

Abstract
Key points
Mutations in the caveolae scaffolding protein, caveolin‐3 (Cav3), have been linked to the long QT type 9 inherited arrhythmia syndrome (LQT9) and the cause of underlying action potential duration prolongation is incompletely understood.
In the present study, we show that LQT9 Cav3 mutations, F97C and S141R, cause mutation‐specific gain of function effects on Cav1.2‐encoded L‐type Ca2+ channels responsible for I Ca,L and also cause loss of function effects on heterologously expressed Kv4.2 and Kv4.3 channels responsible for I to.
A computational model of the human ventricular myocyte action potential suggests that the major ionic current change causing action potential duration prolongation in the presence of Cav3‐F97C is the slowly inactivating I Ca,L but, for Cav3‐S141R, both increased I Ca,L and increased late Na+ current contribute equally to action potential duration prolongation.
Overall, the LQT9 Cav3‐F97C and Cav3‐S141R mutations differentially impact multiple ionic currents, highlighting the complexity of Cav3 regulation of cardiac excitability and suggesting mutation‐specific therapeutic approaches.
Abstract
Mutations in the CAV3 gene encoding caveolin‐3 (Cav3), a scaffolding protein integral to caveolae in cardiomyocytes, have been associated with the congenital long‐QT syndrome (LQT9). Initial studies demonstrated that LQT9‐associated Cav3 mutations, F97C and S141R, increase late sodium current as a potential mechanism to prolong action potential duration (APD) and cause LQT9. Whether these Cav3 LQT9 mutations impact other caveolae related ion channels remains unknown. We used the whole‐cell, patch clamp technique to characterize the effect of Cav3‐F97C and Cav3‐S141R mutations on heterologously expressed Cav1.2+Cavβ2cN4 channels, as well as Kv4.2 and Kv4.3 channels, in HEK 293 cells. Expression of Cav3‐S141R increased I Ca,L density without changes in gating properties, whereas expression of Cav3‐F97C reduced Ca2+‐dependent inactivation of I Ca,L without changing current density. The Cav3‐F97C mutation reduced current density and altered the kinetics of I Kv4.2 and I Kv4.3 and also slowed recovery from inactivation. Cav3‐S141R decreased current density and also slowed activation kinetics and recovery from inactivation of I Kv4.2 but had no effect on I Kv4.3. Using the O'Hara–Rudy computational model of the human ventricular myocyte action potential, the Cav3 mutation‐induced changes in I to are predicted to have negligible effect on APD, whereas blunted Ca2+‐dependent inactivation of I Ca,L by Cav3‐F97C is predicted to be primarily responsible for APD prolongation, although increased I Ca,L and late I Na by Cav3‐S141R contribute equally to APD prolongation. Thus, LQT9 Cav3‐associated mutations, F97C and S141R, produce mutation‐specific changes in multiple ionic currents leading to different primary causes of APD prolongation, which suggests the use of mutation‐specific therapeutic approaches in the future.
Keywords: arrhythmia, cardiac electrophysiology, cardiac potassium current, voltage‐dependent calcium channel, caveola, computer modeling
Key points
Mutations in the caveolae scaffolding protein, caveolin‐3 (Cav3), have been linked to the long QT type 9 inherited arrhythmia syndrome (LQT9) and the cause of underlying action potential duration prolongation is incompletely understood.
In the present study, we show that LQT9 Cav3 mutations, F97C and S141R, cause mutation‐specific gain of function effects on Cav1.2‐encoded L‐type Ca2+ channels responsible for I Ca,L and also cause loss of function effects on heterologously expressed Kv4.2 and Kv4.3 channels responsible for I to.
A computational model of the human ventricular myocyte action potential suggests that the major ionic current change causing action potential duration prolongation in the presence of Cav3‐F97C is the slowly inactivating I Ca,L but, for Cav3‐S141R, both increased I Ca,L and increased late Na+ current contribute equally to action potential duration prolongation.
Overall, the LQT9 Cav3‐F97C and Cav3‐S141R mutations differentially impact multiple ionic currents, highlighting the complexity of Cav3 regulation of cardiac excitability and suggesting mutation‐specific therapeutic approaches.
Introduction
Caveolae are flask‐shaped membrane microdomains enriched in cholesterol and sphingolipids that are present in many cell types. Caveolae serve as signalling hubs localizing a number of different ion channels and signalling proteins (Balijepalli & Kamp, 2008). In cardiomyocytes, a variety of ion channels and transporters have been localized to caveolae, including voltage‐gated Na+ channels (Nav1.5) (Yarbrough et al. 2002), L‐type Ca2+ channels (Cav1.2) (Balijepalli et al. 2006), a subset of voltage‐gated K+ channels (Kv4.2, and Kv4.3) (Alday et al. 2010), inward rectifier channels K+ channels (Kir2.1) (Vaidyanathan et al. 2013), Na+/Ca2+ exchanger (NCX) (Bossuyt et al. 2002) and hyperpolarization activated channels (HCN4) (Barbuti et al. 2007; Ye et al. 2008). However, the extent to which cardiac ion channels are localized to caveolae, the dynamics of that localization, and the specific regulation of caveolar ion channels represent critical issues that need to be addressed order to advance the understanding of cardiac excitability and arrhythmias.
Caveolin‐3 (Cav3) is a muscle‐specific isoform of the caveolin scaffolding protein family required for caveolae formation in muscle cells (Song et al. 1996). Mutations in CAV3 can lead to a variety of pathological conditions impacting primarily cardiac and skeletal muscle (Balijepalli & Kamp, 2008). For example, F97C and S141R mutations in CAV3 have been linked to the congenital long QT syndrome 9 (LQT9) (Vatta et al. 2006), which is characterized by a prolongation of the QT interval on the electrocardiogram and predisposition of affected individuals to ventricular tachycardia, syncope and sudden cardiac death (Schwartz et al. 2012). Cav3‐F97C and Cav3‐S141R increased the late sodium channel current in heterologous expression studies in HEK293 cells, providing a potential mechanism for delayed repolarization and the associated LQT9 (Vatta et al. 2006). The increase in late sodium current induced by the Cav3‐F97C mutation was further demonstrated in cultured rat ventricular myocytes and induced pluripotent stem cell‐derived cardiomyocytes (Cheng et al. 2013; Vaidyanathan et al. 2016). In addition, the Cav3‐F97C mutation was shown to decrease the inward rectifier K+ current encoded by Kir2.1 when expressed in HEK293 cells (Vaidyanathan et al. 2013). However, whether the F97C and S141R Cav3 mutations affect other currents impacting repolarization of the cardiomyocyte action potential (AP) remains unknown.
In the present study, we aimed to determine the impact of LQT9‐associated Cav3 mutations, F97C and S141R, on the function of Kv4.2, Kv4.3 and Cav1.2 channels. We focused on these channels because there is evidence that they are localized to caveolae in ventricular cardiomyocytes (Balijepalli et al. 2006; Alday et al. 2010) and thus may be impacted by the LQT9‐associated mutations. Using heterologous expression of the channels and Cav3 mutations in HEK293 cells, we found that the Cav3‐S141R mutation increased I Ca,L density without an effect on current kinetics, whereas Cav3‐F97C slowed Ca2+‐dependent inactivation (CDI) of I Ca,L with no effect on the current density. Both Cav3‐F97C and Cav3‐S141R mutations decrease I Kv4.2 density and slow recovery from inactivation, although the mutations differentially impact I Kv4.3. The Cav3‐F97C mutation decreased I Kv4.3 and slowed the recovery from inactivation, whereas the Cav3‐S141R mutation had no effect on I Kv4.3. By use of a human ventricular cardiomyocyte computational model and integrating our heterologous expression data, we found that, for both Cav3 mutations, the changes to I Kv4.3 had a negligible effect on the AP duration (APD). In the Cav3‐F97C model, the APD prolongation was dominated by the contribution from I Ca,L; however, in the Cav3‐S141R model, I Ca,L and I Na,L contributed equally to APD prolongation. Altogether, our findings demonstrate that Cav3‐F97C and Cav3‐S141R mutations lead to loss‐of‐function effects on I Kv4.2/Kv4.3 and gain‐of‐function effects on I Ca,L, although these effects on ionic currents are Cav3 mutation‐specific contributing to distinct ionic current alterations that can produce APD prolongation and promote ventricular arrhythmogenesis in LQT9. Furthermore, the distinct effects of the Cav3 mutations on Cav1.2 channels, altering current density or CDI specifically, suggest that Cav3 can impact the regulation of channel function in multiple ways.
Methods
Chemical reagents and antibodies
All chemicals and reagents were procured from Sigma (St Louis, MO, USA) unless otherwise stated.
Plasmid construction
The cDNA for Kv4.2 (human KCND2, Genbank accession no. BC110449) and Kv4.3 (human KCND3, Genbank accession no. BC113475) were obtained from Open Biosystems (GE Life Sciences, Lafayette, CO, USA) and subcloned into pcDNA3.1 plasmid vector (Invitrogen, Carlsbad, CA, USA), as described previously (Vatta et al. 2006). Plasmids encoding the human pore‐forming α1C77 (Cav1.2), auxiliary β2cN4 L‐type Ca2+ channel and haemagglutinin (HA)‐Cav1.2 subunits have been described previously (Soldatov et al. 1995; Foell et al. 2004; Best et al. 2011). Preparation and subcloning of wild‐type (WT) human Cav3 (GenBank accession no. NM_033337), Cav3‐F97C and Cav3‐S141R mutations have been reported previously (Vatta et al. 2006).
HEK293 cell maintenance and transfections
HEK293 cells were grown and maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Gibco, Gaithersburg, MD, USA), 100 U mL–1 penicillin, 100 g mL−1 streptomycin and 2 mm l‐glutamine. Prior to transfection, HEK293 cells were plated on 35 mm tissue culture dishes, at densities of between 2 and 2.5 × 104 cells/dish. The Cav3‐WT, Cav3‐F97C and Cav3‐S141R (1 μg each) were then transiently co‐transfected with expression vectors containing Cav1.2 and Cavβ2cN4 L‐type Ca2+ channel subunits with pSV40Tag (Foell et al. 2004), Kv4.2 or Kv4.3 at a ratio of 1:1, respectively, using Lipofectamine 2000 (Invitrogen) in accordance with the manufacturer's instructions. In all cases, 0.15 μg of plasmid DNA encoding green fluorescent protein (GFP) was included to identify transfected cells. Two days after transfection, GFP‐positive HEK293 cells were selected for functional studies.
Immunofluorescence confocal microscopy
Immunolabelling was performed on transfected HEK293 cells using a mouse monoclonal antibody to Cav3 (catalogue number 610420; BD Biosciences, Rockville, MD, USA) or a rabbit polyclonal antibody to Cav3 (catalogue number PA1‐066; ThermoFisher Scientific, Waltham, MA, USA). HEK293 cells were transfected with an HA‐tagged Cav1.2 (labelled with rat anti‐HA antibody; Clone 3F10; Roche Applied Science, Indianapolis, IN, USA), Kv4.2 (labelled with rabbit anti‐Kv4.2 polyclonal antibody; catalogue number PA1‐12660; ThermoFisher Scientific) and Kv4.3 (labelled with mouse anti‐Kv4.3 monoclonal antibody; catalogue number 75‐017; Neuromab, Davis, CA, USA). HEK293 cells were fixed 2 days after transfection with 2% buffered paraformaldehyde for 10 min, permeabilized with Triton X‐100 (0.1%) for 10 min and then quenched for aldehyde groups in 0.75% glycine buffer for 10 min. After being washed with TBS (2 × 10 min), cells were incubated with 1 mL of blocking solution (2% BSA and 2% goat serum, 0.05% NaN3 in Tris‐buffered saline) for 2 h in 35 mm tissue culture dishes with gentle agitation at 4°C to block the non‐specific binding sites. Subsequently, cells were incubated overnight with respective primary antibodies in blocking solution at 4°C. Antibody dilution for primary antibodies was 1:250 for anti‐HA; 1:200 for the polyclonal anti‐Kv4.2, anti‐Kv4.3 and rabbit polyclonal anti‐Cav3; and 1:500 for the mouse monoclonal anti‐Cav3. Excess primary antibody was washed off with blocking solution (3 × 1 h). Cells were then incubated for 1 h with Alexa‐conjugated secondary antibodies (2 mg ml−1; Molecular Probes, Inc. Eugene, OR, USA) diluted 1:200 in blocking solution. Highly cross‐absorbed Alexa 568 goat anti‐rabbit IgG (H+L) (catalogue number A‐11011; Molecular Probes, Inc.), Alexa 430 goat anti‐mouse IgG (H+L) (catalogue number A‐11063; Molecular Probes, Inc.) and Alexa 488 goat anti‐rat IgG (H+L) (catalogue number A‐11006; Invitrogen) were used for immunostaining of transfected HEK293 cells. Cells were then washed with blocking solution (3 × 2 h). After the final wash, cells were mounted using ProLong® Gold Antifade Mountant (ThermoFisher Scientific). To determine non‐specific immunolabelling by the secondary antibody, control experiments were routinely performed with secondary antibody without the primary antibody, and these experiments demonstrated a minimal fluorescence signal (data not shown). To determine the specificity of the primary antibodies for the transfected channel constructs, control immunolabelling experiments were also performed using non‐transfected cells, which demonstrated no specific immunolabelling (data not shown). HEK293 cells were imaged with a SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany) using 63× oil objective and filters. Excitation and emission were set at 415/525 ± 20 nm for Alexa 430, 488/525 ± 20 nm for Alexa 488 and 561/525 ± 20 nm for Alexa 568. A sequencing pattern and 256 line averaging was applied during imaging.
Western blot
Transiently transfected HEK293 cells grown on 35 mm diameter tissue culture dishes were harvested, suspended in lysis buffer (50 mm Tris‐HCl, pH 7.40, 150 mm NaCl, 0.4% deoxycholate and 1% Triton X‐100 supplemented with 2 mm phenylmethylsulphonyl fluoride, 12 μg ml−1 aprotinin, 1 μm benzamidin, 21.5 μm leupeptin, 5 μm pepstatin A and calpain inhibitors from Sigma) and sonicated 48 h after transfection. Then, 50 μg of input protein was resolved by SDS‐PAGE and transferred to 0.2 μm polyvinylidene fluoride membrane. To detect multiple proteins, blots were cut and incubated with anti‐Cav3 (catalogue number 610421; Becton‐Dickinson Biosciences, Franklin Lakes, NJ, USA) or anti‐GAPDH (MAB374; Millipore Sigma, Burlington, MA, USA) and then incubated with HRP‐conjugated secondary antibodies.
Whole‐cell, patch clamp electrophysiology
Electrophysiological experiments were carried out in the whole‐cell configuration of the patch clamp technique at room temperature using the Axopatch 200B amplifier with pCLAMP, version 10.2 (Axon Instruments, Foster City, CA, USA). Recording pipettes were pulled from thin walled borosilicate glass capillaries (World Precision Instruments, Inc., Sarasota, FL, USA) with a pipette resistance of 1.5–2.5 MΩ. The recordings were filtered at 5 kHz and digitized at 50 kHz. All solutions and buffers are indicated in mmol L–1. For I to current measurements, external bath solution contained 136 NaCl, 4 KCl, 2 MgCl2, 1 CaCl2, 5 CoCl2, 10 glucose and 10 Hepes, pH 7.4 (NaOH). Intracellular pipette solution contained 137 KCl, 1 MgCl2, 4 Mg‐ATP, 1 Li‐GTP, 10 EGTA and 10 Hepes, pH 7.2 (KOH). Whole‐cell currents were recorded from a holding potential −60 mV with 2.5 s test pulses from −40 to 60 mV in increments of 10 mV. A small endogenous non‐inactivated K+ current of ∼15 pA pF–1 was present in non‐transfected HEK293 cells. To avoid contamination by this endogenous K+ current in HEK293, the current amplitudes of Kv4.2 or Kv4.3 were measured from steady‐state level to the current peak. Recovery from the inactivation was investigated using a conventional two‐depolarizing pulse protocol: an inactivating pulse depolarizing to 40 mV for 2 s (P1) followed by a variable recovery interval steps of 50 ms (∆0.05–3 s) and subsequent 40 mV test pulse (P2). The interpulse potential was set at −60 mV. The ratio of test pulse current/inactivating pulse current amplitudes (P2/P1) was used to evaluate I to recovery from inactivation, which was best fit with a single exponential equation: y = A1*exp(–x/t 1) + y 0.
For currents measurements through L‐type Ca2+ channels, external bath solution contained 142 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 0.33 Na2HPO4, 5 Hepes and 5 glucose (pH 7.4) and the pipette solution: 100 CsOH‐H2O, 100 glutamic acid, 40 CsCl, 10 Hepes, 0.5 CaCl2 and 240 mg ml−1 amphotericin B (pH 7.2). Recordings were performed using amphotericin perforated whole‐cell, patch configuration in two different solutions: 10 Ba2+ (in mm): 10 BaCl2, 133 CsCl and 10 Hepes (pH 7.4); 10 Ca2+ (mm): 10 CaCl2, 133 CsCl and 10 Hepes (pH 7.4). The holding potential was −80 mV. The inactivation curves were fit to a Boltzmann distribution with the V 1/2 as the voltage midpoint using the equation I = (I max – I min)/[1 + e (V – V1/2)/k] + I min. To evaluate the relative strength of CDI compared to voltage‐dependent inactivation (VDI), the f value was calculated as the difference between r100 values for I Ba,L and I Ca,L (Peterson et al. 1999). Data were analysed using Microcal Origin (OriginLab Corp., Northampton, MA USA).
Computational modelling
A mathematical model of the human ventricular AP (O'Hara et al. 2011) was used to further investigate the effects of Cav3 mutations on I Ca,L, I to and I Na,L. The model consisted of a coupled system of 41 ordinary differential equations that together describe the ionic currents and intracellular Ca2+ cycling of a cardiac myocyte. The Cav3‐WT version of the model corresponds to the O'Hara–Rudy model as originally published (O'Hara et al. 2011). To simulate the effect of the Cav3‐F97C and Cav3‐S141R mutations, currents were adjusted to match the whole‐cell, patch clamp data. Significant differences in channel gating or current density were incorporated into the Cav3 mutant models. When there were discrepancies between measured values and the baseline model parameters, relative changes were applied. For the Cav3‐F97C model, I Ca,L, I to and I Na,L were modified. For the Cav3‐S141R model, changes were made to I Ca,L and I Na,L. All modified model parameters are listed in the Appendix. Additionally, an 8.7 mV liquid junction potential was adjusted for in the I Ca,L I–V experimental data. Virtual knockout of the individual effect for each channel was performed to identify their overall contribution to the changes in APD. All simulations were paced to steady‐state (1000 beats) and were solved using the ode15s algorithm in MATLAB (The MathWorks, Inc., Natick, MA, USA). Simulations used the epicardial parameterization of the O'Hara–Rudy model, unless otherwise stated. Finally, the WT and mutant models were also compared after applying the channel conductance scaling factors proposed by Mann et al. (2016) to more accurately reproduce APD prolongation for clinical LQTS1, 2 and 3.
Statistical analysis
Data are reported as the mean ± SEM. Determinations of statistical significance were performed using Student's t test for comparisons of two means or using ANOVA followed by post hoc Bonferroni test for comparisons of multiple groups. P < 0.05 was considered statistically significant. Statistical analysis was performed using Origin, version 7.5 (OriginLab Corp.).
Results
Co‐localization of mutant Cav3 proteins and Cav1.2 channels in HEK293 cells
To determine whether Cav3 mutations impact trafficking or membrane localization of Cav1.2, HEK293 cells were transfected with Cav3‐WT, Cav3‐F97C or Cav3‐S141R together with HA tagged‐Cav1.2 and Cavβ2cN4, and then immunolabelled for Cav3 and HA‐Cav1.2. Anti‐Cav3 immunolabelling of HEK293 cells transfected with either Cav3‐WT, F97C or S141R and Cav1.2 + Cavβ2cN4 showed a similar predominant plasma membrane staining for WT and mutations (Fig. 1 A, D and G). Similarly, HA‐tagged Cav1.2 channels were primarily localized to the plasma membrane and showed regions of co‐localization with Cav3 for all three groups (Fig. 1 B, C, E, F, H and I). We performed western blotting experiments using HEK293 cells transiently expressing Cav1.2 and Cavβ2cN4 subunits with Cav3‐WT, Cav3‐F97C or Cav3‐S141R and assessed the expression of Cav3 protein. Our findings demonstrate that there are no significant differences in Cav3 protein expression when comparing Cav3‐WT, Cav3‐F97C or Cav3‐S141R coexpression with the calcium channel subunits. As a negative control, we also used lysates from HEK293 cells not transfected with any of our plasmid constructs, which did not demonstrate any positive signal for Cav3 (Fig. 1 J and K). These results confirm that Cav3‐F97C, Cav3‐S141R and Cav3 proteins are similarly expressed at the plasma membrane and comparably co‐localize with Cav1.2 channels.
Figure 1.
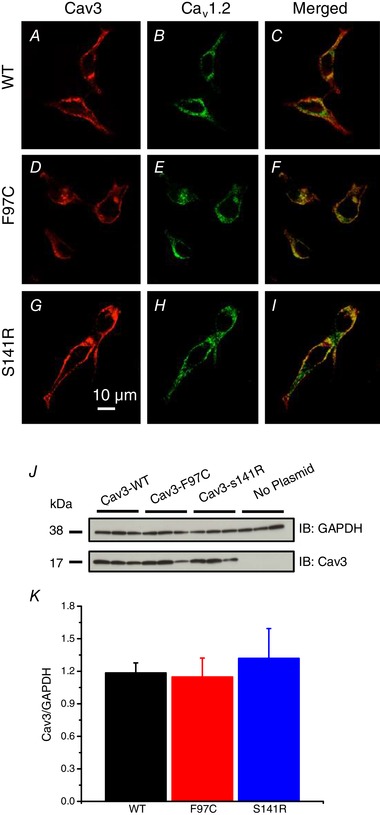
Expression of Cav3‐WT, ‐F97C or ‐S141R with Cav1.2 channels in HEK293 cells
HEK293 cells were transfected with Cav3‐WT, Cav3‐F97C or Cav3‐S141R with Cav1.2 + Cavβ2cN4 subunits. Cells were immunolabelled with anti‐Cav‐3 (red) and anti‐HA (green) antibodies and imaged using confocal microscopy (A–I). J, representative western blot from HEK293 cells expressing CaV1.2 and Cavβ2CN4 subunits with Cav3‐WT, F97C or S141R plasmids probed for Cav3 protein expression with GAPDH loading control. Lysates from HEK293 cells with no plasmid transfected served as a negative control. K, normalized Cav3 protein signals (Cav3/GAPDH) were analysed using ANOVA and were not statistically different between transfected groups.
Lack of effect of Cav3‐F97C and Cav3‐S141R on I Ba,L
To test the effect of the LQT9 Cav3 mutations on Cav1.2 L‐type calcium channels, HEK293 cells were transfected with Cav1.2, Cavβ2cN4 and either Cav3‐WT, Cav3‐F97C or Cav3‐S141R. Initial experiments were performed using 10 mm Ba2+ as the charge carrier to examine effects on voltage‐dependent gating. As shown in Fig. 2, expression of Cav3‐F97C or Cav3‐S141R compared with Cav3‐WT did not significantly affect peak I Ba,L density or alter the I Ba,L–V relationships for Cav1.2 + Cavβ2cN4 channels.
Figure 2.
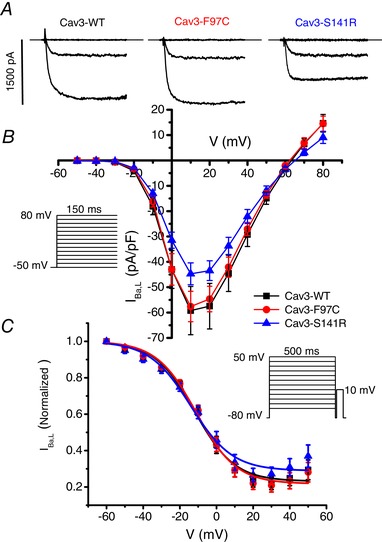
Lack of the effect of Cav3 mutations F97C and S141R on IBa,L density and steady‐state inactivation
A, representative I Ba,L traces resulting from test pulses to −50, −10 and +10 mV from HEK293 cells transfected with Cav1.2 + Cavβ2cN4 and Cav3‐WT, Cav3‐F97C or Cav3‐S141R. B, I–V relationship of I Ba,L recorded from HEK293 cells transfected with Cav1.2 + Cavβ2cN4 and Cav3‐WT (n = 7), Cav3‐F97C (n = 9) or Cav3‐S141R (n = 7). C, steady‐state inactivation of I Ba,L measured using a 500 ms prespulse (shown in inset) voltage protocol from HEK293 cells transfected with Cav1.2 + Cavβ2cN4 and Cav3‐WT (n = 7), Cav3‐F97C (n = 9) or Cav3‐S141R (n = 7).
Previous reports have shown that LQT8 mutations in the Cav1.2 gene greatly reduce VDI of Cav1.2 channels, which leads to delayed repolarization (Splawski et al. 2004) and so we examined the effect of Cav3‐F97C and Cav3‐S141R on VDI using Ba2+ as the charge carrier in transfected HEK293 cells. Using a double pulse protocol, we measured the availability of I Ba,L following 500 ms prepulses over a range of voltages in Cav1.2 + Cavβ2cN4 transfected HEK293 cells. Steady‐state inactivation data were fit by Boltzmann distributions and there were no significant differences in the normalized fit parameters for k, V 1/2 or I min between the groups expressing Cav3‐WT, Cav3‐F97C or Cav3‐S141R (Fig. 2 C).
Cav3‐F97C and ‐S141R differentially modulate I Ca,L
Under physiological conditions, Ca2+ is the primary charge carrier for Cav1.2 channels; therefore, we performed a similar series of experiments in HEK293 cells using 10 mm Ca2+ instead of Ba2+. As shown in Fig. 3 A and B, co‐expression of Cav1.2 + Cavβ2cN4 with Cav3‐S141R significantly increased I Ca,L density compared to Cav3‐WT with a shift in the peak of the I–V in the hyperpolarizing direction. By contrast, Cav3‐F97C did not change I Ca,L density. Using a double pulse inactivation protocol with a 500 ms prepulse, no differences between Cav3‐WT, F97C and S141R were observed on the inactivation of I Ca,L (Fig. 3 C).
Figure 3.
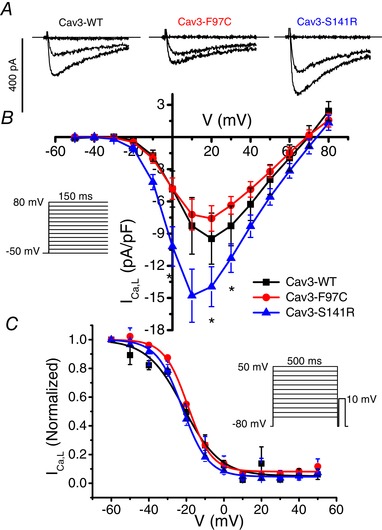
Cav3‐S141R but not Cav3‐F97 increases ICa,L density
A, representative ICa,L traces elicited by test pulses to −50, 0 and +20 mV from HEK293 cells transfected with Cav1.2 + Cavβ2cN4 and Cav3‐WT, Cav3‐F97C or Cav3‐S141R. B, the I–V relationship of I Ca,L measured in HEK293 cells transfected with Cav1.2 + Cavβ2cN4 and Cav3‐WT (n = 5), Cav3‐F97C (n = 5) or Cav‐3 S141R (n = 6; * P < 0.05). C, steady‐state inactivation of I Ca,L measured using a 500 ms prespulse (shown in inset) voltage protocol from HEK293 cells transfected with Cav1.2 + Cavβ2cN4 and Cav3‐WT (n = 7), Cav3‐F97C (n = 9) or Cav3‐S141R (n = 7).
Cav3‐F97C delays Ca2+‐dependent inactivation of I Ca,L
Despite a lack of effect of the Cav3 mutations on steady‐state inactivation of Cav1.2 channels measured after 500 ms prepulses, we examined the kinetics of inactivation of L‐type Ca2+ channels to investigate changes specifically in VDI and CDI using Ba2+ and Ca2+ as charge carriers. The rate of current decay was determined by measuring the ratio of current present after 100 ms of a voltage step relative to the peak current (r100). As shown in Fig. 4 A, no differences were observed in r100 for I Ba,L at test potentials from −20 to 60 mV comparing Cav3‐WT, F97C and S141R. This suggests that Cav3 mutations do not impact VDI. At the same time, in the presence of Ca2+, current decay was delayed in Cav3‐F97C expressing cells compared to Cav3‐WT and Cav3‐S141R as assessed with r100 (Fig. 4 B). We determined the relative strength of CDI by the f value calculated as the difference between r100 for I Ba,L minus r100 for I Ca,L (Peterson et al. 1999). At test potentials of −20, −10 and 0 mV, Cav3‐F97C resulted in a significantly decreased f value consistent with reduced CDI compared to Cav3‐WT. By contrast, Cav3‐S141R mutation did not alter the f value and hence CDI relative to Cav3‐WT (Fig. 4 C).
Figure 4.
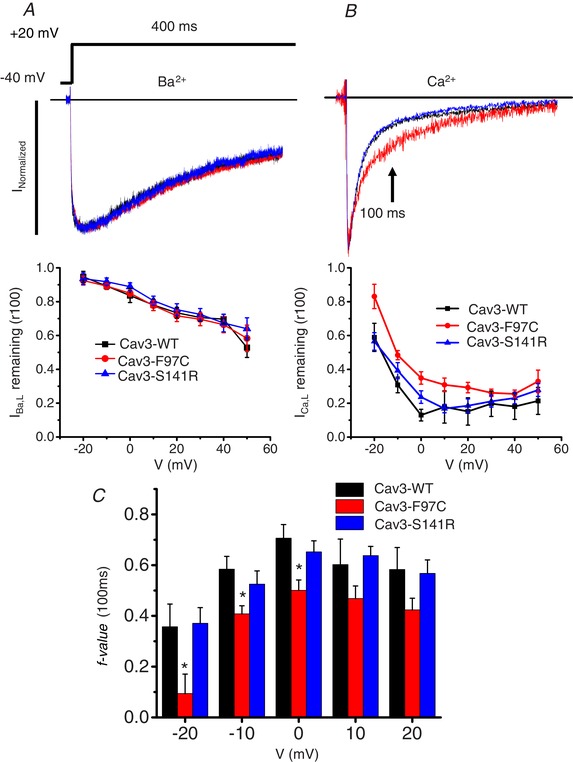
Cav3‐F97C slows Ca2+‐dependent inactivation of Cav1.2 channels
A, representative I Ba,L traces resulting from test pulses at 20 mV from HEK293 cells transfected with Cav1.2 + Cavβ2cN4 and Cav3‐WT (n = 7), Cav3‐F97C (n = 9) or Cav3‐S141R (n = 7). Current decay of I Ba,L was determined by measuring the ratio of the current present after 100 ms of a voltage step relative to the peak current (r100). B, representative I Ca,L traces resulting from test pulses at 20 mV from HEK293 cells transfected with Cav1.2 + Cavβ2cN4 and Cav3‐WT (n = 3), Cav3‐F97C (n = 5) or Cav3‐S141R (n = 5). Current decay of I Ca,L was determined by measuring the ratio of the current present after 100 ms of a voltage step relative to the peak current (r100). C, the f value is plotted as an index of Ca2+‐dependent inactivation by eliminating the effect of VDI at test potentials from −20 to 20 mV, between Cav3‐WT (n = 3–7), Cav3‐F97C (n = 5–9) and Cav3‐S141R (n = 5–7), as described in the Methods.
Co‐localization of LQT9 Cav3 mutations and Kv4.2 channels in HEK293 cells
To determine whether LQT9 Cav3 mutations impact the trafficking and subcellular localization of Kv4.2, HEK293 cells were transfected with Cav3‐WT, Cav3‐F97C or Cav3‐S141R and Kv4.2. The transfected cells were then immunolabelled for Cav3 and Kv4.2. Anti‐Cav3 immunolabelling of HEK293 cells transfected with Cav3‐WT, F97C or S141R and Kv4.2 all show a similar prominent plasma membrane immunolabelling (Fig. 5 A, D and G). Kv4.2 channels were primarily localized to the plasma membrane and showed a similar pattern of co‐localization with Cav3 for all three groups (Fig. 5 B, C, E, F, H and I). These results demonstrate that Cav3‐F97 and Cav3‐S141R proteins are expressed in HEK293 cells like Cav3‐WT and comparably co‐localize with Kv4.2 at the plasma membrane.
Figure 5.
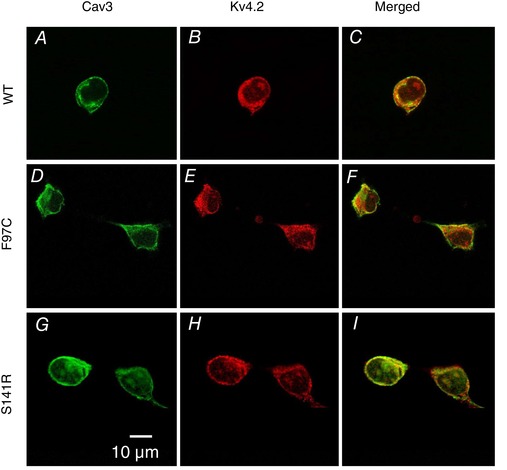
Co‐localization of Cav3‐WT, Cav3‐F97C and Cav3‐S141R with Kv4.2 channels expressed in HEK293 cells
HEK293 cells transfected with Cav3‐WT (A‐C), Cav3‐F97C (D‐F), or Cav3‐S141R (G‐I) + Kv4.2. Cells were immunolabelled with anti‐Cav3 (green) and anti‐Kv4.2 (red) antibodies and imaged using confocal microscopy.
Effect of Cav3‐F97C on Kv4.2 current
To investigate whether the Cav3‐F97C mutation impacts Ito, we first co‐expressed Kv4.2 with Cav3‐WT or Cav3‐F97C transfected in a 1:1 ratio in HEK293 cells. The peak I Kv4.2 normalized to cell capacitance was significantly smaller in cells transfected with Cav3‐F97C (30.1 ± 5.7 pA pF–1 at 60 mV, n = 13, P < 0.005) compared to Cav3‐WT (70.5 ± 8.8 pA pF–1 at 60 mV, n = 23) (Fig. 6 A and B).
Figure 6.
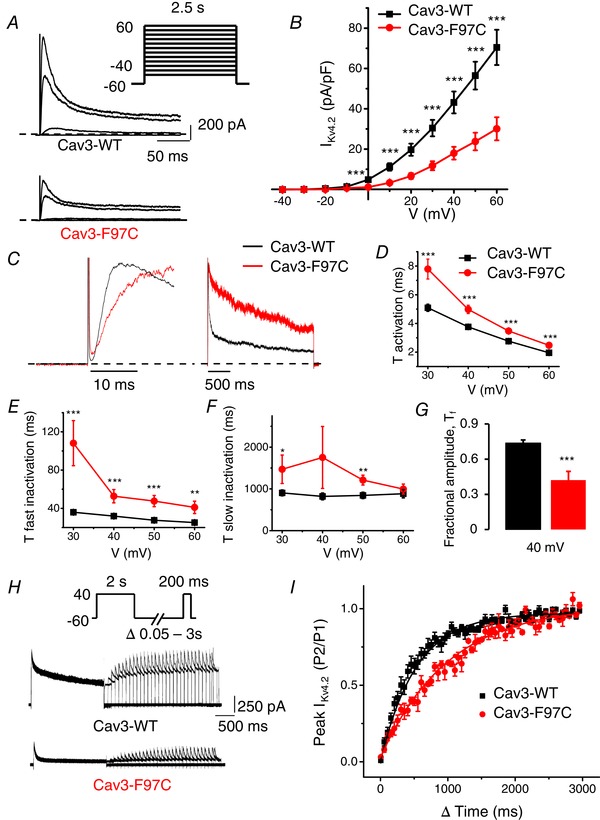
Cav3‐F97C coexpression decreases IKv4.2 density, alters kinetics and slows recovery from inactivation
A, representative I Kv4.2 current traces measured at −40, 0, 40 and 60 mV in HEK293 cells transfected with Kv4.2 and Cav3‐WT or Cav3‐F97C. Whole‐cell currents were recorded using the protocol indicated in the inset. B, mean I–V relationships for peak I Kv4.2 density in transfected HEK293 cells with Kv4.2 and Cav3‐WT or Cav3‐F97C (mean ± SEM, n = 13–23 cells, **P < 0.01, *P < 0.05). C, representative I Kv4.2 traces normalized to the peak current measured at 40 mV from cells expressing Cav3‐WT or Cav3‐F97C. D, average τ activation for I Kv4.2 from Cav3‐WT and Cav3‐F97C expressing cells as a function of test voltage (mean ± SEM, n = 13–23 cells, ***P < 0.005, **P < 0.01, *P < 0.05). τ activation determined by fitting a single exponential function to the rising phase (activation) of I Kv4.2. E and F, average τfast inactivation and τslow inactivation for I Kv4.2 as determined by fitting a double exponential function to the decaying phase of I Kv4.2 traces (mean ± SEM, n = 13–23 cells, ***P < 0.005, **P < 0.01, *P < 0.05). G, average fractional amplitude of fast inactivating (τfast) I Kv4.2 at 40 mV comparing Cav3‐WT and Cav3‐F97C (mean ± SEM, n = 13‐23, P < 0.005). H, representative I Kv4.2 traces demonstrating recovery from inactivation following a 2 s depolarization to 40 mV with co‐expression of Cav3‐WT or Cav3‐F97C. I, time course of recovery from inactivation for I Kv4.2 currents in Cav3‐WT and Cav3‐F97C expressing cells. Ratio between the current amplitude obtained with both pulses of the protocol is plotted against the interpulse interval. Recovery time constant for Cav3‐WT was 331 ± 40 ms (n = 6) compared to 809 ± 83 ms (n = 5) Cav3‐F97C (P < 0.001).
The kinetics of voltage‐dependent activation and inactivation contribute to the determination of peak I to. Therefore, we analysed activation and inactivation of I Kv4.2 from the groups of cells co‐transfected with Kv4.2 and either Cav3‐WT or Cav3‐F97C. Figure 6 C displays the time course of activation (left) and inactivation (right) of IKv4.2 comparing cells transfected with Cav3‐WT and Cav3‐F97C at a test potential of 40 mV. The activation of I Kv4.2 was analysed by fitting the rising phase of current traces to a single exponential fit at test voltages from 30 to 60 mV. As shown in Fig. 6 D, the activation time constants for Cav3‐F97C were significantly greater than those for Cav3‐WT from 30 to 60 mV. The fast (τfast inactivation) (Fig. 6 E) and slow inactivation (τslow inactivation) (Fig. 6 F) time constants were determined for I Kv4.2 by fitting the decaying phase of the current traces to a double exponential function at test voltages from 30 to 60 mV. The τfast and τslow inactivation time constants were significantly greater for Cav3‐F97C compared to Cav3‐WT (Fig. 6 E and F) and the fractional amplitude of the fast component for Cav3‐F97C was significantly decreased (Fig. 6 G). These data demonstrate that co‐expression of Cav3‐F97C results in both slower activation and inactivation kinetics of Kv4.2 channel currents relative to coexpression with Cav3‐WT.
Recovery from inactivation is another important feature of I Kv4. Prolonged recovery from inactivation of Kv4 channels could lead to accumulation of inactivated channels at physiological heart rates, thus reducing I to during an AP and consequently causing a delay in cardiac repolarization. Therefore, we investigated the effect of the Cav3 mutations on the time course of recovery of Kv4.2 channel currents. We used a double‐pulse protocol as shown (Fig. 6 H) by applying an inactivating pulse depolarizing to 40 mV for 2 s (P1). This was followed by a variable recovery interpulse interval (Δt; 0.05–3 s) and second pulse depolarizing to 40 mV for 200 ms (P2) (Bahring et al. 2001; Colinas et al. 2006). Analysis of recovery from inactivation of Kv4.2 current revealed a significantly slower recovery of I Kv4.2 when the channel was co‐expressed with Cav3‐F97C with a recovery time constant of 809.1 ± 83.7 ms; (n = 5, P < 0.001), compared to the recovery time constant for Cav3‐WT (331.7 ± 40.4 ms; n = 6) (Fig. 6 I and H). The above results demonstrate that the LQT9 Cav3‐F97C mutation slows recovery of I Kv4.2, which could cause a reduction of I to,f at physiological heart rates.
Effect of Cav3‐S141R on Kv4.2 current
We then examined the impact of another LQT9 associated Cav3 mutation, S141R, on I Kv4.2 in an independent series of experiments. HEK293 cells were transiently transfected with Kv4.2 and either Cav3‐WT or Cav3‐S141R in a 1:1 ratio. The peak I Kv4.2 normalized to cell capacitance was significantly smaller in cells transfected with Cav3‐S141R (41.6 ± 9.3 pA pF–1 at 60 mV, n = 11, P < 0.05) compared to cells transfected with Cav3‐WT (69.1 ± 7.9 pA pF–1 at 60 mV, n = 12) (Fig. 7 A and B). Analysis of activation and inactivation properties of I Kv4.2 revealed a difference between Cav3‐WT and Cav3‐S141R for activation but not for inactivation. Figure 7 C displays the time course of activation (left) and inactivation (right) of I Kv4.2 comparing cells transfected with Cav3‐WT and Cav3‐S141R at a test potential of 40 mV. As shown in Fig. 7 D, the activation time constants for Cav3‐S141R were significantly greater than those for Cav3‐WT at 30 and 40 mV. The fast (τfast) (Fig. 7 E) and slow inactivation (τslow) (Fig. 7 F) time constants were not different between Cav3‐WT and Cav3‐S141R. The fractional amplitude of the fast component was decreased for Cav3‐S141R (Fig. 7 G). Therefore, co‐expression of Cav3‐S141R mutation resulted in a slower activation but not inactivation of Kv4.2 channel currents. We also investigated the effect of Cav3‐S141R mutation on the time course of recovery of Kv4.2 channel currents. Analysis of recovery from inactivation of Kv4.2 channel current revealed a significantly slower recovery of I Kv4.2 when the channel was co‐expressed with Cav3‐S141R compared to Cav3‐WT, recovery time constants of 518.9 ± 94.2 ms (n = 8, P < 0.05) and 177.5 ± 24.5 ms (n = 5), respectively (Fig. 7 H and I).
Figure 7.
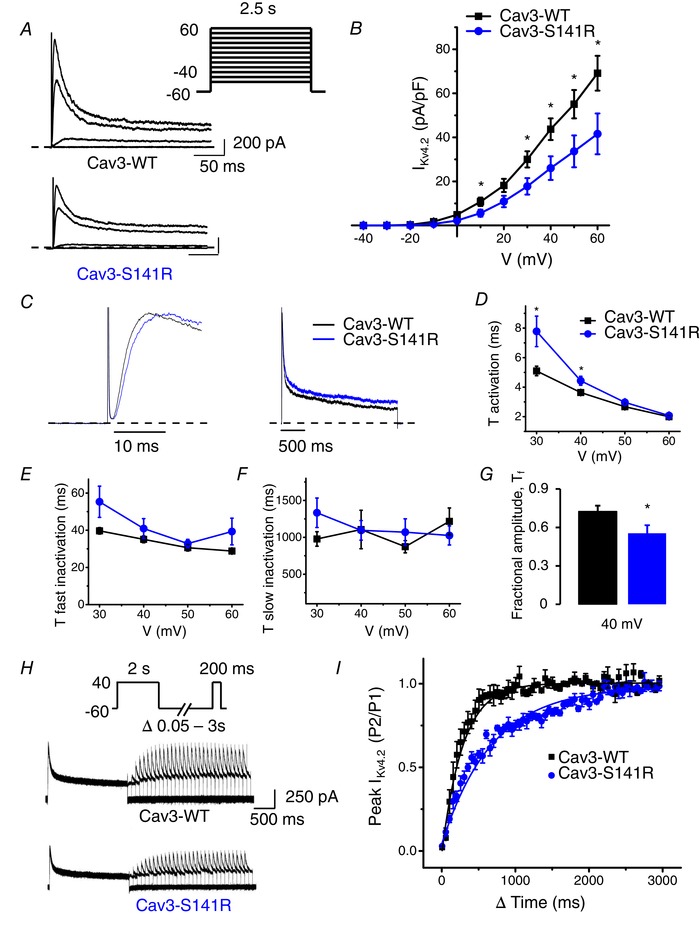
Cav3‐S141R coexpression decreases Kv4.2 current density, alters current kinetics and slows recovery from inactivation
A, representative current traces illustrating Kv4.2 currents measured at −40, 0, 40 and 60 mV in HEK293 cells transfected with Kv4.2 and Cav3‐WT or Cav3‐S141R. Whole‐cell currents were recorded using the protocol indicated in the inset. B, mean I–V relationships of peak Kv4.2 current density in HEK293 cells transfected with Kv4.2 and Cav3‐WT or Cav3‐S141R (mean ± SEM, n = 11 or 12 cells, *P < 0.05). C, representative I Kv4.2 traces normalized by peak transfected with Cav3‐WT and Cav3‐S141R measured at 40 mV showing the kinetic changes. D, average τ activation for I Kv4.2 from Cav3‐WT and Cav3‐ S141R expressing cells as a function of test voltage (mean ± SEM, n = 11 cells, *P < 0.05). τ activation determined by fitting a single exponential function to the rising phase (activation) of I Kv4.2. E and F, average τfast inactivation and τslow inactivation for I Kv4.2 as determined by fitting a double exponential function to the decaying phase of I Kv4.2 traces (mean ± SEM, n = 11 cells, *P < 0.05). G, average fractional amplitude of fast inactivating (τfast) I Kv4.2 at 40 mV comparing Cav3‐WT and Cav3‐S141R expressing cells (mean ± SEM, n = 11 cells, *P < 0.05). H, representative I Kv4.2 traces demonstrating recovery from inactivation following a 2 s depolarization to 40 mV with co‐expression of Cav3‐WT or Cav3‐S141R. I, time course of recovery from inactivation for I Kv4.2 currents in Cav3‐WT and Cav3‐S141R expressing cells. Ratio between the current amplitude obtained with both pulses of the protocol is plotted against the interpulse interval. Recovery time constant for Cav3‐WT was 177.5 ± 24.5 ms (n = 5) compared to 518.9 ± 94.2 ms for Cav3‐S141R (n = 8; P < 0.05).
Co‐localization of LQT9 Cav3 mutations and Kv4.3 channels in HEK293 cells
To determine whether LQT9 Cav3 mutations impact the trafficking and subcellular localization of Kv4.3, HEK293 cells were transfected with Cav3‐WT, Cav3‐F97C or Cav3‐S141R, as well as Kv4.3, and then immunolabelled for Cav3 and Kv4.3. Anti‐Cav3 immunolabelling of HEK293 cells transfected with Cav3‐WT, F97C or S141R and Kv4.3, all show a similar predominant plasma membrane staining (Fig. 8 A, D and G). The Kv4.3 channels were primarily localized to the plasma membrane and showed similar regions of co‐localization with Cav3 for all three groups (Fig. 8 B, C, E, F, H and I). These results demonstrate that Cav3‐F97 and Cav3‐S141R proteins are expressed in HEK293 cells such as Cav3‐WT and comparably co‐localize with Kv4.3 at the plasma membrane.
Figure 8.
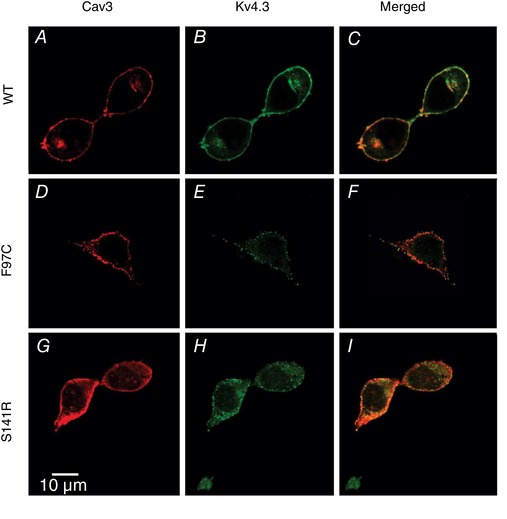
Co‐localization of WT Cav3 and LQT9‐associated Cav3 mutations, F97C and S141R, with Kv4.3 channels in HEK293 cells
HEK293 cells transfected with Kv4.3 and Cav3‐WT (A‐C), Cav3‐F97C (D‐F), or Cav3‐S141R (G‐I). Cells were immunolabelled with anti‐Cav3 (red) and anti‐Kv4.3 (green) antibodies and imaged using confocal microscopy.
Effect of F97C and S141R Cav3 mutations on Kv4.3 current
We next examined whether Cav3‐F97C and Cav3‐S141R are able to affect I Kv4.3, the second component of I to,f. HEK293 cells were transiently transfected with Kv4.3 and either Cav3‐WT, Cav3‐F97C or Cav3‐S141R at a 1:1 ratio. Co‐transfection of the Cav3‐F97C caused a significant reduction in the peak I Kv4.3 density (181.20 ± 30.6 pA pF–1 at 60 mV, n = 12, P < 0.01) compared to Cav3‐WT (327.8 ± 35.7 pA pF–1 at 60 mV, n = 18) (Fig. 9 A).
Figure 9.
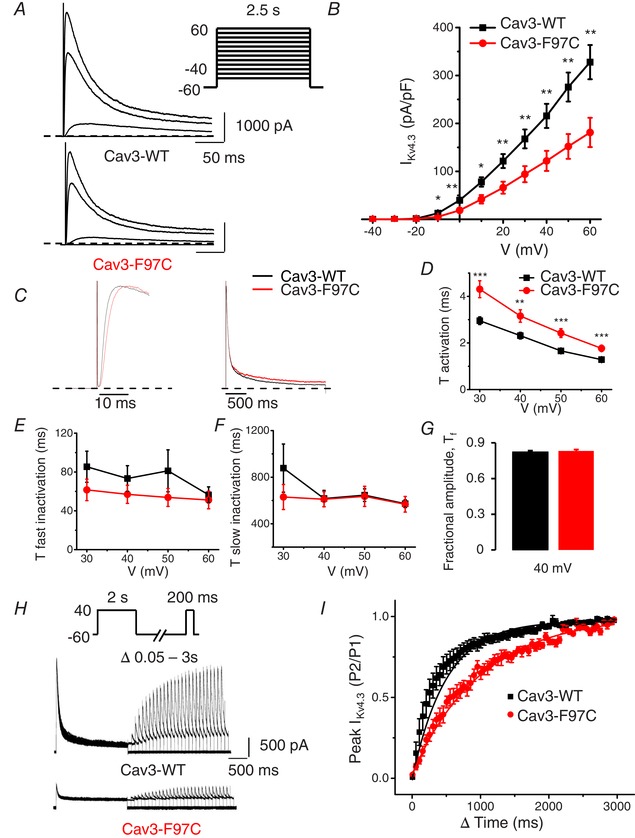
Cav3 mutation F97C coexpression decreases IKv4.3 density, alters current kinetics and slows recovery from inactivation
A, representative current traces illustrating Kv4.3 currents measured at −40, 0, 40 and 60 mV in HEK293 cells transfected with Cav3‐WT and Cav3‐F97C. Whole‐cell currents were recorded using the protocol indicated in the inset. B, mean I–V relationships of peak I Kv4.3 density in transfected HEK293 cells with Kv4.3 and Cav3‐WT or Cav3‐F97C (mean ± SEM, n = 12–18 cells; **P < 0.01, *P < 0.05). C, representative I Kv4.3 traces normalized by peak current in cells transfected with Cav3‐WT and Cav3‐F97C measured at 40 mV showing the current kinetics differences. D, average τ activation for I Kv4.3 from Cav3‐WT and Cav3‐F97C expressing cells as a function of test voltage (mean ± SEM, n = 12 or 13 cells; ***P < 0.005, **P < 0.01). τ activation determined by fitting a single exponential function to the rising phase (activation) of I Kv4.3. E and F, average τfast inactivation and τslow inactivation for I Kv4.3 as determined by fitting a double exponential function to the decaying phase of I Kv4.3 traces (mean ± SEM, n = 12 or 13 cells, no significant differences). G, average fractional amplitude of fast inactivating (τfast) I Kv4.3 at 40 mV comparing Cav3‐WT and Cav3‐F97C (mean ± SEM, n = 12 or 13 cells, no significant difference). H, representative I Kv4.3 traces demonstrating recovery from inactivation following a 2 s depolarization to 40 mV with co‐expression of Cav3‐WT or Cav3‐F97C. I, time course of recovery from inactivation for I Kv4.3 currents in Cav3‐WT and Cav3‐F97C expressing cells. Recovery time constant for Cav3‐WT was 289.4 ± 62.1 ms (n = 8) compared to 698.7 ± 130.5 ms (n = 6) for Cav3‐F97C (P < 0.005).
We then analysed time constants of activation and inactivation of Kv4.3 channel when co‐expressed with Cav3‐WT or Cav3‐F97C. Figure 9 C shows the time course of activation (left) and inactivation (right) of I Kv4.3 for cells transfected with Cav3‐WT or Cav3‐S141R measured at 40 mV. As shown in Fig. 9 D, the activation time constant of I Kv4.3 was significantly increased for the Kv4.3 channel co‐expressed with Cav3‐F97C compared to Cav3‐WT at membrane voltages from 30 to 60 mV. Analysis of fast and slow inactivation time constants revealed that Cav3‐F97C did not affect the inactivation kinetics of I Kv4.3 (Fig. 9 E and F) and analysis of the fractional amplitude of the fast component did not reveal any difference between Cav3‐F97C and Cav3‐WT (Fig. 9 G). These data indicate that Cav3‐F97C altered the I Kv4.3 kinetics by slowing the channel activation.
We also analysed recovery from inactivation for Kv4.3 channel currents co‐expressed with Cav3‐WT or Cav3‐F97C. Similar to the Kv4.2 channel, the recovery from inactivation of Kv4.3 was significantly slower when co‐expressed with Cav3‐F97C (698.7 ± 130.5 ms; n = 6, P < 0.005) compared to the recovery time constant for Cav3‐WT (289.4 ± 62.1 ms; n = 8) (Fig. 9 H and I). Interestingly, peak I Kv4.3 density in cells transfected with Cav3‐S141R (287 ± 34.1 pA pF–1 at 60 mV, n = 15) was not significantly different compared to Cav3‐WT (331.8 ± 30.7 pA pF–1 at 60 mV, n = 16). Time constants of activation or inactivation were also not different between two groups (data not shown). These data suggest that the two different LQT9‐linked Cav3 mutations have different effects on the I Kv4.3 in the HEK293 cells.
Computation models of Cav3‐F97C and Cav3‐S141R predict prolonged APD
The changes in current density and kinetics described above were combined with previously published experimental data on I Na,L (Vatta et al. 2006; Cheng et al. 2013) to create AP models meant to test the electrophysiological effects of the Cav3 mutations. To recapitulate the Cav3‐F97C behaviour, the I Ca,L I–V relationship was left unchanged (Fig. 10 A) at the same time as incorporating slowed CDI (Fig. 10 B). Because Kv4.3 is expressed much more abundantly in human ventricular tissue compared to Kv4.2 (Dixon et al. 1996; Gaborit et al. 2007), only the significant changes seen in I Kv4.3 were incorporated into the modelled I to. Slowing the I to activation time constant was insufficient to explain the observed decrease in current density (Fig. 10 C, dotted line) and therefore the maximal conductance was reduced to reproduce the experimental data (Fig. 10 C). Finally, the I Na,L conductance was doubled (Fig. 10 D) in accordance with previously published data (Vatta et al. 2006; Cheng et al. 2013). For the Cav3‐S141R model, the maximal I Ca,L conductance was increased, the steady‐state I Ca,L I–V relationship was shifted to become more negative (Fig. 10 E) and the I Na,L conductance was increased (Fig. 10 F).
Figure 10.
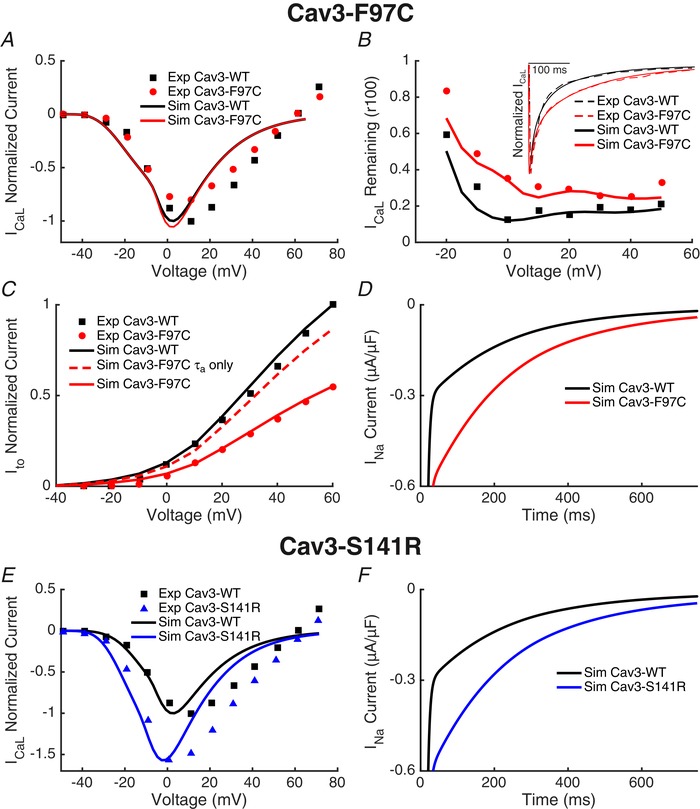
Reparameterization of the O'Hara–Rudy model to reproduce Cav3 mutant effects on ionic currents
A, because the I Ca,L I–V relationship was not significantly different between the Cav3‐WT and Cav3‐F97C, the peak conductance, steady‐state activation and steady‐state inactivation were not altered between the Cav3‐WT and Cav3‐F97C models. B, current decay of I Ca,L determined by measuring the ratio of current present after 100 ms of a voltage step relative to the peak current (r100) was similar between experimental Cav3‐WT results and the Cav3‐WT model. I Ca,L CDI time constants were slowed to recapitulate experimental Cav3‐F97C results in the Cav3‐F97C model. The inset shows a representative I Ca,L traces experimental and with the computational model response to the same test voltage with the Cav3‐WT and Cav3‐F97C models. C, slowing the Ito activation time constant resulted in a small decrease in current density. Current density was further reduced by scaling the channel conductance to fit the observed reduction in IKv4.3 with Cav3‐F97C. D, INa,L conductance was doubled in the Cav3‐F97C model. E, for the Cav3‐S141R model, ICa,L was altered by increasing the peak current and shifting the steady state inactivation cure more negative compared to the Cav3‐WT model. F, INa,L conductance was doubled in the Cav3‐S141R model.
The resulting simulated AP for Cav3‐F97C and ‐S141R both have prolonged APD at 90% of repolarization (APD90) (Fig. 11). APD90 prolongation at 1000 ms cycle length was greater for the Cav3‐F97C mutation (+53.5 ms) (Fig. 11 A) compared to the Cav3‐S141R (+21.7 ms) (Fig. 11 B). For Cav3‐F97C mutation, these APD effects were consistent across pacing rates and similar for endocardial and epicardial parameterizations of the O'Hara–Rudy model (Fig. 11 C). For the Cav3‐S141R mutation, APD90 prolongation increased in the endocardial version of the cell model at slow rates (Fig. 11 D). Virtually ablating the effect of one of the currents at the same time as leaving the others intact can allow the relative contributions to APD90 prolongation to be identified. The AP prolongation at 1000 ms cycle length in the Cav3‐F97C model was dominated by the I Ca,L effect, whereas the decrease in I to density had minimal impact (Fig. 11 E). For the Cav3‐S141R model, however, the I Ca,L and I Na,L contributed more equally (Fig. 11 F). Finally, similar results were obtained when using the conductance scaling factors proposed by Mann et al. (2016). At a cycle length of 1000 ms, the Cav3‐F97 ADP90 was prolonged by 47.8 ms and the Cav3‐S141R APD90 was prolonged by 30.2 ms.
Figure 11.
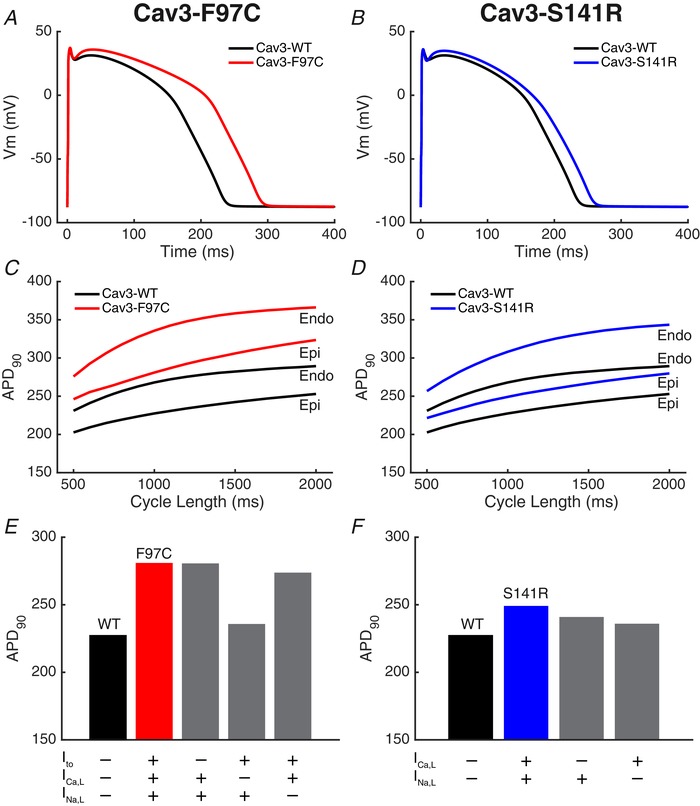
Cav3 mutant AP models
A and B, models of the Cav3‐F97C mutation (A) and the Cav3‐S141R mutation (B) had longer APs compared to the Cav3‐WT model (continuous lines). APs are shown for 1000 ms cycle length after 1000 beats simulated pacing. C and D, effect of pacing cycle length on APD90 is shown for endocardial (Endo) and epicardial (Epi) parameterizations of the O'Hara–Rudy model (right). E and F, relative contribution of each altered current to the APD90 prolongation in the Cav3 mutant models is shown by virtually ablating the effect of the mutation on one channel at the same time as retaining the effect on the other. The absence (–) or presence (+) of the simulated current mutations are shown (below) with the WT model corresponding to the absence of all the mutations and the mutant corresponding to the presence of all the mutant current effects.
Discussion
The original description of the LQT9 syndrome mutations in CAV3 described an increase in I Na,L as a cause of APD prolongation and associated QT prolongation (Vatta et al. 2006), and the present study identifies two additional currents, I to and I Ca,L, impacted by the Cav3‐F97C and Cav3‐S141R LQT9 mutations. The Cav3‐F97C mutation reduces the amplitude of both I Kv4.2 and I Kv4.3, and also slows the recovery from inactivation, suggesting an important effect of this mutation on I to in the native heart. By contrast, the Cav3‐S141R mutations reduced only I Kv4.2 without exerting any significant effect on I Kv4.3. The different effects of the two mutations were even more striking on I Ca,L in which Cav3‐F97C did not change current density but slowed CDI, in contrast to Cav3‐S141R, which caused an increase in I Ca,L density without changes in kinetics. Furthermore, all of the effects on the Cav1.2 expressed L‐type Ca2+ channels required Ca2+ as the charge carrier and, in Ba2+, no effects of the mutations were observed. Importantly, extrapolating our results to a human ventricular AP model demonstrated that the changes in I to had little impact on APD, although the slowed CDI induced by the F97C mutation is the major contributor to APD prolongation with little contribution by increased I Na,L, whereas the S141R mutation increased I Ca,L density and increased I Na,L contribute almost equally to APD prolongation. These results further unmask the complexity of Cav3 regulation of cardiac ion channels and the remarkably different ion channel‐specific effects of LQT9 associated mutations in CAV3.
LQT9 Cav3 mutations and I to
In ventricular myocytes, I to is responsible for rapid initial repolarization manifest as phase 1 of the AP. Thus, I to generates the notch morphology in the ventricular AP and helps set the plateau potential. There is also a prominent transmural gradient of I to with the current showing higher density in the epicardium and midmyocardium than endocardium (Furukawa et al. 1990; Liu et al. 1993). In human ventricular myocardium, Kv4 α subunits provide the pore forming subunit for I to, and Kv4.3 is the dominant isoform compared to Kv4.2 in the human heart. In addition, accessory subunits for Kv4 channels have been identified. The cytoplasmic Kv channel interacting protein 2 (KChIP2) was demonstrated to be the most essential auxiliary subunit for cardiac I to (Kuo et al. 2001) and a transmural gradient of this subunit has been suggested to be responsible for the transmural gradient of I to density (Rosati et al. 2001). The diaminopeptidyl transferase‐like protein 6 (DPP6) was reported to associate with Kv4 α subunits regulating surface density and the gating properties of the channels (Radicke et al. 2005). The MinK‐related peptide (MiRP) family of transmembrane proteins have also been identified as subunits for Kv4 channels (McCrossan & Abbott, 2004). Reduced I to,f density was observed in failing human hearts (Beuckelmann et al. 1993), as well as in canine animal models of heart failure (Kaab et al. 1996). Gain of function mutations in the MiRP2 subunits (KCNE3 and KCNE5) and Kv4.3 (KCND3) have been identified that produce increased I to and can cause the inherited arrhythmia syndrome, Brugada syndrome (Delpon et al. 2008; Giudicessi et al. 2011; Ohno et al. 2011). However, no prior studies have linked changes in I to with LQTS. Furthermore, it is controversial regarding whether changes in I to density directly contribute to APD in large animal hearts and humans in contrast to its dominant role in rodent hearts (Niwa & Nerbonne, 2010). Our computational modelling did not reveal a significant effect of I to reduction on APD. Nevertheless, changes in I to can importantly impact the activation of voltage‐gated L‐type Ca2+ channels, as well as the balance between the inward and outward currents during the plateau phase, leading to changes in overall contractility (Gussak et al. 2000; Sah et al. 2002). Thus, the impact of Cav3 mutations on Ito may provide a unique aspect of the LQT9 phenotype that involves more than APD prolongation.
How do Cav3‐F97C and Cav3‐S141R alter the properties of I to? The Cav3 mutations reduce I Kv4.2 density and slow the activation and inactivation kinetics, as well as slow recovery from inactivation. Although Cav3‐F97C exerts similar effects on I Kv4.3, the Cav3‐S141R mutation does not impact I Kv4.3. These results show that the Cav3 mutations fundamentally regulate the gating of the channels and suggest several possible mechanisms. Cav3 and its mutants may directly interact with the Kv4 α subunit or accessory subunits to modulate gating. Alternatively, because, similar to most ion channels, Kv4 channels are subject to post‐translation modification regulating their function such as phosphorylation, it is possible that the Cav3 mutants alter this regulation. For example, Kv4.3 channels are known to be subject to α1‐adrenergic‐mediated inhibition acting through protein kinase C, which differentially impacts different splice variants of the Kv4.3 (Po et al. 2001). Calcium‐dependent calmodulin kinase II (CaMKII) also has been suggested to regulate the channels with inhibition of CaMKII slowing inactivation of I to in cardiomyocytes (Colinas et al. 2006) but, interestingly, the effects of CaMKII are distinct on Kv4.3 and Kv4.2 expressed channels. This highlights the possibility that the differential effects of the Cav3‐S141R mutation on Kv4.2 and Kv4.3 channels may reflect differences in the downstream regulation by kinases such as CaMKII. There are a variety of other kinases and regulatory pathways linked to modulation of I to or gene expression of its key subunits that are less well studied but might contribute to the observed effects of the Cav3 mutations (Niwa & Nerbonne, 2010). Future studies are needed to define the mechanistic pathways responsible for altered function of Kv4.2 and Kv4.3 in the presence of Cav3‐F97C and S141R mutations.
LQT9 Cav3 mutations and I Ca,L
I Ca,L provides the influx of Ca2+ that triggers intracellular Ca2+ release and resulting contraction. I Ca,L occurs largely during the plateau phase of the AP. The main pore forming subunit for L‐type Ca2+ channels in the human ventricular myocardium is the α1C or Cav1.2 subunit, and there are auxiliary subunits that importantly regulate the density of channels and gating, including Cavβ, Cavα2‐δ and Cavγ subunits (Hofmann et al. 2014). There are multiple splice variants for all of the channel subunits expressed in the heart, with at least 18 different Cavβ isoforms identified in the human ventricle (Foell et al. 2004). There are other proteins besides the classical subunits which associate with Cav1.2 channels to fine tune channel function, including calmodulin (CaM), which plays an essential role in mediating CDI, and the more recently recognized cardiac‐specific LRRC10, which modulates channel gating (Peterson et al. 1999; Woon et al. 2017). Cav3 has also been shown to interact with a subpopulation of the L‐type Ca2+ channels localized to caveolae, and these channels are distinct from the subpopulation of L‐type Ca2+ channels present in the dyadic regions in ventricular myocytes that play a critical role in excitation–contraction coupling (Balijepalli et al. 2006).
A severe form of LQTS has been linked to impaired CDI of I Ca,L as a result of mutations in CaM that cause a reduced binding affinity for Ca2+ (Crotti et al. 2013; Limpitikul et al. 2014; Yin et al. 2014; Boczek et al. 2016). Although CaM has a variety of cellular targets, including multiple ion channels, a subset of disease‐associated CaM associated mutations exert their dominant effect by blunting CDI of I Ca,L (Limpitikul et al. 2014; Yin et al. 2014; Boczek et al. 2016). There are three distinct CaM genes that encode identical CaM proteins (CALM1, CALM2 and CALM3) and a mutation in only one of these genes can produce disease showing a strong dominant negative effect. In the case of the Cav3‐F97C mutation examined in the present study, a similar blunting of CDI is observed that is comparably linked to the LQTS phenotype. This finding raises new questions about how CDI can be impacted by Cav3 and the Cav3‐F97C mutation. Presumably, even a subset of L‐type calcium channels localized to caveolae is adequate to produce an arrhythmic phenotype with altered CDI, consistent with a strong non‐linear effect of CDI on arrhythmia phenotype. Future studies are required that investigate the precise mechanism of Cav3 regulation of CDI.
LQTS9 Cav3 mutations, APD prolongation and arrhythmias
Initial experiments evaluating the LQT9‐associated Cav3 mutations, Cav3‐F97C and Cav3‐S141R demonstrated an increase in sodium channel late current (I Na,L) as a potential cause of APD prolongation and associated QT prolongation (Vatta et al. 2006). The present study demonstrates that the Cav3‐F97C and S141R variants can also cause loss of function effects on Kv4.2 and Kv4.3 channels responsible for I to and gain of function effects on Cav1.2 channels increasing I Ca,L as contributors to the disease phenotype. Other studies have suggested that the LQTS9 Cav3 mutations can also cause a decrease in Kir2.1 current density, which can contribute to the arrhythmia phenotype (Vaidyanathan et al. 2013; Vaidyanathan et al. 2018). Furthermore, Cav3‐F97C was shown to differentially regulate current density and cell surface expression of Kir2.x homomeric and heteromeric channels, which can lead to delayed afterdepolarizations as a possible arrhythmia mechanism (Vaidyanathan et al. 2018). Intriguingly, the two Cav3 mutations investigated can have distinct effects on the sensitive ionic currents, with Cav3‐F97C causing a reduction in I Kv4.3, whereas Cav3‐S141R had no effect on I Kv4.3. Even more striking is the distinct effects of the Cav3 mutations on I Ca,L, with Cav3‐F97C specifically slowing CDI, whereas Cav3‐S141R caused a general gain of function increasing I Ca,L. Integrating the heterologous expression data into a computational model of the human ventricular AP showed that APD prolongation can result from the effects of both Cav3‐F97C and Cav3‐S141R, although the major driving conductance for APD prolongation differs with Cav3‐F97C APD prolongation dominated by the blunted CDI of I Ca,L and the Cav3‐S141R APD prolongation equally contributed to by increases in I Ca,L and I Na,L. Thus, Cav3 mutation‐specific therapy may be optimal for reducing APD prolongation and arrhythmia risk with therapies for Cav‐F97C aimed at specifically reducing CDI, whereas therapies for Cav3‐S141R can target overall I Ca,L and/or I Na,L. In addition, the overall effects of both mutations will impact Ca2+ cycling in myocytes with increased Ca2+ influx through I Ca,L, as well as increased I Na,L, leading to Ca2+ overload by activating reverse mode NCX activity. The propensity for Ca2+ overload introduced by these mutations may also contribute to arrhythmogenesis. Finally, although changes in I to are not predicted to directly impact APD, the delayed recovery of I to induced by the Cav3 mutations may produce an exaggerated loss of I to at faster heart rates that have an impact on the plateau phase of the AP and hence on I Ca,L and Ca2+ cycling.
Limitations
The present study took advantage of heterologous expression in HEK293 cells to enable study of specific channel isoform encoded ionic currents; however, the behaviour of these channels, as well as the subunit composition, may differ in native human cardiomyocytes. The regulatory pathways impacting the ionic currents under investigation are also probably different in native cardiomyocytes compared to the heterologous expression system. Furthermore, additional effects of Cav3 mutations on other ionic currents present in cardiomyocytes cannot be excluded.
Conclusions
The present study provides a new understanding with respect to how two Cav3 mutations (F97C and S141R) cause APD prolongation and are associated with LQT9. Adding to the previously demonstrated increase in I Na,L by the Cav3 variants, we find a loss of function of I Kv4.2 and I Kv4.3, as well as gain of function effects on I Ca,L. Remarkably different effects on I Ca,L are observed, with the two mutations with Cav3‐F97C causing a specific decrease in CDI of I Ca,L and Cav3‐S141R causing an overall increase in I Ca,L without specific effects on the kinetics of the currents. These distinct effects of Cav3 LQT9 mutations have implications for therapeutic strategies aiming to prevent life threatening ventricular arrhythmias and also highlight the need for further investigations into the complexities of regulation of ion channels in the heart by Cav3 and disease causing mutations in the gene.
Additional information
Competing interests
A.D. Mcculloch is a co‐founder of and has an equity interest in Insilicomed and Vektor Medical. He serves on the scientific advisory board of Insilicomed and as scientific advisor to both companies. Some of his research grants acknowledged here have been identified for conflict of interest management based on the overall scope of the project and its potential benefit to these companies, neither of which had any involvement in this research.
Author contributions
LT, JDF and KPV collected, assembled, analysed and interpreted the data, designed the experiments and drafted the manuscript. MTW, WTM, DL and JMB collected, assembled, analysed and interpreted the data. ADM and MJA conceived the work. TJK, LT, AVG and RCB conceived the experiment and revised the manuscript critically for important intellectual content. All authors approved the final version of the manuscript submitted for publication and qualify for authorship.
Funding
This work was supported by National Institutes of Health grants R01 HL078878 (TJK), P50 GM094503 (TJK, ADM) and R01 HL141214‐01 (AVG), R01 HL105242 (ADM), R01 HL137100 (ADM), R01 HL122384 (ADM), HL126273 (ADM) and P41 GM103426 (ADM) as well as American Heart Association grants SDG 16SDG29120011 (AVG) and postdoctoral fellowship 17POST33370089 (DL).
Biographies
Leonid Tyan is an electrophysiologist who has a strong interest in ion channels and their involvement in disease. He obtained his PhD in Biology in 2012 at the Eberhard Karls University of Tuebingen, Germany. He is currently an assistant scientist in Department of Medicine at the University of Wisconsin–Madison, USA. His current research interest is the impact of caveolin‐3 on ionic currents and its role in atrial and ventricular arrhythmias.

Jason D. Foell has worked at the University of Wisconsin's Cellular and Molecular Arrhythmia Research Program where he focused on identifying the essential role of caveolae in β2‐AR regulation of cardiac L‐type Ca2+ channels and characterizing the impact on L‐type Ca2+ channels of caveolin‐3 mutations from long QT patients. Other work focused on the elucidation of the cardiac L‐type Ca2+ channels β subunits in normal and heart failure patients.
Ventricular AP modelling
The 2011 O'Hara–Rudy human ventricular AP model was altered to account for changes in channel density and kinetics as a result of Cav3 mutations. The model code was obtained from the website of the original author (http://rudylab.wustl.edu) and the Cav3‐WT model corresponds to the original formulation of the model. The equations shown below were altered for one or both of the Cav3 mutant models. Parameters indicated in bold were adjusted in accordance with the experimental results, and the specific changes are listed in Table A1.
Table A1.
Parameter changes in computational model of Cav3‐F97C and Cav3‐S141R mutation
| Cav3‐WT | Cav3‐F97C | Cav3‐S141R | ||
|---|---|---|---|---|
|
|
1.0 | 1.41 | 1.0 | |
|
|
0.08 | 0.0508 | 0.08 | |
| a 1 | 3.940 | 3.940 | 8.940 | |
| b 1 | 0.04 | 0.003 | 0.04 | |
| b 2 | 4.0 | –6.0 | 4.0 | |
| b 3 | 7.0 | 16.0 | 7.0 | |
| b 4 | 0.04 | 0.0045 | 0.04 | |
| b 5 | 4.0 | –2.0 | 4.0 | |
| b 6 | 7.0 | 5.0 | 7.0 | |
| c 1 | 0.00012 | 0.0012 | 0.00012 | |
| c 2 | 0.0 | 25.0 | 0.0 | |
| c 3 | 3.0 | 5.0 | 3.0 | |
| τ j, Ca | 75.0 | 150.0 | 75.0 | |
| P Ca | 0.00012 | 0.00012 | 0.00016 | |
|
|
0.0045 | 0.009 | 0.009 |
Transient outward current (I to)
L‐type calcium current (I CaL)
Late sodium current (I Na,L)
Edited by: Don Bers & Bjorn Knollmann
References
- Alday A, Urrutia J, Gallego M & Casis O. (2010). alpha1‐adrenoceptors regulate only the caveolae‐located subpopulation of cardiac K(V)4 channels. Channels (Austin) 4, 168–178. [DOI] [PubMed] [Google Scholar]
- Bahring R, Boland LM, Varghese A, Gebauer M & Pongs O. (2001). Kinetic analysis of open‐ and closed‐state inactivation transitions in human Kv4.2 A‐type potassium channels. J Physiol 535, 65–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balijepalli RC, Foell JD, Hall DD, Hell JW & Kamp TJ. (2006). Localization of cardiac L‐type Ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)‐adrenergic regulation. Proc Natl Acad Sci U S A 103, 7500–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balijepalli RC & Kamp TJ. (2008). Caveolae, ion channels and cardiac arrhythmias. Prog Biophys Mol Biol 98, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbuti A, Terragni B, Brioschi C & DiFrancesco D. (2007). Localization of f‐channels to caveolae mediates specific beta2‐adrenergic receptor modulation of rate in sinoatrial myocytes. J Mol Cell Cardiol 42, 71–78. [DOI] [PubMed] [Google Scholar]
- Best JM, Foell JD, Buss CR, Delisle BP, Balijepalli RC, January CT & Kamp TJ. (2011). Small GTPase Rab11b regulates degradation of surface membrane L‐type Cav1.2 channels. Am J Physiol Cell Physiol 300, C1023–C1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuckelmann DJ, Nabauer M & Erdmann E. (1993). Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res 73, 379–385. [DOI] [PubMed] [Google Scholar]
- Boczek NJ, Gomez‐Hurtado N, Ye D, Calvert ML, Tester DJ, Kryshtal D, Hwang HS, Johnson CN, Chazin WJ, Loporcaro CG, Shah M, Papez AL, Lau YR, Kanter R, Knollmann BC & Ackerman MJ. (2016). Spectrum and prevalence of CALM1‐, CALM2‐, and CALM3‐encoded calmodulin variants in long QT syndrome and functional characterization of a novel long QT syndrome‐associated calmodulin missense variant, E141G. Circ Cardiovasc Genet 9, 136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossuyt J, Taylor BE, James‐Kracke M & Hale CC. (2002). The cardiac sodium‐calcium exchanger associates with caveolin‐3. Ann NY Acad Sci 976, 197–204. [DOI] [PubMed] [Google Scholar]
- Cheng J, Valdivia CR, Vaidyanathan R, Balijepalli RC, Ackerman MJ & Makielski JC. (2013). Caveolin‐3 suppresses late sodium current by inhibiting nNOS‐dependent S‐nitrosylation of SCN5A. J Mol Cell Cardiol 61, 102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colinas O, Gallego M, Setien R, Lopez‐Lopez JR, Perez‐Garcia MT & Casis O. (2006). Differential modulation of Kv4.2 and Kv4.3 channels by calmodulin‐dependent protein kinase II in rat cardiac myocytes. Am J Physiol Heart Circ Physiol 291, H1978–H1987. [DOI] [PubMed] [Google Scholar]
- Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M, Papagiannis J, Feldkamp MD, Rathi SG, Kunic JD, Pedrazzini M, Wieland T, Lichtner P, Beckmann BM, Clark T, Shaffer C, Benson DW, Kaab S, Meitinger T, Strom TM, Chazin WJ, Schwartz PJ & George AL, Jr. (2013). Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 127, 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpon E, Cordeiro JM, Nunez L, Thomsen PE, Guerchicoff A, Pollevick GD, Wu Y, Kanters JK, Larsen CT, Hofman‐Bang J, Burashnikov E, Christiansen M & Antzelevitch C. (2008). Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol 1, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JE, Shi W, Wang HS, McDonald C, Yu H, Wymore RS, Cohen IS & McKinnon D. (1996). Role of the Kv4.3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circ Res 79, 659–668. [DOI] [PubMed] [Google Scholar]
- Foell JD, Balijepalli RC, Delisle BP, Yunker AM, Robia SL, Walker JW, McEnery MW, January CT & Kamp TJ. (2004). Molecular heterogeneity of calcium channel beta‐subunits in canine and human heart: evidence for differential subcellular localization. Physiol Genomics 17, 183–200. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Myerburg RJ, Furukawa N, Bassett AL & Kimura S. (1990). Differences in transient outward currents of feline endocardial and epicardial myocytes. Circ Res 67, 1287–1291. [DOI] [PubMed] [Google Scholar]
- Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S & Demolombe S. (2007). Regional and tissue specific transcript signatures of ion channel genes in the non‐diseased human heart. J Physiol 582, 675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudicessi JR, Ye D, Tester DJ, Crotti L, Mugione A, Nesterenko VV, Albertson RM, Antzelevitch C, Schwartz PJ & Ackerman MJ. (2011). Transient outward current (I(to)) gain‐of‐function mutations in the KCND3‐encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 8, 1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gussak I, Chaitman BR, Kopecky SL & Nerbonne JM. (2000). Rapid ventricular repolarization in rodents: electrocardiographic manifestations, molecular mechanisms, and clinical insights. J Electrocardiol 33, 159–170. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Flockerzi V, Kahl S & Wegener JW. (2014). L‐type CaV1.2 calcium channels: from in vitro findings to in vivo function. Physiol Rev 94, 303–326. [DOI] [PubMed] [Google Scholar]
- Kaab S, Nuss HB, Chiamvimonvat N, O'Rourke B, Pak PH, Kass DA, Marban E & Tomaselli GF. (1996). Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing‐induced heart failure. Circ Res 78, 262–273. [DOI] [PubMed] [Google Scholar]
- Kuo HC, Cheng CF, Clark RB, Lin JJ, Lin JL, Hoshijima M, Nguyen‐Tran VT, Gu Y, Ikeda Y, Chu PH, Ross J, Giles WR & Chien KR. (2001). A defect in the Kv channel‐interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell 107, 801–813. [DOI] [PubMed] [Google Scholar]
- Limpitikul WB, Dick IE, Joshi‐Mukherjee R, Overgaard MT, George AL, Jr. & Yue DT. (2014). Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L‐type Ca(2+) currents and promote proarrhythmic behavior in ventricular myocytes. J Mol Cell Cardiol 74, 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DW, Gintant GA & Antzelevitch C. (1993). Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ Res 72, 671–687. [DOI] [PubMed] [Google Scholar]
- Mann SA, Imtiaz M, Winbo A, Rydberg A, Perry MD, Couderc JP, Polonsky B, McNitt S, Zareba W, Hill AP & Vandenberg JI. (2016). Convergence of models of human ventricular myocyte electrophysiology after global optimization to recapitulate clinical long QT phenotypes. J Mol Cell Cardiol 100, 25–34. [DOI] [PubMed] [Google Scholar]
- McCrossan ZA & Abbott GW. (2004). The MinK‐related peptides. Neuropharmacology 47, 787–821. [DOI] [PubMed] [Google Scholar]
- Niwa N & Nerbonne JM. (2010). Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J Mol Cell Cardiol 48, 12–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara T, Virag L, Varro A & Rudy Y. (2011). Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol 7, e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S, Zankov DP, Ding WG, Itoh H, Makiyama T, Doi T, Shizuta S, Hattori T, Miyamoto A, Naiki N, Hancox JC, Matsuura H & Horie M. (2011). KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ Arrhythm Electrophysiol 4, 352–361. [DOI] [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Adelman JP & Yue DT. (1999). Calmodulin is the Ca2+ sensor for Ca2+‐dependent inactivation of L‐type calcium channels. Neuron 22, 549–558. [DOI] [PubMed] [Google Scholar]
- Po SS, Wu RC, Juang GJ, Kong W & Tomaselli GF. (2001). Mechanism of alpha‐adrenergic regulation of expressed hKv4.3 currents. Am J Physiol Heart Circ Physiol 281, H2518–H2527. [DOI] [PubMed] [Google Scholar]
- Radicke S, Cotella D, Graf EM, Ravens U & Wettwer E. (2005). Expression and function of dipeptidyl‐aminopeptidase‐like protein 6 as a putative beta‐subunit of human cardiac transient outward current encoded by Kv4.3. J Physiol 565, 751–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati B, Pan Z, Lypen S, Wang HS, Cohen I, Dixon JE & McKinnon D. (2001). Regulation of KChIP2 potassium channel beta subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. J Physiol 533, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah R, Ramirez RJ & Backx PH. (2002). Modulation of Ca(2+) release in cardiac myocytes by changes in repolarization rate: role of phase‐1 action potential repolarization in excitation‐contraction coupling. Circ Res 90, 165–173. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Crotti L & Insolia R. (2012). Long‐QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol 5, 868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldatov NM, Bouron A & Reuter H. (1995). Different voltage‐dependent inhibition by dihydropyridines of human Ca2+ channel splice variants. J Biol Chem 270, 10540–10543. [DOI] [PubMed] [Google Scholar]
- Song KS, Scherer PE, Tang Z, Okamoto T, Li S, Chafel M, Chu C, Kohtz DS & Lisanti MP. (1996). Expression of caveolin‐3 in skeletal, cardiac, and smooth muscle cells. Caveolin‐3 is a component of the sarcolemma and co‐fractionates with dystrophin and dystrophin‐associated glycoproteins. J Biol Chem 271, 15160–15165. [DOI] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager‐Flusberg H, Priori SG, Sanguinetti MC & Keating MT. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. [DOI] [PubMed] [Google Scholar]
- Vaidyanathan R, Markandeya YS, Kamp TJ, Makielski JC, January CT & Eckhardt LL. (2016). IK1‐enhanced human‐induced pluripotent stem cell‐derived cardiomyocytes: an improved cardiomyocyte model to investigate inherited arrhythmia syndromes. Am J Physiol Heart Circ Physiol 310, H1611–H1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan R, Van Ert H, Haq KT, Morotti S, Esch S, McCune EC, Grandi E & Eckhardt LL. (2018). Inward rectifier potassium channels (Kir2.x) and caveolin‐3 domain‐specific interaction: implications for purkinje cell‐dependent ventricular arrhythmias. Circ Arrhythm Electrophysiol 11, e005800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan R, Vega AL, Song C, Zhou Q, Tan BH, Berger S, Makielski JC & Eckhardt LL. (2013). The interaction of caveolin 3 protein with the potassium inward rectifier channel Kir2.1: physiology and pathology related to long qt syndrome 9 (LQT9). J Biol Chem 288, 17472–17480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ & Towbin JA. (2006). Mutant caveolin‐3 induces persistent late sodium current and is associated with long‐QT syndrome. Circulation 114, 2104–2112. [DOI] [PubMed] [Google Scholar]
- Woon MT, Long PA, Reilly L, Evans JM, Keefe AM, Lea MR, Bellinger CJ, Balijepalli RC, Lee Y, Olson TM, & Kamp TJ. (2017). Pediatric dilated cardiomyopathy‐associated LRRC10 (Leucine‐rich repeat‐containing 10) variant reveals LRRC10 as an auxiliary subunit of cardiac L‐type Ca2+ channels. J Am Heart Assoc 6, e006428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarbrough TL, Lu T, Lee HC & Shibata EF. (2002). Localization of cardiac sodium channels in caveolin‐rich membrane domains: regulation of sodium current amplitude. Circ Res 90, 443–449. [DOI] [PubMed] [Google Scholar]
- Ye B, Balijepalli RC, Foell JD, Kroboth S, Ye Q, Luo YH & Shi NQ. (2008). Caveolin‐3 associates with and affects the function of hyperpolarization‐activated cyclic nucleotide‐gated channel 4. Biochemistry 47, 12312–12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin G, Hassan F, Haroun AR, Murphy LL, Crotti L, Schwartz PJ, George AL & Satin J. (2014). Arrhythmogenic calmodulin mutations disrupt intracellular cardiomyocyte Ca2+ regulation by distinct mechanisms. J Am Heart Assoc 3, e000996. [DOI] [PMC free article] [PubMed] [Google Scholar]