

Abstract
In the early 80s, renal microperfusion studies led to the identification of a basolateral K+‐Cl− cotransport mechanism in the proximal tubule, thick ascending limb of Henle and collecting duct. More than ten years later, this mechanism was found to be accounted for by three different K+‐Cl− cotransporters (KCC1, KCC3 and KCC4) that are differentially distributed along the renal epithelium. Two of these isoforms (KCC1 and KCC3) were also found to be expressed in arterial walls, the myocardium and a variety of neurons. Subsequently, valuable insights have been gained into the molecular and physiological properties of the KCCs in both the mammalian kidney and cardiovascular system. There is now robust evidence indicating that KCC4 sustains distal renal acidification and that KCC3 regulates myogenic tone in resistance vessels. However, progress in understanding the functional significance of these transporters has been slow, probably because each of the KCC isoforms is not identically distributed among species and some of them share common subcellular localizations with other KCC isoforms or sizeable conductive Cl− pathways. In addition, the mechanisms underlying the process of K+‐Cl− cotransport are still ill defined. The present review focuses on the knowledge gained regarding the roles and properties of KCCs in renal and cardiovascular tissues.
Keywords: K+‐Cl− cotransporter, Cation‐Cl− cotransporter, Renal tubular acidosis, Animal models, Cardiovascular system, Systemic hypertension
Introduction
The K+‐Cl− cotransporters (KCCs) consist of cell surface membrane‐embedded glycoproteins that come as four isoforms termed KCC1, KCC2, KCC3 and KCC4 (Garneau et al. 2017; Marcoux et al. 2017). They belong to the cation‐Cl− cotransporter (CCC) protein family along with five additional members: Na+‐K+‐Cl− cotransporter (NKCC2; SLC12A1), NKCC1 (SLC12A2), Na+‐Cl− cotransporter (NCC; SLC12A3), CCC9 (SLC12A8) and CCC8 (SLC12A9). Except for CCC8 and CCC9, the main role of CCC family members is to mediate the electroneutral translocation of Cl− with Na+ and/or K+ across the membrane. For the KCCs, the transport cycle is Na+‐independent.
The amino acid sequences of the KCCs were uncovered during the ’90s (Gillen et al. 1996; Payne et al. 1996; Hiki et al. 1999; Mount et al. 1999; Race et al. 1999) just after those of the Na+‐dependent CCCs and 15 years after the identification of a K+‐Cl− cotransport mechanism in red blood cells (Kregenow, 1971; Dunham et al. 1980; Lauf & Theg, 1980). During and subsequent to their initial characterization, the Na+‐independent CCCs were shown to be widely distributed and three of them were detected in the kidney (Pollak et al. 1996; Simon et al. 1996; Mount et al. 1999; Di Stefano et al. 2001; Boettger et al. 2003; Velazquez & Silva, 2003). Based on localization studies or indirect evidence, KCC3 was also detected in vascular smooth muscle cells (VSMCs) (Di Fulvio et al. 2001a , 2003; Rust et al. 2006; Garneau et al. 2016), cardiomyocytes (Garneau et al. 2016) and dorsal root ganglion neurons (Pearson et al. 2001; Boettger et al. 2003; Le Rouzic et al. 2006; Mizuno et al. 2011; Lucas et al. 2012), all of which are involved in blood pressure control.
This review provides an overview of the knowledge gained over the years on the molecular and functional features of K+‐Cl− cotransport in the kidney and cardiovascular system, with an emphasis on the effects of Kcc ablation in animal models. Although progress in understanding such features has been substantial, further research to link loss‐ or gain‐of‐function mutations in the KCCs with renal salt handling and blood pressure regulation is needed, particularly in humans.
Molecular characteristics of the KCCs
Splice variants
Each of the KCC genes can be processed into several splice variants (Garneau et al. 2017; Marcoux et al. 2017). However, not all of the predicted mRNAs have been identified thus far in animal tissues according to databanks and expression studies. The mouse and human variants predicted for KCC1, KCC3 and KCC4 are listed in Table 1, along with their loci of origin, lengths and characteristics. Those indicated with the letter κ have been identified in the kidney and those with the letter ε are the ones found in extrarenal as well as extravascular tissues.
Table 1.
Splice variants
| Splice variants predicted and expressed | |||||||
|---|---|---|---|---|---|---|---|
| Isoform | Gene locus | Official name | Names in Garneau and Marcoux reviews | Reference NCBI | n of E | Protein length | Presence in tissues |
| Human | 16q22.1 | 1 | E1Ais2 | NM_005072.4 | 24 | 1085 | κ, ε |
| KCC1 | 2 | E1Ais2/E20* | NM_001145961.1 | 24 | 1079 | ‐ | |
| 3 | E1Ais1/E2∆ | NM_001145962.1 | 23 | 1087 | ε | ||
| 4 | E1C | NM_001145963.1 | 24 | 1079 | ε | ||
| 5 | E1B | NM_001145964.1 | 24 | 1054 | ε | ||
| Mouse | 8D3.8 | 1 | E1is2/E1* | NM_001253804.1 | 24 | 1087 | ε |
| KCC1 | 2 | E1is2 | NM_009195.3 | 24 | 1085 | κ, ε | |
| X1 | E1is1/E2∆ | XM_006530792 | 23 | 1080 | κ, ε | ||
| 3 | E1is2/E15* | NR_045594 | 24 | 692 | ε | ||
| Human | 15q14 | 1 | E1Ais1 | NM_133647.1 | 25 | 1150 | ε |
| KCC3 | 5 | E1Ais2 | NM_001042496.1 | 26 | 1141 | ε | |
| 3 | E1Ais3 | NM_001042494.1 | 26 | 1091 | ε | ||
| 4 | E1Ais3 | NM_001042495.1 | 26 | 1091 | ε | ||
| 6 | E1Ais1/E2∆ | NM_001042497.1 | 24 | 1135 | ε | ||
| 2 | E1B | NM_005135.2 | 25 | 1099 | εa | ||
| ‐ | E1B/E2∆ | ‐ | 24 | ‐ | εa | ||
| Mouse | 2E3 | 1 | E1Bis2 | NM_133648.2 | 25 | 1099 | κ |
| KCC3 | 2 | E1Ais2 | NM_133649.2 | 25 | 1150 | ε | |
| X1 | E1Ais1/E2∆ | XM_006498545.3 | 24 | 1135 | ε | ||
| X2 | E1Bis1/E2∆ | XM_006498546.3 | 24 | 1084 | ε | ||
| Human | 5p15.33 | X1 | E1A | XM_005248231.3 | 25 | 1088 | ε |
| KCC4 | mRNA | E1A/E23∆ | NM_006598.2 | 24 | 1083 | ε | |
| X2 | E1B | XM_011513941.2 | 25 | 1111 | ‐ | ||
| X3 | E1C/E1D | XM_011513939.2 | 26 | 1029 | ‐ | ||
| X4 | E1E | XM_011513940.2 | 25 | 1074 | ‐ | ||
| X5 | E1F | XM_017008958.1 | 25 | 1020 | ‐ | ||
| CRA_b | E1F/E23∆ | EAX08179.1 | 24 | 1015 | ‐ | ||
| CRA_a | E1F/E10*/I10+*/E23∆ | EAX08178.1 | 24 | 1014 | ‐ | ||
| Mouse | 13C1.13 | X3 | E1A | XM_006517172.3 | 25 | 1088 | κ, ε |
| KCC4 | CRA_b | E1A/E23∆ | NM_011390.2 | 24 | 1083 | κ, ε | |
| CRA_a | E1B/I9+* | EDL37071.1 | 25b | 528 | ε | ||
| X1 | B | XM_006517170.3 | 25 | 1117 | ε | ||
| X2 | E1B/E23∆ | XM_006517171.2 | 24 | 1112 | ε | ||
| X6 | E1C/E1D | XM_006517175.2 | 26 | 1020 | ε | ||
| X4 | E1E | XM_006517173.3 | 25 | 1063 | ‐ | ||
| X5 | E1F | XM_006517174.3 | 25 | 1055 | ‐ | ||
The mRNA sequences are designated according to the nomenclature of GenBank and to that of previous reviews (Garneau et al. 2017; Marcoux et al. 2017). Unless indicated, all of the exons beyond the first one are included in the mRNA.
expressed in kidney of orangutan, but not of mouse and human.
if exon 23 is present.
Abbreviations: ∆, deleted; E, exon; ε expression in extrarenal tissues; I, intron; is, initiation site; κ, expression in kidney; +, inserted; *, present only in part. Note that several of the cDNA sequences identified, those of KCC3 in heart, vascular tissues and adipose depots more specifically, were incomplete so that the variants to which they belonged could not be determined.
Structural aspects
The KCC isoforms share high levels of identity in amino acid sequence among each other (∼70% overall), but moderate levels of identity with the Na+‐dependent CCCs, CCC8 and CCC9 (∼30% overall). For these reasons, and as illustrated in the cladogram of Fig. 1, the KCCs fall into a distinct phylogenetic clade within the family. Among the various CCCs, importantly, the central domain and C‐terminus are more conserved than the N‐terminus (Garneau et al. 2017; Marcoux et al. 2017).
Figure 1.
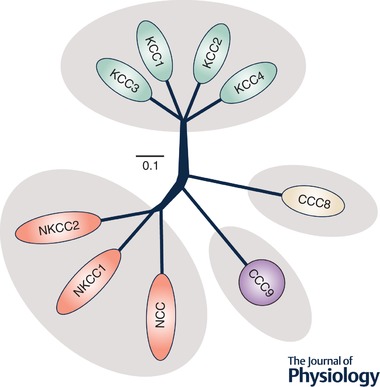
Cladogram of human CCC family members
The tree was generated with Clustal Omega and FigTree v1.4.3. Amino acid sequences included in the phylogenetic analysis were from the most abundant CCC variants in human. Accession numbers used: NKCC1, NP_001037.1; NKCC2, NP_000329.2; NCC, NP_000330.2; KCC1, NP_005063.1; KCC2, NP_001128243.1; KCC3, NP_598408.1; KCC4, NP_006589.2; CCC8, NP_064631.2; CCC9, NP_078904.3.
The topological configuration of the KCCs is predicted to be nearly identical to that of NKCC1, NKCC2 and NCC (Garneau et al. 2017; Marcoux et al. 2017), i.e. to consist of a central core of 12 α‐helicoidal transmembrane domains flanked by cytosolic extremities (see Fig. 2). As for the higher‐order structure of the KCCs, it consists mainly of homodimers that are assembled through self‐interacting domains within the C‐terminus (Simard et al. 2004, 2007; Bergeron et al. 2011). Using the Xenopus laevis oocyte system, Simard et al. (2007) have also found that KCC isoforms can associate with each other or with NKCC1 to form heterodimers. If such structures were to be relevant in vivo, they would bring about substantial diversity to the functional properties of cation‐Cl− cotransport along the renal epithelium and in the cardiovascular system.
Figure 2.
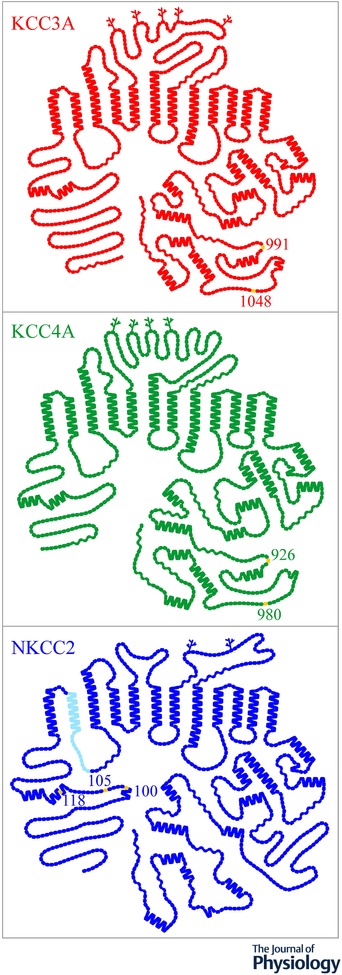
Topological models of NKCC1, KCC3 and KCC4
The models were generated with PLOT (Biff Forbush, Yale University). Each glycosylation site is illustrated through a branched line and each residue through a single symbol. Yellow is used to designate well‐characterized phosphoacceptor residues and light blue a protein segment that comes in three variants from alternative splicing of the primary transcript.
Characteristics and mechanisms of ion movement
There is strong evidence to suggest that during each transport cycle by a KCC, K+ and Cl− ions are translocated simultaneously across the membrane in a stoichiometric ratio of 1 cation:1 anion (Piwnica‐Worms et al. 1985; Kaji, 1993; Jennings & Adame, 2001). Carrier activity is thus electroneutral. Although many investigators assume that there is only one binding site for each substrate (Payne, 1996), other investigators have observed that the dependence of certain KCCs on [Cl−]e was described more adequately by a two‐ or even three‐binding site model based on the data presented (see data in Mercado et al. 2000, 2005; Bergeron et al. 2003).
In vivo, K+‐Cl− cotransport is typically outwardly directed given that the [K+]i‐to‐[K+]e chemical gradient is at least 2.0 times higher than the [Cl−]e‐to‐[Cl−]i chemical gradient. As such, the outwardly directed K+ chemical gradient drives Cl− against its own chemical gradient. All KCCs can also act as efficient NH4 +‐Cl− cotransporters by translocating NH4 + through the K+ binding site (Bergeron et al. 2003, 2009). However, NH4 +‐Cl− cotransport by these proteins should be inwardly directed given that the [NH4 +]e‐to‐[NH4 +]i chemical gradient is well above 0 in most tissues (Prosser, 1991; Wall et al. 1995; Evans & Turner, 1998; Glanville et al. 2001). By facilitating the uptake of NH4 +, the KCCs are thus likely to be involved in pHi regulation and transepithelial NH4 + transport. Even though inward NH4 +‐Cl− cotransport is probably less important quantitatively than outward K+‐Cl− cotransport under physiological conditions, it should also reduce the net amount of water that follows the direction of K+.
Aside from only a few exceptions, the KCCs have been found to be expressed basolaterally in epithelial cells (Sasaki et al. 1988; Liapis et al. 1998; Sangan et al. 2000; Pearson et al. 2001; Boettger et al. 2002, 2003; Velazquez & Silva, 2003; Gamba, 2005; Mercado et al. 2005; Fujii et al. 2009; Melo et al. 2013a). They should thus promote net Cl− and fluid reabsorption or net Cl− and NH4 + secretion in tubular and alveolar glands (Wall et al. 1995; Bergeron et al. 2003; Garneau et al. 2017; Marcoux et al. 2017). In certain epithelia such as those of the proximal nephron and gastric glands, they probably play a minor role in these processes given the transported ions are recycled back to the cytosol once they have exited the cell (Boettger et al. 2002; Mobasheri et al. 2003; Fujii et al. 2008, 2009). In all likelihood, the purpose of K+‐Cl− or NH4 +‐Cl− cotransport would then be to sustain the activity of other transport systems by providing them with a constant supply of substrates.
So far, the isoform‐specific functional characteristics of the KCCs have been examined by a few research groups in Xenopus laevis oocytes and HEK‐293 cells (Gillen et al. 1996; Payne et al. 1996; Race et al. 1999; Song et al. 2002; Bergeron et al. 2003; Gamba, 2005; Rinehart et al. 2009). It was found that compared to the other isoforms, KCC1 tended to exhibit lower transport capacities and ion affinities, and KCC4 to exhibit much lower sensitivity to furosemide and greater activity at low pHi (Bergeron et al. 2003). As for the native properties of K+‐Cl− cotransport, the data reported are difficult to interpret given that most cell types express more than one KCC isoform in addition to NKCC1 (Lauf et al. 2001; Crable et al. 2005; Rust et al. 2007) and given that CCC family members cannot be distinguished easily from one another based on functional and pharmacological studies. Otherwise, K+‐Cl− cotransport activity was seen in several studies to be increased by cell swelling, high [Cl−]i and N‐ethylmaleimide (NEM).
Mechanisms of regulation
According to the widely held view, the KCCs are activated by cell swelling through their dephosphorylation and inactivated under basal conditions through their phosphorylation. This view is supported by a large body of indirect evidence (Jennings & Schulz, 1991; Mercado et al. 2000, 2001, 2016; Song et al. 2002; Garzon‐Muvdi et al. 2007; Agalakova & Gusev, 2009; Gusev & Agalakova, 2010) and the identification of conserved threonine residues that undergo dephosphorylation during cell swelling (Rinehart et al. 2009; Melo et al. 2013b; de Los Heros et al. 2014). The localization of two such residues is shown in Fig. 2. Frenette‐Cotton et al. (2017) have observed more recently that KCC4 activation under hypotonic conditions was accompanied by an increase rather than a decrease in overall carrier phosphorylation, indicating that the mechanisms of regulation by kinases and phosphatases are much more complex than implied by the current view.
A number of the signalling intermediates involved have now been identified. They include the ‘with no‐lysine’ protein kinases (WNKs) of which four isoforms have been identified (Garzon‐Muvdi et al. 2007; de Los Heros et al. 2014; Mercado et al. 2016; Frenette‐Cotton et al. 2017). Two of them drew much attention a few years ago when they were linked to pseudohypoaldosteronism type II (PHAII), a rare hereditary disorder of renal electrolyte handling characterized by high blood pressure, metabolic acidosis and hyperkalaemia (Wilson et al. 2001, 2003; Golbang et al. 2005). The mutations uncovered, loss‐of‐function in WNK4 and gain‐of‐function in WNK1, probably lead to increased Na+ and Cl− absorption by a nephron segment that is proximal to the site of active K+ and H+ secretion. It is thought that NCC plays an important pathogenic role in PHAII given that the electrolyte abnormalities and blood pressure can be normalized through the administration of thiazides. In addition, WNK1 has been shown to increase NCC expression by downregulating WNK4 (Wilson et al. 2001; Mayan et al. 2002; Yang et al. 2003, 2005, 2007; Cai et al. 2006; Lalioti et al. 2006).
The KCCs could also play a role in PHAII given that all of the WNKs have been found to inhibit K+‐Cl− cotransport (Garzon‐Muvdi et al. 2007; Rinehart et al. 2009; Melo et al. 2013b; de Los Heros et al. 2014; Kahle et al. 2016; Mercado et al. 2016; Frenette‐Cotton et al. 2017) and that loss‐of‐function mutations in WNK4 should therefore increase K+‐Cl− cotransport in the renal epithelium. Curiously, however, the KCCs are also inhibited by WNK1 while gain‐of‐function mutations in this enzyme lead to increased Na+‐Cl− cotransport by relieving the inhibitory effect of WNK4 on NCC. Presumably, the ability of WNK1 to affect WNK4 could vary as a function of the target protein. Many other transport systems are affected by these enzymes and could thus play a role in disease expression as well (see reviews by Garneau et al. 2017; Marcoux et al. 2017).
Role and distribution of the KCCs in the kidney
Preamble
During the early characterization of KCC1, KCC3 and KCC4, Northern or Western blot analyses showed that all three isoforms were expressed in human kidney at high levels (Gillen et al. 1996; Hiki et al. 1999; Mount et al. 1999; Race et al. 1999). Message abundance inferred from the current EST databanks is still in keeping with these findings. As for KCC2, its presence in renal cells has not been actively looked for as it was initially determined to be neuron‐specific (Payne et al. 1996; Williams et al. 1999). Yet, it could be expressed in epithelial fibre lens cells based on a recent study (Frederikse & Kasinathan, 2015) and is represented in the human kidney EST databank by a few transcripts. Of note, the KCCs are all present on the basolateral side of the renal epithelium (Liapis et al. 1998; Boettger et al. 2002, 2003; Velazquez & Silva, 2003; Melo et al. 2013a). Table 2 summarizes the renal distribution of KCC1, KCC3 and KCC4 in two species.
Table 2.
Localization of the KCCs in the nephron
| Renal segment | KCC1 | KCC3 | KCC4 |
|---|---|---|---|
| PT | h‐IHc | m‐ILb | m‐ILa |
| ‐S1 | m‐ILe | ||
| ‐S2 | m‐ILe | ||
| ‐S3 | m‐ILe | ||
| TAL | |||
| ‐MTAL | r‐ILf | ||
| ‐CTAL | |||
| DT | h‐IHc | r‐ILf | |
| CT | r‐ILf | ||
| CD | h‐IHc | ||
| ‐CCD | m‐ILa, d | ||
| ‐OMCD | r‐ILe |
The table was constructed from multiple data sources as indicated by the references. KCC1, KCC3 and KCC4 were detected predominantly or exclusively in the nephron segments listed. Abbreviations: h‐IH, based on in situ hybridization in human (3); m‐IL, based on immunolocalization studies in mouse (1, 2, 4 and 5); r‐IL, based on immunolocalization studies in rabbit (6). Other abbreviations: CCD, cortical collecting duct; CD, collecting duct; CT, connecting tubule; CTAL, cortical thick ascending limb; DT, distal tubule; MTAL, medullary thick ascending limb; OMCD, outer medullary collecting duct; PT, proximal tubule.
Boettger et al. 2002
Boettger et al. 2003
Liapis et al. 1998
Melo et al. 2013a
Mercado et al. 2005
Velazquez & Silva, 2003
Even though the renal epithelium is endowed with sizeable K+‐Cl− cotransport activity (Greger, 1985; Avison et al. 1988; Sasaki et al. 1988) and several mouse models of Kcc inactivation have been characterized (Boettger et al. 2002, 2003; Wang et al. 2003; Garneau et al. 2016), the overall contribution of this transport function to renal electrolyte and water handling remains unsettled. As alluded to previously, there are various reasons for this: the renal phenotype of the animal models generated has not been the object of exhaustive reports; the KCCs have still not been linked to Mendelian forms of renal disorders; the basolateral step of K+ and Cl− reabsorption is already ensured through conductive pathways in many nephron segments; and the question of whether renal K+‐Cl− cotransport could fulfil a more important role during adaptive responses or pathological conditions has not been addressed either.
In the following sections, the role and distribution of KCCs along the nephron will be discussed ‘segment’ by ‘segment’ in view of the evidence available. The potential effect of K+‐Cl− cotransport on the activity of other transport systems and overall transepithelial salt movement will also be presented through illustrative models. The role of KCC2 will not be discussed as there are no data available.
Proximal tubule
KCC1, KCC3 and KCC4 have been detected in the proximal tubule (PT): KCC1 by in situ hybridization (Liapis et al. 1998), KCC3 and KCC4 by immunolocalization (Boettger et al. 2002, 2003; Velazquez & Silva, 2003; Melo et al. 2013a) and KCC4 by RT‐PCR (Velazquez & Silva, 2003). In two additional reports (Pearson et al. 2001; Mercado et al. 2005), Northern blot analyses revealed the presence of KCC3A and KCC3B in mouse kidney, with KCC3B being more abundant than KCC3A. In these reports, however, the segmental distribution of the splice variants was not assessed and their detection was achieved through different probes. As for KCC4, immunofluorescence studies showed differences in segmental distribution among species, that is, the PT was the site of maximal expression in mouse kidney while it was the lowest in rabbit kidney (Boettger et al. 2002; Velazquez & Silva, 2003). Such differences could also suggest that the antibody used differed in specificity.
Before the localization data were reported, a K+‐Cl− cotransport system had already been identified through microperfusion studies in the basolateral PT of rabbit (Avison et al. 1988; Sasaki et al. 1988). Based on the current knowledge, it was probably accounted for by KCC1, KCC3 and/or KCC4. In one of the microperfusion studies, carrier activity was also found to be higher in the presence of glucose (Avison et al. 1988). Although interesting, this observation did not provide insight into the functional meaning of K+‐Cl− cotransport in the proximal epithelium, nor did it illuminate the role and contribution of the individual isoforms.
Among the Kcc3‐null mouse models that have been characterized since the early 2000s, two of them (termed Howard‐Kcc3 −/− 129/Sv × C57BL/6 and Garneau‐Kcc3 −/− C57BL/6J in this review) were reported to exhibit polyuria (Wang et al. 2003; Garneau et al. 2016). In one model, this abnormality was associated with lower fluid reabsorption in microperfused PT (Wang et al. 2003), and in the other, with higher food and water intake (Garneau et al. 2016). Based on these findings, and as illustrated in Fig. 3 A, KCC3 could thus play a role in the serosal step of Cl− and fluid reabsorption in the proximal tubule. Polyuria in either of the models could have also been of dietary origin given that it was associated with widespread lesions of the central nervous system.
Figure 3.
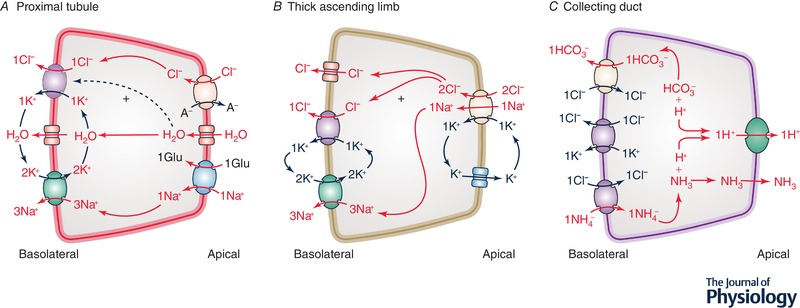
Role of K+‐Cl− cotransport in the mammalian nephron
A, proximal nephron. Ion transport systems shown correspond to a KCC, AQP1 and the Na+/K+‐ATPase on the basolateral membrane and to a Cl−‐dependent carrier (such as SLC26A6), AQP1 and a Na+‐dependent carrier (such as SGLT2) on the apical membrane. B, thick ascending limb of Henle. Ion transport systems shown correspond to a Cl− channel, a KCC and the Na+/K+‐ATPase on the basolateral membrane and to NKCC2 and KCNJ1 on the apical membrane. C, collecting duct (α‐IC cell). Ion transport systems shown correspond to SLC4A1 (Cl−/HCO3 − exchanger) or SLC26A7 (Cl−/A− exchanger) and two KCCs on the basolateral membrane and to vacuolar H+‐ATPase on the apical membrane. Note that a H+/K+‐ATPase is also present on the apical membrane but is not shown. Red is used to indicate net secretion or absorption of the substrates. Abbreviations: A−, anion including HCO3 −, OH−, SO4 −2 and oxalate−2; Glu, glucose.
In keeping with previous findings, Melo et al. (2013a) observed that hyperglycaemia increased KCC3 expression in rodent kidney ∼2‐fold while Na+ or K+ deprivation led to no change in carrier abundance. On this basis, they suggested that the role of KCC3 in the PT was to prevent SGLT2 activity from swelling the epithelium through the apical uptake of glucose or to sustain SGLT2 activity by promoting the basolateral efflux of Na+ through the Na+ pump (see Fig. 3 A). At that point, however, Melo et al. (2013a) provided no data on the precise localization of KCC3 or SGLT2 in the kidney and no data either on the expression of SGLT2 or the pump in response to hyperglycaemia.
A role for KCC3 in the proximal nephron was suggested more convincingly by the studies of Boettger et al. (2003) on a third mouse model of Kcc3 inactivation, here referred to as Boettger‐Kcc3 −/− 129/Sv × C57BL/6. Indeed, straight PTs isolated from this model were found to exhibit excessive cell swelling under hypotonic condition. Intriguingly, however, the Boettger‐Kcc3 −/− 129/Sv × C57BL/6 model was not reported to exhibit polyuria as were the two other models. In this regard, neurological abnormalities were also present in the Boettger model, but the structures affected differed from those of the other models to some extent (Garneau et al. 2017; Marcoux et al. 2017). Such differences suggest that the genetic backgrounds, generation numbers or experimental approaches exploited, all of which varied among the studies, affected the expression or detection of certain traits.
Melo et al. (2013a) have also studied the effect of hyperglycaemia and Na+ deprivation on KCC4 expression. For this isoform, abundance increased under both challenges. Yet the changes reported were modest (less than ∼1.5‐fold), they were not localized precisely through imaging studies and they could have been unrelated to glucose transport in tubular cells as polyuria of any cause is known to alter the expression of ion transporters along the nephron (Capasso et al. 1995). Otherwise, the PT of another mouse model called Boettger‐Kcc4 −/− 129/Sv × C57BL/6 was also characterized by an impaired RVD response (Boettger et al. 2002), albeit to a lesser extent than the PT of the Boettger‐Kcc3 −/− 129/Sv × C57BL/6 mouse model (Boettger et al. 2003).
Taken together, the evidence reviewed does not clarify the physiological role of K+‐Cl− cotransport in the PT. A clearer picture should emerge once the renal phenotype of the Kcc‐null mice is fully characterized under normal and pathological conditions. A clearer picture could also emerge through the identification of mutations in the human Kccs. Such mutations have already been uncovered in KCC3 (Howard et al. 2002; Uyanik et al. 2006; Rudnik‐Schoneborn et al. 2009; Lourenco et al. 2012; Kahle et al. 2016), but their clinical repercussions on salt and water handling by the kidney remain to be determined.
Thick ascending limb of Henle
KCC4 was detected in the thick ascending limb of Henle (TAL) of rabbit by Velazquez & Silva (2003) through immunofluorescence studies (where carrier abundance was seen to be higher in the medullary TAL than in the cortical TAL) and through single nephron RT‐PCR studies (where distribution was seen to be equal). In other studies (Boettger et al. 2002; Melo et al. 2013a), KCC4 was not reported to be present in this nephron segment, but the localization data were obtained from rodent rather than rabbit tissues. It should be mentioned, in addition, that the specificity of the antibodies used does not appear to have been validated against Kcc4‐null tissues in all of the studies.
A K+‐Cl− cotransport system had also been identified through earlier microperfusion studies at the basolateral side of rabbit TAL (Greger & Schlatter, 1983; Greger, 1985). It was suspected to be a KCC because K+ efflux at this location was higher than K+ conductance and because a Cl−‐coupled electroneutral efflux pathway would be more efficient than an electrogenic Cl− efflux pathway at sustaining Na+/K+‐ATPase activity (Fig. 3 B). In these studies, however, no other experiments were conducted to confirm that the functional signature of the transport system belonged to a KCC. In addition, Cl− efflux across the basolateral TAL is also known to be driven by a large intracellular negative voltage so that it is probably mediated via Cl− channels to a substantial degree (Hebert & Andreoli, 1984; Greger, 1985; Yoshitomi et al. 1988; Mount, 2014).
As discussed earlier, Melo et al. (2013a) have found that Na+ deprivation increased KCC4 expression in the kidney but did not report whether the TAL was affected by such changes. They concluded that an increase in KCC4 activity on the basolateral side of this nephron segment could still maximize transepithelial salt reabsorption under conditions of decreased extracellular fluid volume. Considering mathematical models by another research group (Weinstein, 2010), KCC4 was also suspected to play an important role in the TAL based on the prediction that it would have the required capacity to modulate the activity of luminal NKCC2 by altering cytosolic [Cl−] based on serosal [K+].
To date, it is thus not clear if KCC4 is expressed in the mammalian TAL and would even be functionally relevant at this location. If so, the Boettger‐Kcc4 −/− 129/Sv × C57BL/6 mouse model would have been expected to develop a Bartter‐like phenotype (Boettger et al. 2002) as did the Nkcc2 −/−, Kcnj1 −/− or Clcnkb −/− mouse models (Takahashi et al. 2000; Lorenz et al. 2002; Grill et al. 2016). Yet, no such phenotype has been reported in the various animal models of Kcc inactivation or in association with pathogenic mutations at the human Kcc4 chromosomal locus.
Collecting duct
KCC4 has been detected in the renal collecting duct (CD) of mouse and rabbit (Boettger et al. 2002; Velazquez & Silva, 2003). Based on immunolocalization studies in mouse using Cl−/HCO3 − exchanger type 1 (SLC4A1) and the β subunit of vacuolar H+/K+‐ATPase as comarkers, expression was seen at the basolateral side of cortical α‐intercalated (α‐IC) cells exclusively (Boettger et al. 2002). Based on the immunolocalization studies in rabbit, however, expression was higher in other nephron segments, albeit still higher in the cortical CD. KCC4 was also detected through single nephron RT‐PCR in rabbit CD, where it was seen to be diffusively distributed (Velazquez & Silva, 2003). As for KCC1, it was also present in the CD based on in situ hybridization of human kidney sections, but at higher levels in the renal medulla (Liapis et al. 1998).
A role for KCC4 in ion transport along the CD was confirmed most persuasively through the phenotype exhibited by the Boettger‐Kcc4 −/− 129/Sv × C57BL/6 model (Boettger et al. 2002). This phenotype was in fact consistent with a distal acidification defect as serum pH was lower and urinary pH higher than in WT littermates. Given that [Cl−]i was also higher in α‐IC cells than in any other cell types along the nephron, it was concluded that the ultimate consequence of KCC4 inactivation in the CD was to decrease the driving force for Cl−/HCO3 − exchange by SLC4A1 and decrease net HCO3 − reabsorption secondarily (see Fig. 3 C).
It should be noted that [Cl−]i measurements in the Kcc4‐null model were obtained by energy dispersive X‐ray microanalysis (Boettger et al. 2002) rather than electrophysiological determinations or imaging studies with Cl−‐sensitive probes. In addition, no data were provided on the renal clearance of Na+, K+, NH4 +, Cl− and citrate2− to confirm the distal acidification defect and determine whether TAL dysfunction could have been masked by the preponderant action of KCC4 in α‐IC cells. Of importance, lastly, ablation of KCC4 in the CD could also have accounted for the acidification defect by limiting the basolateral uptake of NH4 +. Under such circumstances, the apical secretion of NH3 would be expected to decrease secondarily along with the capacity of urine to buffer free H+ ions distally. An illustration of this hypothesis is provided in Fig. 3 C.
In the work of Melo et al. (2013a), the effect of systemic acidification on KCC4 expression had been examined as well, but through immunofluorescence studies. Although the microscopic fields presented were limited, α‐IC cells in NH4Cl‐loaded animals exhibited higher signal intensities. This finding was thus consistent with a role for KCC4 in renal acidification. It was also in keeping with an earlier observation that KCC4 is more active at low pHi (Bergeron et al. 2003) and would then react to systemic acidosis by decreasing [Cl−]i to increase HCO3 − reabsorption. Curiously, Melo et al. found that KCC4 expression in α‐IC cells was increased through hyperglycaemia as well, even though SGLT2 is not present in the distal nephron.
Other nephron segments and renal structures
According to immunolocalization studies in rabbit kidney, KCC4 was also found on the serosal side of both the rabbit distal tubule (DT) and connecting tubule. It was in fact maximally abundant at these locations of the nephron (Velazquez & Silva, 2003) despite the absence of documented K+‐Cl− cotransport activity between the macula densa and collecting duct. The antibody used could have therefore lacked specificity, all the more so that it does not appear to have been tested in Kcc4‐null tissues. Two of the KCCs were localized in the renal circulation as well: KCC1 in the endothelial layer of human vasa recta (Liapis et al. 1998) and KCC3 in the medial layer of mouse arterioles (Rust et al. 2006).
Cardiovascular system
Preamble
In the early 2000s, a recessive neurodegenerative disorder called agenesis of the corpus callosum with peripheral neuronopathy or Andermann syndrome was linked to inactivating mutations in KCC3 (Howard et al. 2002). Because of a founder effect, this disorder was unusually common in the Northern Appalachian front a few years ago (De Braekeleer et al. 1993; Dupre et al. 2006). After linkage was established, several mouse models of Kcc3 inactivation were phenotyped to understand the molecular mechanisms involved in the development of this disease.
Somewhat unexpectedly, three of the models generated were found to exhibit high blood pressures in addition to the neurological defects observed in human (Boettger et al. 2003; Adragna et al. 2004; Rust et al. 2006; Garneau et al. 2016). Further studies provided some clues as to why a cardiovascular phenotype was present in these models. However, the mechanisms at play are still uncertain as many different cell types could probably affect blood pressure control through changes in K+‐Cl− cotransport. As for the other isoforms, they do not appear to play an important role in the cardiovascular system.
In the following section, we will discuss the role of KCC3‐mediated K+‐Cl− cotransport in tissues that are known to be of cardiometabolic relevance. Such tissues include the adipose mass, the vascular wall and the nervous system in particular.
Localization of KCC3
Based on the human EST databank, KCC3 appears to be distributed in many of the tissues that are key in ensuring proper cardiometabolic function. Indeed, this isoform is represented by more than 11 messages per million in adipose tissue, vessels, brain, kidney and heart (by decreasing order of abundance). Previous multiple tissue Northern and Western blot analyses are also consistent with robust expression of KCC3 in human brain, heart and kidney (Hiki et al. 1999; Mount et al. 1999; Race et al. 1999). In many of these tissues, however, the nature of the variants at play cannot be determined from the data available (Table 1).
Additional localization studies in the tissues of interest have revealed that KCC3 was found more specifically in hippocampus, dorsal root ganglion neurons, cardiomyocytes, VSMCs (in both conductive and resistance arteries), medullary adrenal gland and, as mentioned, renal proximal tubular cells (Pearson et al. 2001; Rahmouni et al. 2002; Boettger et al. 2003; Gao & Wang, 2010; Mao et al. 2012; Shekarabi et al. 2012; Melo et al. 2013a; Garneau et al. 2016). Studies in single cell types have also revealed expression in rat VSMCs (Di Fulvio et al. 2001b), VEGF‐stimulated HUVEC (Hiki et al. 1999) and hippocampal neurons (Pearson et al. 2001; Boettger et al. 2003; Byun & Delpire, 2007).
Role of KCC3 in blood pressure control
High blood pressure was first reported in the Boettger‐Kcc3 −/− 129/Sv × C57BL/6 mice (Boettger et al. 2003) and was subsequently confirmed by another group in the Howard‐Kcc3 −/− 129/Sv × C57BL/6 mice (Adragna et al. 2004). In both animal models, light or dark phase mean arterial blood pressures (MAPs) were measured at 3 to 6 months of age through intra‐arterial catheterization and found to be at least 18 mmHg higher than in WT littermates.
Based on a subsequent study, Rust et al. (2006) concluded that the Boettger‐Kcc3 −/− 129/Sv × C57BL/6 mice were affected by a neurogenic form of systemic hypertension. Compared to WT mice, in particular, MAPs reacted similarly to α1 agonists, β1 antagonists or nitric oxide, but were more sensitive to ganglionic blockers or α2 antagonists. Additionally, the response of isolated third‐order saphenous arteries to vasoactive interventions did not differ between null and WT mice. In the null mice, however, the saphenous arteries were characterized by higher [Cl−]i and medial hypertrophy, implying that Kcc3 inactivation led to reduced K+‐Cl− cotransport in the vascular wall itself and that it could have affected VSMC growth as well as peripheral resistance for this reason (Klausen et al. 2010; Matchkov et al. 2013).
By studying the physiological effects of Kcc3 inactivation in a background that is genetically prone to cardiometabolic disorders (Simon et al. 2013), Garneau et al. (2016) confirmed that other or additional mechanisms could account for the hypertensive phenotype. Compared to WT mice, for instance, isolated thoracic aortas exhibited decreased wall thickness and reactivity to α1‐adrenoreceptor stimulation even after denervation, while diastolic blood pressure and left ventricular mass were increased. A number of these abnormalities had not been reported previously, perhaps because they went unnoticed or were too discrete in the background exploited. They suggest nonetheless that Kcc3 ablation has the potential to alter the arterial wall both functionally and pathologically.
KCC3 is known to be expressed in VSMCs along the arterial circulation and in cardiomyocytes (Di Fulvio et al. 2001a, 2003; Rust et al. 2006; Garneau et al. 2016). Interestingly, the contractile and trophic responses of these cell types are reduced at lower levels of [Cl−]i as occurs when the activity of NKCC1 is inhibited (Akar et al. 1999; Meyer et al. 2002). It is thus tempting to postulate that these responses will go in the opposite direction at higher levels of [Cl−]i, as occurs when the activity of KCC3 is inhibited. Based on Fig. 4, however, various effectors would be involved for changes in [Cl−]i to exert physiological effects (Boettger et al. 2002; Woo et al. 2002; Alberts et al. 2015).
Figure 4.
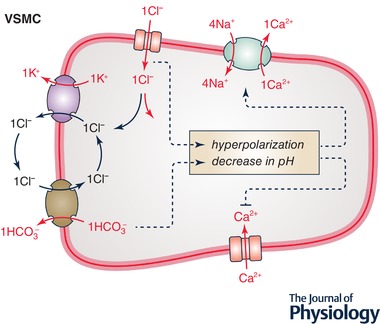
Role of K+‐Cl− cotransport in arterial VSMCs
Ion transport systems shown correspond to KCC3, Ca2+‐activated Cl− channel (CACC), SLC8A1 (Na+/Ca2+ exchanger or NCX type 1), L‐type voltage‐sensitive Ca2+ channel (Cav1) and SLC4A1 or SLC26A7. Increased KCC3 activity promotes Cl− entry (or decrease Cl− exit) through CACC and, secondarily, anion exchange by SLC4A1 or SLC26A7. It should therefore decrease membrane potential as well as intracellular pH, and thereby stimulate Na+/Ca2+ exchange by NCX1 and inhibit Ca2+ movement through Cav1. The end‐result of increased KCC3 activity should thus be a decrease in [Ca2+]i and, secondarily, in myogenic tone.
Role of KCC3 in metabolism
It is widely accepted that the endocrine and metabolic function of adipose tissues and skeletal muscles can play a major role in the development of cardiovascular diseases (Tatemoto et al. 2001; Okamoto et al. 2002; Marsh et al. 2003; Lee & Pratley, 2007; Denroche et al. 2012; Jiang et al. 2016). In morbid obesity, for instance, changes in the circulating profile of certain adipokines have been associated with insulin resistance, decreased metabolic activity in skeletal muscles, high blood pressure and inflammation (Yamauchi et al. 2001; Simonds et al. 2014; Jaganathan et al. 2018). There is also evidence to suggest that severe leanness can affect the cardiovascular system adversely through changes in adipocyte function (Campos et al. 2008; Resnyk et al. 2013).
Several of the Kcc3‐null mice that have been phenotyped since the early 2000s were also characterized by low body weights at adult age (Shekarabi et al. 2012; Garneau et al. 2016), but as one could have expected, it was in the C57BL/6J background that this trait was the most pronounced, with a 4.5‐fold lower gonadal fat weight compared to WT mice (Garneau et al. 2016). Additional experiments by Garneau et al. showed that the lean phenotype was associated with higher chow intake, glucose utilization and energy expenditure, as well as with a more favorable plasma adipokine profile (A. P. Garneau, unpublished data).
KCC3 could thus play an important role in adipocyte function and energy homeostasis. However, inactivation of the encoding gene could exert both detrimental and beneficial cardiometabolic effects depending on the cell type targeted. As for the mechanisms of low adiposity, whether they involve deficient K+‐Cl− cotransport in adipocytes or in the nervous system is still unknown. Even though one of the recently characterized neuron‐specific Kcc3‐null mouse was found to exhibit low body weight at 2 months of age (Shekarabi et al. 2012), it was also affected by a severe phenotype, suggesting that gene inactivation was not entirely neuron specific in this model.
Conclusion
K+‐Cl− cotransport could play at least three different roles in the renal epithelium: (1) ensuring cell volume maintenance during transepithelial solute transport; (2) promoting vectorial movement of Cl− along some of the nephron segments; and (3) sustaining the activity of other transport systems through changes in intracellular or extracellular ion concentration. Even if the primary purpose of K+‐Cl− cotransport in the kidney is to sustain RVD responses, cell swelling could still result in substantial transepithelial Cl− and water movement.
The role of KCC1 and KCC3 in the kidney has not been investigated in great detail (Boettger et al. 2003; Wang et al. 2003; Rust et al. 2007; Garneau et al. 2016). However, it does not appear to be of major importance under basal conditions according to the data available. As for KCC4, this isoform was found to sustain renal acidification in mouse, but its distribution along the nephron could differ among species.
There may be other reasons why the function of K+‐Cl− cotransport in the renal epithelium is still uncertain. In particular, the in vivo characteristics and organization of these transporters are largely unknown so that it is not possible to make predictions regarding their contribution to solute reabsorption. In addition, isoform‐specific inhibitors and relevant tissue‐specific floxed Kcc‐null mouse models are still unavailable.
As for the role of K+‐Cl− cotransport in cardiometabolic homeostasis, it has been demonstrated convincingly through the characterization of several mouse models of Kcc3 inactivation (Boettger et al. 2003; Adragna et al. 2004; Rust et al. 2006; Shekarabi et al. 2012; Garneau et al. 2016). The cell types responsible for this role have not been identified precisely, but they could very well include VSMCs. Lastly, KCC3 could become a beneficial therapeutic target if its activity is altered in certain tissues specifically.
Additional information
Competing interests
The authors have no competing interests and conflict of interests to declare.
Author contributions
The manuscript was prepared at the Centre de recherche du CHU de Québec (Laval University) and the Centre de recherche du CHUM (Montreal University). Conception and design of the work: A.P.G., P.I.; acquisition, analysis or interpretation of data for the work: A.P.G., P.I.; drafting of the work and critical revising of the work for intellectual content: all authors. All persons designated as authors qualify for authorship and have approved the final version of the manuscript. They have also agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This work was funded by the Canadian Institutes of Health Research, the Kidney Foundation of Canada, le Fonds de recherche du Québec – Santé and la Fondation des jumelles Coudé. Underlying research materials, such as data, samples or models can be accessed on demand through the corresponding author.
Biographies
Paul Isenring obtained a PhD degree in Molecular Physiology at Yale University. He is currently a Full Professor of Medicine at Laval University and Clinician Scientist in Nephrology at the Centre de recherche du CHU de Québec. His main research interest is to elucidate the molecular mechanisms of cation Cl− cotransport in the kidney and cardiovascular system through structure‐function studies and animal models.

Alexandre Garneau obtained a Master's degree in Experimental Medicine at Laval University and is pursuing PhD studies under the supervision of Dr Isenring at Laval University and Dr Julie Lavoie in the School of Kinesiology and Physical Activity Sciences at Montreal University. His main research project has been to characterize the cardiovascular and metabolic phenotype of a Kcc3‐null mouse model.
Edited by: Ole Petersen & Dennis Brown
References
- Adragna NC, Chen Y, Delpire E, Lauf PK & Morris M (2004). Hypertension in K‐Cl cotransporter‐3 knockout mice. Adv Exp Med Biol 559, 379–385. [DOI] [PubMed] [Google Scholar]
- Agalakova NI & Gusev GP (2009). Effects of phorbol 12‐myristate 13‐acetate on potassium transport in the red blood cells of frog Rana temporaria . J Comp Physiol B 179, 443–450. [DOI] [PubMed] [Google Scholar]
- Akar F, Skinner E, Klein JD, Jena M, Paul RJ & O'Neill WC (1999). Vasoconstrictors and nitrovasodilators reciprocally regulate the Na+‐K+‐2Cl− cotransporter in rat aorta. Am J Physiol 276, C1383–C1390. [DOI] [PubMed] [Google Scholar]
- Alberts B, Johnson A, Lewis J, Morgan DO, Raff MC, Roberts K, Walter P, Wilson JH & Hunt T (2015). Molecular Biology of the Cell. Garland Science, New York. [Google Scholar]
- Avison MJ, Gullans SR, Ogino T & Giebisch G (1988). Na+ and K+ fluxes stimulated by Na+‐coupled glucose transport: evidence for a Ba2+‐insensitive K+ efflux pathway in rabbit proximal tubules. J Membr Biol 105, 197–205. [DOI] [PubMed] [Google Scholar]
- Bergeron MJ, Boggavarapu R, Meury M, Ucurum Z, Caron L, Isenring P, Hediger MA & Fotiadis D (2011). Frog oocytes to unveil the structure and supramolecular organization of human transport proteins. PLoS One 6, e21901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron MJ, Frenette‐Cotton R, Carpentier GA, Simard MG, Caron L & Isenring P (2009). Phosphoregulation of K+‐Cl− cotransporter 4 during changes in intracellular Cl− and cell volume. J Cell Physiol 219, 787–796. [DOI] [PubMed] [Google Scholar]
- Bergeron MJ, Gagnon E, Wallendorff B, Lapointe JY & Isenring P (2003). Ammonium transport and pH regulation by K+‐Cl− cotransporters. Am J Physiol Renal Physiol 285, F68–F78. [DOI] [PubMed] [Google Scholar]
- Boettger T, Hubner CA, Maier H, Rust MB, Beck FX & Jentsch TJ (2002). Deafness and renal tubular acidosis in mice lacking the K‐Cl co‐transporter Kcc4. Nature 416, 874–878. [DOI] [PubMed] [Google Scholar]
- Boettger T, Rust MB, Maier H, Seidenbecher T, Schweizer M, Keating DJ, Faulhaber J, Ehmke H, Pfeffer C, Scheel O, Lemcke B, Horst J, Leuwer R, Pape HC, Volkl H, Hubner CA & Jentsch TJ (2003). Loss of K‐Cl co‐transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J 22, 5422–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun N & Delpire E (2007). Axonal and periaxonal swelling precede peripheral neurodegeneration in KCC3 knockout mice. Neurobiol Dis 28, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Cebotaru V, Wang YH, Zhang XM, Cebotaru L, Guggino SE & Guggino WB (2006). WNK4 kinase regulates surface expression of the human sodium chloride cotransporter in mammalian cells. Kidney Int 69, 2162–2170. [DOI] [PubMed] [Google Scholar]
- Campos DB, Palin MF, Bordignon V & Murphy BD (2008). The ‘beneficial’ adipokines in reproduction and fertility. Int J Obes (Lond) 32, 223–231. [DOI] [PubMed] [Google Scholar]
- Capasso G, Mollica F, Saviano C & De Santo NG (1995). Tubule effects of glomerular hyperfiltration: an integrated view. Semin Nephrol 15, 419–425. [PubMed] [Google Scholar]
- Crable SC, Hammond SM, Papes R, Rettig RK, Zhou GP, Gallagher PG, Joiner CH & Anderson KP (2005). Multiple isoforms of the KC1 cotransporter are expressed in sickle and normal erythroid cells. Exp Hematol 33, 624–631. [DOI] [PubMed] [Google Scholar]
- De Braekeleer M, Dallaire A & Mathieu J (1993). Genetic epidemiology of sensorimotor polyneuropathy with or without agenesis of the corpus callosum in northeastern Quebec. Hum Genet 91, 223–227. [DOI] [PubMed] [Google Scholar]
- de Los Heros P, Alessi DR, Gourlay R, Campbell DG, Deak M, Macartney TJ, Kahle KT & Zhang J (2014). The WNK‐regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+‐Cl− co‐transporters. Biochem J 458, 559–573. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Denroche HC, Huynh FK & Kieffer TJ (2012). The role of leptin in glucose homeostasis. J Diabetes Investig 3, 115–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fulvio M, Lauf PK & Adragna NC (2001a). Nitric oxide signaling pathway regulates potassium chloride cotransporter‐1 mRNA expression in vascular smooth muscle cells. J Biol Chem 276, 44534–44540. [DOI] [PubMed] [Google Scholar]
- Di Fulvio M, Lauf PK & Adragna NC (2003). The NO signaling pathway differentially regulates KCC3a and KCC3b mRNA expression. Nitric Oxide 9, 165–171. [DOI] [PubMed] [Google Scholar]
- Di Fulvio M, Lincoln TM, Lauf PK & Adragna NC (2001b). Protein kinase G regulates potassium chloride cotransporter‐4 [corrected] expression in primary cultures of rat vascular smooth muscle cells. J Biol Chem 276, 21046–21052. [DOI] [PubMed] [Google Scholar]
- Di Stefano A, Jounier S & Wittner M (2001). Evidence supporting a role for KCl cotransporter in the thick ascending limb of Henle's loop. Kidney Int 60, 1809–1823. [DOI] [PubMed] [Google Scholar]
- Dunham PB, Stewart GW & Ellory JC (1980). Chloride‐activated passive potassium transport in human erythrocytes. Proc Natl Acad Sci U S A 77, 1711–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre N, Howard HC & Rouleau GA (2006). Hereditary motor and sensory neuropathy with agenesis of the corpus callosum In GeneReviews(R), eds Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH. & Stephens K. Seattle, WA, USA. [PubMed] [Google Scholar]
- Evans RL & Turner RJ (1998). Evidence for a physiological role of NH4+ transport on the secretory Na+‐K+‐2Cl− cotransporter. Biochem Biophys Res Commun 245, 301–306. [DOI] [PubMed] [Google Scholar]
- Frederikse PH & Kasinathan C (2015). KCC2 expression supersedes NKCC1 in mature fiber cells in mouse and rabbit lenses. Mol Vis 21, 1142–1150. [PMC free article] [PubMed] [Google Scholar]
- Frenette‐Cotton R, Marcoux AA, Garneau AP, Noel M & Isenring P (2017). Phosphoregulation of K+‐Cl− cotransporters during cell swelling: novel insights. J Cell Physiol 233, 396–408. [DOI] [PubMed] [Google Scholar]
- Fujii T, Takahashi Y, Ikari A, Morii M, Tabuchi Y, Tsukada K, Takeguchi N & Sakai H (2009). Functional association between K+‐Cl− cotransporter‐4 and H+,K+‐ATPase in the apical canalicular membrane of gastric parietal cells. J Biol Chem 284, 619–629. [DOI] [PubMed] [Google Scholar]
- Fujii T, Takahashi Y, Itomi Y, Fujita K, Morii M, Tabuchi Y, Asano S, Tsukada K, Takeguchi N & Sakai H (2008). K+‐Cl− cotransporter‐3a up‐regulates Na+,K+‐ATPase in lipid rafts of gastric luminal parietal Cells. J Biol Chem 283, 6869–6877. [DOI] [PubMed] [Google Scholar]
- Gamba G (2005). Molecular physiology and pathophysiology of electroneutral cation‐chloride cotransporters. Physiol Rev 85, 423–493. [DOI] [PubMed] [Google Scholar]
- Gao F & Wang DH (2010). Impairment in function and expression of transient receptor potential vanilloid type 4 in Dahl salt‐sensitive rats: significance and mechanism. Hypertension 55, 1018–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau AP, Marcoux AA, Frenette‐Cotton R, Mac‐Way F, Lavoie JL & Isenring P (2017). Molecular insights into the normal operation, regulation, and multisystemic roles of K+‐Cl− cotransporter 3 (KCC3). Am J Physiol Cell Physiol 313, C516–C532. [DOI] [PubMed] [Google Scholar]
- Garneau AP, Marcoux AA, Noel M, Frenette‐Cotton R, Drolet MC, Couet J, Lariviere R & Isenring P (2016). Ablation of potassium‐chloride cotransporter type 3 (Kcc3) in mouse causes multiple cardiovascular defects and isosmotic polyuria. PLoS One 11, e0154398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon‐Muvdi T, Pacheco‐Alvarez D, Gagnon KB, Vazquez N, Ponce‐Coria J, Moreno E, Delpire E & Gamba G (2007). WNK4 kinase is a negative regulator of K+‐Cl− cotransporters. Am J Physiol Renal Physiol 292, F1197–F1207. [DOI] [PubMed] [Google Scholar]
- Gillen CM, Brill S, Payne JA & Forbush B 3rd (1996). Molecular cloning and functional expression of the K‐Cl cotransporter from rabbit, rat, and human. A new member of the cation‐chloride cotransporter family. J Biol Chem 271, 16237–16244. [DOI] [PubMed] [Google Scholar]
- Glanville M, Kingscote S, Thwaites DT & Simmons NL (2001). Expression and role of sodium, potassium, chloride cotransport (NKCC1) in mouse inner medullary collecting duct (mIMCD‐K2) epithelial cells. Pflugers Arch 443, 123–131. [DOI] [PubMed] [Google Scholar]
- Golbang AP, Murthy M, Hamad A, Liu CH, Cope G, Van't Hoff W, Cuthbert A & O'Shaughnessy KM (2005). A new kindred with pseudohypoaldosteronism type II and a novel mutation (564D>H) in the acidic motif of the WNK4 gene. Hypertension 46, 295–300. [DOI] [PubMed] [Google Scholar]
- Greger R (1985). Ion transport mechanisms in thick ascending limb of Henle's loop of mammalian nephron. Physiol Rev 65, 760–797. [DOI] [PubMed] [Google Scholar]
- Greger R & Schlatter E (1983). Properties of the basolateral membrane of the cortical thick ascending limb of Henle's loop of rabbit kidney. A model for secondary active chloride transport. Pflugers Arch 396, 325–334. [DOI] [PubMed] [Google Scholar]
- Grill A, Schiessl IM, Gess B, Fremter K, Hammer A & Castrop H (2016). Salt‐losing nephropathy in mice with a null mutation of the Clcnk2 gene. Acta Physiol (Oxf) 218, 198–211. [DOI] [PubMed] [Google Scholar]
- Gusev GP & Agalakova NI (2010). Regulation of K‐Cl cotransport in erythrocytes of frog Rana temporaria by commonly used protein kinase and protein phosphatase inhibitors. J Comp Physiol B 180, 385–391. [DOI] [PubMed] [Google Scholar]
- Hebert SC & Andreoli TE (1984). Control of NaCl transport in the thick ascending limb. Am J Physiol 246, F745–F756. [DOI] [PubMed] [Google Scholar]
- Hiki K, D'Andrea RJ, Furze J, Crawford J, Woollatt E, Sutherland GR, Vadas MA & Gamble JR (1999). Cloning, characterization, and chromosomal location of a novel human K+‐Cl− cotransporter. J Biol Chem 274, 10661–10667. [DOI] [PubMed] [Google Scholar]
- Howard HC, Mount DB, Rochefort D, Byun N, Dupre N, Lu J, Fan X, Song L, Riviere JB, Prevost C, Horst J, Simonati A, Lemcke B, Welch R, England R, Zhan FQ, Mercado A, Siesser WB, George AL Jr, McDonald MP, Bouchard JP, Mathieu J, Delpire E & Rouleau GA (2002). The K‐Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet 32, 384–392. [DOI] [PubMed] [Google Scholar]
- Jaganathan R, Ravindran R & Dhanasekaran S (2018). Emerging role of adipocytokines in type 2 diabetes as mediators of insulin resistance and cardiovascular disease. Can J Diabetes 42, 446–456.e441. [DOI] [PubMed] [Google Scholar]
- Jennings ML & Adame MF (2001). Direct estimate of 1:1 stoichiometry of K+‐Cl− cotransport in rabbit erythrocytes. Am J Physiol Cell Physiol 281, C825–C832. [DOI] [PubMed] [Google Scholar]
- Jennings ML & Schulz RK (1991). Okadaic acid inhibition of K+‐Cl− cotransport. Evidence that protein dephosphorylation is necessary for activation of transport by either cell swelling or N‐ethylmaleimide. J Gen Physiol 97, 799–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Lu L, Hu Y, Li Q, An C, Yu X, Shu L, Chen A, Niu C, Zhou L & Yang Z (2016). Resistin induces hypertension and insulin resistance in mice via a TLR4‐dependent pathway. Sci Rep 6, 22193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Flores B, Bharucha‐Goebel D, Zhang J, Donkervoort S, Hegde M, Hussain G, Duran D, Liang B, Sun D, Bonnemann CG & Delpire E (2016). Peripheral motor neuropathy is associated with defective kinase regulation of the KCC3 cotransporter. Sci Signal 9, ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji DM (1993). Effect of membrane potential on K‐Cl transport in human erythrocytes. Am J Physiol 264, C376–C382. [DOI] [PubMed] [Google Scholar]
- Klausen TK, Preisler S, Pedersen SF & Hoffmann EK (2010). Monovalent ions control proliferation of Ehrlich Lettre ascites cells. Am J Physiol Cell Physiol 299, C714–C725. [DOI] [PubMed] [Google Scholar]
- Kregenow FM (1971). The response of duck erythrocytes to nonhemolytic hypotonic media. Evidence for a volume‐controlling mechanism. J Gen Physiol 58, 372–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson‐Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS & Lifton RP (2006). Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet 38, 1124–1132. [DOI] [PubMed] [Google Scholar]
- Lauf PK & Theg BE (1980). A chloride dependent K+ flux induced by N‐ethylmaleimide in genetically low K+ sheep and goat erythrocytes. Biochem Biophys Res Commun 92, 1422–1428. [DOI] [PubMed] [Google Scholar]
- Lauf PK, Zhang J, Delpire E, Fyffe RE, Mount DB & Adragna NC (2001). K‐Cl co‐transport: immunocytochemical and functional evidence for more than one KCC isoform in high K and low K sheep erythrocytes. Comp Biochem Physiol A Mol Integr Physiol 130, 499–509. [DOI] [PubMed] [Google Scholar]
- Lee YH & Pratley RE (2007). Abdominal obesity and cardiovascular disease risk: the emerging role of the adipocyte. J Cardiopulm Rehabil Prev 27, 2–10. [DOI] [PubMed] [Google Scholar]
- Le Rouzic P, Ivanov TR, Stanley PJ, Baudoin FM, Chan F, Pinteaux E, Brown PD & Luckman SM (2006). KCC3 and KCC4 expression in rat adult forebrain. Brain Res 1110, 39–45. [DOI] [PubMed] [Google Scholar]
- Liapis H, Nag M & Kaji DM (1998). K‐Cl cotransporter expression in the human kidney. Am J Physiol Cell Physiol 275, C1432–C1437. [DOI] [PubMed] [Google Scholar]
- Lorenz JN, Baird NR, Judd LM, Noonan WT, Andringa A, Doetschman T, Manning PA, Liu LH, Miller ML & Shull GE (2002). Impaired renal NaCl absorption in mice lacking the ROMK potassium channel, a model for type II Bartter's syndrome. J Biol Chem 277, 37871–37880. [DOI] [PubMed] [Google Scholar]
- Lourenco CM, Dupre N, Riviere JB, Rouleau GA, Marques VD, Genari AB, Santos AC, Barreira AA & Marques W Jr (2012). Expanding the differential diagnosis of inherited neuropathies with non‐uniform conduction: Andermann syndrome. J Peripher Nerv Syst 17, 123–127. [DOI] [PubMed] [Google Scholar]
- Lucas O, Hilaire C, Delpire E & Scamps F (2012). KCC3‐dependent chloride extrusion in adult sensory neurons. Mol Cell Neurosci 50, 211–220. [DOI] [PubMed] [Google Scholar]
- Mao S, Garzon‐Muvdi T, Di Fulvio M, Chen Y, Delpire E, Alvarez FJ & Alvarez‐Leefmans FJ (2012). Molecular and functional expression of cation‐chloride cotransporters in dorsal root ganglion neurons during postnatal maturation. J Neurophysiol 108, 834–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcoux AA, Garneau AP, Frenette‐Cotton R, Slimani S, Mac‐Way F & Isenring P (2017). Molecular features and physiological roles of K+‐Cl− cotransporter 4 (KCC4). Biochim Biophys Acta 1861, 3154–3166. [DOI] [PubMed] [Google Scholar]
- Marsh AJ, Fontes MA, Killinger S, Pawlak DB, Polson JW & Dampney RA (2003). Cardiovascular responses evoked by leptin acting on neurons in the ventromedial and dorsomedial hypothalamus. Hypertension 42, 488–493. [DOI] [PubMed] [Google Scholar]
- Matchkov VV, Secher Dam V, Bødtkjer DM & Aalkjær C (2013). Transport and function of chloride in vascular smooth muscles. J Vasc Res 50, 69–87. [DOI] [PubMed] [Google Scholar]
- Mayan H, Vered I, Mouallem M, Tzadok‐Witkon M, Pauzner R & Farfel Z (2002). Pseudohypoaldosteronism type II: marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J Clin Endocrinol Metab 87, 3248–3254. [DOI] [PubMed] [Google Scholar]
- Melo Z, Cruz‐Rangel S, Bautista R, Vazquez N, Castaneda‐Bueno M, Mount DB, Pasantes‐Morales H, Mercado A & Gamba G (2013a). Molecular evidence for a role for K+‐Cl− cotransporters in the kidney. Am J Physiol Renal Physiol 305, F1402–F1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo Z, de los Heros P, Cruz‐Rangel S, Vazquez N, Bobadilla NA, Pasantes‐Morales H, Alessi DR, Mercado A & Gamba G (2013b). N‐terminal serine dephosphorylation is required for KCC3 cotransporter full activation by cell swelling. J Biol Chem 288, 31468–31476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado A, de Los Heros P, Melo Z, Chavez‐Canales M, Murillo‐de‐Ozores AR, Moreno E, Bazua‐Valenti S, Vazquez N, Hadchouel J & Gamba G (2016). With no lysine L‐WNK1 isoforms are negative regulators of the K+‐Cl− cotransporters. Am J Physiol Cell Physiol 311, C54–C66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado A, de los Heros P, Vazquez N, Meade P, Mount DB & Gamba G (2001). Functional and molecular characterization of the K‐Cl cotransporter of Xenopus laevis oocytes. Am J Physiol Cell Physiol 281, C670–C680. [DOI] [PubMed] [Google Scholar]
- Mercado A, Song L, Vazquez N, Mount DB & Gamba G (2000). Functional comparison of the K+‐Cl− cotransporters KCC1 and KCC4. J Biol Chem 275, 30326–30334. [DOI] [PubMed] [Google Scholar]
- Mercado A, Vazquez N, Song L, Cortes R, Enck AH, Welch R, Delpire E, Gamba G & Mount DB (2005). NH2‐terminal heterogeneity in the KCC3 K+‐Cl− cotransporter. Am J Physiol Renal Physiol 289, F1246–F1261. [DOI] [PubMed] [Google Scholar]
- Meyer JW, Flagella M, Sutliff RL, Lorenz JN, Nieman ML, Weber CS, Paul RJ & Shull GE (2002). Decreased blood pressure and vascular smooth muscle tone in mice lacking basolateral Na+‐K+‐2Cl− cotransporter. Am J Physiol Heart Circ Physiol 283, H1846–H1855. [DOI] [PubMed] [Google Scholar]
- Mizuno M, Murphy MN, Mitchell JH & Smith SA (2011). Antagonism of the TRPv1 receptor partially corrects muscle metaboreflex overactivity in spontaneously hypertensive rats. J Physiol 589, 6191–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobasheri A, Pestov NB, Papanicolaou S, Kajee R, Cozar‐Castellano I, Avila J, Martin‐Vasallo P, Foster CS, Modyanov NN & Djamgoz MB (2003). Expression and cellular localization of Na,K‐ATPase isoforms in the rat ventral prostate. BJU Int 92, 793–802. [DOI] [PubMed] [Google Scholar]
- Mount DB (2014). Thick ascending limb of the loop of Henle. Clin J Am Soc Nephrol 9, 1974–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount DB, Mercado A, Song L, Xu J, George AL Jr Delpire E & Gamba G (1999). Cloning and characterization of KCC3 and KCC4, new members of the cation‐chloride cotransporter gene family. J Biol Chem 274, 16355–16362. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Kihara S, Ouchi N, Nishida M, Arita Y, Kumada M, Ohashi K, Sakai N, Shimomura I, Kobayashi H, Terasaka N, Inaba T, Funahashi T & Matsuzawa Y (2002). Adiponectin reduces atherosclerosis in apolipoprotein E‐deficient mice. Circulation 106, 2767–2770. [DOI] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ & Donaldson LF (1996). Molecular characterization of a putative K‐Cl cotransporter in rat brain. A neuronal‐specific isoform. J Biol Chem 271, 16245–16252. [DOI] [PubMed] [Google Scholar]
- Pearson MM, Lu J, Mount DB & Delpire E (2001). Localization of the K+‐Cl− cotransporter, KCC3, in the central and peripheral nervous systems: expression in the choroid plexus, large neurons and white matter tracts. Neuroscience 103, 481–491. [DOI] [PubMed] [Google Scholar]
- Piwnica‐Worms D, Jacob R, Horres CR & Lieberman M (1985). Potassium‐chloride cotransport in cultured chick heart cells. Am J Physiol Cell Physiol 249, C337–C344. [DOI] [PubMed] [Google Scholar]
- Pollak MR, Delaney VB, Graham RM & Hebert SC (1996). Gitelman's syndrome (Bartter's variant) maps to the thiazide‐sensitive cotransporter gene locus on chromosome 16q13 in a large kindred. J Am Soc Nephrol 7, 2244–2248. [DOI] [PubMed] [Google Scholar]
- Prosser CL (1991). Environmental and Metabolic Animal Physiology. Wiley‐Liss, New York. [Google Scholar]
- Race JE, Makhlouf FN, Logue PJ, Wilson FH, Dunham PB & Holtzman EJ (1999). Molecular cloning and functional characterization of KCC3, a new K‐Cl cotransporter. Am J Physiol Cell Physiol 277, C1210–C1219. [DOI] [PubMed] [Google Scholar]
- Rahmouni K, Sibug RM, De Kloet ER, Barthelmebs M, Grima M, Imbs JL & De Jong W (2002). Effects of brain mineralocorticoid receptor blockade on blood pressure and renal functions in DOCA‐salt hypertension. Eur J Pharmacol 436, 207–216. [DOI] [PubMed] [Google Scholar]
- Resnyk CW, Carre W, Wang X, Porter TE, Simon J, Le Bihan‐Duval E, Duclos MJ, Aggrey SE & Cogburn LA (2013). Transcriptional analysis of abdominal fat in genetically fat and lean chickens reveals adipokines, lipogenic genes and a link between hemostasis and leanness. BMC Genomics 14, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinehart J, Maksimova YD, Tanis JE, Stone KL, Hodson CA, Zhang J, Risinger M, Pan W, Wu D, Colangelo CM, Forbush B, Joiner CH, Gulcicek EE, Gallagher PG & Lifton RP (2009). Sites of regulated phosphorylation that control K‐Cl cotransporter activity. Cell 138, 525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnik‐Schoneborn S, Hehr U, von Kalle T, Bornemann A, Winkler J & Zerres K (2009). Andermann syndrome can be a phenocopy of hereditary motor and sensory neuropathy – report of a discordant sibship with a compound heterozygous mutation of the KCC3 gene. Neuropediatrics 40, 129–133. [DOI] [PubMed] [Google Scholar]
- Rust MB, Alper SL, Rudhard Y, Shmukler BE, Vicente R, Brugnara C, Trudel M, Jentsch TJ & Hubner CA (2007). Disruption of erythroid K‐Cl cotransporters alters erythrocyte volume and partially rescues erythrocyte dehydration in SAD mice. J Clin Invest 117, 1708–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MB, Faulhaber J, Budack MK, Pfeffer C, Maritzen T, Didie M, Beck FX, Boettger T, Schubert R, Ehmke H, Jentsch TJ & Hubner CA (2006). Neurogenic mechanisms contribute to hypertension in mice with disruption of the K‐Cl cotransporter KCC3. Circ Res 98, 549–556. [DOI] [PubMed] [Google Scholar]
- Sangan P, Brill SR, Sangan S, Forbush B 3rd & Binder HJ (2000). Basolateral K‐Cl cotransporter regulates colonic potassium absorption in potassium depletion. J Biol Chem 275, 30813–30816. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Ishibashi K, Yoshiyama N & Shiigai T (1988). KCl co‐transport across the basolateral membrane of rabbit renal proximal straight tubules. J Clin Invest 81, 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekarabi M, Moldrich RX, Rasheed S, Salin‐Cantegrel A, Laganiere J, Rochefort D, Hince P, Huot K, Gaudet R, Kurniawan N, Sotocinal SG, Ritchie J, Dion PA, Mogil JS, Richards LJ & Rouleau GA (2012). Loss of neuronal potassium/chloride cotransporter 3 (KCC3) is responsible for the degenerative phenotype in a conditional mouse model of hereditary motor and sensory neuropathy associated with agenesis of the corpus callosum. J Neurosci 32, 3865–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MR, Chou CY & Ellory JC (2000). Volume‐sensitive KCI cotransport associated with human cervical carcinogenesis. Pflugers Arch 440, 751–760. [DOI] [PubMed] [Google Scholar]
- Simard CF, Bergeron MJ, Frenette‐Cotton R, Carpentier GA, Pelchat ME, Caron L & Isenring P (2007). Homooligomeric and heterooligomeric associations between K+‐Cl− cotransporter isoforms and between K+‐Cl− and Na+‐K+‐Cl− cotransporters. J Biol Chem 282, 18083–18093. [DOI] [PubMed] [Google Scholar]
- Simard CF, Brunet GM, Daigle ND, Montminy V, Caron L & Isenring P (2004). Self‐interacting domains in the C terminus of a cation‐Cl− cotransporter described for the first time. J Biol Chem 279, 40769–40777. [DOI] [PubMed] [Google Scholar]
- Simon DB, Nelson‐Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ & Lifton RP (1996). Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide‐sensitive Na‐Cl cotransporter. Nat Genet 12, 24–30. [DOI] [PubMed] [Google Scholar]
- Simon MM, Greenaway S, White JK, Fuchs H, Gailus‐Durner V, Wells S, Sorg T, Wong K, Bedu E, Cartwright EJ, Dacquin R, Djebali S, Estabel J, Graw J, Ingham NJ, Jackson IJ, Lengeling A, Mandillo S, Marvel J, Meziane H, Preitner F, Puk O, Roux M, Adams DJ, Atkins S, Ayadi A, Becker L, Blake A, Brooker D, Cater H, Champy MF, Combe R, Danecek P, di Fenza A, Gates H, Gerdin AK, Golini E, Hancock JM, Hans W, Holter SM, Hough T, Jurdic P, Keane TM, Morgan H, Muller W, Neff F, Nicholson G, Pasche B, Roberson LA, Rozman J, Sanderson M, Santos L, Selloum M, Shannon C, Southwell A, Tocchini‐Valentini GP, Vancollie VE, Westerberg H, Wurst W, Zi M, Yalcin B, Ramirez‐Solis R, Steel KP, Mallon AM, de Angelis MH, Herault Y & Brown SD (2013). A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol 14, R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonds SE, Pryor JT, Ravussin E, Greenway FL, Dileone R, Allen AM, Bassi J, Elmquist JK, Keogh JM, Henning E, Myers MG Jr, Licinio J, Brown RD, Enriori PJ, O'Rahilly S, Sternson SM, Grove KL, Spanswick DC, Farooqi IS & Cowley MA (2014). Leptin mediates the increase in blood pressure associated with obesity. Cell 159, 1404–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Mercado A, Vazquez N, Xie Q, Desai R, George AL Jr, Gamba G & Mount DB (2002). Molecular, functional, and genomic characterization of human KCC2, the neuronal K‐Cl cotransporter. Brain Res Mol Brain Res 103, 91–105. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Chernavvsky DR, Gomez RA, Igarashi P, Gitelman HJ & Smithies O (2000). Uncompensated polyuria in a mouse model of Bartter's syndrome. Proc Natl Acad Sci U S A 97, 5434–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatemoto K, Takayama K, Zou MX, Kumaki I, Zhang W, Kumano K & Fujimiya M (2001). The novel peptide apelin lowers blood pressure via a nitric oxide‐dependent mechanism. Regul Pept 99, 87–92. [DOI] [PubMed] [Google Scholar]
- Uyanik G, Elcioglu N, Penzien J, Gross C, Yilmaz Y, Olmez A, Demir E, Wahl D, Scheglmann K, Winner B, Bogdahn U, Topaloglu H, Hehr U & Winkler J (2006). Novel truncating and missense mutations of the KCC3 gene associated with Andermann syndrome. Neurology 66, 1044–1048. [DOI] [PubMed] [Google Scholar]
- Velazquez H & Silva T (2003). Cloning and localization of KCC4 in rabbit kidney: expression in distal convoluted tubule. Am J Physiol Renal Physiol 285, F49–F58. [DOI] [PubMed] [Google Scholar]
- Wall SM, Trinh HN & Woodward KE (1995). Heterogeneity of NH+4 transport in mouse inner medullary collecting duct cells. Am J Physiol 269, F536–F544. [DOI] [PubMed] [Google Scholar]
- Wang T, Delpire E, Giebisch G, Hebert S & Mount D (2003). Impaired fluid and bicarbonate absorption in proximal tubules (PT) of KCC3 knockout mice. FASEB J 17, A464–A464. [Google Scholar]
- Weinstein AM (2010). A mathematical model of rat ascending Henle limb. I. Cotransporter function. Am J Physiol Renal Physiol 298, F512–F524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JR, Sharp JW, Kumari VG, Wilson M & Payne JA (1999). The neuron‐specific K‐Cl cotransporter, KCC2. Antibody development and initial characterization of the protein. J Biol Chem 274, 12656–12664. [DOI] [PubMed] [Google Scholar]
- Wilson FH, Disse‐Nicodeme S, Choate KA, Ishikawa K, Nelson‐Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X & Lifton RP (2001). Human hypertension caused by mutations in WNK kinases. Science 293, 1107–1112. [DOI] [PubMed] [Google Scholar]
- Wilson FH, Kahle KT, Sabath E, Lalioti MD, Rapson AK, Hoover RS, Hebert SC, Gamba G & Lifton RP (2003). Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na‐Cl cotransporter is inhibited by wild‐type but not mutant WNK4. Proc Natl Acad Sci U S A 100, 680–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo NS, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM & Delpire E (2002). Hyperexcitability and epilepsy associated with disruption of the mouse neuronal‐specific K‐Cl cotransporter gene. Hippocampus 12, 258–268. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama‐Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P & Kadowaki T (2001). The fat‐derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 7, 941–946. [DOI] [PubMed] [Google Scholar]
- Yang CL, Angell J, Mitchell R & Ellison DH (2003). WNK kinases regulate thiazide‐sensitive Na‐Cl cotransport. J Clin Invest 111, 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SS, Morimoto T, Rai T, Chiga M, Sohara E, Ohno M, Uchida K, Lin SH, Moriguchi T, Shibuya H, Kondo Y, Sasaki S & Uchida S (2007). Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab 5, 331–344. [DOI] [PubMed] [Google Scholar]
- Yang SS, Yamauchi K, Rai T, Hiyama A, Sohara E, Suzuki T, Itoh T, Suda S, Sasaki S & Uchida S (2005). Regulation of apical localization of the thiazide‐sensitive NaCl cotransporter by WNK4 in polarized epithelial cells. Biochem Biophys Res Commun 330, 410–414. [DOI] [PubMed] [Google Scholar]
- Yoshitomi K, Kondo Y & Imai M (1988). Evidence for conductive Cl− pathways across the cell membranes of the thin ascending limb of Henle's loop. J Clin Invest 82, 866–871. [DOI] [PMC free article] [PubMed] [Google Scholar]