

Abstract
Key points
Potassium‐chloride co‐transporter 2 (KCC2) plays a critical role in regulating chloride homeostasis, which is essential for hyperpolarizing inhibition in the mature nervous system.
KCC2 interacts with many proteins involved in excitatory neurotransmission, including the GluK2 subunit of the kainate receptor (KAR).
We show that activation of KARs hyperpolarizes the reversal potential for GABA (E GABA) via both ionotropic and metabotropic signalling mechanisms.
KCC2 is required for the metabotropic KAR‐mediated regulation of E GABA, although ionotropic KAR signalling can hyperpolarize E GABA independent of KCC2 transporter function.
The KAR‐mediated hyperpolarization of E GABA is absent in the GluK1/2−/− mouse and is independent of zinc release from mossy fibre terminals.
The ability of KARs to regulate KCC2 function may have implications in diseases with disrupted excitation: inhibition balance, such as epilepsy, neuropathic pain, autism spectrum disorders and Down's syndrome.
Abstract
Potassium‐chloride co‐transporter 2 (KCC2) plays a critical role in the regulation of chloride (Cl−) homeostasis within mature neurons. KCC2 is a secondarily active transporter that extrudes Cl− from the neuron, which maintains a low intracellular Cl− concentration [Cl−]. This results in a hyperpolarized reversal potential of GABA (E GABA), which is required for fast synaptic inhibition in the mature central nervous system. KCC2 also plays a structural role in dendritic spines and at excitatory synapses, and interacts with ‘excitatory’ proteins, including the GluK2 subunit of kainate receptors (KARs). KARs are glutamate receptors that display both ionotropic and metabotropic signalling. We show that activating KARs in the hippocampus hyperpolarizes E GABA, thus strengthening inhibition. This hyperpolarization occurs via both ionotropic and metabotropic KAR signalling in the CA3 region, whereas it is absent in the GluK1/2−/− mouse, and is independent of zinc release from mossy fibre terminals. The metabotropic signalling mechanism is dependent on KCC2, although the ionotropic signalling mechanism produces a hyperpolarization of E GABA even in the absence of KCC2 transporter function. These results demonstrate a novel functional interaction between a glutamate receptor and KCC2, a transporter critical for maintaining inhibition, suggesting that the KAR:KCC2 complex may play an important role in excitatory:inhibitory balance in the hippocampus. Additionally, the ability of KARs to regulate chloride homeostasis independently of KCC2 suggests that KAR signalling can regulate inhibition via multiple mechanisms. Activation of kainate‐type glutamate receptors could serve as an important mechanism for increasing the strength of inhibition during periods of strong glutamatergic activity.
Keywords: KCC2, Kainate receptor, hippocampus, GABA, Chloride transport, electrophysiology, metabotropic, ionotropic
Key points
Potassium‐chloride co‐transporter 2 (KCC2) plays a critical role in regulating chloride homeostasis, which is essential for hyperpolarizing inhibition in the mature nervous system.
KCC2 interacts with many proteins involved in excitatory neurotransmission, including the GluK2 subunit of the kainate receptor (KAR).
We show that activation of KARs hyperpolarizes the reversal potential for GABA (E GABA) via both ionotropic and metabotropic signalling mechanisms.
KCC2 is required for the metabotropic KAR‐mediated regulation of E GABA, although ionotropic KAR signalling can hyperpolarize E GABA independent of KCC2 transporter function.
The KAR‐mediated hyperpolarization of E GABA is absent in the GluK1/2−/− mouse and is independent of zinc release from mossy fibre terminals.
The ability of KARs to regulate KCC2 function may have implications in diseases with disrupted excitation: inhibition balance, such as epilepsy, neuropathic pain, autism spectrum disorders and Down's syndrome.
Introduction
In the central nervous system, fast hyperpolarizing GABAergic inhibition depends upon the neuronal Cl− gradient (Kaila, K., 1994), which is primarily maintained by members of the cation chloride co‐transporter (CCC) family (Blaesse et al. 2009, Kaila, Kai et al. 2014). Of these transporters, potassium‐chloride co‐transporter 2 (KCC2) is the primary extruder of Cl− in mature neurons, undergoing a developmental upregulation that contributes to the establishment and maintenance of hyperpolarizing inhibition (Rivera et al. 1999). KCC2 is a secondarily active transporter that uses the existing potassium gradient generated by the Na+/K+‐ATPase to extrude Cl− from the neuron, resulting in relatively low concentration of Cl− inside mature neurons (Kaila, Kai et al. 2014, Doyon et al. 2016). This allows Cl− to flow down its electrochemical gradient and into the neuron when the GABAA receptor is activated, comprising a necessary condition for hyperpolarizing inhibition. Fast and strong GABAergic inhibition is critical for proper neurophysiological function, including the regulation of spike timing (Pouille and Scanziani, 2001, Kopp‐Scheinpflug et al. 2011, D′amour and Froemke, 2015).
Because of its key role in maintaining the Cl− gradient that is critical to inhibition, KCC2 has been considered historically as an ‘inhibitory protein’. However, recent evidence suggests that KCC2 is also important for excitatory neurotransmission. KCC2 is highly expressed at excitatory synapses (Gulyás et al. 2001, Li et al. 2007, Llano et al. 2015), where it plays an important role in dendritic spine formation and maintenance, and influences AMPA receptor (AMPAR) content and diffusion out of the synapse (Li et al. 2007, Gauvain et al. 2011, Llano et al. 2015). KCC2 has also been shown to physically interact with several excitatory receptors and proteins (Mahadevan et al. 2017), including the GluK2 subunit of kainate receptors (KARs) (Mahadevan et al. 2014), and the KAR auxiliary subunit Neto‐2 (Ivakine et al. 2013, Mahadevan et al. 2015). KCC2 also functionally interacts with several other glutamate receptors, including NMDA receptors (Lee et al. 2011), and metabotropic glutamate receptors (mGluRs) (Banke and Gegelashvili, 2008).
KARs are classed as ionotropic glutamate receptors but are unusual in their signalling properties in that they can signal both in an ionotropic (canonical) and metabotropic (non‐canonical) fashion (Lerma and Marques, 2013). Similar to other ionotropic glutamate receptors, the ionic current of KARs is largely carried by Na+; however, KAR currents are typically smaller and possess slower decay kinetics compared to other glutamate receptors (Castillo et al. 1997) as a result of their interaction with auxiliary subunits (Straub and Tomita, 2012). Activation of the KAR current produces a long lasting depolarization that increases action potential firing via increased summation (Frerking, Matthew and Ohliger‐Frerking, 2002, Sachidhanandam et al. 2009, Pinheiro et al. 2013). In addition to their ability to produce an ionotropic current, KARs also signal metabotropically via a G‐protein coupled mechanism (Rodríguez‐Moreno and Lerma, 1998, Rozas et al. 2003, Lerma and Marques, 2013). Metabotropic KAR signalling has been shown to regulate presynaptic neurotransmitter release and neuronal excitability (Rodrigues and Lerma, 2012). In particular, KAR activation decreases the amplitude of the slow afterhyperpolarization (I SAHP), which leads to a subsequent increase in neuronal firing (Melyan et al. 2002, Melyan et al. 2004, Fisahn et al. 2005).
KCC2 interacts with the KAR subunit GluK2 and this physical interaction is required to maintain normal surface expression and oligomerization of the KCC2 protein (Mahadevan et al. 2014, Pressey et al. 2017). However, whether KAR‐mediated ionotropic current and/or metabotropic signalling can also regulate KCC2 function is unknown. To determine whether KAR signalling regulates KCC2 function, we made electrophysiological recordings from pyramidal neurons in the CA3. The CA3 is one of the few regions in the brain, and the only region in the hippocampus, where postsynaptic kainate ionotropic currents and KAR‐metabotropic signalling have been recorded in pyramidal neurons (Castillo et al. 1997, Vignes and Collingridge, 1997, Fisahn et al. 2005, Lerma and Marques, 2013). We demonstrate that independently activating either the ionotropic or metabotropic KAR signalling pathways produces a hyperpolarization of the reversal potential for GABA (E GABA). Interestingly, pharmacological blockade of KCC2 prevents KAR‐mediated regulation of E GABA when low concentrations of kainic acid (KA) are applied, but not when higher concentrations of KA are applied, suggesting that the two modes of KAR signalling regulate chloride homeostasis via two independent mechanisms. KAR‐mediated hyperpolarization of E GABA is not observed in GluK1/2−/− CA3‐pyramidal cells and occurs independently from zinc release from mossy fibre (MF) terminals. This represents a novel mechanism for glutamatergic activity to modulate the strength of inhibitory synaptic transmission in the hippocampus.
Methods
Ethical approval
All animal procedures were approved by the University of Toronto Animal Care Committee in accordance with the Canadian Council for Animal Care guidelines (Animal Protocol #20012022) and conformed with the principles and standards as described by Grundy (2015). All efforts were made to minimize animal suffering and to reduce the number of animals used.
Animals
Experiments were performed on male and female mice. C57Bl/6 mice were obtained from a colony in the Faculty of Arts and Sciences Biosciences Facility (originally obtained from Jackson Laboratories, Bar Harbor, ME, USA). GluK1/2−/− mice were originally obtained from Dr Chris McBain (NIH, Bethesda, MD, USA) (Mulle et al. 1998, Contractor et al. 2000, Mulle et al. 2000) and were used to establish a colony in the Faculty of Arts and Sciences Biosciences Facility. All mice were housed under a 12:12 h light/dark photocycle with access to food and water available ad libitum. Mice were anaesthetized with open‐drop isoflurane prior to decapitation with a guillotine.
Electrophysiology
P19–P26 C57Bl/6 mice were anaesthetized with isoflurane and brains were rapidly removed after decapitation and placed into a cutting solution containing (in mm): 205 sucrose, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 0.4 ascorbic acid, 1 CaCl2, 2 MgCl2 and 3 sodium pyruvate (pH 7.4), osmolality ∼295 mosmol kg–1. Coronal slices (300 μm) containing the hippocampus were prepared and recovered at 32°C in a 50 : 50 mixture composed of cutting saline + artificial CSF (aCSF) for 30 min and then placed in aCSF alone for 30 min. During experimentation, slices were perfused at a rate of ∼ 2 mL min–1 in aCSF. The aCSF solution consisted of (in mm): 123 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 2 CaCl2 and 1 MgCl2 in double‐distilled water and saturated with 95% O2/5% CO2 (pH 7.4), osmolarity ∼295 mosmol L–1. Recording pipettes were pulled from thin‐walled borosilicate (TW‐150F, World Precision Industries; Sarasota, FDL, USA) to resistances of 5–12 MΩ with a P‐87 micropipette (Sutter Instruments, Novato, CA, USA).
Gramicidin perforated patch clamp recordings from hippocampal slices were made in oxygenated aCSF at 35–37°C from putative pyramidal cells in the CA3. Recording pipettes were filled with an internal solution containing 150 mm KCl, 10 mm HEPES and 50 μg mL–1 gramicidin (pH 7.4, 300 mosmol kg−1). Slices were first perfused with aCSF containing GYKI‐52466 (30 μm) to block AMPAR‐mediated transmission followed by perfusion with aCSF containing both GYKI‐52466 (30 μm) and UBP 310 (5 μm) to block both AMPAR and KAR‐mediated transmission.
Whole‐cell, patch clamp recordings were obtained from putative pyramidal cells in the CA3 in oxygenated aCSF containing the antagonists: GYKI 52466 (10 μm), dl‐APV (50 μm), DL‐AP3 (300 μm) and CGP55845 (3 μm). In a subset of experiments, aCSF contained additional antagonists NEM (50 μm), VU 0463271 (1 μm) or ZX1 (100 μm). KARs were activated with KA (1 μm or 0.1 μm) in addition to these antagonists; control cells were exposed to antagonists alone. All experiments were performed at 22–24°C. Pipettes were filled with an internal solution containing (in mm): 130 potassium gluconate, 10 KCl, 10 HEPES, 0.2 EGTA, 4 ATP, 0.3 GTP and 10 phosphocreatine (pH 7.4), osmolality ∼285 mosmol kg–1. In a subset of experiments, GDP‐β‐S (300 μm) was also included in the internal solution. Recordings began 5 min after entering whole‐cell configuration, with this time point taken as t = 0. E GABA was determined in voltage clamp mode by evoking IPSCs when step depolarizing the membrane potential. During each current step, an IPSC was evoked by a stimulation electrode placed in the stratum lucidum/radiatum. A linear regression of the IPSC amplitudes was then used and the intercept of this line with the abscissa was taken as E GABA; synaptic conductance was taken as the slope of this regression line, expressed in pS. Driving force was calculated by subtracting E GABA from the resting membrane potential (RMP) and is expressed in mV. Paired‐pulse ratio (PPR) was determined by stimulating two IPSCs 50 ms apart, and then dividing the peak amplitude of the second IPSC by the first.
Chemicals
The sources of the agonists and antagonists used in the experiments were: KA (Sigma‐Aldrich, St Louis, MO, USA), NEM (Sigma‐Aldrich), GDP‐β‐S (Sigma‐Aldrich), dl‐APV (Tocris Bioscience, St Louis, MO, USA), GYKI 52466 (Tocris Bioscience), CGP55845 (Tocris Bioscience), dl‐AP3 (Tocris Bioscience), ZX1 (Strem Chemicals, Newbury Port, MA, USA) and VU 0463271 (Tocris Bioscience). All chemicals were diluted in aCSF with the exception of GDP‐β‐S, which was added to pipette internal solution; CGP55845, which was diluted in DMSO; and VU 0463271, which was also diluted in DMSO resulting in a concentration of 0.0002% DMSO in aCSF (all experiments) and 0.0003% (VU experiments) in the electrophysiological recordings.
Statistical analysis
All population data are expressed as the mean ± SD, unless otherwise specified. Full time course data was analysed using two‐way repeated measures ANOVA. All other data were analysed using a paired Student's t test to examine the statistical significance of the differences. Each n value represents an individual neuron. Only one n value was obtained from each acute slice. multiple n values were obtained from the same animal. The number of mice (N) used for each experiment was: Fig. 1 A and B (N = 3), Fig. 1 C–I (control, N = 7; KA, N = 7), Fig. 2 (control, N = 7; KA, N = 7), Fig. 3 (control, N = 10; KA, N = 5), Fig. 4 A–G (control, N = 6; KA, N = 4) Fig. 4 H–J (control, N = 6; KA, N = 9), Fig. 5 (control, N = 5; 1 μm KA, N = 6; 0.1 μm KA, N = 4), Fig. 6 (control, N = 4; 1 μm KA, N = 4; 0.1 μm KA, N = 3), Fig. 7 (control, N = 4; KA, N = 3), Fig. 8 (control, N = 4; KA, N = 6).
Figure 1.
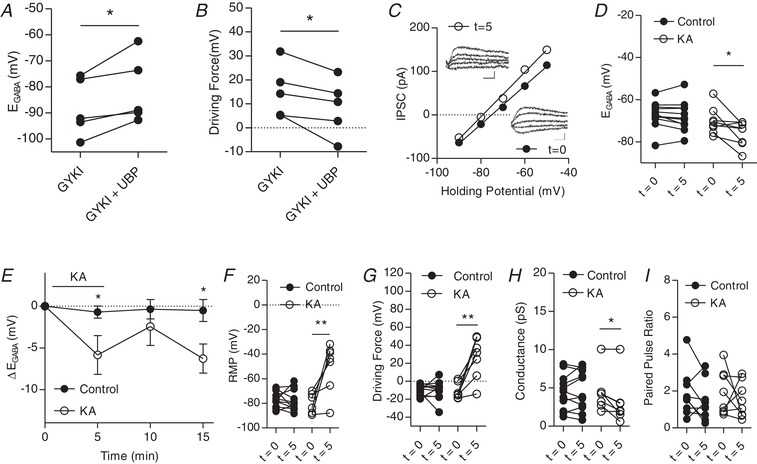
Kainate receptors modulate IPSCs in CA3 pyramidal cells
A, group data showing E GABA before and after KAR blockade with UBP310 (n = 5). B, group data showing driving force for Cl– before and after KAR blockade with UBP310 (n = 5). C, plot of voltage–current curve for a cell at t = 0 (E GABA = −75.1 mV) and t = 5(E GABA = −79.1 mV). Inset: examples of evoked IPSCs (scale bar = 60 pA, 10 ms). D, group data showing E GABA at t = 0 and t = 5 (n = 11 control, 8 KA). E, plot of group data showing the effect of 1 μm KA application on E GABA over time (n = 11 control, 8 KA) F, group data showing resting membrane potential at t = 0 and t = 5 (n = 11 control, 8 KA). G, group data showing driving force for Cl– at t = 0 and t = 5 (n = 11 control, 8 KA). H, group data showing C– conductance at t = 0 and t = 5 (n = 11 control, 8 KA). I, group data showing paired pulse ratio at t = 0 and t = 5 (n = 9 control, 8 KA) (* P < 0.05, ** P < 0.01).
Figure 2.
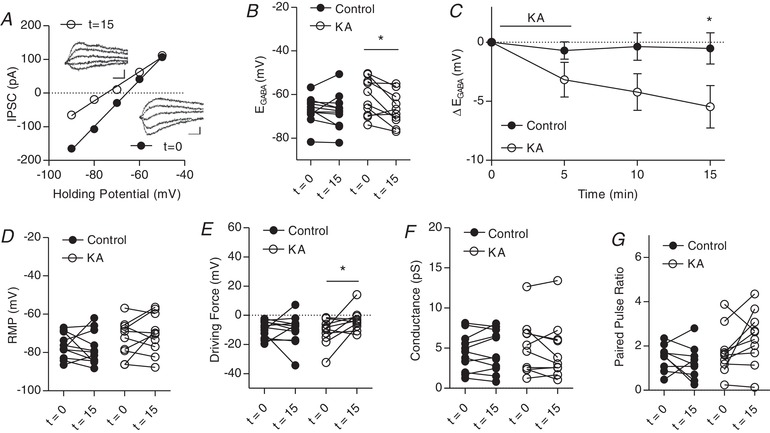
Kainate receptors activated by 0.1 μm KA modulate IPSCs
A, plot of voltage–current curve for a cell at t = 0 (E GABA = −65.6 mV) and t = 15 (E GABA = −74.6 mV). Inset: examples of evoked IPSCs (scale bar = 60 pA, 10 ms). B, group data showing E GABA at t = 0 and t = 15. C, plot of group data showing the effect of 0.1 μm KA on E GABA over time. D, group data showing resting membrane potential at t = 0 and t = 15. E, group data showing driving force for Cl– at t = 0 and t = 15. F, group data showing Cl– conductance at t = 0 and t = 15. G, group data showing paired pulse ratio at t = 0 and t = 15 (n = 9 control, n = 10 KA, all experiments) (* P < 0.05).
Figure 3.
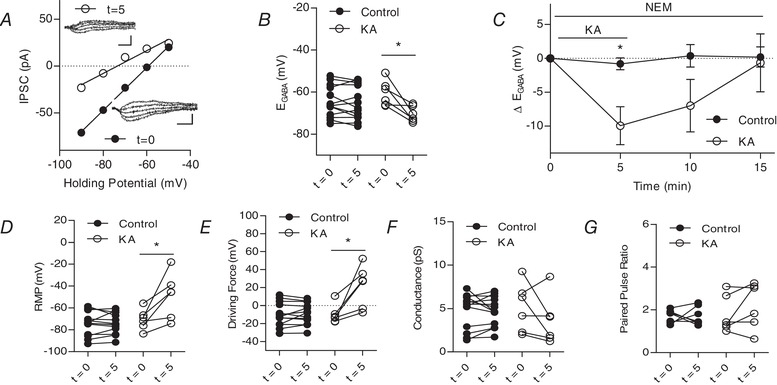
Kainate receptors activated by 1 μM kainic acid modulate IPSCs in the presence of metabotropic signalling blocker NEM
A, plot of voltage–current curve for a cell at t = 0 (E GABA = −59.2 mV) and t = 5 (E GABA = −73.5 mV). Inset, examples of evoked IPSCs (scale bar: 60 pA, 10 ms). B, group data showing E GABA at t = 0 and t = 5 (n = 13 control, n = 6 KA). C, plot of group data showing the effect of 1 μm KA on E GABA over time (n = 13 control, n = 6 KA). D, group data showing resting membrane potential at t = 0 and t = 5 (n = 13 control, n = 6 KA). E, group data showing driving force for Cl− at t = 0 and t = 5 (n = 13 control, n = 6 KA). F, group data showing Cl− conductance at t = 0 and t = 5 (n = 11 control, 8 KA). G, group data showing paired pulse ratio at t = 0 and t = 5 (n = 7 control, 8 KA) (* P < 0.05) [Correction made on 9 February 2019, after first online publication: Figure 3 was replaced with the current version].
Figure 4.
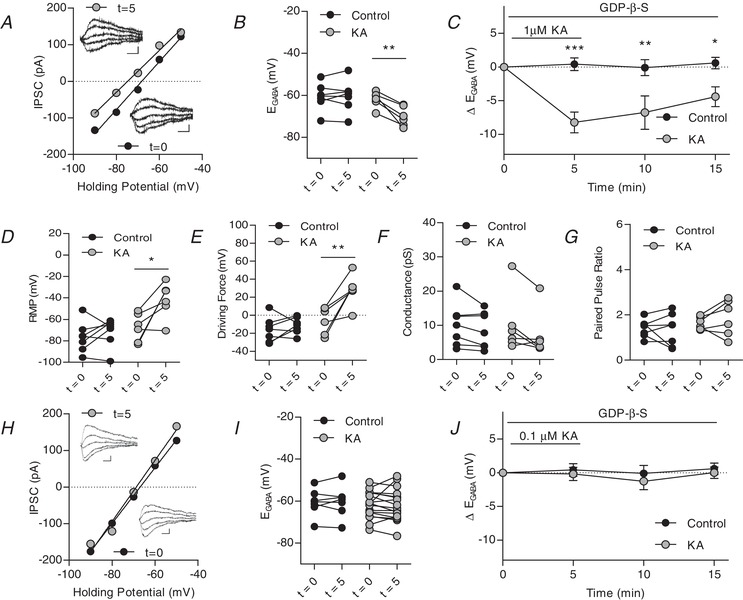
Kainate receptors activated by 1 μM kainic acid modulate IPSCs in the presence of metabotropic signalling blocker GDP‐β‐S
A, plot of voltage–current curve for a cell at t = 0 (E GABA = −68.6 mV) and t = 5 (E GABA = −74.2 mV). Inset, examples of evoked IPSCs (scale bar: 60 pA, 10 ms). B, group data showing E GABA at t = 0 and t = 5. C, plot of group data showing the effect of 1 μM KA on E GABA over time. D, group data showing resting membrane potential at t = 0 and t = 5. with 1 μM KA application E, group data showing driving force for Cl− at t = 0 and t = 5 (n = 13 control, n = 6 KA). F, group data showing Cl− conductance at t = 0 and t = 5 with 1 μM KA application. G, group data showing paired pulse ratio at t = 0 and t = 5 with 1 μM KA application (n = 7 control, n = 6 KA, all 1 μM KA experiments). H, plot of voltage‐current curve for a cell at t = 0 (E GABA = −67.3 mV) and t = 15 (E GABA = −69.1 mV) with 0.1 μM KA application. Inset, examples of evoked IPSCs (scale bar: 60 pA, 10 ms). I, group data showing E GABA at t = 0 and t = 5 with 0.1 μM KA. J, plot of group data showing the effect of 0.1 μM KA on E GABA over time. (n = 7 control, n = 17 KA, all 0.1 μM KA, experiments) (* P < 0.05, ** P < 0.01, *** P < 0.001) [Correction made on 9 February 2019, after first online publication: Figure 4 was replaced with the current version].
Figure 5.
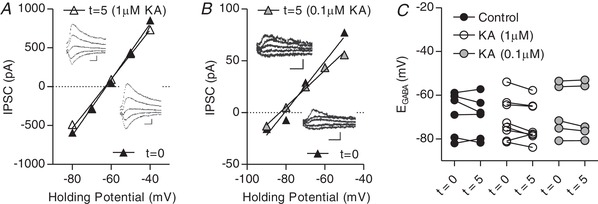
1 μm or 0.1 μm KA does modulate IPSCs in GluK1/2–/– CA3 pyramidal cells
A, plot of voltage–current curve for a GluK1/2–/– neuron at t = 0 (E GABA = −64.4 mV) and t = 5 with 1 μm KA applied (E GABA = −65.1 mV). Inset: examples of evoked IPSCs (scale bar = 200 pA, 10 ms). B, plot of voltage–current curve for a GluK1/2–/– neuron at t = 0 (E GABA = −80.7 mV) and t = 5 with 0.1 μm KA applied (E GABA = −83.1 mV). Inset: examples of evoked IPSCs (scale bar = 60 pA, 10 ms). C, group data showing E GABA at t = 0 and t = 5 (n = 6 control, 7 KA 1 μm, 5 KA 0.1 μm).
Figure 6.
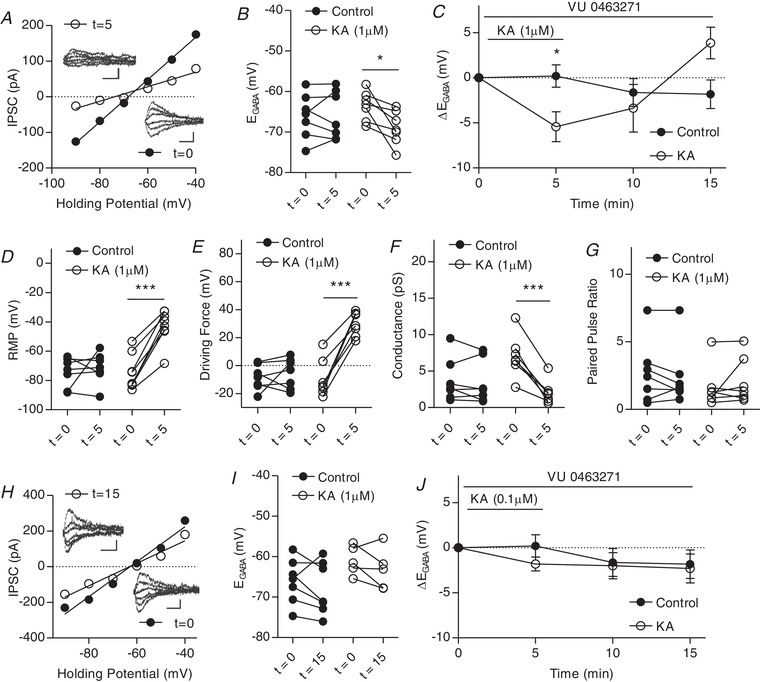
Kainate receptor mediated modulation of IPSCs activated by 0.1 μm but not 1 μm KA is dependent on KCC2 transporter function
A, plot of voltage–current curve for a cell at t = 0 (E GABA = −68.6 mV) and t = 5 (E GABA = −71.9 mV). Inset: examples of evoked IPSCs (scale bar = 100 pA, 20 ms). B, group data showing E GABA at t = 0 and t = 5. C, plot of group data showing the effect of 1 μm KA on E GABA over time in the presence of specific KCC2 antagonist VU0463271. D, group data showing resting membrane potential at t = 0 and t = 5. E, group data showing driving force for Cl– at t = 0 and t = 5. F, group data showing Cl– conductance at t = 0 and t = 5. G, group data showing paired pulse ratio at t = 0 and t = 15. H, plot of voltage‐current curve for a cell at t = 0 (E GABA = −62.8 mV) and t = 15 (E GABA = −63.1 mV). Inset, examples of evoked IPSCs (scale bar: 100 pA, 20 ms). I, group data showing E GABA at t = 0 and t = 15. J, plot of group data showing the effect of 0.1 μM KA on E GABA over time in the presence VU0463271. (n = 7 control, n = 7 1 μm KA, n = 5 1 μm KA, all experiments) (* P < 0.05, *** P < 0.001).
Figure 7.
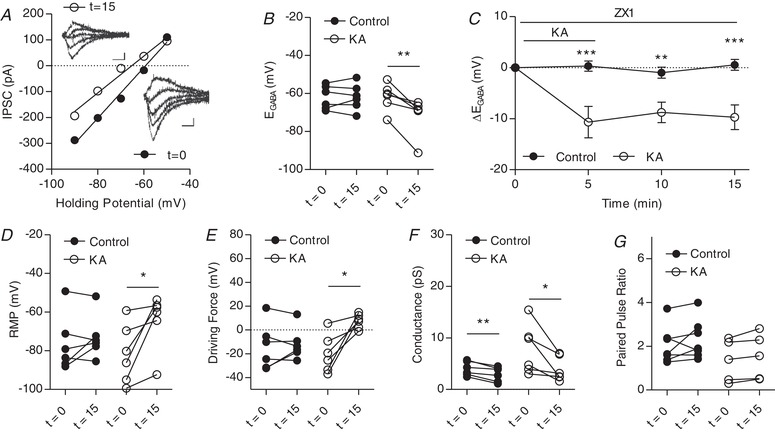
Kainate receptors activated by 1 μm KA modulate IPSCs in CA3 pyramidal cells in the presence of zinc chelator ZX1
A, plot of voltage–current curve for a cell at t = 0 (E GABA = −59.3 mV) and t = 15 ( = −65.2 mV). Inset: examples of evoked IPSCs (scale bar = 60 pA, 10 ms). B, group data showing E GABA at t = 0 and t = 5 (n = 6 control, n = 6 KA). C, plot of group data showing the effect of 1 μm KA on E GABA over time (n = 6 control, n = 6 KA). D, group data showing resting membrane potential at t = 0 and t = 15 (n = 6 control, n = 6 KA). E, group data showing driving force for Cl– at t = 0 and t = 5 (n = 6 control, n = 6 KA). F, group data showing Cl– at t = 0 and t = 15. G, group data showing paired pulse ratio at t = 0 and t = 15 (n = 6 control, n = 5 KA) (* P < 0.05, ** P < 0.01).
Figure 8.
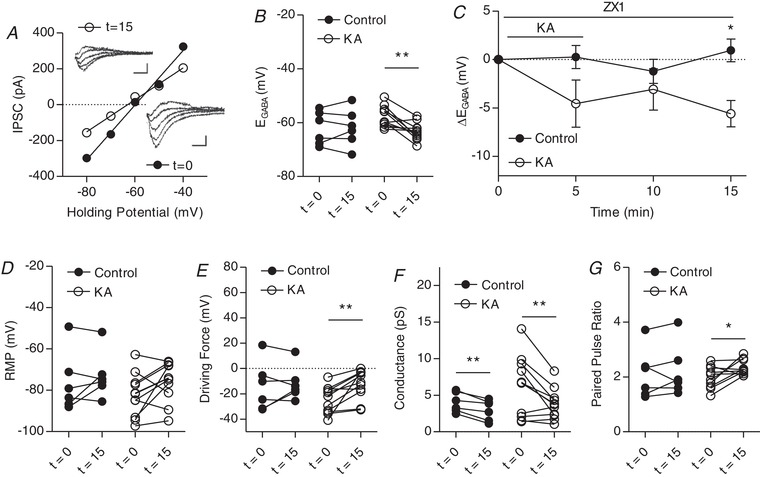
Kainate receptors activated by 0.1 μm KA modulate IPSCs in CA3 pyramidal cells in the presence of zinc chelator ZX1
A, plot of voltage–current curve for a cell at t = 0 (E GABA = −59.7 mV) and t = 15 (E GABA = −63.0 mV). Inset: examples of evoked IPSCs (scale bar = 60 pA, 10 ms). B, group data showing E GABA at t = 0 and t = 5. C, plot of group data showing the effect of 1 μm KA on E GABA over time. D, group data showing resting membrane potential at t = 0 and t = 15. E, group data showing driving force for Cl– at t = 0 and t = 5. F, group data showing Cl– at t = 0 and t = 15. G, group data showing paired pulse ratio at t = 0 and t = 15 (n = 6 control, 10 KA, experiments) (* P < 0.05, ** P < 0.01).
Results
To determine whether activation of KARs regulate KCC2 function and therefore GABAergic inhibition, we recorded the reversal potential for GABA (E GABA) from putative pyramidal neurons in the CA3 region in acute hippocampal slices. KCC2 function cannot be tested directly in neurons, and thus E GABA is used as the standard measure of the efficacy of this transporter in the brain (Woodin, 2013).
We first recorded EGABA using the gramicidin perforated patch clamp technique to preserve the intracellular Cl− concentration at the same time as blocking AMPA receptors with GYKI‐52466. We then examined the effect of blocking KARs using UBP 310, which blocks postsynaptic mossy‐fibre CA3 KAR transmission (Pinheiro et al. 2013). Upon UBP application, E GABA depolarized from −87.9 ± 4.1 mV to −81.5 ± 4.7 mV (Fig. 1 A). This was accompanied by a significant decrease in the driving force for Cl− from 15.1 ± 9.9 mV to −8.7 ± 10.5 mV (Fig. 1 B). These results suggest that KAR activity may tonically regulate KCC2 function.
Next, we performed experiments to determine whether KAR activation could regulate KCC2 function GABAergic currents were isolated by bath applying antagonists to block NMDA receptors (dl‐APV), AMPARs (GYKI 52466), GABABRs (CGP55845) and Group I mGluRs (DL‐AP3). IPSCs were evoked with a stimulating electrode placed in the stratum lucidum/radiatum when recording in whole‐cell mode with a holding potential of −70 mV. Although gramicidin‐perforated patch clamp recordings are often used to record E GABA, including in our own studies (Acton et al. 2012, Ivakine et al. 2013, Mahadevan et al. 2014, Mahadevan et al. 2015), we have previously demonstrated that changes in E GABA can be detected using whole‐cell recordings (Ormond and Woodin, 2009, Ormond and Woodin, 2011, Takkala and Woodin, 2013, Mahadevan et al. 2017). The use of the whole‐cell technique was necessary in the present study as a result of the longer‐term nature of the recordings, which required the washing‐in and ‐out of pharmacological agents, and the need to include a pharmacological agent in the recording pipette.
To determine the effects of KAR activation on KCC2 function, we bath applied the KAR agonist KA (1 μm) for 5 min (t = 0 to t = 5 min). KAR activation produced a significant hyperpolarization of E GABA (t = 5 min) compared to control E GABA measurements that were made in aCSF containing only the inhibitors described above. Following KA washout, E GABA transiently depolarized (t = 10 min) and was not significantly different from controls, significantly hyperpolarizing again after further washout (t = 15 min). Upon KA application (t = 5), E GABA hyperpolarized from −70.1 ± 6.4 mV to −76.0 ± 5.8 mV (Fig. 1 C–E). A significant hyperpolarization of E GABA to −76.4 ± 5.9 mV persisted after KA washout (t = 15). This change in E GABA was accompanied by a significant depolarization of the RMP (Fig. 1 F), which, when combined, resulted in a significant change in the driving force for Cl− (Fig. 1 G). This effect was accompanied by a significant decrease in synaptic conductance but not paired pulse ratio (Fig. 1 H and I).
Next, we aimed to determine whether metabotropic KAR activation alone could independently regulate KCC2 function. Nanomolar concentrations of KA are sufficient to activate metabotropic signalling (Fernandes et al. 2009), as well as to produce much smaller or no kainate receptor currents (Castillo et al. 1997). Therefore, we bath applied 0.1 μm KA to investigate the effects of metabotropic KAR signalling on KCC2 function. At this concentration of KA, there was no significant hyperpolarization of E GABA upon KA application at t = 5, although a significant hyperpolarization of E GABA occurred after washout at t = 15 (Fig. 2 A–C). This resulted in a significant change in the driving force for Cl− (Fig. 2 E), although no changes in RMP, conductance or PPR were observed after 10 min of washout (Fig. 2 D, F and G). Taken together, these findings suggest that metabotropic KAR signalling can regulate E GABA.
To determine whether the KAR‐mediated hyperpolarization of E GABA can result from ionotropic signalling, we isolated the ionotropic signalling pathway using NEM, which is commonly used to inhibit KAR‐mediated metabotropic signalling (Frerking et al. 2001, Melyan et al. 2002). When we activated KARs with 1 μm KA during bath application of NEM (50 μm), E GABA significantly hyperpolarized (t = 5 min) (Fig. 3 C); however, this hyperpolarization did not persist following KA washout. This change in E GABA was again accompanied by a significant depolarization of the RMP, which resulted in a significant change in the driving force for Cl− (Fig. 3 D and E); similar to E GABA, these changes in RMP and the driving force did not persist through washout. No significant changes in conductance or paired pulse ratio were observed upon KA application (Fig. 3 F and G). These results suggest that KAR‐mediated ionotropic can regulate E GABA independent of metabotropic signalling. Although NEM is a commonly used antagonist for metabotropic KAR signalling, it is also known to alter KCC2 function and surface expression via an interaction with a WNK kinase that alters KCC2 phosphorylation (Conway et al. 2017). Although control cells in our NEM experiments did not show any changes in E GABA as a result of bath application of NEM over the course of the experiments, we repeated the above experiments using GDP‐β‐S to isolate the ionotropic signalling pathway to mitigate any potential concerns regarding known effects of NEM on KCC2. GDP‐β‐S is a GDP analogue that interferes with G‐protein binding, and has been found to inhibit the ability of KARs to reduce the amplitude of the I SAHP (Ruiz et al. 2005, Segerstråle et al. 2010).
Similar to our experiments with NEM, activation of KARs with 1 μm KA with GDP‐β‐S (300 μm) in the patch pipette led to a significant hyperpolarization of E GABA compared to controls, which had GDP‐β‐S introduced via the patch pipette but did not have KA bath applied (Fig. 4 A–C). E GABA remained hyperpolarized throughout the washout period, which was accompanied by a depolarization of the resting membrane potential, again resulting in a significant change in the driving force for chloride (Fig. 4 D and E). We also briefly repeated these experiments using 0.1 μm KA application with inclusion of GDP‐β‐S in the patch pipette. At this low concentration of KA, no significant changes in E GABA were observed throughout the experiment (Fig. 4 H–J), suggesting that G‐protein signalling is necessary for the effect.
To determine whether the KAR‐mediated hyperpolarization of E GABA was a result of GluK1 or Gluk2‐containing KAR activation, we performed KA‐application experiments in GluK1/2−/− mice (Mulle et al. 1998, Contractor et al. 2000, Mulle et al. 2000) at both previously used KA concentrations. No significant differences were observed between control and cells with either 1 μm or 0.1 μm KA applied in these slices (Fig. 5). These results suggest that GluK1/GluK2‐containing KAR activation can increase Cl− driving force via a combination of depolarizing the membrane potential and hyperpolarizing E GABA.
Next, we aimed to determine whether the KAR‐mediated hyperpolarization observed was the result of a change in KCC2 transporter function. Although KCC2 function is the primary determinant of E GABA, other transporters such as NKCC1 are also present in the neuron and maybe influence E GABA. We repeated our previous experiments with the addition of selective KCC2 antagonist VU 0463271 (VU) (1 μm) to the bath to parse out the contribution of KCC2. Interestingly, when 1 μm KA was applied, a hyperpolarization of E GABA was observed at t = 5 (Fig. 6 A–C), although this hyperpolarization did not persist through washout as previously observed in experiments without VU. This was accompanied by a significant depolarization of the resting membrane potential and a significant increase in the driving force for Cl− (Fig. 6 D and E). A significant decrease in conductance was observed upon KA application, although the paired pulse ratio did not change significantly (Fig. 6 F and G). By contrast, bath application of 0.1 μm KA in the presence of VU did not produce any significant changes in E GABA throughout the experiment (Fig. 6 H–J). These results suggest that KARs can regulate E GABA in both a KCC2 independent and KCC2‐dependent manner.
Finally, we aimed to determine whether zinc release of MF‐CA3 plays a role in KAR‐mediated regulation of E GABA. It has been shown previously that, at MF‐CA3 synapses, zinc release and subsequent metabotropic zinc receptor (mZnR) activation can enhance KCC2 function (Chorin et al. 2011). Kainate‐induced zinc release at mossy fibre synapses has also been shown to increase activity and surface expression of KCC2, an effect that was prevented with application of a broad divalent cation chelator (Gilad et al. 2015). To test whether the KAR‐mediated hyperpolarization of E GABA resulted from mZnR activation, we used a highly specific Zn2+ chelator, ZX1 (Pan et al. 2011). Upon 1 μm KA application in the presence of ZX1, E GABA significantly hyperpolarized (Fig. 7 A–C) and this hyperpolarization persisted throughout washout. This was accompanied by a significant depolarization of the resting membrane potential, as well as an increase in the driving force for Cl− (Fig. 7 D and E), as previously observed in the absence of the chelator. This shift was not accompanied by a change in PPR, although a change in conductance over time was observed both in control and in KA groups (Fig. 7 F and G).
When repeated with 0.1 μm KA in the presence of ZX1, E GABA significantly hyperpolarized after 10 min of washout, producing a significant change in the driving force for GABA despite an absence of significant changes in resting membrane potential (Fig. 8 A–E). This was accompanied by a change in conductance, which was also observed in control cells, as well as a change in paired pulse ratio (Fig. 8 F and G). These results further indicate that the primary locus of the KA‐mediated effect on E GABA is metabotropic KAR signalling and that this is independent of mZnR activation.
Discussion
In the present study, we have shown that activation of KARs with KA hyperpolarizes the reversal potential of GABA in the CA3 region of the hippocampus via both ionotropic and metabotropic signalling mechanisms. This hyperpolarization of E GABA, combined with the depolarization of the resting membrane potential, results in a large driving force for Cl−. At a concentration of 1 μm KA, this also resulted in a decreased conductance, probably because of altered presynaptic GABA release, which is a well documented effect of KAR activation (Rodríguez‐Moreno et al. 1997, Vignes et al. 1998). This suggests that activation of kainate‐type glutamate receptors could serve as an important homeostatic mechanism for increasing the strength of inhibition during periods of strong glutamatergic activity.
Activation of the metabotropic signalling pathway of KARs was able to hyperpolarize E GABA. This effect was found to be dependent both on GluK1/2‐containing KARs and KCC2 transporter activity because it was not present in GluK1/2−/− neurons or when KCC2 transporter activity was blocked. It was also abolished via blockade of metabotropic KAR signalling with GDP‐β‐S, suggesting that G‐proteins are necessary for this process, and was also shown to be independent of zinc release at MF‐CA3 terminals. This finding is in line with a growing body of literature suggesting that KCC2 activity can be regulated by a number of different receptors that signal via G‐proteins, including mGluRs and mZnRs (Banke and Gegelashvili, 2008, Chorin et al. 2011, Gilad et al. 2015, Mahadevan and Woodin, 2016). KARs, similar to mGluRs and mZnRs, activate a G‐protein signalling pathway that increases PKC activity (Melyan et al. 2002, Lerma and Marques, 2013). It is well known that PKC increases KCC2 activity via phosphorylation at serine 940 (Lee et al. 2007) and so it possible that all three of these metabotropic receptors could increase KCC2 activity via signalling pathways that ultimately result in the activation of PKC. If KARs, mGluRs, and mZnRs were activated by high amounts of glutamatergic activity and zinc release at the MF‐CA3 synapse of the hippocampus, they could work in a concerted effort to strengthen inhibition, which could subsequently put a brake on excitatory activity.
Conversely, activation of the ionotropic signalling pathway of KARs was demonstrated to hyperpolarize E GABA even when KCC2 transporter function was blocked by VU 0463271, suggesting that KARs can regulate chloride homeostasis in both a KCC2‐dependent (metabotropic) and KCC2 independent (ionotropic) manner. This effect was also observed using two different methods of blockade for metabotropic KAR signalling (i.e. application of NEM or GDP‐β‐S), suggesting that metabotropic KAR signalling is not involved in this process. However, the mechanism of action for how ionotropic KAR signalling could regulate intracellular Cl− remains to be determined. Although KCC2 is the primary extruder of Cl− in mature neurons, NKCC1, which normally imports Cl− into the cell, also plays a role in Cl− homeostasis. The hyperpolarization of E GABA could possibly indicate a reduction in NKCC1 transporter function, although this is probably not the case given that KA application has been implicated in increased NKCC1 expression and function in models of epilepsy (Dzhala et al. 2005, Dzhala et al. 2010, Nogueira et al. 2015). Further experiments involving pharmacological blockade of NKCC1 with bumetanide would allow us to explore this possibility.
In both cases, ionotropic and metabotropic KAR signalling acted in a homeostatic manner: an increase in postsynaptic KAR activity results in a hyperpolarization of E GABA, below the resting membrane potential, which would ensure robust inhibition upon activation of the GABAA receptor. This contrasts with pre‐synaptic KARs, which have been shown, in most cases, to inhibit GABA release from presynaptic terminals (Sihra and Rodríguez‐Moreno, 2011). Postsynaptic KARs existing as part of a larger macromolecular complex that includes KCC2 (Mahadevan et al. 2014) would be well positioned to influence inhibitory activity during periods of high glutamate activity via their metabotropic signalling mode. KCC2 is highly expressed at excitatory synapses, where it is known to serve an important role in structural maintenance and osmotic regulation of dendritic spines (Gulyás et al. 2001, Li et al. 2007, Llano et al. 2015). Metabotropic KAR regulation of KCC2 may be particularly relevant in areas where GABAergic inputs converge with excitatory inputs (Higley, 2014) or at locations where KARs could potentially detect glutamate spillover from nearby excitatory synapses.
The two signalling modes of KARs may serve different functional roles for chloride homeostasis, allowing the cell to regulate the strength of GABAergic inhibition in response to excitatory activity in both a KCC2‐dependent and KCC2 independent manner. The metabotropic signalling mode, which we have shown regulates E GABA and is dependent on KCC2 transporter activity, places KARs on a growing list of proteins that are able to regulate KCC2 via G‐protein signalling (Banke and Gegelashvili, 2008, Chorin et al. 2011, Mahadevan and Woodin, 2016). Two previous studies have reported an increase in transporter activity and expression of KCC2 upon KA application (Khirug et al. 2010, Gilad et al. 2015), although the possibility that KARs themselves might serve as the locus of action has not been examined. We have shown that KA application hyperpolarizes the reversal potential of GABA via the activation of two independent KAR signalling mechanisms, and that this effect can occur independently from zinc release and subsequent mZnR activation, revealing a novel role for KARs in the hippocampus. It is well known that disruptions in the Cl− gradient are implicated in several neurological diseases and disorders that result from excessive glutamatergic excitation, including epilepsy and neuropathic pain (Coull et al. 2003, Kahle et al. 2014), marking metabotropic regulation of KCC2 as a potential target for the treatment of these diseases. These results represent a novel mechanism for the regulation of excitatory:inhibitory balance in the mature nervous system and could have important implications for both the normal physiological function of neurons and disease states.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
All experiments were performed in the laboratory of Melanie Woodin in the Department of Cell and Systems Biology at the University of Toronto. DG and VM conducted the experiments. DG analysed the data. DG, VM and MAW contributed to the experimental design. DG and MAW wrote the manuscript. All authors approved the final version of this manuscript submitted for publication and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by an NSERC Discovery grant to MAW.
Acknowledgements
We thank Dr Chris McBain (NIH, Bethesda) for the GluK1/2−/− mice; Dr Jessica Pressey (University of Toronto, Ontario) for her helpful comments on the manuscript; and Simon Bedard for technical assistance.
Biography
Danielle Garand is a PhD candidate in the laboratory of Melanie A Woodin in the Department of Cell and Systems Biology at the University of Toronto. Danielle is an electrophysiologist whose PhD thesis research in neuroscience focuses on kainate receptor‐mediated regulation of chloride homeostasis in the hippocampus. She collaborates on research projects demonstrating the important role of synaptic inhibition and chloride regulation in the healthy brain, as well as in neurological disorders and diseases.

Edited by: Ian Forsythe & David MacLean
References
- Acton BA, Mahadevan V, Mercado A, Uvarov P, Ding Y, Pressey J, Airaksinen MS, Mount DB & Woodin MA (2012). Hyperpolarizing GABAergic transmission requires the KCC2C‐terminal ISO domain. J Neurosci 32, 8746–8751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke TG & Gegelashvili G (2008). Tonic activation of group I mGluRs modulates inhibitory synaptic strength by regulating KCC2 activity. J Physiol (Lond) 586, 4925–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C & Kaila K (2009). Cation‐chloride cotransporters and neuronal function. Neuron 61, 820–838. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC & Nicoll RA (1997). Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature 388, 182–186. [DOI] [PubMed] [Google Scholar]
- Chorin E, Vinograd O, Fleidervish I, Gilad D, Herrmann S, Sekler I, Aizenman E & Hershfinkel M (2011). Upregulation of KCC2 activity by zinc‐mediated neurotransmission via the mZnR/GPR39 receptor. J Neurosci 31, 12916–12926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Swanson GT, Sailer A, O'Gorman S & Heinemann SF (2000). Identification of the kainate receptor subunits underlying modulation of excitatory synaptic transmission in the CA3 region of the hippocampus. J Neurosci 20, 8269–8278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway LC, Cardarelli RA, Moore YE, Jones K, McWilliams LJ, Baker DJ, Burnham MP, Bürli RW, Wang Q, Brandon NJ, Moss SJ & Deeb TZ (2017). N‐Ethylmaleimide increases KCC2 cotransporter activity by modulating transporter phosphorylation. J Biol Chem 292, 21253–21263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JAM, Boudreau D, Bachand K, Prescott SA, Nault F, Sík A, De Koninck P & De Koninck Y (2003). Trans‐synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424, 938–942. [DOI] [PubMed] [Google Scholar]
- D'amour JA & Froemke RC (2015). Inhibitory and excitatory spike‐timing‐dependent plasticity in the auditory cortex. Neuron 86, 514–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon N, Vinay L, Prescott SA & De Koninck Y (2016). Chloride regulation: a dynamic equilibrium crucial for synaptic inhibition. Neuron 89, 1157–1172. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Kuchibhotla KV, Glykys JC, Kahle KT, Swiercz WB, Feng G, Kuner T, Augustine GJ, Bacskai BJ & Staley KJ (2010). Progressive NKCC1‐dependent neuronal chloride accumulation during neonatal seizures. J Neurosci 30, 11745–11761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE & Staley KJ (2005). NKCC1 transporter facilitates seizures in the developing brain. Nat Med 11, 1205–1213. [DOI] [PubMed] [Google Scholar]
- Fernandes HB, Catches JS, Petralia RS, Copits BA, Xu J, Russell TA, Swanson GT & Contractor A (2009). High‐affinity kainate receptor subunits are necessary for ionotropic but not metabotropic signaling. Neuron 63, 818–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisahn A, Heinemann SF & McBain CJ (2005). The kainate receptor subunit GluR6 mediates metabotropic regulation of the slow and medium AHP currents in mouse hippocampal neurones. J Physiol (Lond) 562, 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M, Schmitz D, Zhou Q, Johansen J & Nicoll RA (2001). Kainate receptors depress excitatory synaptic transmission at CA3→CA1 synapses in the hippocampus via a direct presynaptic action. J Neurosci 21, 2958–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M & Ohliger‐Frerking P (2002). AMPA receptors and kainate receptors encode different features of afferent activity. J Neurosci 22, 7434–7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauvain G, Chamma I, Chevy Q, Cabezas C, Irinopoulou T, Bodrug N, Carnaud M, Lévi S & Poncer JC (2011). The neuronal K‐Cl cotransporter KCC2 influences postsynaptic AMPA receptor content and lateral diffusion in dendritic spines. Proc Natl Acad Sci U S A 108, 15474–15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad D, Shorer S, Ketzef M, Friedman A, Sekler I, Aizenman E & Hershfinkel M (2015). Homeostatic regulation of KCC2 activity by the zinc receptor mZnR/GPR39 during seizures. Neurobiol Dis 81, 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyás AI, Sík A, Payne JA, Kaila K & Freund TF (2001). The KCl cotransporter, KCC2, is highly expressed in the vicinity of excitatory synapses in the rat hippocampus. Eur J Neurosci 13, 2205–2217. [DOI] [PubMed] [Google Scholar]
- Higley MJ (2014). Localized GABAergic inhibition of dendritic Ca2+ signalling. Nat Rev Neurosci 15, 567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivakine EA, Acton BA, Mahadevan V, Ormond J, Tang M, Pressey JC, Huang MY, Ng D, Delpire E, Salter MW, Woodin MA & McInnes RR (2013). Neto2 is a KCC2 interacting protein required for neuronal Cl− regulation in hippocampal neurons. Proc Natl Acad Sci U S A 110, 3561–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Merner ND, Friedel P, Silayeva L, Liang B, Khanna A, Shang Y, Lachance‐Touchette P, Bourassa C, Levert A, Dion PA, Walcott B, Spiegelman D, Dionne‐Laporte A, Hodgkinson A, Awadalla P, Nikbakht H, Majewski J, Cossette P, Deeb TZ, Moss SJ, Medina I & Rouleau GA (2014). Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep 15, 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K (1994). Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol 42, 489–537. [DOI] [PubMed] [Google Scholar]
- Kaila K, Price TJ, Payne JA, Puskarjov M & Voipio J (2014). Cation‐chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci 15, 637–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirug S, Ahmad F, Puskarjov M, Afzalov R, Kaila K & Blaesse P (2010). A single seizure episode leads to rapid functional activation of KCC2 in the neonatal rat hippocampus. J Neurosci 30, 12028–12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp‐Scheinpflug C, Tozer AJB, Robinson SW, Tempel BL, Hennig MH & Forsythe ID (2011). The sound of silence: ionic mechanisms encoding sound termination. Neuron 71, 911–925. [DOI] [PubMed] [Google Scholar]
- Lee HHC, Deeb TZ, Walker JA, Davies PA & Moss SJ (2011). NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor‐mediated currents. Nat Neurosci 14, 736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HHC, Walker JA, Williams JR, Goodier RJ, Payne JA & Moss SJ (2007). Direct protein kinase C‐dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J Biol Chem 282, 29777–29784. [DOI] [PubMed] [Google Scholar]
- Lerma J & Marques JM (2013). Kainate receptors in health and disease. Neuron 80, 292–311. [DOI] [PubMed] [Google Scholar]
- Li H, Khirug S, Cai C, Ludwig A, Blaesse P, Kolikova J, Afzalov R, Coleman SK, Lauri S, Airaksinen MS, Keinänen K, Khiroug L, Saarma M, Kaila K & Rivera C (2007). KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron 56, 1019–1033. [DOI] [PubMed] [Google Scholar]
- Llano O, Smirnov S, Soni S, Golubtsov A, Guillemin I, Hotulainen P, Medina I, Nothwang HG, Rivera C & Ludwig A (2015). KCC2 regulates actin dynamics in dendritic spines via interaction with β‐PIX. J Cell Biol 209, 671–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan V, Dargaei Z, Ivakine EA, Hartmann A, Ng D, Chevrier J, Ormond J, Nothwang HG, McInnes RR & Woodin MA (2015). Neto2‐null mice have impaired GABAergic inhibition and are susceptible to seizures. Front Cell Neurosci 9, 368 10.3389/fncel.2015.00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan V, Khademullah CS, Dargaei Z, Chevrier J, Uvarov P, Kwan J, Bagshaw RD, Pawson T, Emili A, De Koninck Y, Anggono V, Airaksinen M & Woodin MA (2017). Native KCC2 interactome reveals PACSIN1 as a critical regulator of synaptic inhibition. Elife 6 pii: e28270 10.7554/eLife.28270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan V, Pressey J, Acton B, Uvarov P, Huang M, Chevrier J, Puchalski A, Li C, Ivakine E, Airaksinen M, Delpire E, McInnes R & Woodin M (2014). Kainate receptors coexist in a functional complex with KCC2 and regulate chloride homeostasis in hippocampal neurons. Cell Reports 7, 1762–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan V & Woodin MA (2016). Regulation of neuronal chloride homeostasis by neuromodulators. J Physiol 594, 2593–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melyan Z, Lancaster B & Wheal HV (2004). Metabotropic regulation of intrinsic excitability by synaptic activation of kainate receptors. J Neurosci 24, 4530–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melyan Z, Wheal HV & Lancaster B (2002). Metabotropic‐mediated kainate receptor regulation of IsAHP and excitability in pyramidal cells. Neuron 34, 107–114. [DOI] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Pérez‐Otaño I, Dickinson‐Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B & Heinemann SF (1998). Altered synaptic physiology and reduced susceptibility to kainate‐induced seizures in GluR6‐deficient mice. Nature 392, 601–605. [DOI] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Swanson GT, Brana C, O'Gorman S, Bettler B & Heinemann SF (2000). Subunit composition of kainate receptors in hippocampal interneurons. Neuron 28, 475–484. [DOI] [PubMed] [Google Scholar]
- Nogueira GS, Santos LE, Rodrigues AM, Scorza CA, Scorza FA, Cavalheiro EA & de Almeida AC (2015). Enhanced nonsynaptic epileptiform activity in the dentate gyrus after kainate‐induced status epilepticus. Neuroscience 303, 59–72. [DOI] [PubMed] [Google Scholar]
- Ormond J & Woodin MA (2011). Disinhibition‐mediated LTP in the hippocampus is synapse specific. Front Cell Neurosci 5, 17 10.3389/fncel.2011.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormond J & Woodin MA (2009). Disinhibition mediates a form of hippocampal long‐term potentiation in area CA1. PLoS ONE 4(9), e7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan E, Zhang X, Huang Z, Krezel A, Zhao M, Tinberg C, Lippard S & McNamara J (2011). Vesicular zinc promotes presynaptic and inhibits postsynaptic long‐term potentiation of mossy fiber‐CA3 synapse. Neuron 71, 1116–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro PS, Lanore F, Veran J, Artinian J, Blanchet C, Crépel V, Perrais D & Mulle C (2013). Selective block of postsynaptic kainate receptors reveals their function at hippocampal mossy fiber synapses. Cereb Cortex 23, 323–331. [DOI] [PubMed] [Google Scholar]
- Pouille F & Scanziani M (2001). Enforcement of temporal fidelity in pyramidal cells by somatic feed‐forward inhibition. Science 293, 1159–1163. [DOI] [PubMed] [Google Scholar]
- Pressey JC, Mahadevan V, Khademullah CS, Dargaei Z, Chevrier J, Ye W, Huang M, Chauhan AK, Meas SJ, Uvarov P, Airaksinen MS & Woodin MA (2017). A kainate receptor subunit promotes the recycling of the neuron‐specific K+‐Cl– co‐transporter KCC2 in hippocampal neurons. J Biol Chem 292, 6190‐6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M & Kaila K (1999). The K+/Cl– co‐transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. [DOI] [PubMed] [Google Scholar]
- Rodrigues RJ & Lerma J (2012). Metabotropic signaling by kainate receptors. Wiley Interdiscip Rev Membr Transp Signal 1, 10.1002/wmts.35. [DOI] [Google Scholar]
- Rodríguez‐Moreno A, Herreras O & Lerma J (1997). Kainate receptors presynaptically downregulate GABAergic inhibition in the rat hippocampus. Neuron 19, 893–901. [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Moreno A & Lerma J (1998). Kainate receptor modulation of GABA release involves a metabotropic function. Neuron 20, 1211–1218. [DOI] [PubMed] [Google Scholar]
- Rozas JL, Paternain AV & Lerma J (2003). Noncanonical signaling by ionotropic kainate receptors. Neuron 39, 543–553. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Sachidhanandam S, Utvik JK, Coussen F & Mulle C (2005). Distinct subunits in heteromeric kainate receptors mediate ionotropic and metabotropic function at hippocampal mossy fiber synapses. J Neurosci 25, 11710–11718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachidhanandam S, Blanchet C, Jeantet Y, Cho YH & Mulle C (2009). Kainate receptors act as conditional amplifiers of spike transmission at hippocampal mossy fiber synapses. J Neurosci 29, 5000–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segerstråle M, Juuri J, Lanore F, Piepponen P, Lauri SE, Mulle C & Taira T (2010). High firing rate of neonatal hippocampal interneurons is caused by attenuation of afterhyperpolarizing potassium currents by tonically active kainate receptors. J Neurosci 30, 6507–6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sihra TS & Rodríguez‐Moreno A (2011). Metabotropic actions of kainate receptors in the control of GABA release. Adv Exp Med Biol 717, 1–10. [DOI] [PubMed] [Google Scholar]
- Straub C & Tomita S (2012). The regulation of glutamate receptor trafficking and function by TARPs and other transmembrane auxiliary subunits. Curr Opin Neurobiol 22, 488–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takkala P & Woodin MA (2013). Muscarinic acetylcholine receptor activation prevents disinhibition‐mediated LTP in the hippocampus. Front Cell Neurosci 7, 10.3389/fncel.2013.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignes M, Clarke VR, Parry MJ, Bleakman D, Lodge D, Ornstein PL & Collingridge GL (1998). The GluR5 subtype of kainate receptor regulates excitatory synaptic transmission in areas CA1 and CA3 of the rat hippocampus. Neuropharmacology 37, 1269–1277. [DOI] [PubMed] [Google Scholar]
- Vignes M & Collingridge GL (1997). The synaptic activation of kainate receptors. Nature 388, 179–182. [DOI] [PubMed] [Google Scholar]
- Woodin MA (2013). Electrophysiological methods for investigating inhibitory synaptic plasticity In Multidisciplinary Tools for Investigating Synaptic Plasticity, ed. Nguyen Peter V., pp. 209–221. Humana Press, Totowa, NJ. [Google Scholar]