S. aureus to fine tune the expression of genes according to essential nutrient availability and their possible consequences on metabolism adaptation and virulence.
Results
The expression of RsaI is inhibited by glucose and by the catabolite control protein A
We have previously shown that the synthesis of RsaI is high at the stationary phase of growth in BHI medium while its expression was constitutive in MHB medium (Geissmann et al, 2009). These data suggested that RsaI expression is regulated in a manner dependent on nutrient or biosynthetic intermediate availability. A major difference between BHI and MHB composition is their carbon source, glucose, and starch, respectively. We wondered whether the expression of RsaI might be dependent on the available carbon source. For this purpose, Northern blot experiments were performed on total RNAs prepared from HG001 (wild‐type) strain grown in MHB medium at various time points (Fig 1A). In MHB, where the glucose is not immediately available as the carbon source, RsaI was constitutively and highly expressed (Fig 1A). Conversely, when glucose was added, either at the beginning of the culture or after 3 h of growth, the steady‐state level of RsaI was immediately dropped, indicating the repressing effect of this sugar (Fig 1A).
Figure 1. RsaI responds to glucose through the transcriptional factor CcpA.
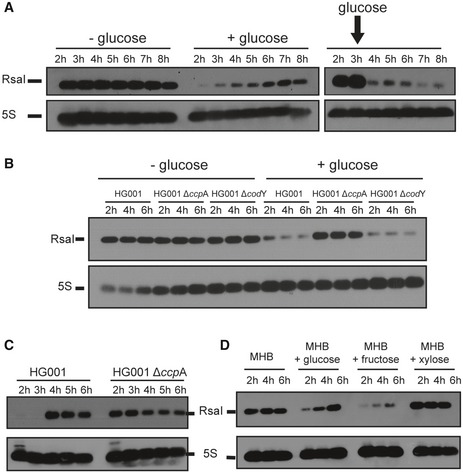
- Northern blot experiments show the expression of RsaI during growth phase in the HG001 strain in MHB medium with or without the addition of 1.5 g/l of d‐glucose. Glucose was added either at the beginning of growth (+ glucose, left panel) or after 3 h of growth (right panel).
- Northern blot experiment shows the expression of RsaI during growth phase in the HG001, ∆ccpA mutant strain, and ∆codY mutant strain, in MHB medium with (+) or without (−) the addition of 1.5 g/l of d‐glucose.
- Northern blot experiment shows the expression of RsaI in the HG001 and ∆ccpA mutant strains. Total RNA was prepared after 2, 3, 4, 5, and 6 h of culture in BHI medium at 37°C.
- Northern blot analysis of RsaI in the HG001 strain grown in MHB medium with or without the addition of 1 g/l of glucose, fructose, or xylose. For all the experiments, loading controls were done using the expression of 5S rRNA (5S) as revealed after hybridization of the membranes with a specific probe. However for these controls, we used aliquots of the same RNA preparations, but the migration of the samples was performed in parallel to the experiments on a separate agarose gel because RsaI and 5S rRNA have very similar sizes.
Source data are available online for this figure.
As CcpA sensed the intracellular concentration of glucose (Seidl et al, 2006), we analyzed whether the expression of RsaI was CcpA‐dependent. Northern blot analysis was performed on total RNA extracts prepared from HG001 strain and an isogenic mutant deleted of ccpA gene (∆ccpA). In parallel, we have also tested CodY, another global regulator of metabolism and virulence, which was shown as a direct regulator of amino acid biosynthesis and transport of macromolecules (Pohl et al, 2009; Majerczyk et al, 2010). The data showed that in the absence of glucose, the yield of RsaI was similar in all strains. However, in MHB medium supplemented with glucose, the yield of RsaI declined dramatically in the WT and ∆codY strains, whereas it was still high in ∆ccpA strain (Fig 1B). In BHI medium, RsaI was expressed after 4 h of growth in the WT strain, while its expression became constitutive in the mutant ∆ccpA strain (Figs 1C and EV1A).
Figure EV1. Analysis of the RsaI expression.
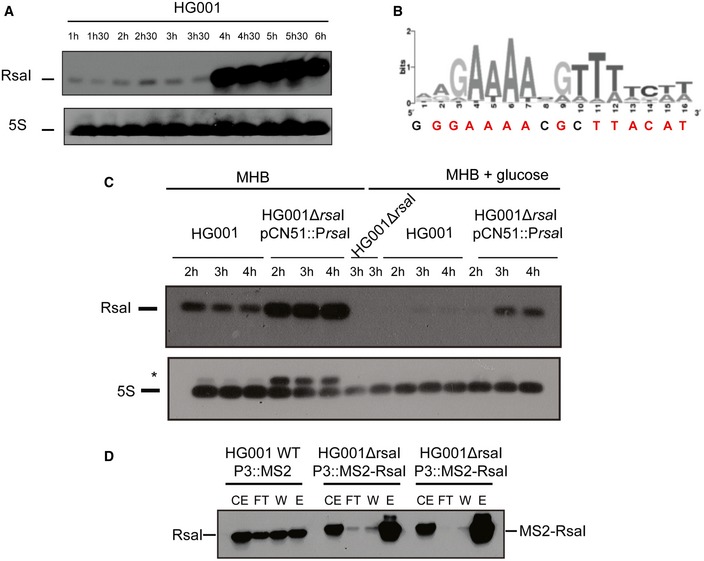
- Northern blot experiments showing the expression of RsaI in HG001 strain during growth phase. Total RNA was prepared from samples taken after various time points of growth in BHI medium at 37°C. Hybridization against 5S rRNA was used as loading control on the same samples which have been migrated on another agarose gel because RsaI and 5S rRNA have very similar sizes.
- The cre‐site consensus was defined in Bacillus cereus (van der Voort et al, 2008). The sequence found in the 5′ region of RsaI is represented below the graph, the conserved residues are in red.
- Northern blot experiments showing the expression of RsaI in HG001 strain, the isogenic HG001∆rsaI mutant strain, and the same mutant strain complemented with a plasmid expressing RsaI under the control of its own promoter (HG001∆rsaI pCN51::PrsaI). Total RNA was prepared from samples taken after various time points of growth in MHB medium in the absence or presence of 1% glucose. For the mutant HG001∆rsaI strain (used as a negative control), total RNA was prepared after 4 h of growth in MHB medium in the absence or in the presence of glucose. Hybridization against 5S rRNA was used as loading control on the same membrane. *Traces of RsaI signal after re‐hybridization of the membrane with the 5S probe were still observed.
- MS2‐RsaI is specifically retained by affinity chromatography containing the MS2‐MBP protein. The Northern blot was performed using a DIG‐labeled RsaI probe (Table EV3) to visualize RsaI and MS2‐RsaI following the MS2 chromatography affinity. CE is for crude extract, FT for flow‐through, W for washing, and E for elution. For CE/FT/W samples, 5 μg of total RNA was loaded on a 1.5% agarose gel while for E sample, only 0.5 μg of total RNA was used. The two replicates are shown for the RNA purified from the strain HG001ΔrsaI::MS2‐RsaI.
Source data are available online for this figure.
We also analyzed the expression of RsaI in MHB medium supplemented with various sugars such as fructose and xylose (Fig 1D). The data showed that expression of RsaI was very low at the beginning of growth in MHB supplemented with either glucose or fructose but not with xylose. These data show that CcpA senses both glucose and fructose (Fig 1).
Overall, we showed that CcpA represses RsaI expression in the presence of glucose. In accordance with this observation, a conserved cre (GGAAAcGcTTACAT) sequence was found at position –30 upstream the transcriptional start site of RsaI (Fig EV1B). This region was sufficient to confer repression by glucose in the complemented strain containing pCN51::PrsaI (Fig EV1C).
The targetome of RsaI as revealed by the MAPS approach
The MAPS approach (“MS2 affinity purification coupled with RNA sequencing”) was used to purify in vivo regulatory complexes involving RsaI. MAPS has been successful to identify the RNA targets of any sRNAs in Escherichia coli (Lalaouna et al, 2015) and more recently of RsaA sRNA in S. aureus (Tomasini et al, 2017). Briefly, the MS2‐tagged version of RsaI was expressed from a plasmid under the control of the agr‐dependent P3 promoter, allowing an accumulation of RsaI at the stationary phase of growth in the ∆rsaI mutant strain. RsaI was detected by Northern blot using total RNAs extracted at 2, 4, and 6 h of growth in BHI medium. Using a DIG‐labeled RsaI probe, we showed that the steady‐state levels of MS2‐RsaI were very similar to the wild‐type (WT) RsaI and that MS2‐RsaI was specifically and strongly retained by the MS2‐MBP fusion protein after the affinity chromatography (Fig EV1D). The RNAs were then extracted from the eluted fraction to be sequenced. The data were analyzed using Galaxy (Afgan et al, 2016), and the sequencing reads were mapped, counted per feature, and normalized using the HG001 genome as reference (Herbert et al, 2010; Caldelari et al, 2017). The enrichment of putative targets was measured by comparing the number of reads obtained from the MS2‐RsaI purification and the MS2 alone as control. Since the MS2 tag alone was produced in the WT HG001 background, the untagged RsaI was also expressed under the conditions of growth but was poorly retained on the affinity chromatography (Fig EV1D). The data were sorted by a decreasing fold change. In the following study, we have considered as RsaI targets, the RNAs that were enriched at least fourfold and were reproducibly detected in two independent experiments (Tables EV1 and EV2).
Two classes of RNAs were co‐purified with RsaI, including mRNAs and sRNAs. On the one hand, the most enriched mRNA encodes IcaR, the repressor of the icaADBC operon, which encodes the enzymes required for the synthesis of the poly‐β‐1,6‐N‐acetylglucosamine polymer (PIA‐PNAG), the main staphylococcal exopolysaccharide constituting biofilms. In addition, several mRNAs encoding proteins linked to sugar utilization and transport, such as glcU_2 encoding a major transporter of glucose, and fn3K encoding fructosamine 3‐kinase were significantly enriched. It is noteworthy that other less enriched mRNAs (< 4‐fold) encode proteins related to sugar metabolism such as the trehalose‐specific PTS transporter (TreB), a sugar phosphatase (YidA), and a maltose transport system permease (Table EV2). Finally, in addition to icaR, several mRNAs express transcriptional regulators (Xre type, the maltose regulatory protein GlvR, SlyA, SigS). On the other hand, we found the RsaD, RsaE, and RsaG sRNAs as putative RsaI targets (Table EV1), which all contained at least one conserved C‐rich motif (Geissmann et al, 2009). RsaH could be also included in this group though it was less enriched (< 4‐fold; Table EV2).
The MAPS data suggested that RsaI is involved in networks of RNA pairings.
RsaI contains two distinct regulatory domains
Following the MAPS experiments, we searched for intermolecular base‐pairing interactions between RsaI and its potential RNA targets using IntaRNA. Stable interactions were predicted for most of the enriched RNAs (Table EV1). The CU‐rich unpaired region of RsaI was predicted to form base‐pairings with most of the mRNAs. They were located close or at the ribosome‐binding site of most of the mRNAs except for icaR and isaA, which involves nucleotides in their 3′ untranslated regions (Table EV1). A second domain of interaction corresponds to the G‐track sequences located in the first hairpin domain of RsaI and the C‐rich sequences of the sRNAs RsaD, RsaE, RsaG, and RsaH (Fig 2A).
Figure 2. RsaI binds to icaR, glcU_2, fn3K mRNA, and the sRNA RsaG.
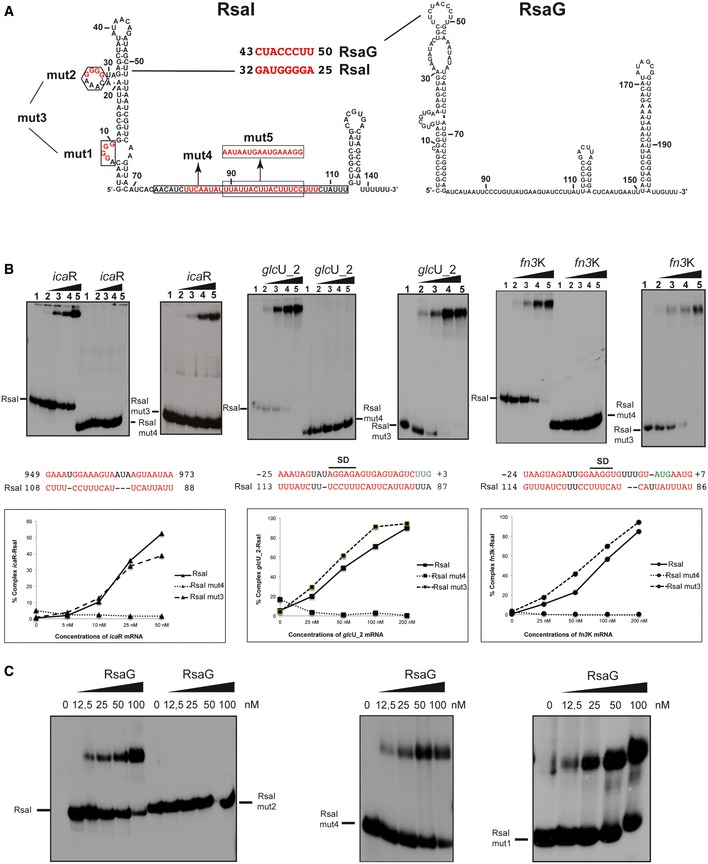
- Secondary structure model of RsaI. In red, are the nucleotides deleted in the RsaI mutants (mut1 to mut4), and the nucleotides, which were substituted (mut5). The potential base‐pairings between RsaI and RsaG are shown. Squared nucleotides are conserved sequences in RsaI.
- Gel retardation assays show the formation of the complex between RsaI and icaR, glcU_2 and fn3K mRNAs. The 5′ end‐labeled wild‐type RsaI (RsaI), RsaI mutant 3 (RsaI mut3, deletion of the two G‐track motifs), and RsaI mutant 4 (RsaI mut4, deletion of the C/U‐rich unpaired interhelical region) were incubated with increasing concentrations of mRNAs: lane 1, 0 nM; lane 2, 25 nM; lane 3, 50 nM; lane 4, 100 nM; and lane 5, 200 nM. Below the gels, the predicted interactions between RsaI and its targets are shown. Translation start codons are in green, and SD is for Shine and Dalgarno sequence. Graphs represented the % of complex formed between either RsaI or its two mutant forms (RsaI mut3 and RsaI mut4) and the target mRNA (icaR, glcU_2, fn3K) as the function of mRNA concentrations.
- Gel retardation assays show the formation of the complex between RsaI and RsaG. The 5′ end‐labeled wild‐type RsaI (RsaI), RsaI mutant 2 (RsaI mut2), RsaI mutant 4 (RsaI mut4), and RsaI mutant 1 (RsaI mut1) were incubated with increasing concentrations of RsaG given in nM on the top of the autoradiographies.
Source data are available online for this figure.
These results suggested that RsaI contains at least two functional domains, the unpaired CU‐rich region dedicated to mRNA pairing and the G‐track used to interact with other sRNAs. To validate whether RsaI effectively binds to the RNA candidates identified by MAPS through these motifs, we performed gel retardation assays using WT and mutated RsaI molecules (Figs 2B and C, and EV2). For that, in vitro 5′ end‐labeled RsaI was first incubated with increasing concentrations of various mRNAs encoding proteins involved in biofilm formation (IcaR), sugar uptake and metabolism (GlcU_2 and Fn3K), and several sRNAs (RsaG, RsaE, and RsaD). For these experiments, we used the full‐length mRNAs and sRNAs (Table EV3). Complex formation was performed with RNAs, which were renatured separately in a buffer containing magnesium and salt. As expected, the data showed that RsaI formed complexes with high affinity (between 20 and 100 nM, Table EV1) with many RNAs such as icaR, glcU_2, and fn3K mRNAs, and RsaG sRNA (Fig 2B). The stability of other complexes (e.g., treB mRNA and RsaD sRNA) was significantly lowered (> 250 nM) (Fig EV2).
Figure EV2. RsaI interacts with several RNA species.
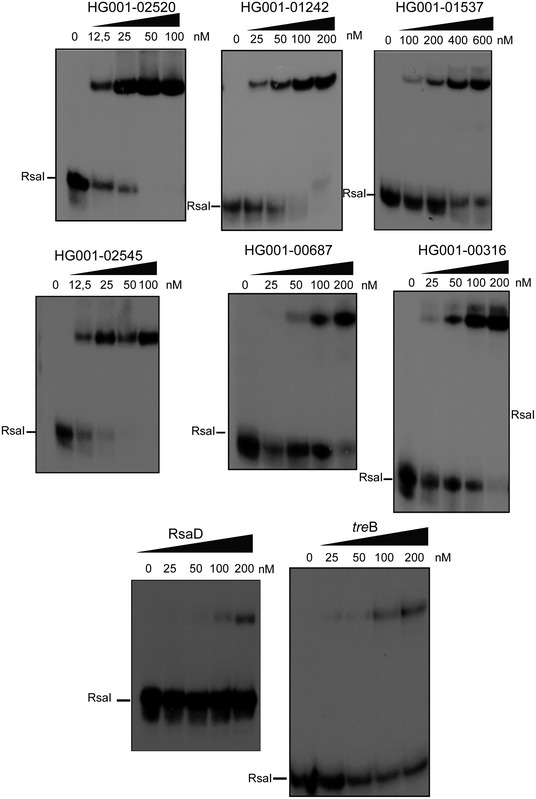
Gel retardation assays showing the formation of complexes between RsaI and several mRNAs, and the sRNA RsaD. The 5′ end‐labeled RsaI was incubated with increasing concentrations of target mRNAs or RsaD as shown on the top of the autoradiographies. Quantification of the autoradiographies showed that RsaI forms stable complexes with many of the mRNAs (Kd < 50 nM) except for TreB and RsaD (Kd > 200 nM).Source data are available online for this figure.
Based on the base‐pairing predictions, mutations have been introduced into rsaI to map its functional regulatory regions. The two conserved G‐track sequences were mutated separately (RsaI mut1: ∆G7‐G10, RsaI mut2: ∆G26‐G29) or together (RsaI mut3: ∆G7‐G10/∆G26‐G29), while several nucleotides (RsaI mut4: ∆U81‐U107) were deleted in the conserved interhelical unpaired sequence (Fig 2A). We analyzed the ability of mutated RsaI derivatives to form complexes with glcU_2, fn3K, and icaR mRNAs, and RsaG sRNA (Fig 2B). Quantification of the data showed that RsaI mut3 binds to all three mRNAs similarly to the WT RsaI, while complex formation was completely abolished with RsaI mut4. Only the mutation in the second G‐track sequences (RsaI mut2) strongly altered the binding to RsaG, while the two other mutated RsaI (mut1 and mut4) recognized RsaG with an equivalent binding affinity as the WT RsaI (Fig 2C).
To demonstrate that the RNA‐RNA interactions were also occurring in vivo, MAPS approach was used to monitor the effect of the RsaI mutations on its target RNAs. The RsaI mut2 and mut4, which independently affected in vitro binding of sRNAs and mRNAs, respectively, were tagged with MS2 and expressed in the HG001∆rsaI mutant strain. As described above, the enrichment of putative targets was calculated using a similar procedure (Table EV4). For this analysis, we have used the data coming from three independent experiments performed with the WT MS2‐RsaI, the two MS2 controls and the MS2‐RsaI mutants (mut2 and mut4) taking into account that the MAPS was done in two distinct sets of experiments (see Supplementary Material and Methods). This allowed us to directly compare the fold changes between the WT and the mutant RsaI versions (Table EV5). In the fraction containing MS2‐RsaI mut4, most of the mRNA targets including icaR, fn3K, and glcU_2 were strongly reduced while RsaG was still significantly enriched. Conversely, we observed that the three mRNAs were still co‐purified together with the MS2‐RsaI mut2 at a level close to the WT RsaI while RsaG was strongly reduced in the fraction containing MS2‐RsaI mut2 (Table EV5). In addition, with the two MS2‐RsaI mutants, RsaE was almost lost while the enrichment of the transcriptional regulator XRE was strongly reduced. Finally, dck mRNA remained enriched at the same level as for the WT MS2‐RsaI (Table EV5). These data strongly suggested that mutations at the G‐track motif of RsaI had a major effect on RsaG/RsaE sRNAs binding, whereas the deletion of the C/U‐rich sequence in RsaI mut4 had a major impact on the recognition of many mRNAs. The lost of RsaE in the MAPS performed with MS2‐RsaI mut4 suggested that the sRNA might also be associated with one of the RsaI‐dependent mRNA target. In support to the hypothesis, base‐pairings have been recently predicted between RsaE and the 5′UTR of icaR mRNA (Rochat et al, 2018).
Taken together, our data reveal that RsaI has at least two distinct regulatory domains that directly interact either with mRNAs or with sRNAs.
RsaI inhibits ribosome binding by masking the Shine and Dalgarno sequence of mRNAs
Because the C/U unpaired region of RsaI was predicted to form base‐pairings with the Shine and Dalgarno (SD) sequence of several mRNAs (Figs 2 and 3A), we analyzed whether RsaI would have the ability to compete with the ribosome for binding to glcU_2 and fn3K mRNAs. Using toe‐printing assays, we analyzed the effect of RsaI on the formation of the ternary initiation complex constituting of mRNA, the initiator tRNA, and the 30S subunit. Binding of the 30S on the two mRNAs is illustrated by the presence of a toe‐print signal at position +16 (+1 being the initiation codon). For fn3K mRNA, two toe‐print signals were observed at +16 and at +20, most probably corresponding to the presence of two AUG codons distant of 5 nucleotides. However, only the first AUG is used in vivo (Gemayel et al, 2007). The addition of increasing concentrations of RsaI together with the 30S strongly decreased the toe‐print signals for both mRNAs showing that RsaI is able to form a stable complex with the mRNA sufficient to prevent the binding of the 30S subunit (Fig 3A).
Figure 3. RsaI inhibits glcU_2 and fn3K mRNA translation.
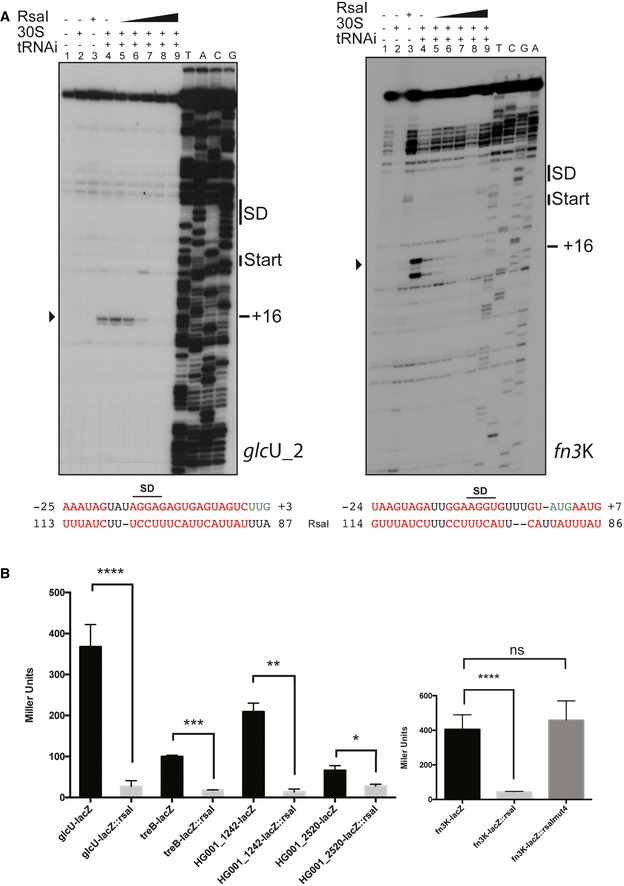
- Toe‐print assays showing the effect of RsaI on the formation of the ribosomal initiation complex of glcU_2 and fn3K mRNAs, respectively. Lane 1: incubation control of mRNA alone; lane 2: incubation control of mRNA with 30S subunits; lane 3: incubation control of mRNA with RsaI; lane 4: formation of the ribosomal initiation complex containing mRNA, 30S, and the initiator tRNAfMet (tRNAi); lanes 5–9: formation of the initiation complex in the presence of increasing concentrations of RsaI, respectively: 50 nM (lane 5), 100 nM (lane 6), 150 nM (lane 7), 300 nM (lane 8), and 400 nM (lane 9). Lanes T, A, C, G: sequencing ladders. The Shine and Dalgarno (SD) sequence, the start site of translation (START), and the toe‐printing signals (+16) are indicated. At the bottom of the gels are shown the predicted interactions between RsaI and its targets. Translation start codons are in green, and the Shine and Dalgarno (SD) sequence is underlined, and the arrowheads depict the toe‐printing signals.
- The β‐galactosidase activity (Miller Units) has been measured from various fusions expressed from a plasmid which also carries rsaI gene under its own promoter: PrpoB::glcU::lacZ::rsaI, PrpoB::fn3K::lacZ::rsaI, PrpoB::treB::lacZ::rsaI, PrpoB::HG001_1242::lacZ::rsaI, and PrpoB::HG001_2520::lacZ::rsaI expressed in the mutant strain HG001‐ΔrsaI. The same constructs were made in the absence of rsaI gene. For the fn3K‐lacZ fusion, we also used an additional construct PrpoB::fn3K::lacZ::rsaImut4 expressing both the fusion FN3K‐LacZ protein and RsaI mut4. The β‐galactosidase activity was normalized for bacterial density, and the results represented the mean of four independent experiments. The error bars are standard deviations, and the statistical significance was determined using the Student's t‐test. *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001, ns is for not significant.
Source data are available online for this figure.
The in vivo relevance of RsaI‐dependent repression of several mRNAs (glcU_2, fn3K, treB, HG001_01242, HG001_02520) was then analyzed using gene reporter assays. The whole leader regions of the mRNAs (54 nts in glcU_2, 34 nts in fn3K, 24 nts in treB, 35 nts in HG001_01242, and 72 nts in HG001_02520) including 100 nucleotides of their coding sequences were cloned in frame with lacZ under the control of the strong promoter PrpoB. The synthesis of β‐galactosidase was analyzed in the ∆rsaI mutant strain and in the same strain transformed with the plasmid carrying the lacZ reporter and RsaI under its own promoter. For the fn3K‐lacZ fusion, we also analyzed its expression in the mutant strain expressing RsaI mut4. The β‐galactosidase activity was reduced almost eight times in cells expressing wild‐type RsaI for all reporter constructs (Fig 3B). In addition, the expression of RsaI mut4 did not cause any repression of the β‐galactosidase expressed from fn3K‐lacZ (Fig 3B). Therefore, disrupting the interaction between RsaI mut4 and fn3K alleviated the repression of the reporter gene suggesting that the regulation of fn3k is occurring at the translational level through specific RNA‐RNA interactions.
These data showed that RsaI hinders ribosome binding on glcU_2 and fn3K mRNAs in agreement with the fact that the C/U‐rich region of RsaI would bind to the SD sequence of the mRNAs. We propose a similar mechanism for treB, HG001_01242, and HG001_02520 mRNAs considering that the predicted base‐pairings include the RBS of these mRNAs (Tables EV1 and EV2).
RsaI interacts with the 3′UTR of icaR mRNA and affects PIA‐PNAG synthesis
The MAPS approach revealed that icaR mRNA, which encodes the repressor of the main exopolysaccharidic compound of S. aureus biofilm matrix, is the most enriched mRNA co‐purified with MS2‐RsaI. This mRNA is of particular interest because it contains a large 3′UTR that is able to bind to its own SD sequence through the anti‐SD UCCCCUG motif (Ruiz de los Mozos et al, 2013). Consequently, the long‐range interaction provokes an inhibitory effect on translation and generates a cleavage site for RNase III (Ruiz de los Mozos et al, 2013). RsaI is predicted to form base‐pairings with the 3′UTR of icaR downstream of the anti‐SD sequence (Table EV1, Fig 4A). We first monitored whether the long‐range interaction might be critical for RsaI binding. Previous work showed that the substitution of the anti‐SD UCCCCUG sequence by AGGGGAC significantly destabilized the long‐range interaction to enhance icaR translation (Ruiz de los Mozos et al, 2013). However, gel retardation assays showed that the WT and the mutant icaR mRNAs bind to RsaI with an equivalent binding affinity (Fig 4B), suggesting that the anti‐SD sequence is not required for RsaI binding. Instead, the 3′UTR of icaR retained the full capacity to efficiently bind RsaI (Fig 4B, Table EV1). We then analyzed whether RsaI binding to the 3′UTR would alter the circularization of icaR mRNA. Gel retardation assay was performed with radiolabelled 3′UTR in the presence of the 5′UTR at a concentration of 500 nM leading to almost 50% of binding (Fig EV3A, lane 2). The addition of increasing concentrations of RsaI led to the formation of a 3′UTR‐RsaI binary complex but also of a ternary complex involving the three RNA species (Fig EV3A, lanes 4–8). Hence, RsaI does not prevent the formation of the interaction between the two UTRs of icaR mRNA in vitro.
Figure 4. Interaction of RsaI to icaR mRNA induces biofilm production.
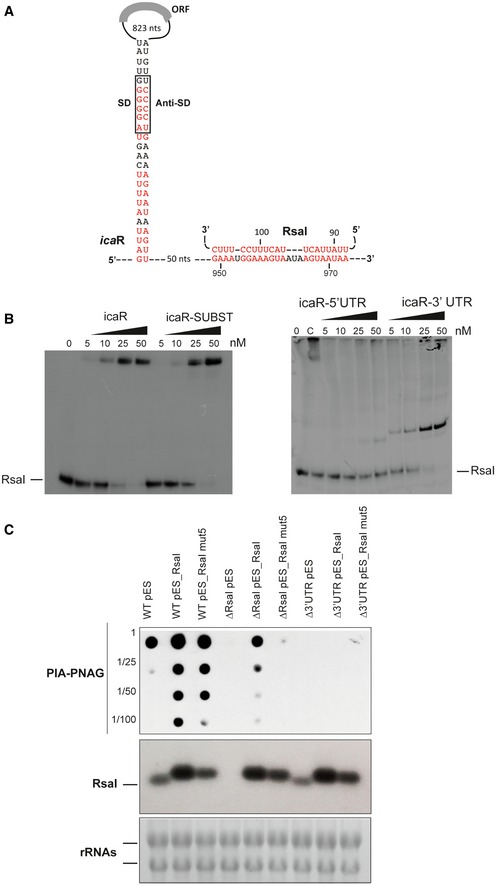
- Secondary structure model of icaR mRNA. The nucleotides in red are involved in the long‐range interaction between the 3′ and 5′UTRs or with RsaI. SD and anti‐SD are, respectively, for the Shine and Dalgarno sequence and for the sequence complementary to the SD sequence.
- Gel retardation assays show the formation of the complex between RsaI and icaR full‐length, icaR SUBST, icaR‐5′UTR, and icaR‐3′UTR. The 5′ end‐labeled of RsaI was incubated with increasing concentrations of the various mRNAs. UTR is for untranslated region, and SUBST stands for the substitution of UCCCCUG sequence by AGGGGAC. This mutation, which is located in the 3′UTR of icaR, significantly destabilizes the long‐range interaction and enhances icaR translation. Lane C represents binding between radiolabelled RsaI and full‐length icaR mRNA (50 nM). Due to the rather short migration of the gel, the full‐length mRNA is still observed in the pocket.
- In vivo effect of RsaI on PIA‐PNAG synthesis in the S. aureus wild‐type (WT) 132 strain, the ΔrsaI mutant, and the strain carrying a deletion of icaR 3′UTR (Δ3′UTR). This last mutant strain has been transformed with the pES plasmids expressing rsaI or rsaI mut5. The PIA‐PNAG exopolysaccharide biosynthesis was quantified using dot‐blot assays. Serial dilutions (1/5) of the samples were spotted onto nitrocellulose membranes, and PIA‐PNAG production was detected with specific anti‐PIA‐PNAG antibodies. RsaI was detected in the same samples by Northern blot using a probe directed against RsaI. Ethidium bromide staining of rRNA was used as loading controls of the same gel.
Source data are available online for this figure.
Figure EV3. RsaI interacts with the 3′UTR of icaR mRNA .
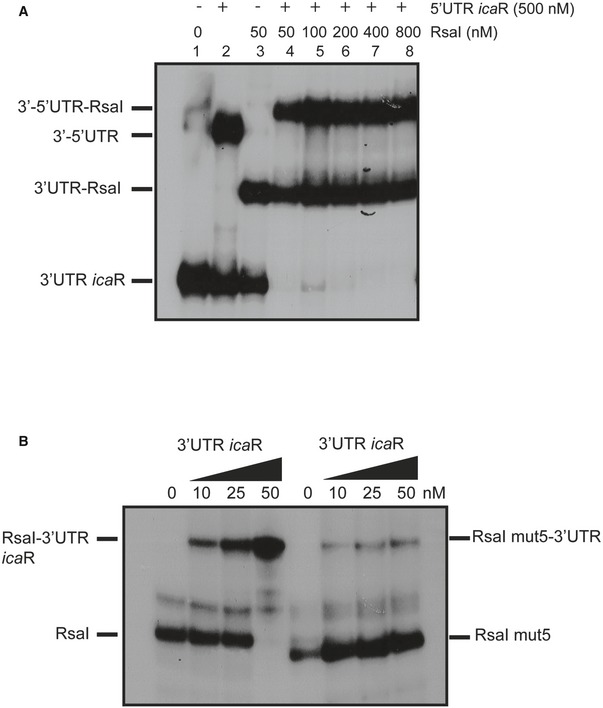
- Analysis of the effect of RsaI on the interaction between the 5′UTR and the 3′UTR of icaR. The 5′ end‐labeled of the 3′UTR of icaR was incubated with 500 nM of the 5′UTR of icaR alone or in the presence of increasing concentrations of RsaI, as shown on the top of the autoradiography.
- Gel retardation assays showing the formation of complexes between RsaI or RsaI mut5 and the 3′UTR of icaR. RsaI or RsaI mut5 labeled at their 5′ ends was incubated with increasing concentrations of the 3′UTR of icaR (10, 25, and 50 nM). In both gels, the various RNA species present in the different bands are given on one side of the autoradiographies.
Source data are available online for this figure.
Then, to analyze the in vivo effect of the RsaI interaction with the icaR mRNA, we monitored the production of PIA‐PNAG as a natural reporter, assuming that variations on IcaR expression would be directly reflected on the production of this exopolysaccharide. For that, dot‐blot assays were performed with anti‐PIA‐PNAG‐specific antibodies to monitor the levels of PIA‐PNAG in the strain 132, which expresses rsaI and produces high levels of this exopolysaccharide compared to HG001 in our experimental conditions. We measure the PIA‐PNAG levels in WT 132 strain and the isogenic mutant strains ∆rsaI and ∆3′UTR icaR, which carried a deletion of the 3′UTR of icaR. Bacteria were grown for 6 h in TSB containing NaCl, which is required to enhance PIA‐PNAG synthesis in 132 strain (Vergara‐Irigaray et al, 2009) (Fig 4C). The results showed that both RsaI and the 3′UTR of icaR are required for efficient production of PIA‐PNAG because only the WT strain produces significant levels of exopolysaccharides. In addition, the three strains were transformed with a plasmid overexpressing RsaI or RsaI mut5 carrying a substitution of nucleotides 88–103 (UUAUUACUUACUUUCC to AAUAAUGAAUGAAAGG) under the control of a strong and constitutive promoter. This mutation decreased the stability of RsaI‐icaR duplex (Fig EV3B). Northern blots confirmed RsaI overexpression in these strains (Fig 4C). Dot‐blot results revealed that production of PIA‐PNAG could be restored in ∆rsaI strain when RsaI is constitutively expressed. However, complementation is observed neither in the ∆rsaI expressing the RsaI mut5 nor in the Δ3′UTR mutant strains overexpressing RsaI or RsaI mut5. These results suggested that the interaction of RsaI with 3′UTR is required to promote PIA‐PNAG synthesis (Fig 4C). Interestingly, overexpression of RsaI in the WT strain considerably increased PIA‐PNAG production. Although this effect is less pronounced in the WT expressing RsaI mut5, it suggested the presence of additional regulatory pathways.
Taken together, these data suggest that RsaI would contribute to PIA‐PNAG synthesis by at least reducing the IcaR repressor protein levels through a specific interaction with the 3′UTR of icaR mRNA.
Two sRNAs responded to sugar uptake
The MAPS experiments revealed that RsaG was the most highly co‐purified sRNA together with RsaI (Table EV1), and the complex formed between RsaI and RsaG is very stable (Fig 2C). Interestingly, rsaG is localized just downstream uhpT encoding the hexose phosphate transporter (Fig EV4A), whose transcription is activated by the two‐component system HptRS in response to extracellular glucose‐6 phosphate (G‐6P), another major carbon source produced by host cells (Park et al, 2015). We therefore analyzed whether RsaG expression would also respond to the cellular concentration of G‐6P. Northern blot experiments were performed on total RNA extracts produced from the HG001 strain grown in BHI medium supplemented with G‐6P. Under these conditions, the synthesis of RsaG is strongly enhanced (Fig EV4B, left panel), in contrast to RsaI, which is completely inhibited under the same conditions (Fig EV4B, right panel). The deletion of hptRS considerably reduced the levels of RsaG (Fig EV4C). Therefore, these data indicated that RsaG is activated by HptRS upon G‐6P signaling together with uhpT and that RsaG sRNA might be derived from the uhpT 3′UTR.
Figure EV4. Analysis of the expression of RsaG and RsaD in response to stress.
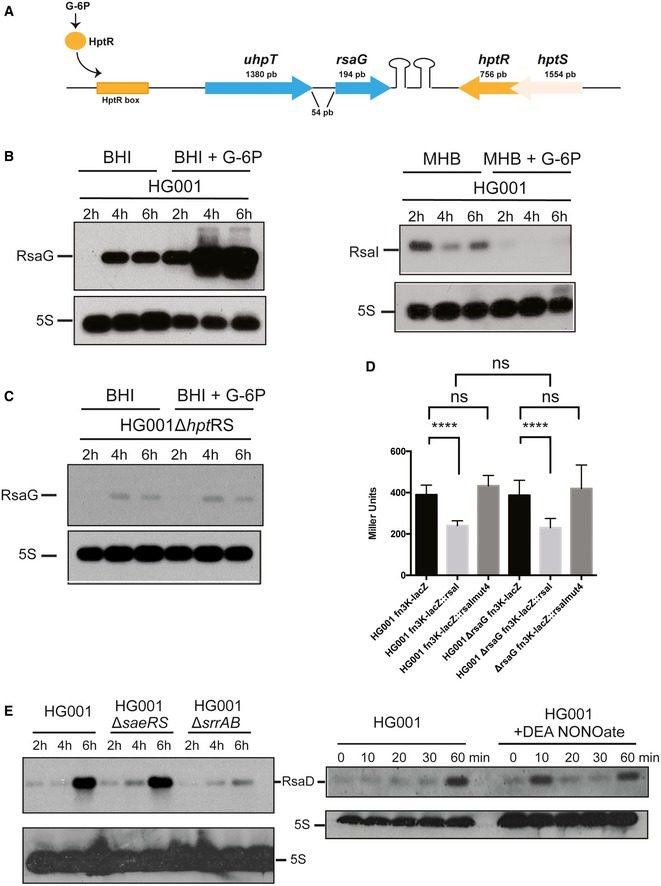
- Genomic organization of the uhpT locus in HG001 strain. This locus is conserved in all S. aureus strains (Geissmann et al, 2009). The transcription of uhpT containing RsaG is induced by the two‐component system HptRS, which senses the extracellular concentration of glucose‐6 phosphate (G‐6P).
- Northern blot experiment showing RsaG (left panel) or RsaI (right panel) expression in the HG001 strain. Total RNA was extracted at 2, 4, and 6 h of growth in BHI or MHB with or without addition of glucose‐6 phosphate (+G‐6P). 5S rRNA was used as loading control using the same samples, which have been migrated on another agarose gel.
- Northern blot experiment showing RsaG expression in the HG001∆hptRS deleted mutant. Total RNA was extracted at 2, 4, and 6 h of growth in BHI with or without addition of glucose‐6 phosphate (+G‐6P). 5S rRNA was used as loading control using the same samples, which have been migrated on another agarose gel.
- The β‐galactosidase activity (Miller Units) have been measured from PrpoB::fn3K::lacZ, PrpoB::fn3K::lacZ::rsaI, and PrpoB::HG001_fn3K::lacZ::rsaI mut4 expressed in HG001 and HG001ΔrsaG strains. The β‐galactosidase activity was normalized for bacterial density and the results represented the mean of four independent experiments. ****P < 0.0001, ns: not significant.
- Northern blot experiment showing the steady‐state level of RsaD. On the left, RsaD expression was analyzed at 2, 4, and 6 h of growth, in HG001 wild‐type strain and in ∆saeRS and ∆srrAB mutant strains. On the right, the expression of RsaD was monitored in response to nitric oxide (NO) stress. The culture performed in BHI medium until OD600 = 0.2 at 37°C was then treated with the addition of 100 μM Na‐diethylamine (+DEA NONOate). 5S rRNA was used as loading control using the same samples, which have been migrated on another agarose gel.
Source data are available online for this figure.
We then addressed the consequences of RsaI‐RsaG pairings on target recognition. Using gel retardation assays, we analyzed whether RsaG is able to form a ternary complex with RsaI and one of its target mRNA or whether RsaG competes with the mRNA for RsaI binding. These experiments were conducted for three mRNA targets (glcU_2, HG001_1242, and HG001_0942), which all formed stable complexes with the 5′ end‐labeled RsaI (Figs 2, 5A and EV2, Table EV1). On the opposite, RsaG is not able to interact with glcU_2 and HG001_1242 as shown the gel retardation assays performed with 5′ end‐labeled RsaG (Fig 5B). We then performed competition experiments between RsaI and RsaG for mRNA binding. The experiments were performed in the presence of a single concentration of unlabeled RsaG (50 nM) sufficient to bind most of 5′ end‐labeled RsaI in the presence of increasing concentrations of mRNA (Fig 5A). For the three mRNAs, the addition of RsaG causes the formation of a high molecular weight complex most likely formed by RsaG, RsaI, and the mRNA (Fig 5A). Hence, RsaG does not interfere with the mRNA binding to RsaI.
Figure 5. Regulatory function of RsaI is not impaired by binding of RsaG.
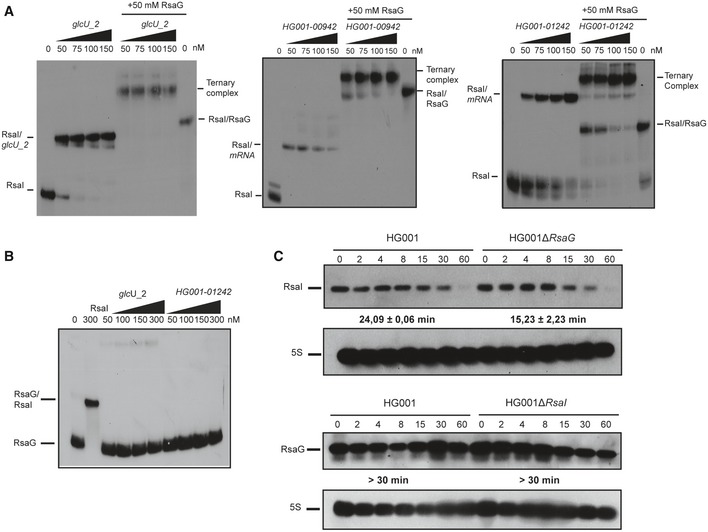
- Ternary complex formation between RsaI, various mRNAs (glcU_2, HG001_00942 and HG001_1242), and RsaG. The 5′ end‐labeled RsaI was incubated with increasing concentrations of the target mRNA alone or in the presence of 50 nM of RsaG. The various complexes are notified on the sides of the autoradiographies.
- RsaG does not form stable complexes with glcU_2 and HG001_01242. Binding assays were done in the presence of 5′ end‐labeled RsaG and either cold RsaI (300 nM) or increasing concentrations of glcU_2 and HG001_01242.
- Measurements of the half‐lives of RsaI and RsaG in HG001‐∆rsaG or HG001‐∆rsaI mutant strains using Northern blot experiments. The cells were treated with rifampicin at 4 h of growth, and total RNAs were extracted after 2, 4, 8, 15, 30, and 60 min at 37°C in BHI. 5S rRNA was probed to quantify the yield of RNAs in each lane using the same samples, which were however run on two different gels. Calculated half‐lives are shown beneath the autoradiographies and are the average of 2 experiments. The data were normalized to 5S rRNA.
Source data are available online for this figure.
Furthermore, in vivo, the synthesis of the β‐galactosidase expressed from the fn3K‐lacZ fusion was measured in the WT and ∆rsaG mutant strains. The expression of the fusion was strictly identical in both strains. The overexpression of RsaI WT from a plasmid causes a weak but specific inhibition of β‐galactosidase synthesis because the RsaI‐dependent repression was alleviated in the strain transformed with a plasmid expressing RsaI mut4 (Fig EV4D). This experiment showed that the expression of RsaG does not significantly impact the synthesis of one of the RsaI target.
Using rifampicin treatment, the half‐lives of RsaI and RsaG were measured in the WT and the mutant strains (Fig 5C). Quantification of the autoradiographies showed that the two RNAs are highly stable as it was previously shown (Geissmann et al, 2009). However, the stability of RsaI was reproducibly higher in the WT strain (24 min) than in the ∆rsaG mutant strain (15 min; Fig 5C).
Taken together, the data show that the synthesis of RsaG and RsaI is regulated in response to different but related carbon sources, G‐6P and glucose, respectively. In addition, their binding has no major consequences on the RsaI target recognition and regulation, although RsaG has a slight effect on RsaI stability.
RsaD, the other sRNA target candidate of RsaI, responds to nitric oxide
The signaling pathway of RsaD, another potential sRNA partner of RsaI, has been studied. RsaD binds to RsaI in vitro albeit with a lower affinity than RsaG. Although RsaD was significantly enriched in the MAPS experiments using the wild‐type MS2‐RsaI (Table EV2), the data were unfortunately not reliable with the mutant MS2‐RsaI mut2 and mut4. This is most probably due to the fact that RsaD is weakly expressed at the stationary phase of growth in BHI medium as revealed by the transcriptomic analysis (Table EV6).
Upstream the rsaD gene, we identified a conserved motif AGTGACAA that has been described as a binding site for the response regulator SrrA (Pragman et al, 2004; Kinkel et al, 2013). SrrAB is a major two‐component system affecting the temporal expression of the virulence factors (Pragman et al, 2004) while it promotes resistance to nitrosative stress and hypoxia (Richardson et al, 2006; Kinkel et al, 2013). We demonstrated that the expression of RsaD drops considerably in a ∆srrAB mutant strain (Fig EV4E) while its expression is not under the control of the two‐component system SaeRS, another global regulator of virulence (Fig EV4E; Liu et al, 2016). Because SrrAB is able to sense and respond to nitric oxide (NO) and hypoxia (Kinkel et al, 2013), we tested the effect of NO on RsaD synthesis by adding diethylamine‐NONOate, as it was previously described for RsaE (Durand et al, 2015). We observed a significant and reproducible enhanced expression of RsaD in the WT HG001 strain about 10 min after the addition of diethylamine‐NONOate to BHI medium (Fig EV4E). RsaD expression decreased after 20 min due to the short half‐life of diethylamine‐NONOate. Noteworthy, RsaE, which was significantly enriched together with MS2‐RsaI, was also shown to be under the control of SrrAB (Durand et al, 2015).
These data suggested that through the binding of sRNAs, a network of sRNAs would coordinate sugar metabolism pathways, energy production, and NO stress responses.
The effect of RsaI on global gene expression in Staphylococcus aureus
Before to perform the comparative transcriptomic analysis, we first analyzed whether the deletion or the overexpression of RsaI might cause growth phenotype. Unexpectedly, we did not succeed to overexpress RsaI from a strong promoter in the WT HG001 strain. This result was not due to technical problems as we managed to express RsaI under the control of its own promoter on the same plasmid. Hence, the growth phenotypic experiment was performed in the 132 strain. The WT 132 and the ∆rsaI mutant strains harbor a similar growth curve in BHI medium (Fig EV5A). However, when RsaI was expressed under the control of a strong promoter, a slight but reproducible delay in growth was observed only in the WT 132 strain (Fig EV5A). Northern blot analysis was performed to measure the steady‐state levels of RsaI during growth in the various strains (Fig EV5B). Quantification of the autoradiographies and normalization with 5S rRNA showed that the highest yields of RsaI were observed in the WT 132 strain expressing RsaI from the strong promoter, and only in this context, a significant amount of RsaI was detected at the beginning of growth (Fig EV5B).
Figure EV5. RsaI represents a signature of a metabolic change as the result of glucose consumption.
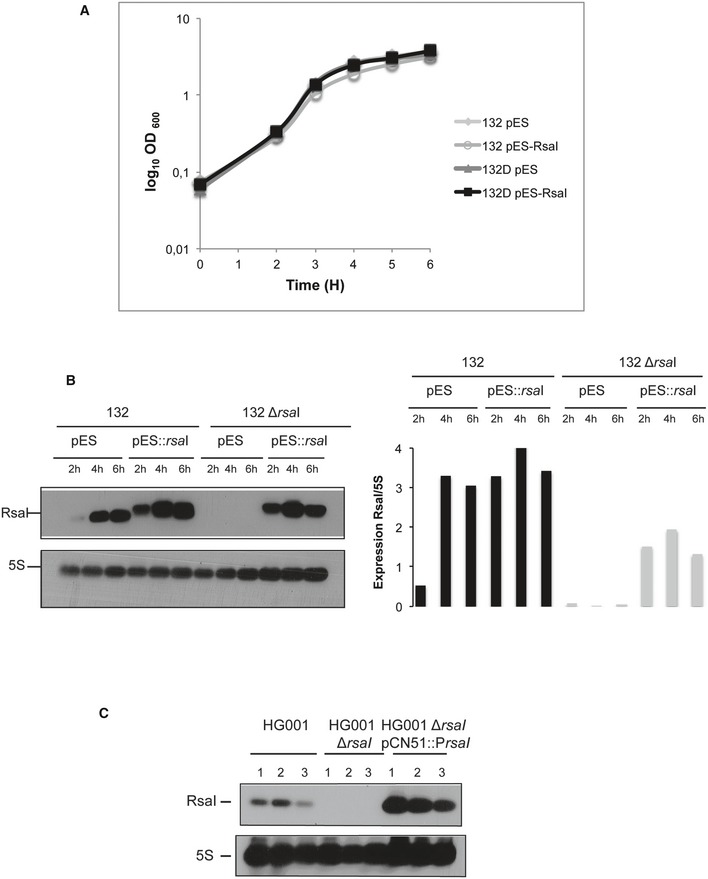
- Growth curves of the wild‐type 132 (132) and mutant ∆rsaI (132D) strains containing the vector pES (empty) or pES::rsaI (expressing RsaI from a constitutive promoter) in BHI.
- Northern blot experiment showing RsaI expression in the wild‐type 132 (132) or mutant ∆rsaI (132∆rsaI) strains containing the vector pES or pES::rsaI. Total RNA was extracted at 2, 4, and 6 h of growth in BHI. 5S rRNA was used as the loading control using the same samples, which have been migrated on another agarose gel. Quantification of RsaI normalized to 5S rRNA was done with ImageQuant TL software (GE Healthcare Life Sciences).
- Northern blot experiments showing the expression of RsaI in the wild‐type HG001 strain, the mutant HG001∆rsaI strain, and the same mutant strain complemented with a plasmid expressing RsaI from its own promoter (HG001∆rsaI pCN51::PrsaI). All the other strains have been transformed with the pCN51 plasmid. Total RNA was prepared from samples taken after 6 h of growth in BHI medium. Hybridization against 5S rRNA was used as loading control using the same membrane. Three replicates were carried out (lanes 1–3), and the same samples were used for the differential transcriptomic analysis.
Source data are available online for this figure.
In order to get a global overview of RsaI impact on gene regulation, a comparative transcriptomic analysis was performed on total RNAs extracted from the WT HG001 strain, the isogenic HG001∆rsaI mutant strain, and the same mutant strain complemented with a plasmid expressing RsaI under the control of its own promoter in order to avoid side effects of a too strong expression of RsaI on growth (Tables EV6 and EV7, and Fig EV5C). The cultures were done in triplicates with high reproducibility in BHI medium at 37°C until 6 h, under the conditions allowing the expression of RsaI (Fig 1). We have considered a gene to be regulated by RsaI if the ratio between two strains was at least twofold. Changes in gene expression were more pronounced between the mutant ∆rsaI versus the same strain expressing RsaI from a plasmid rather than between the mutant ∆rsaI versus the WT strain (Table EV7). This effect is most likely due to the higher expression of RsaI in the complemented strain in comparison with the WT strain (Fig EV5C). The levels of 26 and 50 mRNAs were significantly decreased and enhanced, respectively, when the complemented strain was compared to the mutant ∆rsaI (Table EV7). The complete data set is presented in Table EV6. Beside genes encoding phage‐related proteins, most of the RsaI‐dependent activation was observed for genes involved in fermentation processes, in NO resistance, and in energy‐generating processing (in red in Table EV7). In addition, weaker effects were also observed for mRNAs encoding proteins involved in iron‐sulfur cluster repair (ScdA), in NO detoxification (qoxABCD, hmp), and in various metabolic pathways. The level of RsaG was also enhanced in the complemented strain. Conversely, the overexpression of RsaI caused a reduced expression of genes that are functionally related. Several of them are involved in glycolysis (fba1) and pentose phosphate pathway (gnd, tkt), in thiamine co‐factor synthesis (thiW, tenA), and in arginine catabolism (arcABCD, arcR) (in blue in Table EV7). Additionally, significant repression was also observed for miaB encoding tRNA‐specific modification enzyme, tyrS encoding tryptophanyl‐tRNA synthetase, ebpS encoding the cell surface elastin binding protein, and to a weaker extent for rex encoding redox‐sensing transcriptional regulator. Interestingly, the MAPS approach revealed that several of these mRNAs (i.e., qoxABCD operon, tyrS, and plfA‐plfB) were also enriched together with RsaI (Table EV2). Surprisingly, the most enriched RsaI targets identified by MAPS (i.e., icaR, glcU_2, and fn3K) did not show significant mRNA level variations when RsaI was deleted or overexpressed suggesting that RsaI would primarily regulate their translation (Table EV1).
These data showed that high concentrations of RsaI affected the mRNA levels of several enzymes involved in sugar metabolism, in arginine catabolism, in the pentose phosphate pathway, and in various processes linked to NO detoxification, energy production, and fermentation. However, the small overlap between the two approaches suggested that many of the RsaI‐dependent effects as revealed in the transcriptomic data resulted from indirect effects.
Discussion
In this work, we have investigated the cellular functions of one abundant sRNA, called RsaI (or RsaOG), which is highly conserved among Staphylococcaea (Geissmann et al, 2009; Marchais et al, 2010). In contrast to many sRNAs that contained C‐track motifs, this RNA has the particularity to carry two conserved G‐rich sequences and a large unpaired CU‐rich interhelical region (Fig 2). This RNA was proposed to fold into a pseudoknot structure involving the two conserved regions limiting the access of regulatory regions (Marchais et al, 2010). Combining several global approaches (MAPS, transcriptomic analysis), our data show that RsaI is linked to a large regulon involved in sugar uptake and metabolism, biosynthetic and co‐factor synthesis, cytochrome biosynthesis, anaerobic metabolism, as well as iron‐sulfur cluster repair, and NO detoxification (Fig 6).
Figure 6. Model of RsaI regulation networks in Staphylococcus aureus .
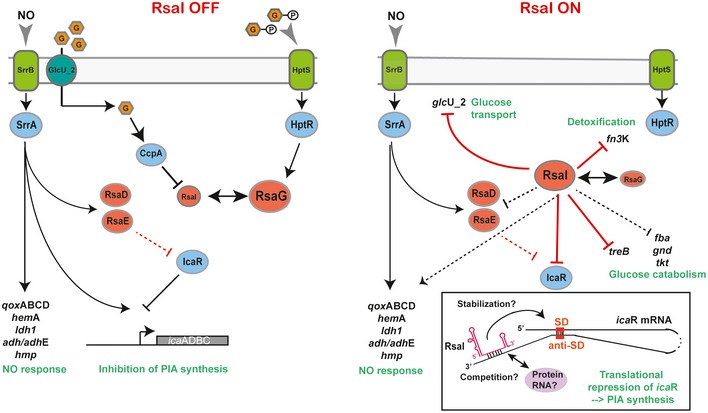
In the presence of glucose or glucose 6‐phosphate (left panel), the expression of RsaI is inhibited, in contrast to RsaG. In the absence of glucose or when glucose is metabolized (right panel), repression of RsaI is alleviated to regulate its RNA targets. In blue, are the transcriptional protein regulators, and in red, the regulatory sRNAs. Gray arrows represent functional links, black arrows are for activation, and bars for repression. Red lines corresponded to post‐transcriptional regulation and black lines to transcriptional regulation. Dotted lines represented the regulatory events for which the regulation is not yet demonstrated. A schematic view of the post‐transcriptional icaR mRNA regulation is represented in the insert. The 3′UTR of icaR contains an anti‐Shine and Dalgarno (anti‐SD) sequence able to bind the SD sequence in the 5′UTR (Ruiz de los Mozos et al, 2013). The CU‐rich unpaired sequence of RsaI (in red) binds to the 3′UTR of icaR mRNA downstream of the anti‐SD and represses the translation indirectly either by stabilizing the interaction between the two UTRs or by preventing the action of trans‐acting activators (protein or RNA).
The expression of RsaI is derepressed when glucose concentration decreases
The cellular level of RsaI is tightly controlled during growth in rich BHI medium. In this study, we showed that RsaI expression is strongly and rapidly repressed at the transcriptional level through the activity of CcpA in response to glucose availability. Carbon catabolite repression is a universal regulatory phenomenon that allows the cells to use the preferred carbon source to produce energy and to provide the building blocks for macromolecules. Concomitantly, it represses genes that are involved in the metabolism of less‐preferred carbon sources. To do so, CcpA has to be activated through a cascade of events involving its co‐regulator histidine‐containing phosphocarrier protein (HPr), which has been phosphorylated by its cognate kinase/phosphorylase HprK/P activated in the presence of glycolytic intermediates. It is thought that binding of the phosphorylated HPr to CcpA enhances its DNA‐binding affinity to the cre‐binding site to repress or activate the target genes. RsaI repression requires the presence of CcpA, most probably through the binding at a cre motif located at −45/−30 (GGAAAACGCTTACAT) from the rsaI transcriptional start site (Figs 1 and EV1B). Interestingly, deletion of ccpA gene affected vancomycin resistance (Seidl et al, 2009) and recent observations showed that sub‐inhibitory treatment of cells with vancomycin led to an enhanced expression of RsaI (Howden et al, 2013). We showed that the repression of RsaI is alleviated as soon as the concentration of glucose is strongly reduced. Therefore, RsaI might be a signature of a metabolic switch of the bacterial population. Using the MAPS approach and gel retardation assays, we could identify several mRNAs that strongly bind to RsaI with its long unpaired and conserved CU‐rich region. Binding of RsaI to glcU_2 encoding a glucose permease and to fn3K encoding the fructosamine 3 kinase hindered the formation of the initiation ribosomal complex. Because their steady‐state yields are not affected, we suggest that both mRNAs are regulated at the translational level. We propose that this mechanism is most likely common to all mRNAs that could form base‐pairings between their ribosome‐binding sites and the CU‐rich region of RsaI. This is the case for mRNAs encoding a transcriptional regulator of the XRE family, the PTS system trehalose‐specific EIIBC component TreB, a peptidase, a cell wall‐binding lipoprotein, and DegV containing protein (Table EV2). Noteworthy, in vivo, most of these mRNAs were no more found enriched using MS2‐RsaI mut4 in which the CU‐rich region has been deleted (Table EV5). Among these proteins, two of them are directly involved in sugar uptake and metabolism (GlcU_2, TreB). In S. aureus, the PTS (phosphotransferase system)‐dependent and PTS‐independent transport systems ensure efficient glucose transport (reviewed in Götz et al, 2006). If a rapidly metabolizable sugar (such as glucose) is used during growth at a rather low concentration, the transport will occur via the PTS system, and concomitantly, the carbon catabolite repression system will be activated through CcpA protein. At high concentration of glucose, it is assumed that the sugar will be transported by the PTS system and in addition by the permease GlcU_2. The glucose transported by the permease will be phosphorylated within the cell by the glucose kinase GlkA. Hence, it is reasonable to propose that RsaI would repress the synthesis of GlcU_2 when the glucose concentration dropped. In addition, RsaI represses the expression of treB mRNA most probably through base‐pairings with its SD sequence. TreB is the enzyme specific for trehalose, a diholoside which is transported exclusively by PTS (Bassias & Brückner, 1998). Transcriptomic analysis also revealed that RsaI strongly repressed various key enzymes involved in glucose catabolism pathway such as fructose‐biphosphate aldolase (fba1), 6‐phosphogluconate dehydrogenase 2C decarboxylase (gnd), and the thiamine‐dependent enzyme transketolase (tkt). Additionally, we showed that RsaI represses the synthesis of fructosamine 3‐kinase (Fn3K), which deglycated products of glycation formed from ribose 5‐phosphate or erythrose 4‐phosphate produced by the pentose phosphate pathway (Gemayel et al, 2007). This enzyme is part of a repair machinery to protect the cells from damages caused by glycation as the results of high glucose concentrations (Deppe et al, 2011). The pentose phosphate pathway is also an alternative route for glucose metabolism and provides the source of pentose phosphates necessary for nucleotide synthesis. Although it is not known whether RsaI regulated their expression in a direct or indirect manner, base‐pairings were predicted between RsaI and the ribosome‐binding site of fba1 and tkt mRNAs. The transketolase is a key enzyme of the pentose phosphate pathway, which requires thiamine diphosphate as a co‐factor. Interestingly, the thiamine pathway is also repressed in strain expressing RsaI (Table EV6). Although, we have observed very similar pathways that are deregulated by RsaI using the MAPS and RNA‐seq approaches, not so many overlaps were identified. It is possible that the conditions of the MAPS approach performed on the WT strain expressing all the ribonucleases have preferentially enriched the mRNAs that are regulated at the translational level. Therefore, it is tempting to deduct that RsaI would inhibit the synthesis of the major permease of glucose uptake, of enzymes involved in the glycolysis, of unnecessary enzymes involved in detoxification of high glucose concentration, and of the pentose phosphate pathway when glucose concentration decreases (Fig 6).
RsaI interacts with sRNA containing C‐rich motifs
The MAPS approach revealed that several sRNAs (RsaG, RsaD, RsaE) were enriched significantly with RsaI. We demonstrated here that the second G‐track sequence located in the first hairpin domain of RsaI is able to form a highly stable complex with RsaG and to a lesser extent with RsaD. Additionally, mRNA targets interacting with the CU‐rich domain of RsaI did not disturb the binding of RsaG suggesting that the two functional domains of RsaI are independent. Conversely, preliminary data suggested that the apical loop of the first hairpin of RsaG contains the C‐rich motif that is recognized by RsaI, but which is also used to regulate the expression of several mRNAs (Desgranges E, Marzi S, Romby P, Caldelari I, unpublished data). RsaG is part of the 3′UTR of uhpT mRNA, and its expression was strongly enhanced by the extracellular concentration of G‐6P. Under these conditions, the levels of RsaI are much lower than for RsaG (Fig EV4B). These two sRNAs are thus involved in pathways related to the use of the preferred carbon sources. Indeed, RsaI negatively controls glucose uptake when glucose is consumed or absent from the medium while RsaG responds to the extracellular concentration of G‐6P. Although the functions of RsaG remain to be addressed, we hypothesized that the sRNA might regulate either the expression of unnecessary genes, or of genes involved in sugar metabolism, or of genes required to protect cells against damages linked to sugar–phosphate uptake and metabolism (Bobrovskyy & Vanderpool, 2016). Noteworthy, RsaI sequence is conserved in the Staphylococcus genus while RsaG is only conserved in the S. aureus species. Our data suggested that RsaG has no major impact on the regulation of RsaI targets. Its slight stabilizing effect on RsaI strongly supports the MAPS data showing that the two sRNAs interact in vivo and potentially would form ternary complexes in the presence of the RsaI targets. Whether RsaI might regulate the functions of RsaG remained to be addressed.
In addition, the overproduction of RsaI induced several changes into the transcriptome of S. aureus that resembled the regulon of the two‐component system SrrAB, which was demonstrated as the essential system responding to both NO and hypoxia (Kinkel et al, 2013). The SrrAB regulon has also been shown to confer to the cells the ability to maintain energy production, to promote repair damages, and NO detoxification (Richardson et al, 2006; Kinkel et al, 2013). We observed here that RsaI enhances the expression of genes encoding cytochrome biosynthesis (qoxABCD), as well as genes involved in anaerobic metabolism (pflAB, ldh1, focA, adh), in iron‐sulfur cluster repair (scdA), and most importantly in NO detoxification (qoxABCD, hmp) and NO resistance (ldh1). These effects are most likely indirect and might be due to the interaction between RsaI and the SrrAB‐dependent sRNAs RsaD and RsaE, which both contained a typical srrA site upstream their genes. These two sRNAs present a C‐rich sequence that can potentially form base‐pairings with the G‐track sequences of RsaI (Table EV1), but the formation of complexes with RsaI is less efficient than with RsaG. It cannot be excluded that an RNA‐binding protein might be required in these cases to stabilize the base‐pairings. We also do not exclude that the two sRNAs were pooled down because they might share similar mRNA targets with RsaI. RsaD and RsaE are both upregulated by the presence of NO in the cellular medium (Durand et al, 2015; Fig EV4D). Although the functions of RsaD require additional studies, S. aureus RsaE was previously shown to coordinate the downregulation of numerous metabolic enzymes involved in the TCA cycle and the folate‐dependent one‐carbon metabolism (Geissmann et al, 2009; Bohn et al, 2010) and of enzymes involved in arginine degradation pathway (Rochat et al, 2018). Additionally, in B. subtilis, the homologue of RsaE called RoxS is under the control of the NADH‐sensitive transcription factor Rex, and the Rex binding site is also conserved in S. aureus rsaE gene (Durand et al, 2017). Hence, it was proposed that RsaE downregulated several enzymes of the central metabolism under non‐favorable conditions and in addition would contribute to readjust the cellular NAD+/NADH balance under stress conditions (Bohn et al, 2010; Durand et al, 2015, 2017).
In addition, the overproduction of RsaI induced several changes into the transcriptome of S. aureus that resembled the regulon of the two‐component system SrrAB, which was demonstrated as the essential system responding to both NO and hypoxia (Kinkel et al, 2013). The SrrAB regulon has also been shown to confer to the cells the ability to maintain energy production, to promote repair damages, and NO detoxification (Richardson et al, 2006; Kinkel et al, 2013). We observed here that RsaI enhances the expression of genes encoding cytochrome biosynthesis (qoxABCD), as well as genes involved in anaerobic metabolism (pflAB, ldh1, focA, adh), in iron‐sulfur cluster repair (scdA), and most importantly in NO detoxification (qoxABCD, hmp) and NO resistance (ldh1). These effects are most likely indirect and might be due to the interaction between RsaI and the SrrAB‐dependent sRNAs RsaD and RsaE, which both contained a typical srrA site upstream their genes. These two sRNAs present a C‐rich sequence that can potentially form base‐pairings with the G‐track sequences of RsaI (Table EV1), but the formation of complexes with RsaI is less efficient than with RsaG. It cannot be excluded that an RNA‐binding protein might be required in these cases to stabilize the base‐pairings. We also do not exclude that the two sRNAs were pooled down because they might share similar mRNA targets with RsaI. RsaD and RsaE are both upregulated by the presence of NO in the cellular medium (Durand et al, 2015; Fig EV4D). Although the functions of RsaD require additional studies, S. aureus RsaE was previously shown to coordinate the downregulation of numerous metabolic enzymes involved in the TCA cycle and the folate‐dependent one‐carbon metabolism (Geissmann et al, 2009; Bohn et al, 2010) and of enzymes involved in arginine degradation pathway (Rochat et al, 2018). Additionally, in B. subtilis, the homologue of RsaE called RoxS is under the control of the NADH‐sensitive transcription factor Rex, and the Rex binding site is also conserved in S. aureus rsaE gene (Durand et al, 2017). Hence, it was proposed that RsaE downregulated several enzymes of the central metabolism under non‐favorable conditions and in addition would contribute to readjust the cellular NAD+/NADH balance under stress conditions (Bohn et al, 2010; Durand et al, 2015, 2017).
Many of the RsaI‐dependent effects, which have been monitored by the transcriptomic analysis, are indirect, and we propose that some of these effects might result from the interaction of RsaI with sRNAs when the preferred carbon source became scarce.
Physiological consequences of RsaI regulation
Our data suggested that several sRNAs in S. aureus are part of intricate regulatory networks to interconnect in a dynamic manner various metabolic pathways following sugar metabolism and uptake. The concept that sRNAs are key actors to coordinate the regulation of metabolic enzymes has been largely demonstrated for Enterobacteriaceae (reviewed in Papenfort & Vogel, 2014). These sRNA‐dependent regulations often induced significant growth phenotypes in response to the availability of carbon sources and nutrient. For instance, E. coli and Salmonella SgrS contribute to stress resistance and growth during glucose–phosphate stress by inhibiting synthesis of sugar transporters or activating dephosphorylation and efflux of sugars (e.g., Vanderpool & Gottesman, 2004, 2007; Kawamoto et al, 2005; Rice et al, 2012; Papenfort et al, 2013). The repertoire of SgrS targets has been recently expanded to mRNAs involved in key metabolic pathways, which allow restoring metabolic homeostasis during sugar–phosphate stress and growth recovery (Richards et al, 2013; Bobrovskyy & Vanderpool, 2016). Salmonella GcvB regulon is required for growth if peptides represent the unique carbon source (Miyakoshi et al, 2015), while E. coli Spot42 is important for optimal utilization of carbon sources and its overproduction inhibited growth on several non‐preferred carbon sources (Møller et al, 2002; Beisel & Storz, 2011). In S. aureus, the overproduction of RsaE induces a growth defect, which was partially alleviated by acetate (Bohn et al, 2010). These examples illustrate how the yields of these sRNAs should be tightly controlled during growth in order to optimize the use of the preferred carbon sources, to restore metabolic homeostasis during stress, and to avoid cell damages or metabolites depletion caused by the intracellular production of glucose–phosphate. Interestingly, in Bacillus subtilis, the sRNA SR1 was found repressed under glycolytic conditions mainly by CcpN and to a lesser extent by CcpA (Licht et al, 2005; Heidrich et al, 2007; Gimpel et al, 2012). SR1 expression is induced by L‐arginine and acts as a main regulator or arginine catabolism through the translational repression of ahrC mRNA (Heidrich et al, 2007). In this case, the deletion or the overexpression of SR1 did not affect growth (Licht et al, 2005). The deletion of rsaI gene does not cause a growth phenotype, and only high levels of RsaI constitutively expressed from a plasmid in the WT strain cause a weak growth delay (Fig EV5A). Additional works will be required to identify the growth conditions under which the function of RsaI can be studied in a more relevant way.
One possible track resides in the fact that RsaI interconnects various metabolic pathways. By acting as a post‐transcriptional regulator, it could play an important role during the infection process. S. aureus has the ability to generate infections through the colonization of many different metabolic host niches. Several studies have shown that both glycolysis and gluconeogenesis systems are mandatory for the infection process, and moreover, S. aureus appears to be resistant to NO radicals that are heavily produced by the macrophages. Interestingly, glycolysis is an important process that contributes to persist within the macrophages and to protect the intracellular bacteria against NO (Vitko et al, 2015). However, if the bacteria escape the macrophages or lyse the host cells, S. aureus is thought to form aggregates at the center of highly inflamed and hypoxic abscesses. Under these conditions, the host cells consumed a large amount of glucose to fight the inflammation. Hence, glucose will become scarce for S. aureus suggesting that lactate and amino acids derived from the host might be used as the major sources of carbon to enter gluconeogenesis (Richardson et al, 2015). These conditions favored the activation of the two‐component system SrrAB, which in turn activates genes required for anaerobic metabolism, cytochrome, and heme biosynthesis, and NO radical detoxification should play an essential role in the survival of the bacteria (Kinkel et al, 2013). Because the CcpA‐dependent repression of RsaI is alleviated under conditions where the glucose is strongly reduced, and because many SrrAB‐dependent genes are also induced when RsaI is expressed at high levels, it is thus expected that RsaI might also contribute to metabolic adaptations of the cells to the dynamic nature of the host immune environment. Besides, it is also tempting to propose that RsaI might be involved in the dormant state of bacterial cells while environmental conditions are unfavorable (Lennon & Jones, 2011). Interestingly enough, we also observed a RsaI‐dependent activation of the expression of the mRNA encoding EsxA, a type VII‐secreted virulence factor required for the release of the intracellular S. aureus during infection (Korea et al, 2014). We also showed here that RsaI activates the synthesis of the PIA‐PNAG exopolysaccharide most probably by decreasing the translation of IcaR, which is the transcriptional repressor of icaADBC operon (Figs 4 and 6). We demonstrated that RsaI base‐pairs icaR mRNA without interfering the interaction between icaR 3′UTR and 5′UTR (Fig EV3). Although the precise molecular mechanism is not yet defined, we propose that RsaI would inhibit the synthesis of IcaR by stabilizing the circularization of the mRNA and/or by counteracting with the binding of an activation factor of icaR mRNA translation (Fig 6). Interestingly, RsaE, which is another RsaI target, also binds icaR mRNA at two distinct positions close to the SD sequence and in the coding sequence through C‐rich motifs (Rochat et al, 2018). These motifs are almost identical to the anti‐SD sequence (UCCCC) comprised in the icaR 3′UTR (Ruiz de los Mozos et al, 2013). This opens the possibility of a competition for hindering the icaR SD by RsaE or the 3′UTR. Thus, RsaI and RsaE might interact individually or simultaneously with icaR mRNA to repress its expression in response to changes in cellular medium of glucose and NO. Though further investigations are needed to decipher the physiological consequences of such complex interactions, it indicates that the regulation of the synthesis of the PIA‐PNAG is rather complexed and multifactorial and is tightly controlled according to the metabolic state of the bacterial population (Cue et al, 2012). The signals modifying the yield of these RNAs would be crucial to determine the levels of IcaR repressor and PIA‐PNAG synthesis. We also do not exclude that RsaI might regulate another regulator of the synthesis of PIA‐PNAG. For instance, inactivation of the TCA cycle resulted in a massive derepression of the PIA‐PNAG biosynthetic enzymes to produce the exopolysaccharide (Sadykov et al, 2008), and glucose enhances PIA‐PNAG‐dependent biofilm formation (You et al, 2014), while SrrAB appears as an inducer of PIA‐PNAG‐dependent biofilm (Ulrich et al, 2007). It remains to be elucidated if the biofilm induction mediated by SrrAB occurs through activation of RsaE that might promote interaction with icaR mRNA and/or RsaI. Hence, icaR can be considered as a hub gene, integrating both transcriptional regulatory networks and sRNA‐mediated post‐transcriptional regulatory signals to control biofilm formation in S. aureus.
Although our study shed light on the regulatory activities of this multifaceted sRNA, there is still much to be learned on how RsaI and other sRNAs can be integrated into the networks regulating the metabolic pathways that are essential for S. aureus biofilm formation, survival, persistence, and invasion within the host.
Materials and Methods
Plasmids and strains constructions
All strains and plasmids constructed in this study are described in Table EV8. The oligonucleotides designed for cloning and for mutagenesis are given in Table EV3. Escherichia coli strain DC10B was used as a host strain for plasmid amplification before electroporation in S. aureus. Plasmids were prepared from transformed E. coli pellets following the NucleoSpin Plasmid kit protocol (Macherey‐Nagel). Transformation of both E. coli and S. aureus strains was performed by electroporation (Bio‐Rad Gene Pulser).
The rsaI deletion mutant was constructed by homologous recombination using plasmid pMAD in S. aureus HG001 and 132 (Arnaud et al, 2004). The deletion comprises nucleotides 2,376,101–2,376,306 according to HG001 genome (Caldelari et al, 2017). Experimental details are given in supplementary materials.
The vector pCN51::PrsaI was constructed by ligating a PCR‐amplified fragment (Table EV3) containing rsaI with 166 pb of its promoter region and digested by SphI and PstI into pCN51 vector digested with the same enzymes. The vector pUC::T7rsaI was constructed by ligating a PCR‐amplified fragment (Table EV3) containing rsaI with the T7 promoter sequence and digested by EcoRI and PstI into pUC18 vector digested with the same enzymes. Mutagenesis of pUC::T7rsaI was performed with QuikChange XL Site‐directed mutagenesis (Stratagene) leading to pUC::T7rsaI mut1, 2, 3, and 4 (Table EV3). To obtain the plasmid pCN51::P3::MS2‐rsaI, a PCR product containing the MS2 tag fused to the 5′ end of rsaI was cloned into pCN51::P3 by digestion of both PCR fragments and of the plasmid by PstI/BamHI (Table EV3). Plasmids from positive clones were sequenced (GATC Biotech) before being transformed in DC10B, extracted, and electroporated into S. aureus strains. The plasmids pCN51::P3::MS2‐rsaI mut2 and mut4 were generated by QuikChange mutagenesis as above. Construction of plasmids pES::rsaI and pES::rsaI mut5 is described in supplementary materials.
Growth conditions
Escherichia coli strains were cultivated in Luria‐Bertani (LB) medium (1% peptone, 0.5% yeast extract, 1% NaCl) supplemented with ampicillin (100 μg/ml) when necessary. Staphylococcus aureus strains were grown in brain‐heart infusion (BHI), tryptic soy broth (TSB), or Muller Hinton broth (MHB) media (Sigma‐Aldrich) supplemented with erythromycin (10 μg/ml) for plasmid selection. When needed, MHB was complemented with either 5 g/l of glucose 6‐phosphate or 1 g/l of glucose, fructose, or xylose (Sigma‐Aldrich). NO production was induced by addition of 100 μM Na‐diethylamine‐NONOate (Sigma‐Aldrich) as previously described (Durand et al, 2015).
Preparation of total RNA extracts
Total RNAs were prepared from S. aureus cultures taken at different times of growth. After centrifugation, bacterial pellets were resuspended in 1 ml of RNA Pro Solution (MP Biomedicals). Lysis was performed with FastPrep and RNA purification followed strictly the procedure described for the FastRNA Pro Blue Kit (MP Biomedicals). Electrophoresis of either total RNA (10 μg) or MS2‐eluted RNA (500 ng) was performed on 1.5% agarose gel containing 25 mM guanidium thiocyanate. After migration, RNAs were vacuum‐transferred on nitrocellulose membrane. Hybridization with specific digoxigenin (DIG)‐labeled probes complementary to RsaI, RsaG, RsaD, 5S, ccpA sequences, followed by luminescent detection, was carried out as previously described (Tomasini et al, 2017).
MAPS experiments, and transcriptomic and RNA‐seq analysis
Crude bacterial extract was prepared and purified by affinity chromatography as previously described (Tomasini et al, 2017). The eluted RNA samples were either used for Northern blot or treated with DNase I prior to RNA‐seq analysis. Isolation of tagged sRNAs and the co‐purified RNAs was performed in duplicates. The experiments were carried out with the tagged wild‐type RsaI and two mutant forms (mut2 and mut4). RNAs were treated to deplete abundant rRNAs, and the cDNA libraries were performed using the ScriptSeq complete kit (bacteria) from Illumina. The libraries were sequenced using Illumina MiSeq with a V3 Reagent kit (Illumina), which preserves the information about the orientation of the transcripts and produces reads of 150 nts, which map on the complementary strand. Each RNA‐seq was performed at least in duplicates. The reads were then processed to remove adapter sequences and poor‐quality reads by Trimmomatic (Bolger et al, 2014), converted to the FASTQ format with FASTQ Groomer (Blankenberg et al, 2010), and aligned on the HG001 genome (Caldelari et al, 2017) using BOWTIE2 (Langmead et al, 2009). Finally, the number of reads mapping to each annotated feature has been counted with HTSeq (Anders et al, 2015) using the interception non‐empty protocol. To estimate the enrichment values for the MAPS experiment or the differential expression analysis for the transcriptomic experiment, we used DEseq2 (Varet et al, 2016). The statistical analysis process includes data normalization, graphical exploration of raw and normalized data, test for differential expression for each feature between the conditions, raw p‐value adjustment, and export of lists of features having a significant differential expression (threshold P‐value = 0.05; fold change threshold = 2) between the conditions. All processing steps were performed using the Galaxy platform (Afgan et al, 2016).
For total RNA extracts and MS2‐eluted RNAs, DNase I (0.1 U/μl) treatment was performed 1 h at 37°C. The reactions mixtures were then purified by phenol::chloroform::isoamylalcohol 25:24:1 (v/v) and subsequent ethanol precipitation. RNA pellets were resuspended in sterile milliQ water. RNA was quantified by Qubit (Life Technologies), and the integrity was assessed with the Bioanalyzer (Agilent Technologies). For transcriptomic, 1 μg of total RNA was ribo‐depleted with the bacterial RiboZero kit from Illumina. The TruSeq total RNA‐stranded kit from Illumina was used for the library preparation. Library quantity was measured by Qubit, and its quality was assessed with a Tapestation on a DNA High sensitivity chip (Agilent Technologies). Libraries were pooled at equimolarity and loaded at 7 pM for clustering. The 50 bases oriented single‐read sequencing was performed using TruSeq SBS HS v3 chemistry on an Illumina HiSeq 2500 sequencer.
Preparation of RNAs for in vitro experiments
Transcription of RsaI, RsaI mutants, and RsaG was performed using linearized pUC18 vectors. PCR fragments containing the 5′UTR of the coding region of selected mRNA targets were directly used as templates for in vitro transcription using T7 RNA polymerase. The RNAs were then purified using a 6% or 8% polyacrylamide‐8 M urea gel electrophoresis. After elution with 0.5 M ammonium acetate pH 6.5 containing 1 mM EDTA, the RNAs were precipitated in cold absolute ethanol, washed with 85% ethanol, and vacuum‐dried. The labeling of the 5′ end of dephosphorylated RNAs (RsaI/RsaI mutants) and DNA oligonucleotides was performed with T4 polynucleotide kinase (Fermentas) and [γ32P] ATP. Before use, cold or labeled RNAs were renaturated by incubation at 90°C for 1 min in 20 mM Tris–HCl pH 7.5, cooled 1 min on ice, and incubated 10 min at 20°C in ToeP+ buffer (20 mM Tris–HCl pH 7.5, 10 mM MgCl2, 60 mM KCl, 1 mM DTT).
Gel retardation assays
Radiolabeled purified RsaI or RsaI mutants (50,000 cps/sample, concentration < 1 pM) and cold mRNAs were renaturated separately as described above. For each experiment, increasing concentrations of cold mRNAs were added to the 5′ end‐labeled wild‐type or RsaI mutants in a total volume of 10 μl containing the ToeP+ buffer. Complex formation was performed at 37°C for 15 min. After incubation, 10 μl of glycerol blue was added and the samples were loaded on a 6% or 8% polyacrylamide gel under non‐denaturing conditions (300 V, 4°C). Under these conditions where the concentration of the labeled RNA is negligible, the KD dissociation constant can be estimated as the concentration of the cold RNA that showed 50% of binding.
Toe‐printing assays
The preparation of E. coli 30S subunits, the formation of a simplified translational initiation complex with mRNA, and the extension inhibition conditions were performed as previously described (Fechter et al, 2009). Increasing concentrations of RsaI were used to monitor their effects on the formation of the initiation complex with glcU_2 and fn3K mRNAs.
In vivo β‐galactosidase assays
Translation fusions were constructed with plasmid pLUG220, a derivative of pTCV‐lac, a low‐copy‐number promoter‐less lacZ vector, containing the constitutive rpoB promoter (Table EV8). The whole leader regions of glcU_2 (−54/+99), fn3K (−33/+99), treB (−23/+99), HG001_01242 (−34/+99), and HG001_02520 (−71/+99) (Table EV3) were cloned downstream the PrpoB in frame with lacZ. The whole gene encoding RsaI or RsaI mut4 (obtained synthetically from IDT) with 166 bp of its promoter region was digested with PstI and ligated at the unique PstI of pLUG220::PrpoB vector. The final constructs were transformed into the strain HG001ΔrsaI. β‐galactosidase activity was measured four times as previously described (Tomasini et al, 2017).
PIA‐PNAG quantification
Cell surface PIA‐PNAG exopolysaccharide levels were monitored according to Cramton et al (1999). Overnight cultures were diluted 1:50 in TSB‐3% NaCl, and bacteria were grown at 37°C. Samples were extracted at 6 h after inoculation. The same number of cells of each strain was resuspended in 50 μl of 0.5 M EDTA (pH 8.0). Then, cells were incubated for 5 min at 100°C and centrifuged 17,000 g for 5 min. Supernatants (40 μl) were incubated with 10 μl of proteinase K (20 mg/ml) (Sigma) for 30 min at 37°C. After the addition of 10 μl of the buffer (20 mM Tris–HCl pH 7.4, 150 mM NaCl, 0.01% bromophenol blue), serial dilutions 1:25 were performed in the same buffer. Then, 10 μl was spotted on a nitrocellulose membrane using a Bio‐Dot microfiltration apparatus (Bio‐Rad). The membrane was blocked overnight with 5% skimmed milk in phosphate‐buffered saline (PBS) with 0.1% Tween 20 and incubated for 2 h with specific anti‐PNAG antibodies diluted 1:20,000 (Maira‐Litrán et al, 2005). Bound antibodies were detected with peroxidase‐conjugated goat anti‐rabbit immunoglobulin G antibodies (Jackson ImmunoResearch Laboratories, Inc., Westgrove, PA) diluted 1:10,000, using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
Author contributions
DB, ED, and IC performed the MAPS experiments and the validation of the data. CJC, LP, AT‐A, and IL constructed several mutant strains and performed the biofilm assays. AC and PF have done the transcriptomic analysis. SM analyzed all the RNA‐Seq analysis and quantification of the data. KM and FV contributed to phenotypic analysis of some of the mutant strains. PR and IC were responsible of the project and wrote the manuscript. All authors contributed to the analysis, interpretation of the experiments, and writing of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Table EV8
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank David Lalaouna for critical reading of the manuscript and helpful discussions, Thomas Geissmann for helpful advices, Marie Beaume for the construction of the mutant strain HG001‐ΔrsaI, and Eve‐Julie Bonetti and Anne‐Catherine Helfer for excellent technical assistance. We are grateful to Joseph Vilardell for the gift of the plasmid expressing the MS2‐MBP, Aurélia Hiron and Tarek Masdek for providing us the HG001‐∆hptRS and ‐∆srrAB mutant strains, and Christiane Wolz for the HG001‐∆ccpA and ‐∆codY mutant strain. RNA‐Seq analyses have been partially done using the Roscoff (France) instance of Galaxy (http://galaxy.sb-roscoff.fr/). This work was supported by the Centre National de la Recherche Scientifique (CNRS) to [P.R.] and by the Agence Nationale de la Recherche (ANR, grant ANR‐16‐CE11‐0007‐01, RIBOSTAPH, to [P.R.]). It has also been published under the framework of the LABEX: ANR‐10‐LABX‐0036 NETRNA to [P.R.], a funding from the state managed by the French National Research Agency as part of the investments for the future program. The work is financed by a “Projet international de coopération scientifique” (PICS) No. PICS07507 between France and Spain to [I.C.]. D. Bronesky was supported by Fondation pour la Recherche Médicale (FDT20160435025). A. T‐A is financed by the Spanish Ministry of Economy and Competitiveness (BFU2014‐56698‐P) and the European Research Council Consolidator Grant (646869‐ReguloBac‐3UTR).
The EMBO Journal (2019) 38:e99363
Contributor Information
Pascale Romby, Email: p.romby@ibmc-cnrs.unistra.fr.
Isabelle Caldelari, Email: i.caldelari@ibmc-cnrs.unistra.fr.
Data availability
The transcriptomics and MAPS data have been deposited in NCBI's Gene Expression Omnibus (Barrett et al, 2013) and are accessible through the GEO Series accession number GSE122092 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122092).
References
- Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Eberhard C, Grüning B, Guerler A, Hillman‐Jackson J, Von Kuster G, Rasche E, Soranzo N, Turaga N, Taylor J, Nekrutenko A, Goecks J (2016) The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res 44: W3–W10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2015) HTSeq‐A Python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud M, Chastanet A, Débarbouillé M (2004) New vector for efficient allelic replacement in naturally nontransformable, low‐GC‐content, gram‐positive bacteria. Appl Environ Microbiol 70: 6887–6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A (2013) NCBI GEO: archive for functional genomics data sets‐update. Nucleic Acids Res 41: D991–D995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassias J, Brückner R (1998) Regulation of lactose utilization genes in Staphylococcus xylosus . J Bacteriol 180: 2273–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisel CL, Storz G (2011) The base‐pairing NA spot42 participates in a multioutput feedforward loop to help enact catabolite repression in Escherichia coli . Mol Cell 41: 286–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff M, Wonnenberg B, Nippe N, Nyffenegger‐Jann NJ, Voss M, Beisswenger C, Sunderkötter C, Molle V, Dinh QT, Lammert F, Bals R, Herrmann M, Somerville GA, Tschernig T, Gaupp R (2017) CcpA affects infectivity of Staphylococcus aureus in a hyperglycemic environment. Front Cell Infect Microbiol 7: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Gordon A, Von Kuster G, Coraor N, Taylor J, Nekrutenko A, Team G (2010) Manipulation of FASTQ data with galaxy. Bioinformatics 26: 1783–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrovskyy M, Vanderpool CK (2016) Diverse mechanisms of post‐transcriptional repression by the small RNA regulator of glucose‐phosphate stress. Mol Microbiol 99: 254–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn C, Rigoulay C, Chabelskaya S, Sharma CM, Marchais A, Skorski P, Borezée‐Durant E, Barbet R, Jacquet E, Jacq A, Gautheret D, Felden B, Vogel J, Bouloc P (2010) Experimental discovery of small RNAs in Staphylococcus aureus reveals a riboregulator of central metabolism. Nucleic Acids Res 38: 6620–6636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldelari I, Chane‐Woon‐Ming B, Noirot C, Moreau K, Romby P, Gaspin C, Marzi S (2017) Complete genome sequence and annotation of the Staphylococcus aureus strain HG001. Genome Announc 5: e00783‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramton SE, Gerke C, Schnell NF, Nichols WW, Gotz F (1999) The intercellular adhesion (ica) locus is present in Staphylococcus aureus and is required for biofilm formation. Infect Immun 67: 5427–5433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cue D, Lei MG, Lee CY (2012) Genetic regulation of the intercellular adhesion locus in Staphylococci. Front Cell Infect Microbiol 2: 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deppe VM, Bongaerts J, O'Connell T, Maurer KH, Meinhardt F (2011) Enzymatic deglycation of Amadori products in bacteria: mechanisms, occurrence and physiological functions. Appl Microbiol Biotechnol 90: 399–406 [DOI] [PubMed] [Google Scholar]
- Durand S, Braun F, Helfer AC, Romby P, Condon C (2017) sRNA‐mediated activation of gene expression by inhibition of 5′‐3′ exonucleolytic mRNA degradation. Elife 6: e23602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand S, Braun F, Lioliou E, Romilly C, Helfer AC, Kuhn L, Quittot N, Nicolas P, Romby P, Condon C (2015) A nitric oxide regulated small RNA controls expression of genes involved in redox homeostasis in Bacillus subtilis . PLoS Genet 11: e1004957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fechter P, Chevalier C, Yusupova G, Yusupov M, Romby P, Marzi S (2009) Ribosomal initiation complexes probed by toeprinting and effect of trans‐acting translational regulators in bacteria. Methods Mol Biol 540: 247–263 [DOI] [PubMed] [Google Scholar]
- Geissmann T, Chevalier C, Cros MJ, Boisset S, Fechter P, Noirot C, Schrenzel J, François P, Vandenesch F, Gaspin C, Romby P (2009) A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res 37: 7239–7257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemayel R, Fortpied J, Rzem R, Vertommen D, Veiga‐da‐Cunha M, Van Schaftingen E (2007) Many fructosamine 3‐kinase homologues in bacteria are ribulosamine/erythrulosamine 3‐kinases potentially involved in protein deglycation. FEBS J 274: 4360–4374 [DOI] [PubMed] [Google Scholar]
- Gimpel M, Pries H, Barth E, Gramzow L, Brantl S (2012) SR1‐a small RNA with two remarkable conserved functions. Nucleic Acids Res 40: 11659–11672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz F, Bannerman T, Schleifer K (2006) The Genera Staphylococcus and Macrococcus In The Prokaryotes, Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E. (eds), pp 5–75. New York, NY: Springer; [Google Scholar]
- Heidrich N, Moll I, Brantl S (2007) In vitro analysis of the interaction between the small RNA SR1 and its primary target ahrC mRNA. Nucleic Acids Res 35: 4331–4346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert S, Ziebandt AK, Ohlsen K, Schäfer T, Hecker M, Albrecht D, Novick R, Götz F (2010) Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect Immun 78: 2877–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden BP, Beaume M, Harrison PF, Hernandez D, Schrenzel J, Seemann T, Francois P, Stinear TP (2013) Analysis of the small RNA transcriptional response in multidrug‐resistant Staphylococcus aureus after antimicrobial exposure. Antimicrob Agents Chemother 57: 3864–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto H, Morita T, Shimizu A, Inada T, Aiba H (2005) Implication of membrane localization of target mRNA in the action of a small RNA: mechanism of post‐transcriptional regulation of glucose transporter in Escherichia coli . Genes Dev 19: 328–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkel TL, Roux CM, Dunman PM, Fang FC (2013) The Staphylococcus aureus SrrAB two‐component system promotes resistance to nitrosative stress and hypoxia. MBio 4: e00696‐13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korea CG, Balsamo G, Pezzicoli A, Merakou C, Tavarini S, Bagnoli F, Serruto D, Unnikrishnan M (2014) Staphylococcal Esx proteins modulate apoptosis and release of intracellular Staphylococcus aureus during infection in epithelial cells. Infect Immun 82: 4144–4153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalaouna D, Carrier MC, Semsey S, Brouard JS, Wang J, Wade JT, Masse E (2015) A 3′ external transcribed spacer in a tRNA transcript acts as a sponge for small RNAs to prevent transcriptional noise. Mol Cell 58: 393–405 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Bowtie: an ultrafast memory‐efficient short read aligner. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon JT, Jones SE (2011) Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev Microbiol 9: 119–130 [DOI] [PubMed] [Google Scholar]
- Li C, Sun F, Cho H, Yelavarthi V, Sohn C, He C, Schneewind O, Bae T (2010) CcpA mediates proline auxotrophy and is required for Staphylococcus aureus pathogenesis. J Bacteriol 192: 3883–3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht A, Preis S, Brantl S (2005) Implication of CcpN in the regulation of a novel untranslated RNA (SR1) in Bacillus subtilis . Mol Microbiol 58: 189–206 [DOI] [PubMed] [Google Scholar]
- Liu Q, Yeo WS, Bae T (2016) The SaeRS two‐component system of Staphylococcus aureus . Genes 7: E81 27706107 [Google Scholar]
- Maira‐Litrán T, Kropec A, Goldmann DA, Pier GB (2005) Comparative opsonic and protective activities of Staphylococcus aureus conjugate vaccines containing native or deacetylated Staphylococcal Poly‐N‐acetyl‐beta‐(1‐6)‐glucosamine. Infect Immun 73: 6752–6762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majerczyk CD, Dunman PM, Luong TT, Lee CY, Sadykov MR, Somerville GA, Bodi K, Sonenshein AL (2010) Direct targets of CodY in Staphylococcus aureus . J Bacteriol 192: 2861–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchais A, Bohn C, Bouloc P, Gautheret D (2010) RsaOG, a new staphylococcal family of highly transcribed non‐coding RNA. RNA Biol 7: 116–119 [DOI] [PubMed] [Google Scholar]
- Miyakoshi M, Chao Y, Vogel J (2015) Cross talk between ABC transporter mRNAs via a target mRNA‐derived sponge of the GcvB small RNA. EMBO J 34: 1478–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller T, Franch T, Udesen C, Gerdes K, Valentin‐Hansen P (2002) Spot 42 RNA mediates discoordinate expression of the E. coli galactose operon. Genes Dev 16: 1696–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick RP (2003) Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol Microbiol 48: 1429–1449 [DOI] [PubMed] [Google Scholar]
- Papenfort K, Sun Y, Miyakoshi M, Vanderpool CK, Vogel J (2013) Small RNA‐mediated activation of sugar phosphatase mRNA regulates glucose homeostasis. Cell 153: 426–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfort K, Vogel J (2014) Small RNA functions in carbon metabolism and virulence of enteric pathogens. Front Cell Infect Microbiol 4: 91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Kim JW, Moon BY, Lee J, Fortin YJ, Austin FW, Yang SJ, Seo KS (2015) Characterization of a novel two‐component regulatory system, HptRS, the regulator for the hexose phosphate transport system in Staphylococcus aureus . Infect Immun 83: 1620–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl K, Francois P, Stenz L, Schlink F, Geiger T, Herbert S, Goerke C, Schrenzel J, Wolz C (2009) CodY in Staphylococcus aureus: a regulatory link between metabolism and virulence gene expression. J Bacteriol 191: 2953–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pragman AA, Yarwood JM, Tripp TJ, Schlievert PM (2004) Characterization of virulence factor regulation by SrrAB, a two‐component system in Staphylococcus aureus . J Bacteriol 186: 2430–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice JB, Balasubramanian D, Vanderpool CK (2012) Small RNA binding‐site multiplicity involved in translational regulation of a polycistronic mRNA. Proc Natl Acad Sci USA 104: 20454–20459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards GR, Patel MV, Llyod CR, Vanderpool CK (2013) Depletion of glycolytic intermediates plays a key role in glucose‐phosphate stress Escherichia coli . J Bacteriol 195: 4816–4825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson AR, Somerville GA, Sonenshein AL (2015) Regulating the intersection of metabolism and pathogenesis in gram‐positive bacteria. Microbiol Spectr 3: 1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson AR, Dunman PM, Fang FC (2006) The nitrosative stress response of Staphylococcus aureus is required for resistance to innate immunity. Mol Microbiol 4: 927–939 [DOI] [PubMed] [Google Scholar]
- Rochat T, Bohn C, Morvan C, Le Lam TN, Razvi F, Pain A, Toffano‐Nioche C, Ponien P, Jacq A, Jacquet E, Fey PD, Gautheret D, Bouloc P (2018) The conserved regulatory RNA RsaE down‐regulates the arginine degradation pathway in Staphylococcus aureus . Nucleic Acids Res 46: 8803–8816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz de los Mozos I, Vergara‐Irigaray M, Segura V, Villanueva M, Bitarte N, Saramago M, Domingues S, Arraiano CM, Fechter P, Romby P, Valle J, Solano C, Lasa I, Toledo‐Arana A (2013) Base pairing interaction between 5′‐ and 3′‐UTRs controls icaR mRNA translation in Staphylococcus aureus . PLoS Genet 9: e1004001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadykov MR, Olson ME, Halouska S, Zhu Y, Fey PD, Powers R, Somerville GA (2008) Tricarboxylic acid cycle‐dependent regulation of Staphylococcus epidermidis polysaccharide intercellular adhesin synthesis. J Bacteriol 190: 7621–7632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl K, Bischoff M, Berger‐Bächi B (2008a) CcpA mediates the catabolite repression of tst in Staphylococcus aureus . Infect Immun 76: 5093–5099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl K, Goerke C, Wolz C, Mack D, Berger‐Bächi B, Bischoff M (2008b) Staphylococcus aureus CcpA affects biofilm formation. Infect Immun 76: 2044–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl K, Müller S, François P, Kriebitzsch C, Schrenzel J, Engelmann S, Bischoff M, Berger‐Bächi B (2009) Effect of a glucose impulse on the CcpA regulon in Staphylococcus aureus . BMC Microbiol 9: 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl K, Stucki M, Ruegg M, Goerke C, Wolz C, Harris L, Berger‐Bächi B, Bischoff M (2006) Staphylococcus aureus CcpA affects virulence determinant production and antibiotic resistance. Antimicrob Agents Chemother 50: 1183–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville GA, Proctor RA (2009) At the crossroads of bacterial metabolism and virulence factor synthesis in staphylococci. Microbiol Mol Biol Rev 73: 233–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasini A, Moreau K, Chicher J, Geissmann T, Vandenesch F, Romby P, Marzi S, Caldelari I (2017) The RNA targetome of Staphylococcus aureus non‐coding RNA RsaA: impact on cell surface properties and defense mechanisms. Nucleic Acids Res 45: 6746–6760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich M, Bastian M, Cramton SE, Ziegler K, Pragman AA, Bragonzi A, Memmi G, Wolz C, Schlievert PM, Cheung A, Döring G (2007) The staphylococcal respiratory response regulator SrrAB induces ica gene transcription and polysaccharide intercellular adhesin expression, protecting Staphylococcus aureus from neutrophil killing under anaerobic growth conditions. Mol Microbiol 65: 1276–1287 [DOI] [PubMed] [Google Scholar]
- Vanderpool CK, Gottesman S (2004) Involvement of a novel transcriptional activator and small RNA in post‐transcriptional regulation of the glucose phosphoenolpyruvate phosphotransferase system. Mol Microbiol 54: 1076–1089 [DOI] [PubMed] [Google Scholar]
- Vanderpool CK, Gottesman S (2007) The novel transcription factor SgrR coordinates the response to glucose‐phosphate stress. J Bacteriol 189: 2238–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varet H, Brillet‐Guéguen L, Coppée JY, Dillies MA (2016) SARTools: a DESeq2‐ and edgeR‐based R pipeline for comprehensive differential analysis of RNA‐Seq data. PLoS ONE 11(6): e0157022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara‐Irigaray M, Valle J, Merino N, Latasa C, Garcia B, Ruiz de Los Mozos I, Solano C, Toledo‐Arana A, Penades JR, Lasa I (2009) Relevant role of fibronectin‐binding proteins in Staphylococcus aureus biofilm‐associated foreign‐body infections. Infect Immun 77: 3978–3991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitko NP, Grosser MR, Khatri D, Lance TR, Richardson AR (2016) Expanded glucose import capability affords Staphylococcus aureus optimized glycolytic flux during infection. MBio 7: e00296‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitko NP, Spahich NA, Richardson AR (2015) Glycolytic dependency of high‐level nitric oxide resistance and virulence in Staphylococcus aureus . MBio 6: e00045‐15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y, Xue T, Cao L, Zhao L, Sun H, Sun B (2014) Staphylococcus aureus glucose‐induced biofilm accessory proteins, GbaAB, influence biofilm formation in a PIA‐dependent manner. Int J Med Microbiol 304: 603–612 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Table EV8
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
The transcriptomics and MAPS data have been deposited in NCBI's Gene Expression Omnibus (Barrett et al, 2013) and are accessible through the GEO Series accession number GSE122092 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122092).