

Abstract
Key points
NMDA receptors are neurotransmitter‐gated ion channels that are critically involved in brain cell communication
Variations in genes encoding NMDA receptor subunits have been found in a range of neurodevelopmental disorders.
We investigated a de novo genetic variant found in patients with epileptic encephalopathy that changes a residue located in the ion channel pore of the GluN2A NMDA receptor subunit.
We found that this variant (GluN2AN615K) impairs physiologically important receptor properties: it markedly reduces Mg2+ blockade and channel conductance, even for receptors in which one GluN2AN615K is co‐assembled with one wild‐type GluN2A subunit.
Our findings are consistent with the GluN2AN615K mutation being the primary cause of the severe neurodevelopmental disorder in carriers.
Abstract
NMDA receptors are ionotropic calcium‐permeable glutamate receptors with a voltage‐dependence mediated by blockade by Mg2+. Their activation is important in signal transduction, as well as synapse formation and maintenance. Two unrelated individuals with epileptic encephalopathy carry a de novo variant in the gene encoding the GluN2A NMDA receptor subunit: a N615K missense variant in the M2 pore helix (GRIN2A C1845A). We hypothesized that this variant underlies the neurodevelopmental disorders in carriers and explored its functional consequences by electrophysiological analysis in heterologous systems. We focused on GluN2AN615K co‐expressed with wild‐type GluN2 subunits in physiologically relevant triheteromeric NMDA receptors containing two GluN1 and two distinct GluN2 subunits, whereas previous studies have investigated the impact of the variant in diheteromeric NMDA receptors with two GluN1 and two identical GluN2 subunits. We found that GluN2AN615K‐containing triheteromers showed markedly reduced Mg2+ blockade, with a value intermediate between GluN2AN615K diheteromers and wild‐type NMDA receptors. Single‐channel conductance was reduced by four‐fold in GluN2AN615K diheteromers, again with an intermediate value in GluN2AN615K‐containing triheteromers. Glutamate deactivation rates were unaffected. Furthermore, we expressed GluN2AN615K in cultured primary mouse cortical neurons, observing a decrease in Mg2+ blockade and reduction in current density, confirming that the variant continues to have significant functional impact in neuronal systems. Our results demonstrate that the GluN2AN615K variant has substantial effects on NMDA receptor properties fundamental to the roles of the receptor in synaptic plasticity, even when expressed alongside wild‐type subunits. This work strengthens the evidence indicating that the GluN2AN615K variant underlies the disabling neurodevelopmental phenotype in carriers.
Keywords: Genetics, intellectual disability, electrophysiology, Human, Epilepsy
Key points
NMDA receptors are neurotransmitter‐gated ion channels that are critically involved in brain cell communication
Variations in genes encoding NMDA receptor subunits have been found in a range of neurodevelopmental disorders.
We investigated a de novo genetic variant found in patients with epileptic encephalopathy that changes a residue located in the ion channel pore of the GluN2A NMDA receptor subunit.
We found that this variant (GluN2AN615K) impairs physiologically important receptor properties: it markedly reduces Mg2+ blockade and channel conductance, even for receptors in which one GluN2AN615K is co‐assembled with one wild‐type GluN2A subunit.
Our findings are consistent with the GluN2AN615K mutation being the primary cause of the severe neurodevelopmental disorder in carriers.
Introduction
The NMDA receptor is an ionotropic voltage‐dependent glutamate receptor with important roles in synaptic signal transduction and plasticity. NMDA receptor dysfunction has been identified in diverse neuropsychiatric disorders (Paoletti et al. 2013). Unlike AMPA‐ and kainate‐type glutamate receptors, it shows high Ca2+ permeability (MacDermott et al. 1986) and voltage‐dependent Mg2+ blockade, which allows it to act as a molecular coincidence detector (Mayer et al. 1984; Nowak et al. 1984). NMDA receptors are heterotetramers comprised of two GluN1 and two GluN2 subunits of which four different subunits have been cloned (GluN2A‐D) (Watanabe et al. 1992; Monyer et al. 1994). The identity of the GluN2 subunits in the receptor impacts on receptor properties important in synaptic plasticity (Wyllie et al. 2013). Diheteromeric GluN1/GluN2B receptors probably form the commonest forebrain receptor composition prenatally but, after GluN2A expression increases postnatally (Bar‐Shira et al. 2015), triheteromeric GluN1/GluN2A/GluN2B receptors comprise a major proportion of NMDA receptors, particularly in the hippocampus (Gray et al. 2011; Rauner & Köhr, 2011; Tovar et al. 2013).
The rapid expansion of whole‐exome sequencing over the last decade has shown that many regions of the gene encoding GluN2A (GRIN2A) are intolerant of variation in healthy controls (Ogden et al. 2017) and, instead, a large number of rare and de novo variants (>100) have been identified in patients with a range of neurodevelopmental disorders, most commonly epilepsy aphasia syndromes or other epileptic disorders, but also intellectual disability, autism, attention deficit hyperactivity disorder and schizophrenia (XiangWei et al. 2018). At the most severe end of the phenotypic spectrum are epileptic encephalopathies, where epileptic activity is considered to contribute to severe cognitive and behavioural impairment (Scheffer et al. 2017). Around half of disease‐associated GRIN2A variants are gene‐disrupting and around half are missense variants, potentially resulting in NMDA receptors with altered function, requiring electrophysiological interrogation for confirmation. This has been a clinically useful strategy, with personalized treatment being offered based on a functional analysis of the variant (Pierson et al. 2014). However, it is improbable that all variants are disease‐causing: some are inherited from phenotypically normal parents and some are in regions of the protein that probably exhibit variation without deleterious functional impact (less highly conserved, or more variation present in healthy controls). Furthermore, of those variants where functional consequences have been assessed, a range of effects have been found, some predominantly ‘gain‐of‐function’, some predominantly ‘loss‐of‐function’ (Swanger et al. 2016) and some with no effect (Marwick et al. 2017). For these reasons, it is important to identify the functional consequences, if any, of a given variant before giving a genetic diagnosis or embarking on targeted treatment.
The large and increasing number of GRIN2A variants (and variants in other genes) found in patients with neurodevelopmental disorders means that some prioritization for functional investigation needs to be applied. The variant that we selected for intensive further study was GluN2AN615K. This variant was selected because it affects a residue that is already known to be crucial in interacting with Mg2+ ions, the ‘N+1’ asparagine (Wollmuth et al. 1998). Missense mutations affecting this residue have a high probability of influencing receptor function. Second, the variant has good genetic evidence of disease‐association: it has arisen de novo in two unrelated individuals with similar phenotypes (Endele et al. 2010; Allen et al. 2016). Both individuals presented with early‐onset epileptic encephalopathy, associated with severe or profound intellectual disability and electroencephalogram abnormalities. Furthermore, so far, the residue has not been found to be mutated in 60 706 people without severe paediatric disease (Exome Aggregation Consortium database, accessed 3 September 2018).
The GluN2AN615K variant was the first missense GRIN2A variant identified as potentially disease causing and some functional work on its consequences has already been performed when expressed as diheteromers: GluN2AN615K has been found to reduce Mg2+ blockade and Ca2+ permeability and impact channel blocker potency but not to affect glutamate or glycine potency (Endele et al. 2010; Pierson et al. 2014). However, no previous work has investigated the impact of this variant when expressed in triheteromers with one wild‐type and one mutated GluN2 subunit. This insight would be clinically relevant, both because the variant is present heterozygously and because the majority of NMDA receptors in key regions of the adolescent and adult brain are probably GluN1/GluN2A/GluN2B triheteromers. Moreover, no previous work has investigated the impact of the variant on NMDA receptors expressed in neurons, where neuron‐specific trafficking and regulation could potentially negate or compensate for the impact of the variant.
In the present study, we employed a recently developed technique to express triheteromeric NMDA receptors containing only one variant subunit in HEK293T cells and assessed the impact of the variant on physiologically relevant receptor properties important for synaptic transmission using electrophysiological recordings. In addition, we expressed the mutant subunit in primary cultured neurons and found that the GluN2AN615K mutation had a marked impact on key physiological properties of NMDA receptors even in the presence of wild‐type subunits, which is consistent with a role in the pathogenesis of the epileptic encephalopathies experienced by its carriers.
Methods
Ethical approval
Experiments conducted during the course of this study received approval from the University of Edinburgh's Animal Welfare Ethical Review Board. Animal breeding and maintenance and experimental procedures were performed in accordance with the UK Animals (Scientific Procedures) Act 1986 under the authority of Project Licence 60/4290 (D.J.A.W.). Animal experiments adhered to the ethical principles required by The Journal of Physiology (Grundy, 2015). Mice were housed under a standard 12:12 h light/dark cycle and received food and water ad libitum. E17.5 CD1 mice (sex not determined) supplied by Charles River (Margate, UK), were culled by decapitation (a Schedule 1 Method) shortly following culling of the dam by cervical dislocation (a Schedule 1 method). Dams were typically 12–22 weeks old.
Mutagenesis
The cDNA for wild‐type human NMDA subunit GluN1‐1a (hereafter GluN1) and GluN2A (GenBank accession codes: NP_015566, NP_000824) (Hedegaard et al. 2012) were gifts from Dr Hongjie Yuan (University of Emory, Atlanta, GA, USA). The GluN2 cDNAs for triheteromer experiments have been described previously: wild‐type rat GluN2A (D13211) and GluN2B (U11419), GluN2AC1, GluN2AC2, GluN2BAC1 and GluN2BAC2 (Hansen et al. 2014). Expression of GluN1 in HEK293T cells was achieved as described previously (Yi et al. 2018) using a plasmid DNA construct with enhanced green fluorescent protein (eGFP) inserted between the cytomegalovirus promoter in pCI‐neo and the open reading frame of rat GluN1 (U08261) (i.e. eGFP and GluN1 were not expressed as a fusion protein). This DNA construct produces high expression of eGFP for identification of transfected cells and maintains a linear relationship between eGFP and GluN1 expression. All cDNAs were in pCI‐neo. Site‐directed mutagenesis was performed via PCR with overlapping mutagenizing oligonucleotides using a thermostable Pfu high fidelity DNA polymerase (New England Biolabs, Ipswich, MA, USA). The PCR product was recircularized into a viable plasmid using an InFusion HD kit (Clontech, Mountain View, CA, USA). The double‐stranded mutant DNA was transformed into TOP10 Competent Cells (Life Tech, Grand Island, NY, USA). Clones were amplified then DNA extracted using QIAPrep Spin MiniPrep Kit (Qiagen, Venlo, The Netherlands) in accordance with the manufacturer's instructions. The mutations were verified by Sanger sequencing through the mutated region.
Preparation and transfection of HEK293T cells
For whole‐cell experiments, human embryonic kidney 293 cells containing the SV40 large T–antigen (HEK293T) cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% dialysed fetal bovine serum, 10 U mL–1 penicillin and 10 mg mL–1 streptomycin). HEK293T cells were chosen for their propensity to grow singly rather than in clumps, facilitating electrophysiological experiments involving rapid solution application. Cells were passaged twice weekly and plated onto cover slips precoated in poly‐d‐lysine (0.1 mg mL–1) to give a density of 10–30% on the day of recording. HEK293T cells were transfected using the calcium phosphate precipitation method. Plasmids containing rGluN1‐1a eGFP and the rGluN2 subunits of interest were mixed in a 1:1 mass ratio and diluted to 200 g L–1 with water. To transfect four wells of a 24‐well plate, 10 μL of cDNA was mixed with 25 μL of 1 m CaCl2 and 100 μL of 2 × Bes (50 mm Bes, 280 mm NaCl and 1.5 mm Na2HPO4, pH 6.95), then mixed by pipetting. After 10–15 min, 50 μL of this mixture was added dropwise to each well. After 4–6 h, the media was replaced with fresh media supplemented with NMDA receptor antagonists [d‐2‐amino‐5‐phosphonopentanoic acid (200 μm) and 7‐chlorokynurenic acid (200 μm)]. Recordings were made ∼24 h post transfection. HEK293T for single‐channel experiments were prepared similarly with minor differences: Dulbecco's modified Eagle's medium was supplemented with Glutamax‐I (Thermo Fisher, Waltham, MA, USA) and 1% antibiotic/antimycotic and the NMDA receptor antagonists used to avoid excitotoxicity post transfection were 100 μm d‐2‐amino‐5‐phosphonopentanoic acid and 10 μm 5,7‐dichlorokynurenic acid.
Whole‐cell, voltage clamp recordings in HEK293T cells
Transfected HEK293T cells were identified by eGFP expression (excitation at 470 nm, coolLED pE‐100; coolLED Ltd, Andover, UK). Whole‐cell, patch clamp recordings were performed using an Axopatch 200B amplifier (Molecular Devices, Sunnydale, CA, USA) at room temperature. The signal was then filtered using an 8 kHz 8‐pole low‐pass filter (–3 dB Bessel; Frequency Devices, Ottawa, IL, USA) and digitized at 20 kHz using a Digidata 1440A analogue‐digital interface (Molecular Devices) using Clampex software (Molecular Devices). Patch pipettes were made from thin‐walled borosilicate glass (TW150F‐4; World Precision Instruments, Sarasota, FL, USA) using a P‐1000 puller (Sutter Instruments, Novato, CA, USA) to give a resistance of 4–5 MΩ when filled with an internal solution containing (in mm): 141 K‐gluconate, 2.5 NaCl, 10 Hepes and 11 EGTA (pH 7.3 with KOH) (300 mosmol L−1). The extracellular solution was composed of (in mm): 150 NaCl, 10 Hepes, 3 KCl, 0.5 CaCl2, 0.01 EDTA and 20 d‐mannitol (pH 7.4 with NaOH). Rapid solution exchange (open tip solution exchange with 10–90% rise times of 0.7–0.9 ms) was achieved using a two‐barrel theta‐glass pipette controlled by a piezoelectric translator (Siskiyou Corporation, Grants Pass, OR, USA). Cells with glutamate‐evoked responses of less than 100 pA or greater than 1000 pA were rejected, to reduce error from noise and to avoid leak of undesired subunit pairings (Hansen et al. 2014). To calculate glutamate deactivation rates, multicomponent exponential decay curves were fitted to macroscopic response time courses in ChanneLab software (Synaptosoft, Fort Lee, NJ, USA).
Single‐channel, voltage clamp recordings in HEK293T cells
Single‐channel, voltage clamp recordings were made at room temperature from cell‐attached patches formed on HEK293T cells placed in a solution that contained (in mm): 150 NaCl, 2.8 KCl, 10 Hepes, 2 CaCl2, 10 glucose and 0.1 glycine (pH 7.35 with NaOH) (300–330 mosmol L−1). Patches were recorded at a range of holding potentials. Transfected cells were identified by eGFP expression as above. Patch pipettes were made from thick‐walled borosilicate glass (GC150F‐7.5; Harvard Apparatus, Cambridge, MA, USA) using a P‐87 puller (Sutter Instruments, Novato, CA, USA) and fire‐polished to give a resistance of 6–12 MΩ when filled with the external solution plus agonist (glutamate 1 mm). Currents were recorded using an Axopatch 200B amplifier (Molecular Devices). Data were filtered at 2 kHz and digitized at 20 kHz via a BNC‐2090A analogue‐digital interface (National Instruments, Newbury, UK) using WinEDR software (Strathclyde Electrophysiology Software, Strathclyde, UK).
WinEDR v3 (Strathclyde Electrophysiology Software) was used to idealize the traces (using a transition threshold of 50% of the predominant conductance level and a 100 μs open and shut resolution) and to fit Gaussian curves to amplitude histograms. The relative proportion of time spent in a given conductance state was calculated by fitting areas under the amplitude histograms. To improve accuracy of amplitude estimates, a 1 ms minimum open time duration was applied. Because the membrane potential was not known, the patch potential was unknown. Therefore, recordings were made at a range of pipette potentials (+40 to +180 mV) and the current plotted against voltage: the slope (fitted by linear regression) gave the conductance. Mean open times were calculated using openings at one pipette potential, with a 1 ms minimum open time duration applied, and fitted using single exponentials in WinEDR. Transitions between subconductance states were manually coded from inspection of all openings with duration >1 ms at a single pipette potential. Traces were excluded if: R 2 < 0.9 for the linear regression, fewer than three pipette potentials were recorded from, or fewer than 20 openings occurred at a given pipette potential.
Preparation and transfection of neurons
Neuronal culturing and transfection was performed as described previously (Marwick et al. 2017). Shortly following decapitation, brains were microdissected in medium containing (in mm): 8.8 Na2SO4, 27 K2SO4, 5.3 MgCl2, 0.23 CaCl2, 0.9 Hepes, 0.001% phenol red, 18 d‐glucose and 0.0005 kynurenic acid (adjusted to pH 7.35 using NaOH). Cortices were incubated for 40 min at 37°C in papain enzyme (36,000 USP units mL–1) (Worthington Biochemical Corporation, Lakewood, NJ, USA) then washed and triturated in NeuroBasal A medium (Thermo Fisher) (supplemented with 1% rat serum; Harlan Laboratories, Indianapolis, IN, USA), 1 × B‐27 supplement, 1% antibacterial/antimycotic and 1 mm glutamine). Opti‐MEM (supplemented with 20 mm glucose and 1% antibacterial/antimycotic) was added to the cell suspension to give an end concentration of 1 hemisphere per 7 mL, and 0.5 mL was plated onto a glass coverslip (diameter 13 mm) precoated with poly‐d‐lysine (1.33% w/v in H2O) and laminin (0.5% w/v) (Roche, Basel, Switzerland) in 24‐well plates. Plates were kept for 2.5 h in a humidified 5% CO2 incubator at 37°C before the cell suspension was removed and replaced with supplemented NeuroBasal A. On days in vitro (DIV) 4, 1 mL well–1 of supplemented NeuroBasal A additionally containing 9.6 mm cytosine β‐d‐arabinofuranoside hydrochloride (a DNA replication inhibitor that prevents glial overproliferation) was added to the cells.
Neurons were transfected on DIV 7 or 8 with plasmids containing cDNA for wild‐type and mutant GluN2A subunits, or the inert control β globin, using Lipofectamine 2000 (Thermo Fisher) (hereafter referred to as lipofectamine) in serum free non‐trophic transfection medium composed of: 10% minimum essential media (+Earles, –l‐glutamine), 88% salt‐glucose‐glycine (comprising in mm 114 NaCl, 26 NaHCO3, 5.3 KCl, 1 MgCl2, 2 CaCl2, 10 Hepes, 1 glycine, 30 d‐glucose and 0.5 sodium pyruvate, with phenol red 0.001%) (Bading et al. 1993) supplemented with 1% antibiotic/antimycotic and 1% insulin–transferrin–selenium supplement. For each coverslip, 575 ng of plasmid DNA comprising a 2:1 mass ratio of GluN2A/GluN2B and eGFP cDNA was mixed with 2.3 μL of lipofectamine. The cotransfection rate was 96% (data not shown). Electrophysiological recordings were performed ∼48 h post transfection.
Whole‐cell, voltage clamp recordings in cultured neurons
Electrophysiological recordings in cultured neurons were performed as described previously (Marwick et al. 2017). Recordings were made at room temperature with neurons superfused (flow rate of 2 mL min–1) with external recording solution composed of (in mm) 150 NaCl, 2.8 KCl, 10 Hepes, 2 CaCl2, 10 glucose, 0.1 glycine and 0.003 TTX (pH 7.35 using NaOH) (300–330 mosmol L−1). Transfected cells were identified using eGFP expression (excitation at 470 nm, coolLED pE‐100; coolLED Ltd). Then, 150 μm NMDA was applied briefly twice to trigger desensitization until a steady response was achieved (∼10 s) at which point 1 mm MgCl2 was co‐applied until the response plateaued. Cells were then perfused with 3 μm ifenprodil for 1 min before the experiments were repeated. Solution application was manually controlled. Patch pipettes were made from thick‐walled borosilicate glass (GC150F‐7.5; Harvard Apparatus) using a P‐87 puller (Sutter Instruments) to give a resistance of 2–4 MΩ when filled with internal solution containing (in mm): 141 K‐gluconate, 2.5 NaCl, 10 Hepes and 11 EGTA (pH 7.3 with KOH) (300–330 mosmol L−1). Currents were recorded using an Axopatch 200B amplifier (Molecular Devices). Data were filtered at 2 kHz and digitized at 20 kHz via a National Instruments BNC‐2090A analogue–digital interface (National Instruments) using WinEDR software. Neurons were voltage clamped at –65 mV, and recordings were rejected if the holding current was greater than 150 pA or if the series resistance was greater than 30 MΩ, or increased by greater than 20% during the course of the recording. Capacitance was calculated by calculating the area under the current response to a 5 mV test pulse plotted against time (giving the charge) and dividing by the voltage of the test pulse. Current density was then calculated as current/capacitance.
Statistical analysis
Data are presented as the mean ± SEM. Graphs depict individual cells (circles), means (columns) and SEM (error bars). N refers to number of cells. R, version 3.1.2 (R Core Team, 2014) was used to perform statistical tests. Comparisons between multiple means are performed by ANOVA, with post hoc tests performed if the F test was significant for a main effect. Comparisons between two means are performed by independent, two‐tailed, Welch t tests (which do not assume equal variance between groups) unless otherwise stated. Correction for multiple comparisons is made using the Bonferroni method. P < 0.05 was considered statistically significant (* P < 0.05, ** P < 0.01 and *** P < 0.001.
Materials
NMDA, NMDA receptor antagonists, ifenprodil and TTX were purchased from Tocris Bioscience (Bristol, UK). Media, media supplements, Lipofectamine 2000, fetal bovine serum, antibiotic/antimycotic and Β‐27 were purchased from Invitrogen (Carlsbad, CA, USA). The remaining substances were purchased from Sigma‐Aldrich (St Louis, MO, USA) unless otherwise stated.
Results
GluN2AN615K reduces Mg2+ blockade in triheteromeric NMDA receptors
We first investigated Mg2+ blockade in GluN1/GluN2AN615K diheteromers expressed in HEK293T cells (Fig. 1). Consistent with previous work (Endele et al. 2010), we found that the GluN2AN615K variant resulted in negligible Mg2+ blockade at approximately physiological concentrations. We next investigated the impact of the GluN2AN615K variant when expressed as part of triheteromeric NMDA receptors containing wild‐type GluN2 subunits (Fig. 1). Accordingly, we employed receptor subunits with modified endoplasmic reticulum retention signals as developed by Hansen et al. (2014) to express receptors containing zero, one or two GluN2AN615K subunits partnered with wild‐type GluN2A or GluN2B subunits. Receptors containing only one GluN2AN615K subunit continued to exhibit markedly reduced Mg2+ blockade, with a blockade intermediate between wild‐type and GluN2AN615K diheteromers (Fig. 1 A and B). Unexpectedly, the identity of the wild‐type subunit partnered with GluN2AN615K impacted on Mg2+ blockade, with a GluN2BWT partner showing lower Mg2+ blockade than a GluN2AWT partner (Fig. 1 A and B). We confirmed that the modified C‐termini did not themselves influence Mg2+ blockade (Fig. 1 C).
Figure 1.
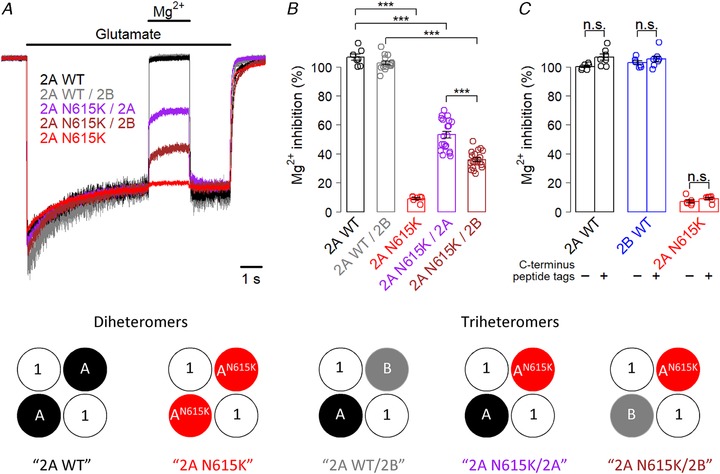
GluN2AN615K reduces Mg2+ blockade in triheteromeric NMDA receptors
A, representative whole‐cell, voltage clamp recordings from HEK293T cells expressing NMDA receptors containing wild‐type GluN2A diheteromers, GluN2A/2B triheteromers and GluN2AN615K‐containing diheteromers and triheteromers, partnered with either GluN2AWT or GluN2BWT. All subunits, including wild‐type, had modified endoplasmic reticulum signals. The traces show responses to glutamate (1 mm) and blockade by Mg2+ (1 mm) in the continuous presence of glycine (100 μm), held at –60 mV. B, summary data showing percentage blockade by Mg2+ for each receptor composition. A one‐way ANOVA showed a significant effect of receptor subunit composition (F 4,64 = 346, P < 2 × 10–16). Post hoc Bonferroni corrected t tests showed that GluN2AN615K diheteromeric receptors had a significantly reduced Mg2+ blockade (9 ± 1%, n = 7) compared to GluN2AWT diheteromers (107 ± 2%, n = 7, t 7.3 = 42, P = 5 × 10–10). Triheteromeric receptors containing only one GluN2AN615K subunit also showed a marked reduction in Mg2+ blockade, whether partnered with one GluN2AWT subunit (53 ± 2%, n = 20), compared with GluN2AWT diheteromers (t 19.9 = 16.5, P = 2 × 10–12) or partnered with one GluN2BWT subunit (36 ± 1%, n = 20), compared with GluN2AWT/2BWT (103 ± 1%, n = 15, t 32.6 = 36.9, P = 9 × 10–16). The reduction in Mg2+ blockade was greater in GluN2AN615K subunits partnered with GluN2BWT than with GluN2AWT (t 29.4 = 6.3, P = 3 × 10–6). Some traces show greater than 100% blockade by Mg2+, reflecting a small contamination of the bath fluid (baseline exposure) by glutamate. C, summary data showing percentage blockade by Mg2+ for NMDA receptors with and without modified C‐termini. A two‐way ANOVA showed a significant influence of C‐terminus tag (F 1,35 = 8.0, P = 0.00785) and a significant effect of receptor subunit composition (F 2,35 = 2635, P < 2 × 10–16), although there was no significant interaction (F 2,35 = 1.3, P = 0.28). However, post hoc Bonferroni corrected t tests showed no significant effect of modifying C‐termini with engineered tags for any subunit.
GluN2AN615K does not influence glutamate deactivation
Previously, GluN2A/2B triheteromer glutamate deactivation rates have been found to be intermediate between those of GluN2A and GluN2B diheteromers (Hansen et al. 2014; Stroebel et al. 2014). We used fast applications of glutamate onto HEK293T cells to replicate this finding, and we also demonstrated that GluN2AN615K does not influence glutamate deactivation rates (Fig. 2 A–C). Current density showed a trend towards lower levels in cells expressing GluN2AN615K‐containing receptors, although this was not statistically significant (Fig. 2 D). We also confirmed that the C‐terminus modifications required for the generation of triheteromers do not influence glutamate deactivation rates in wild‐type or GluN2AN615K‐containing receptors (Fig. 2 E).
Figure 2.
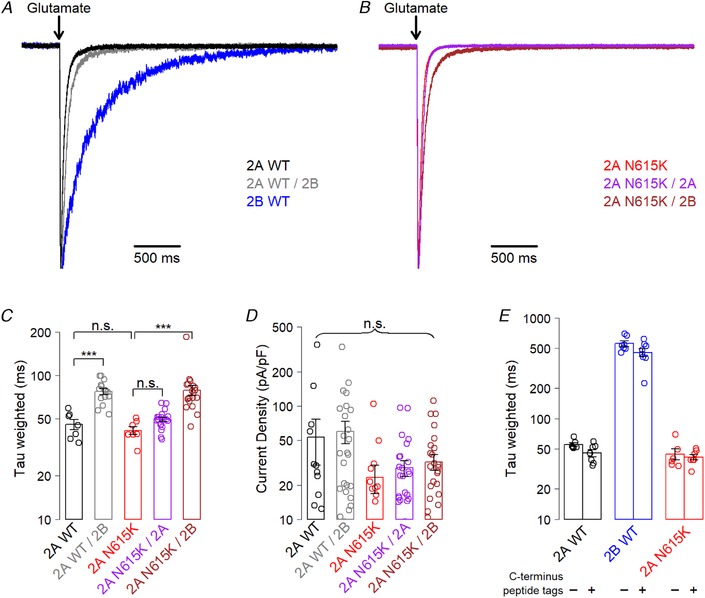
GluN2AN615K does not alter glutamate deactivation
A and B, representative whole‐cell, voltage clamp recordings from HEK293T cells expressing NMDA receptors containing wild‐type GluN2A diheteromers, GluN2A/2B triheteromers and GluN2AN615K containing diheteromers and triheteromers, partnered with either GluN2AWT or GluN2BWT. All subunits, including wild‐type, had modified endoplasmic reticulum signals. The traces show deactivation following 5 ms exposure to glutamate (1 mm) and glycine (100 μm) in the absence of Mg2+ when held at –60 mV. Responses are normalized to their peak amplitudes. C, summary data showing weighted time constants fitted to the glutamate deactivation curves. A one‐way ANOVA showed a significant effect of receptor subunit composition (F 4,64 = 14, P = 3 × 10–8). Post hoc Bonferroni corrected t tests showed that GluN2AWT/2B triheteromers had a slower deactivation time constant (77 ± 4 ms; n = 15), than GluN2AWT diheteromers (45 ± 4 ms; n = 7; t 17 = 6.2, P = 4 × 10–5). GluN2AWT diheteromers were indistinguishable from GluN2AN615K diheteromers (41 ± 3; n = 7; t 11 = 0.9, P ≥ 0.4), with GluN2AN615K and GluN2AN615K/2A (49 ± 2 ms, n = 20) also showing no difference (t 10.9 = 2.6, P = 0.1). GluN2AN615K/2B triheteromers (79 ± 6, n = 20) showed a slower deactivation rate than GluN2AN615K diheteromers (t 10.9 = 2.6, P = 5 × 10–5)). D, summary data showing current density (peak amplitude/capacitance) for cells expressing diheteromeric and triheteromeric receptor combinations (for other experiments, only cells with a current response of between 100 and 1000 pA were used to allow accuracy without escape of unwanted subunit combinations but, here, all traces are included). A one‐way ANOVA showed no effect of receptor subunit composition (F 4,104 = 2.1, P = 0.08). E, summary data showing weighted time constants fitted to glutamate deactivation curves. Modifying the C‐terminus with engineered peptide tags designed to vary retention in the endoplasmic reticulum had no effect on glutamate deactivation in diheteromeric wild‐type receptors containing GluN2AWT or GluN2BWT, or in those containing GluN2AN615K. A two‐way ANOVA showed no significant influence of C terminus tag (F 1,35 = 4.0, P = 0.054), a significant effect of NMDAR subunit composition (F 2,35 = 226, P < 2 × 10–16) and no significant interaction (F 2,35 = 2.5, P = 0.1).
GluN2AN615K reduces single‐channel conductance in diheteromeric and triheteromeric NMDA receptors
The ‘N+1’ residue altered by the GluN2AN615K variant is an ion‐channel lining residue and contributes to form one of the narrowest regions of the receptor pore (Song et al. 2018). We therefore hypothesized that the variant would influence ion permeation in addition to Mg2+ blockade, and investigated single channel conductance. We found that the GluN2AN615K variant lead to a substantial four‐fold reduction in single‐channel conductance in GluN2AN615K‐containing diheteromers (Fig. 3 A, B, E and F). As a control, we confirmed that the modified C‐termini did not influence conductance (Fig. 3 D).
Figure 3.
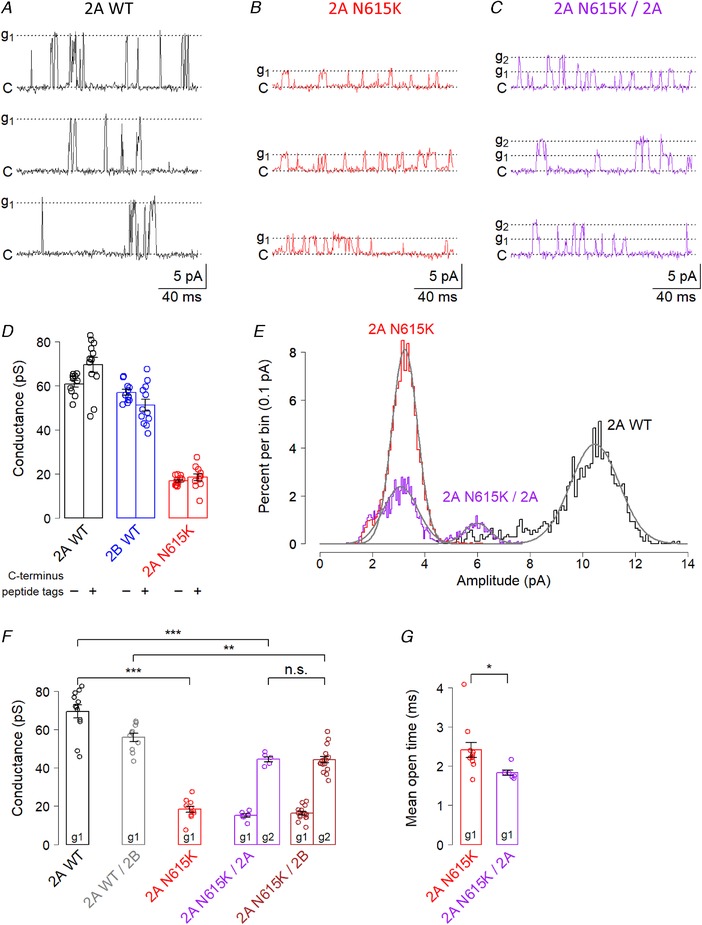
GluN2AN615K reduces single‐channel conductance in triheteromeric NMDA receptors
A–C, representative voltage clamp recordings made from cell‐attached patches from HEK293T cells expressing NMDA receptors containing wild‐type GluN2A diheteromers, GluN2A/2B triheteromers and GluN2AN615K‐containing diheteromers and triheteromers, partnered with either GluN2AWT or GluN2BWT. All subunits, including wild‐type, had modified endoplasmic reticulum signals. The traces show single‐channel currents in the presence of glutamate (1 mm) and glycine (100 μm). The pipette potentials used for the traces illustrated in (A) to (C) were +120, +140 and +140 mV, respectively. ‘C’ = closed; ‘g1’ = first conductance fitted; ‘g2’ = second conductance fitted. A number of transitions between the two conductance states can be seen in (C). D, summary data showing single channel conductance for receptors with and without modified C‐termini. A two‐way ANOVA showed no significant influence of C‐terminus tag (F 1,62 = 0.6, P = 0.42), a significant effect of receptor subunit composition (F 2,62 = 278, P < 2 × 10–16) and a significant interaction (F 2,62 = 5.7, P = 0.006). E, representative amplitude histograms showing fitted normal distributions, superimposed from three different patches. The means of the fitted distributions were used to calculate conductance. For the patch expressing a GluN2AN615K‐containing triheteromer, two peaks can be seen: the prominent subconductance and the less frequent intermediate conductance. F, summary data showing conductances, calculated by plotting current amplitudes against pipette potential (at least three potentials within range + 40 to + 180 mV). A two‐way ANOVA showed a main effect of subunit (F 4,70 = 121, P < 2 × 10–16) and of conductance level (g1 vs. g2; F 1,70 = 196, P < 2 × 10–16), with no significant interaction. Post hoc Bonferroni corrected t tests showed that GluN2AN615K diheteromeric receptors had a significantly lower conductance (18 ± 2 pS; n = 11) than GluN2AWT diheteromers (69 ± 3 pS; n = 12; t 15.4 = 13.7, P = 2 × 10–9). Triheteromeric receptors containing only one GluN2AN615K subunit also showed a marked reduction in intermediate conductance (g2), whether partnered with one GluN2AWT subunit (44 ± 1 pS; n = 6), compared with GluN2AWT diheteromers (t 13.5 = 7.0, P = 3 × 10–5) or with one GluN2BWT subunit (44 ± 1 pS; n = 16), compared with GluN2AWT/2BWT (56 ± 2 pS; n = 10; t 18.4 = 4.2, P = 0.003). The reduction in conductance was no greater in GluN2AN615K subunits partnered with GluN2BWT than with GluN2AWT (t 19.1 = 0.06, P > 0.9). G, summary data showing receptor mean open times calculated by fitting single exponentials to open time distributions from individual patches. Only the lower conductance openings were fitted for GluN2AN615K/2A triheteromers, which showed briefer open times than GluN2AN615K diheteromers (2A N615K/2A: 1.83 ± 0.07 ms; n = 6 vs. 2A N615K 2.42 ± 0.19 ms, n = 11; t 12.5 = 2.9, P = 0.014).
In view of the marked effect of only one GluN2AN615K subunit on Mg2+ blockade in NMDA receptors, we hypothesized that GluN2AN615K containing triheteromeric receptors would also show a conductance intermediate between GluN2AWT and GluN2AN615K diheteromers. Our recordings from cells expressing triheteromeric receptors showed a more complex picture than expected: the vast majority of GluN2AN615K triheteromeric patches showed channels with two conductances (Fig. 3 C, E and F). One conductance was indeed intermediate between that of GluN2AWT and GluN2AN615K diheteromers; the other was a prominent subconductance close in value to the primary conductance of GluN2AN615K diheteromers.
We conducted some additional analyses to investigate two alternative explanations that could mediate what appears to be a prominent subconductance in GluN2AN615K containing triheteromeric NMDA receptors. One alternative explanation is that the higher conductance represents simultaneous openings of two triheteromeric channels. However, the higher conductance was equal to more than double the lower conductance (44 pS vs. 15 pS and 44 pS vs. 16 pS) (Fig. 3 F). We therefore concluded that the upper conductance did not represent the simultaneous opening of two identical lower conductance triheteromeric channels.
Another alternative explanation could be that the patch contains one triheteromeric channel and one diheteromeric channel that has ‘escaped’ retention in the endoplasmic reticulum. To explore this possibility, we compared events attributable to the simultaneous opening and/or closing of two channels in patches expressing triheteromers vs. patches expressing multiple diheteromeric GluN2AN615K receptors and found far fewer such events associated with multiple diheteromers than with triheteromers [GluN2AN615K diheteromers: 4 ± 2% of openings (n = 7) vs. GluN2AN615K/2A triheteromers: 25 ± 1% of openings (n = 6) (t 10.1 = 7.8, P = 2.8 × 10–5) and GluN2AN615K/2B triheteromers: 19 ± 2% of openings (n = 16) (t 17.0 = 4.7, P = 3.9 × 10–4)].
We also assessed mean open times, finding that the GluN2AN615K/2A triheteromeric subconductance openings were briefer than those observed for GluN2AN615K diheteromers (Fig. 3 G). This argues against the possibility that the similar conductance is mediated by GluN2AN615K diheteromeric receptors present in triheteromer‐expressing patches. Such diheteromeric ‘escape currents’ can potentially arise when expression levels are high (Hansen et al. 2014) and, indeed, we did observe a small number of putative triheteromeric receptor patches (five out of 27 patches) in which only one conductance was observed (four with conductances close to wild‐type diheteromers; one with a conductance close to 2A N615K diheteromers). Because these patches presumably contained escaped diheteromeric receptors, they were excluded from the analysis. Overall, our findings suggest that the GluN2AN615K variant results in a marked reduction in NMDA receptor single channel conductance when one or two copies are present. When two copies are present in a receptor, a single low conductance is seen. When one copy is present, two conductances are seen: low and intermediate.
GluN2AN615K reduces Mg2+ blockade and current density in cultured neurons
To complete our investigation, we wished to explore whether the pronounced effects of GluN2AN615K seen in NMDA receptors expressed in a human cell line would also be seen in receptors expressed in primary neurons, subject to neuronal specific trafficking and regulation. Accordingly, we used transient transfection to overexpress GluN2AWT and GluN2AN615K subunits in mouse primary cortical neurons. The resulting NMDA receptor population probably comprised a mixture of diheteromeric and triheteromeric GluN2B and GluN2A‐containing receptors, formed from both endogenous GluN2B subunits and transfected subunits. We first confirmed the expression of transfected GluN2A subunits (wild‐type or GluN2AN615K) by demonstrating a reduction in inhibition by ifenprodil (a selective GluN2B negative allosteric modulator) compared to neurons transfected with an inert control, which express predominantly GluN2B at DIV 9 (McKay et al. 2012) (Fig. 4 A–D). We next assessed the impact of the GluN2AN615K mutation on Mg2+ blockade, in the presence and absence of ifenprodil (Fig. 4 A–C and E). We found that Mg2+ blockade was reduced in neurons transfected with GluN2AN615K and that this effect was more pronounced in the presence of ifenprodil, when a higher proportion of activated receptors contain GluN2A subunits. Third, we assessed the impact of GluN2AN615K on current density (Fig. 4 A–C and F). We found that current density was reduced in neurons transfected with GluN2AN615K and this effect was again more pronounced in the presence of ifenprodil. This reduction in current density is consistent with the reduction in conductance we observed previously (Fig. 3). Taken together, these results show that, when the GluN2AN615K mutation is expressed in cells which endogenously express NMDA receptors, it continues to have a profound influence on Mg2+ blockade and on current density.
Figure 4.
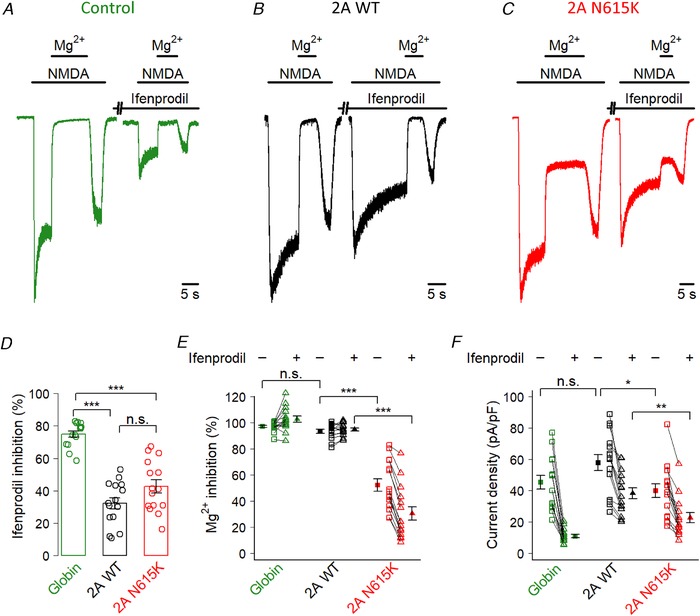
GluN2AN615K reduces Mg2+ blockade and current density in cultured neurons
A–C, representative whole‐cell, voltage clamp recordings made from DIV 9 primary mouse cortical neurons transiently transfected with an inert control: β globin (A), GluN2AWT (B) or GluN2AN615K (C). Traces show the response to saturating NMDA (150 μm) and inhibition by Mg2+ (1 mm), before and after a 1 min application of ifenprodil (3 μm) (a selective GluN2B negative allosteric modulator). D, summary data showing percentage inhibition by ifenprodil. A one‐way ANOVA showed a significant effect of transfected subunit (F 2,42 = 46, P = 2.8 × 10–11), with post hoc Bonferroni corrected t tests indicating that neurons transfected with GluN2AWT showed lower ifenprodil sensitivity than control transfection cells (WT: 32 ± 3; n = 15; globin 75 ± 2; n = 15; t 21.9 = 10.7, P = 1.0 × 10–9), as did neurons transfected with GluN2AN615K (43 ± 4; n = 15; t 20.0 = 7.2, P = 1.9 × 10–6) and with no difference between GluN2A and GluN2AN615K (t 27.3 = 1.9, P = 0.19). These results confirm that the GluN2AN615K subunits have been successfully trafficked to the surface in neurons. E, summary data showing Mg2+ blockade in the presence and absence of ifenprodil. Squares show blockade without ifenprodil; triangles show blockade with ifenprodil. Solid shapes represent means; hollow shapes are individual cells. A two‐way ANOVA showed a significant effect of transfected subunit (F 2,42 = 111, P = 2 × 10–16) and the presence of ifenprodil (F 1,42 = 31, P = 1.9 × 10–6), with a significant interaction (F 2,42 = 90, P = 6.7 × 10–16). Post hoc Bonferroni corrected t tests showed lower Mg2+ blockade in the absence of ifenprodil in neurons transfected with GluN2AN615K compared to GluN2AWT (WT: 93 ± 1%; n = 15; N615K 52 ± 5%; n = 15; t 16.7 = 8.2, P = 9.7 × 10–7), although there was no difference between GluN2AWT and control (globin: 97 ± 1%; n = 15; t 23.3 = 2.2, P = 0.13). In the presence of ifenprodil, the reduction in Mg2+ blockade associated with GluN2AN615K vs. wild‐type was even more marked (WT: 95 ± 1%; n = 15; N615K 31 ± 5%; n = 15; t 15.4 = 12.1, P = 8.4 × 10–9). F, summary data showing current density in the presence and absence of ifenprodil. As with Mg2+ blockade, we found that current density was reduced in neurons transfected with GluN2AN615K, and this effect was more pronounced in the presence of ifenprodil. A two‐way ANOVA showed a significant effect of transfected subunit (F 2,42 = 9.4, P = 0.0004) and the presence of ifenprodil (F 1,42 = 178, P = 2 × 10–16), with a significant interaction (F 2,42 = 9.2, P = 0.0005). Post hoc Bonferroni corrected t tests showed lower current density in the absence of ifenprodil in neurons transfected with GluN2AN615K compared to GluN2AWT (WT: 58 ± 5 pA pF–1; n = 15; N615K 40 ± 4 pA pF–1; n = 15; t 27.3 = 2.7, P = 0.037), although there was no difference between GluN2AWT and control (globin: 45 ± 4 pA pF–1; n = 15; t 27.4 = 1.9, P = 0.22). In the presence of ifenprodil, the reduction in current density associated with GluN2AN615K vs. wild‐type was even more marked (WT: 38 ± 4 pA pF–1; n = 15; N615K 23 ± 3 pA pF–1; n = 15) (t 27.8 = 3.3, P = 0.009).
Discussion
In the present study, we investigated the functional consequences of GluN2AN615K, a heterozygous missense variant found to have arisen de novo in two unrelated people with early onset epileptic encephalopathy. Using heterologous systems, we showed that the GluN2AN615K variant results in major alterations to physiologically crucial aspects of NMDA receptor function: an almost complete loss of Mg2+ blockade and a four‐fold reduction in conductance. Importantly, the variant continues to markedly reduce Mg2+ blockade and conductance when expressed in NMDA receptor triheteromers with one wild‐type GluN2 subunit, and also continues to have effect when expressed in cortical neurons. These findings strengthen the evidence that the GluN2AN615K variant causes the severe neurodevelopmental disorder experienced by its carriers.
GluN2AN615K reduces Mg2+ blockade
The marked reduction in Mg2+ blockade that we observed with the GluN2AN615K mutation is in keeping with previous work identifying the affected residue as important for Mg2+ blockade (Wollmuth et al. 1998) and with the disruptive replacement of an asparagine with a positively‐charged lysine. Our finding is also consistent with previous work reporting minimal Mg2+ blockade in GluN2AN615K diheteromers (Endele et al. 2010). We additionally demonstrated a reduction in Mg2+ blockade when GluN2AN615K is expressed in primary neurons. The smaller magnitude of reduction in Mg2+ blockade seen in neurons probably reflects the presence of endogenous GluN2B subunits. We directly addressed the impact of GluN2AN615K when expressed as part of triheteromeric NMDA receptors with wild‐type partner subunits and found that Mg2+ blockade was reduced to an intermediate extent. This is an important finding because disease‐associated mutations in GRIN2A have so far only been found heterozygously. Observing an effect of the variant despite the presence of wild‐type subunits further supports a role for GluN2AN615K in disease causation. This finding indirectly supports the disease relevance of other NMDA receptor pore mutations where functional consequences have been established in diheteromers (Fedele et al. 2018; Fernández‐Marmiesse et al. 2018).
Interestingly, we observed an impact of partner subunit identity on Mg2+ blockade in GluN2AN615K‐containing triheteromers: receptors with a wild‐type GluN2A partner subunit showed higher Mg2+ blockade than those with a wild‐type GluN2B partner subunit. However, we did not see an impact of partner subunit identity on single‐channel conductance. The mechanism of this intriguing finding and the extent to which it applies to other receptor properties requires additional study.
A reduction in Mg2+ blockade would be hypothesized to have substantial impact on neuronal physiology and synaptic plasticity because voltage‐dependent Mg2+ blockade is essential to the role of the NMDA receptor as a molecular coincidence detector (Huganir & Nicoll, 2013). Alterations to this property could therefore be anticipated to result in deficits in learning and memory, as may be reflected in the severe and profound intellectual disability of carriers. However, plasticity is dependent on many interacting factors: additional receptor properties such as calcium permeability, expression of other NMDA receptor subunits and expression of other receptor types. Furthermore, deficits in plasticity caused by reduced or increased expression of NMDA receptor subunits have been associated with behavioural consequences in several (Sakimura et al. 1995; Kiyama et al. 1998; Roberts et al. 2009) but not all studies (Okabe et al. 1998). Further studies are needed to assess the potential impact of the GluN2AN615K variant on synaptic plasticity, which would be aided by a transgenic animal model.
GluN2AN615K reduces conductance
We found that the GluN2AN615K variant reduced single‐channel conductance by around four‐fold when expressed as diheteromers. Triheteromers containing only one GluN2AN615K subunit showed two conductances: one similar to GluN2AN615K diheteromers and one intermediate conductance equivalent to around two‐thirds of wild‐type. We also found that NMDA‐evoked current density was reduced in neurons expressing GluN2AN615K, suggesting that any acute compensation by other receptor subunits was insufficient to overcome the reduction in conductance associated with the GluN2AN615K variant. Our finding of reduced conductance adds to previous work reporting similarly marked reductions in conductance following mutations of equivalent residues neighbouring the location of the GluN2AN615K variant in GluN1 and GluN2B subunits (Behe et al. 1995; Premkumar et al. 1997; Schneggenburger & Ascher, 1997). In addition to this probable direct impact of the altered residue on ion permeation through the narrow internal region of the pore (Song et al. 2018), it is possible that other charged regions that strongly influence ion permeation (e.g. the DRPEER motif in the GluN1 subunit) (Wollmuth, 2018) may also be indirectly affected by the GluN2AN615K variant, contributing to effects on single‐channel conductance. Our finding that glutamate deactivation rates are unaffected by the mutation shows that the overall time course of receptor openings is similar. A reduction in conductance is of physiological importance because it implies that less charge is passed by activated NMDA receptors containing GluN2AN615K subunits. This would reduce the ability of a receptor to contribute to neuronal excitability, and also potentially alter the receptor's ionotropic signaling pathways.
In summary, the present study has strengthened the evidence indicating that the disease‐associated variant GluN2AN615K is associated with substantial changes in physiologically crucial properties of NMDA receptors. This information can be used to inform genetic counselling. Our work highlights NMDA receptor‐related synaptic transmission as probable candidates for disruption in the pathogenesis of neurodevelopmental disorders. Future work could usefully explore the impact of this mutation in vivo in synaptic plasticity at circuit and behavioural levels.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
KFMM, KBH, PAS, GEH and DJAW were responsible for the conception and design of the experiments. KFMM and KBH were responsible for the collection and assembly of data. KFMM, KBH, PAS, GEH and DJAW were responsible for the analysis and interpretation of data. KFMM, KBH, PAS, GEH and DJAW were responsible for drafting the article or revising it critically for important intellectual content. All authors approved the final version of the manuscript submitted for publication, and all persons designated as authors qualify for authorship and all those who qualify for authorship are listed.
Funding
This work was funded by the Wellcome Trust (WT102838) and National Institutes of Health (GM103546 and NS097536).
Acknowledgements
We thank Dr M. R. Livesey for his help with single‐channel data analysis and critical comments on the manuscript.
Biography
Katie Marwick studied medicine at the University of Cambridge and University of Edinburgh before completing a PhD at the University of Edinburgh, where she is currently a Clinical Lecturer in Psychiatry. Her PhD and postdoctoral work have focused on investigating the functional consequences of disease‐associated variants in NMDA receptors. Future work includes exploring the impact of such variants in transgenic animals. Her goal is to improve the identification and targeting of treatments for NMDA receptor‐associated neurodevelopmental disorders.

Edited by: Kim Barrett & Jean‐Claude Béïque
References
- Allen NM, Conroy J, Shahwan A, Lynch B, Correa RG, Pena SDJ, McCreary D, Magalhães TR, Ennis S, Lynch SA & King MD (2016). Unexplained early onset epileptic encephalopathy: exome screening and phenotype expansion. Epilepsia 57, e12–e17. [DOI] [PubMed] [Google Scholar]
- Bading H, Ginty DD & Greenberg ME (1993). Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260, 181–186. [DOI] [PubMed] [Google Scholar]
- Bar‐Shira O, Maor R & Chechik G (2015). Gene expression switching of receptor subunits in human brain development. PLoS Comput Biol 11, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behe P, Stern P, Wyllie DJA, Nassar M, Schoepfer R & Colquhoun D (1995). Determination of NMDA NR1 subunit copy number in recombinant NMDA receptors. Proc R Soc B Biol Sci 262, 205–213. [DOI] [PubMed] [Google Scholar]
- Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, Milh M, Kortüm F, Fritsch A, Pientka FK, Hellenbroich Y, Kalscheuer VM, Kohlhase J, Moog U, Rappold G, Rauch A, Ropers HH, von Spiczak S, Tönnies H, Villeneuve N, Villard L, Zabel B, Zenker M, Laube B, Reis A, Wieczorek D, Van Maldergem L & Kutsche K (2010). Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet 42, 1–22. [DOI] [PubMed] [Google Scholar]
- Fedele L, Newcombe J, Topf M, Gibb A, Harvey RJ & Smart TG (2018). Disease‐associated missense mutations in GluN2B subunit alter NMDA receptor ligand binding and ion channel properties. Nat Commun 9, 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Marmiesse A, Kusumoto H, Rekarte S, Roca I, Zhang J, Myers SJ, Traynelis SF, Couce ML, Gutierrez‐Solana L & Yuan H (2018). A novel missense mutation in GRIN2A causes a nonepileptic neurodevelopmental disorder. Mov Disord 33, 992–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JA, Shi Y, Usui H, During MJ, Sakimura K & Nicoll RA (2011). Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single‐cell NMDA receptor subunit deletion in vivo. Neuron 71, 1085–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Yuan H & Traynelis SF (2014). Distinct functional and pharmacological properties of triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron 81, 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedegaard MK, Hansen KB, Andersen KT, Brauner‐Osborne H & Traynelis SF (2012). Molecular pharmacology of human NMDA receptors. Neurochem Int 61, 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL & Nicoll RA (2013). AMPARs and synaptic plasticity: the last 25 years. Neuron 80, 704–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyama Y, Manabe T, Sakimura K, Kawakami F, Mori H & Mishina M (1998). Increased thresholds for long‐term potentiation and contextual learning in mice lacking the NMDA‐type glutamate receptor ε1 subunit. J Neurosci 18, 6704–6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermott AB, Mayer ML, Westbrook GL, Smith SJ & Barker JL (1986). NMDA‐receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature 321, 519–522. [DOI] [PubMed] [Google Scholar]
- Marwick KFM, Parker P, Skehel P, Hardingham G & Wyllie DJA (2017). Functional assessment of the NMDA receptor variant GluN2AR586K. Wellcome Open Res 2, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL & Guthrie PB (1984). Voltage‐dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 309, 261–263. [DOI] [PubMed] [Google Scholar]
- McKay S, Griffiths NH, Butters PA, Thubron EB, Hardingham GE & Wyllie DJA (2012). Direct pharmacological monitoring of the developmental switch in NMDA receptor subunit composition using TCN 213, a GluN2A‐selective, glycine‐dependent antagonist. Br J Pharmacol 166, 924–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B & Seeburg PH (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P & Ascher P (1984). Magnesium gates glutamate‐activated channels in mouse central neurones. Nature 307, 472–465. [DOI] [PubMed] [Google Scholar]
- Ogden KK, Chen W, Swanger SA, McDaniel MJ, Fan LZ, Hu C, Tankovic A, Kusumoto H, Kosobucki GJ, Schulien AJ, Su Z, Pecha J, Bhattacharya S, Petrovski S, Cohen AE, Aizenman E, Traynelis SF & Yuan H (2017). Molecular mechanism of disease‐associated mutations in the pre‐M1 helix of NMDA receptors and potential rescue pharmacology. PLoS Genet 13, 1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Collin C, Auerbach JM, Meiri N, Bengzon J, Kennedy MB, Segal M & McKay RD (1998). Hippocampal synaptic plasticity in mice overexpressing an embryonic subunit of the NMDA receptor. J Neurosci 18, 4177–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C & Zhou Q (2013). NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14, 383–400. [DOI] [PubMed] [Google Scholar]
- Pierson TM, Yuan H, Marsh ED, Fuentes‐Fajardo K, Adams DR, Markello T, Golas G, Simeonov DR, Holloman C, Tankovic A, Karamchandani MM, Schreiber JM, Mullikin JC, Tifft CJ, Toro C, Boerkoel CF, Traynelis SF & Gahl WA (2014). GRIN2A mutation and early‐onset epileptic encephalopathy: personalized therapy with memantine. Ann Clin Transl Neurol 1, 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premkumar LS, Qin F & Auerbach A (1997). Subconductance states of a mutant NMDA receptor channel kinetics, calcium, and voltage dependence. J Gen Physiol 109, 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauner C & Köhr G (2011). Triheteromeric NR1/NR2A/NR2B receptors constitute the major N‐methyl‐D‐aspartate receptor population in adult hippocampal synapses. J Biol Chem 286, 7558–7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AC, Díez‐García J, Rodriguiz RM, López IP, Luján R, Martínez‐Turrillas R, Picó E, Henson MA, Bernardo DR, Jarrett TM, Clendeninn DJ, López‐Mascaraque L, Feng G, Lo DC, Wesseling JF, Wetsel WC, Philpot BD & Pérez‐Otaño I (2009). Downregulation of NR3A‐containing NMDARs is required for synapse maturation and memory consolidation. Neuron 63, 342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H & Mishina M (1995). Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature 373, 151–155. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshé SL, Nordli DR, Perucca E, Tomson T, Wiebe S, Zhang YH & Zuberi SM (2017). ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia 58, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R & Ascher P (1997). Coupling of permeation and gating in an NMDA‐channel pore mutant. Neuron 18, 167–177. [DOI] [PubMed] [Google Scholar]
- Song X, Jensen MØ, Jogini V, Stein RA, Lee C‐H, Mchaourab HS, Shaw DE & Gouaux E (2018). Mechanism of NMDA receptor channel block by MK‐801 and memantine. Nature 556, 515–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroebel D, Carvalho S, Grand T, Zhu S & Paoletti P (2014). Controlling NMDA receptor subunit composition using ectopic retention signals. J Neurosci 34, 16630–16636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanger SA, Chen W, Wells G, Burger PB, Tankovic A, Bhattacharya S, Strong KL, Hu C, Kusumoto H, Zhang J, Adams DR, Millichap JJ, Petrovski S, Traynelis SF & Yuan H (2016). Mechanistic insight into NMDA receptor dysregulation by rare variants in the GluN2A and GluN2B agonist binding domains. Am J Hum Genet 99, 1261–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, McGinley MJ & Westbrook GL (2013). Triheteromeric NMDA receptors at hippocampal synapses. J Neurosci 33, 9150–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Inoue Y, Sakimura K & Mishina M (1992). Developmental changes in distribution of NMDA receptor channel subunit mRNAs. Neuroreport 3, 1138–1140. [DOI] [PubMed] [Google Scholar]
- Wollmuth LP (2018). Ion permeation in ionotropic glutamate receptors: still dynamic after all these years. Curr Opin Physiol 2, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmuth LP, Kuner T & Sakmann B (1998). Adjacent asparagines in the NR2‐subunit of the NMDA receptor channel control the voltage‐dependent block by extracellular Mg2+ . J Physiol 506, 13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie DJA, Livesey MR & Hardingham GE (2013). Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology 74, 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XiangWei W, Jiang Y & Yuan H (2018). De novo mutations and rare variants occurring in NMDA receptors. Curr Opin Physiol 2, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Zachariassen LG, Dorsett KN & Hansen KB (2018). Properties of triheteromeric NMDA receptors containing two distinct GluN1 isoforms. Mol Pharmacol 93, 453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]