

Abstract
Autologous induced pluripotent stem cells (iPSCs) constitute an unlimited cell source for patient-specific cell-based organ repair strategies. However, their generation and subsequent differentiation into specific cells or tissues entail cell line-specific manufacturing challenges and form a lengthy process that precludes acute treatment modalities. These shortcomings could be overcome by using prefabricated allogeneic cell or tissue products, but the vigorous immune response against histo-incompatible cells has prevented the successful implementation of this approach. Here we show that both mouse and human iPSCs lose their immunogenicity when major histocompatibility complex (MHC) class I and II genes are inactivated and CD47 is over-expressed. These hypoimmunogenic iPSCs retain their pluripotent stem cell potential and differentiation capacity. Endothelial cells, smooth muscle cells, and cardiomyocytes derived from hypoimmunogenic mouse or human iPSCs reliably evade immune rejection in fully MHC-mismatched allogeneic recipients and survive long-term without the use of immunosuppression. These findings suggest that hypoimmunogenic cell grafts can be engineered for universal transplantation.
Treatment of heart disease with adult multipotent, bone marrow-derived stem cells has shown marginal efficacy in patients with acute myocardial infarction1 or chronic ischemic cardiomyopathy2,3. This has been attributed to the limited plasticity of adult hematopoietic stem cells, which do not differentiate into cardiomyocytes and thus cannot replace contractile elements4. Pluripotent stem cells are more promising cell sources for regenerative strategies as they can produce an unlimited amount of progeny cells that can be differentiated into functional tissue cells. Although reprogramming technology allows the generation of autologous iPSCs for patient-specific treatments, this is laborious, costly, associated with uncertain quality and efficacy of individual cell products and is only practical for chronic diseases5–7. Thus, most regenerative approaches relying on autologous iPSC generation have been abandoned. Allogeneic cell therapies targeting large patient populations could be more economically feasible8,9, but are subject to forceful immune rejection10.The use of allogeneic iPSC-or embryonic stem cell (ESC)-based products would require strong immunosuppression.
We envisioned engineering hypoimmunogenic pluripotent stem cells as a source for universally compatible cell or tissue grafts not requiring any immunosuppression. During pregnancy, the maternal immune system is tolerant of allogeneic paternal antigens although it would reject cells from the baby later in life11. We examined syncytiotrophoblast cells, which form the interface between maternal blood and fetal tissue, and found low MHC class I and II expression (Supplementary Fig. 1) as well as strong expression of CD47, a ubiquitous membrane protein that can interact with several cell surface receptors to inhibit phagocytosis12. We used this knowledge to design hypoimmunogenic mouse iPSCs (miPSCs).
C57BL/6 wild type (WT) miPSCs13 give rise to classical teratomas with ectodermal, mesodermal and endodermal features in SCID-beige mice (Supplementary Fig. 2). To achieve hypoimmunogenicity, these miPSCs underwent a three-step gene-editing process (Supplementary Fig. 3a). First, CRISPR guide RNAs targeting the coding sequence of the mouse β2-microglobulin (B2m) gene were ligated into vectors containing the Cas9 expression cassette and subsequently transfected into miPSCs. B2m is a structural component of MHC class I. Second, B2m−/− miPSCs were transfected with a CRISPR-Cas9 vector targeting Ciita, the master regulator of MHC class II molecules14. Third, the Cd47 gene sequence was synthesized and cloned into a lentivirus with blasticidin resistance, which was used to transduce B2m−/−Ciita−/− miPSC clones followed by antibiotic selection and expansion of B2m−/−Ciita−/− Cd47 transgene (tg)-expressing miPSCs. WT miPSCs had interferon-γ (IFN-γ)-inducible MHC class I surface expression, low but detectable MHC class II expression and negligible Cd47 expression (Supplementary Fig. 3b–d). We confirmed that the miPSC lines we generated lacked MHC class I and II expression, and over-expressed Cd47 roughly 4.5-fold in flow cytometry. All three lines maintained their expression of pluripotency genes (Supplementary Fig. 3e–h).
Next, we transplanted WT miPSCs or engineered miPSCs into syngeneic C57BL/6 (H2b) and allogeneic (H2d) BALB/c recipients without immunosuppression. As expected, WT miPSCs showed 100% teratoma growth in syngeneic recipients, but all cell grafts were rejected in allogeneic BALB/c mice (Fig. 1a,b). After 5 days, splenocytes from allogeneic BALB/c recipients showed a strong IFN-γ and a moderate IL-4 response relative to baseline responder cell activity (not shown); syngeneic mice showed no responsiveness (Fig. 1c). Only allogeneic BALB/c recipients mounted a strong IgM antibody response against the WT miPSCs relative to baseline MFI (not shown) (Fig. 1d). Engineered miPSCs developed comparable teratomas to WT miPSCs in syngeneic recipients, with enhanced survival in allogeneic recipients that depended on their level of hypoimmunogenicity and increased with every engineering step (Supplementary Fig. 4a–h). Our final B2m−/−Ciita−/− Cd47 tg miPSC line showed 100% teratoma formation and induced no IFN-γ or antibody responses (Fig. 1e–h).
Fig. 1 |. Survival of miPSCs and miPSC derivatives.
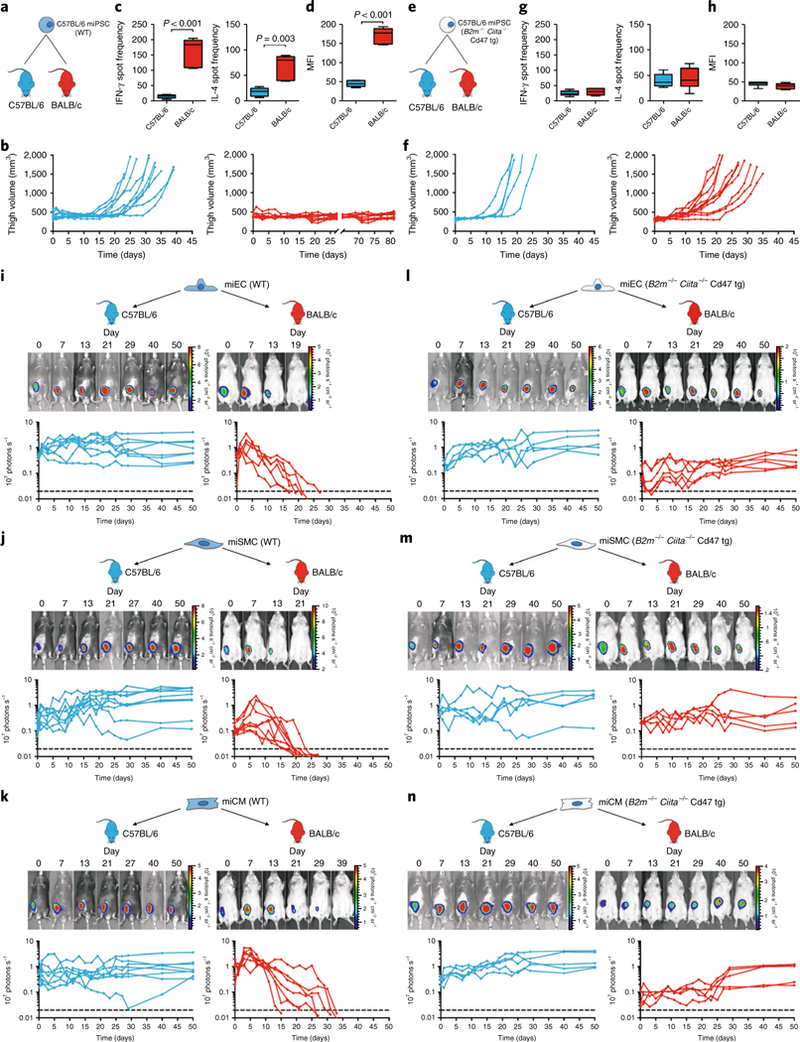
a, WT C57BL/6 miPSCs were injected into the thigh muscle of syngeneic C57BL/6 or allogeneic BALB/c mice. b, Teratoma formation was observed by measuring the thigh muscle (n = 10 per group). c, IFN-γ and IL-4 enzyme-linked immunospots (Elispots) with splenocytes recovered 5 days after the transplantation (box 25th to 75th percentile with median, whiskers min–max, five animals per group, two-tailed Student’s t-test). d, Mean fluorescence imaging (MFI) of IgM binding to WT miPSCs incubated with recipient serum after 5 days (box 25th to 75th percentile with median, whiskers min–max, six animals per group, two-tailed Student’s t-test). e, B2m−/−Ciita−/− Cd47 tg C57BL/6 miPSCs were transplanted into syngeneic C57BL/6 or allogeneic BALB/c recipients. f, Thigh volume C57BL/6 (n = 5) and BALB/c (n = 11) animals. The overall percentage of cell grafts that survived and formed teratomas in BALB/c was 100%. g, IFN-γ and IL-4 Elispots with splenocytes recovered 5 days after the transplantation and B2m−/−Ciita−/− Cd47 tg miPSCs stimulator cells (box 25th to 75th percentile with median, whiskers min–max, n = 6 per group, two-tailed Student’s t-test). h, MFI of IgM binding to B2m−/−Ciita−/− Cd47 tg miPSCs incubated with recipient serum after 5 days (box 25th to 75th percentile with median, whiskers min–max, six animals per group, two-tailed Student’s t-test). i–n, Grafts of Fluc+ C57BL/6 miPSC derivatives in C57BL/6 or BALB/c recipients were longitudinally followed by bioluminescent imaging (BLI). One representative animal is depicted per group and the BLI values of all animals are plotted. All WT miPSC-derived miECs (i, eight animals in C57BL/6 and six animals in BALB/c), miSMCs (j, nine animals in C57BL/6 and eight animals in BALB/c) and miCMs (k, eight animals in C57BL/6 and seven animals in BALB/c) showed long-term survival in syngeneic C57BL/6 recipients but were rejected in allogeneic BALB/c animals. In contrast, all B2m−/−Ciita−/− Cd47 tg miPSC-derived miECs (l, five animals in C57BL/6 and six animals in BALB/c), miSMCs (m, five animals in C57BL/6 and five animals in BALB/c) and miCMs (n, five animals in C57BL/6 and five animals in BALB/c) showed long-term survival in both syngeneic C57BL/6 and allogeneic BALB/c recipients.
We further evaluated the contribution of Cd47 overexpression by comparing B2m−/−Ciita−/− miPSCs to B2m−/−Ciita−/− Cd47 tg miPSCs in natural killer (NK) cell toxicity assays. Gene editing did not enhance the expression of stimulatory ligands for the mouse NK cell NKG2D or NKp46 receptors (Supplementary Fig. 4i), which are constitutively expressed in the NK cell-sensitive target cell line YAC-115. B2m−/−Ciita−/− miPSCs induced IFN-γ release that was significantly elevated when compared to unchallenged NK cells, but lower than IFN-γ release triggered by YAC-1 (Supplementary Fig. 4j). This suggested that Cd47 overexpression completely prevented any miPSC-induced NK cell IFN-γ release in vitro. To further assess innate miPSC clearance in vivo, a 1:1 mixture of CFSE-labeled WT miPSCs and either B2m−/−Ciita−/− miPSCs or B2m−/−Ciita−/− Cd47 tg miPSCs were injected into the innate immune cell-rich peritoneum of syngeneic C57BL/6 mice (Supplementary Fig. 5a). Using a syngeneic host for this assay precluded relevant T cell-mediated cytotoxicity. After 48 h, the peritoneal fluid was aspirated and CFSE-labeled cells were analyzed by flow cytometry. B2m−/−Ciita−/− Cd47 tg miPSCs, but not B2m−/−Ciita−/− miPSCs, were resistant to innate immune clearance and the 1:1 ratio with WT miPSCs could be maintained. We observed the same pattern of cell clearance when mice were pretreated with clodronate to eliminate macrophages (Supplementary Fig. 5b). Notably, a blocking antibody against mouse Cd47 completely abolished the protective effect of Cd47 in macrophage-depleted mice and B2m−/−Ciita−/− Cd47 tg miPSCs were rapidly eliminated (Supplementary Fig. 5c). Collectively, these data suggest an inhibitory effect of Cd47 on NK cells in vivo.
To test whether hypoimmunogenic B2m−/−Ciita−/− Cd47 tg miPSCs could give rise to hypoimmunogenic cardiac tissue, they were differentiated into endothelial cells (miECs), smooth muscle cells (miSMCs) and cardiomyocytes (miCMs) with WT miPSC derivatives serving as controls (Supplementary Fig. 6). All derivatives showed the morphologic appearance, cell marker immunofluorescence and gene expression characteristic of their mature target tissue cell lines, and cultures achieved >90% purity of VE-Cadherin+ miECs, Sma+ miSMCs and troponin I+ miCMs. The expression of MHC class I and II molecules in WT derivatives markedly varied by cell type (Supplementary Fig. 7a–c) but, as expected, miECs had by far the highest expression of IFN-γ induced MHC class I and II, miSMCs had moderate MHC class I and II expression16 and miCMs had moderate MHC class I but very low MHC class II expression17. All B2m−/−Ciita−/− Cd47 tg derivatives appropriately showed a complete lack of MHC class I and II and significantly elevated Cd47 compared to their WT counterparts. None of the B2m−/−Ciita−/− Cd47 tg derivatives showed upregulation of NKG2D or NKp46 ligands (Supplementary Fig. 7d,e).
We next assessed the in vivo immunogenicity of WT and hypoimmunogenic miPSCderivatives. miECs, miSMCs or miCMs derived from WT or B2m−/−Ciita−/− Cd47 tg were transplanted intramuscularly into syngeneic C57BL/6 or allogeneic BALB/c mice and adaptive immune responses were assessed after 5 days. All allogeneic recipients mounted a strong cellular IFN-γ response, as well as a strong IgM antibody response against all differentiated WT cell grafts (Supplementary Fig. 8a–c). In contrast, neither of the corresponding B2m−/−Ciita−/− Cd47 tg derivatives showed detectable increases in IFN-γ Elispot frequencies or IgM antibody production (Supplementary Fig. 8d–f). To assess the efficacy of Cd47 to mitigate the susceptibility to innate immune killing, we performed NK cell Elispot assays with antibody-coated magnetic bead-enriched BALB/c NK cells and B2m−/−Ciita−/− or B2m−/−Ciita−/− Cd47 tg derivatives (Supplementary Fig. 8g–i). While B2m−/−Ciita−/− derivatives triggered NK cell IFN-γ release, none of the B2m−/−Ciita−/− Cd47 tg derivatives produced IFN-γ spot frequencies significantly exceeding those of unchallenged NK cells. Accordingly, in vivo innate immune assays showed rapid clearance of all B2m−/−Ciita−/− derivatives, but confirmed that none of the B2m−/−Ciita−/− Cd47 tg derivatives showed susceptibility to innate elimination (Supplementary Fig. 8j–l). To confirm an inhibitory effect of Cd47 on NK cells, we next performed real-time in vitro killing assays with confluent miECs and highly purified NK cells. Both allogeneic (BALB/c) and syngeneic (C57BL/6) NK cells rapidly killed B2m−/−Ciita−/− miECs, but not WT and B2m−/−Ciita−/− Cd47 tg miECs (Supplementary Fig. 5d,e). However, antibody blocking of mouse Cd47 resulted in the rapid killing of B2m−/−Ciita−/− Cd47 tg miECs (Supplementary Fig. 5f). The effect of CD47 is species-specific as human NK cells rapidly killed both B2m−/−Ciita−/− and B2m−/−Ciita−/− Cd47 tg miECs (Supplementary Fig. 5g).
We next examined survival of WT and hypoimmunogenic miPSC derivates in vivo. WT and B2m−/−Ciita−/− Cd47 tg derivatives were transduced to express firefly luciferase, and Matrigel plugs containing differentiated cells were transplanted into syngeneic C57BL/6 or allogeneic BALB/c mice. All three WT derivatives showed long-term (50 days) survival in syngeneic C57BL/6 recipients, but were rejected in allogeneic mice (Fig. 1i–k). In contrast, all three B2m−/−Ciita−/− Cd47 tg derivatives showed 100% long-term survival in both syngeneic and allogeneic recipients (Fig. 1l–n).
Matrigel plugs containing WT or B2m−/−Ciita−/− Cd47 tg miECs were transplanted into allogeneic BALB/c recipients (Supplementary Fig. 9a). ECs are the most immunogenic cardiac cell type due to their high expression of MHC class I and II, which allows them to function as antigen-presenting cells. We observed infiltrating immune cells containing mostly T and B lymphocytes, but also some NK cells and macrophages in WT miEC plugs. B2m−/−Ciita−/− Cd47 tg miEC-containing plugs had almost no immune cell infiltration (Supplementary Fig. 9b). In the WT plugs, cytokine arrays on day 10 revealed an inflammatory milieu that included upregulated T helper cell (Th)-1 cytokines (IFN-γ and IL-2) and Th-2 cytokines (IL-4, IL-5, IL-10 and IL-13). In contrast, in plugs containing B2m−/−Ciita−/− Cd47 tg miECs, the cytokine milieu was very similar to that of cell-free plugs containing only Matrigel, with no indication of immune activation (Supplementary Fig. 9c). Over time, transplanted B2m−/−Ciita−/− Cd47 tg miECs organized in circular structures and formed primitive vessels that contained erythrocytes (Supplementary Fig. 10a). Similarly transplanted B2m−/−Ciita−/− Cd47 tg miSMCs (Supplementary Fig. 10b) or miCMs (Supplementary Fig. 10c) did not form three-dimensional structures, and their in vivo maturation and integration potential in cardiac tissue remains to be studied.
We next applied our engineering strategy to human iPSCs (hiP-SCs) using a human episomal iPSC line derived from CD34+ cord blood that showed a normal human XX karyotype and features of pluripotency (Supplementary Fig. 11a–c,g–h). The gene-editing process included two steps (Fig. 2a). First, both the human B2M and human CIITA genes were simultaneously targeted for CRISPR/Cas9-mediated disruption. Second, these edited hiPSCs were transduced with a lentivirus carrying human CD47 complementary DNA with an EFS promotor and puromycin resistance. Antibiotic-resistant B2m−/−Ciita−/− CD47 tg hiPSC colonies maintained their normal human karyotype and pluripotency (Supplementary Fig. 11d–f,i–j) and successful depletion of HLA I and II surface expression, along with overexpression of CD47, was confirmed by flow cytometry (Fig. 2b). Both WT hiPSCs and B2M−/−CIITA−/− CD47 tg hiPSCs were differentiated into endothelial-like cells (hiECs) and cardiomyocyte-like cells (hiCMs) (Fig. 2c). All derivatives showed the morphologic features and protein expression of the differentiated target cells and lost their pluripotency genes (Supplementary Fig. 12a,b). Cultures showed >95% purity for VE-Cadherin+ hiECs and troponin I+ hiCMs. There were no alterations in the expression of stimulatory NK cell ligands with gene engineering (Supplementary Fig. 12c–i). WT hiECs and hiCMs upregulated IFN-γ induced HLA I expression roughly three-and two-fold, respectively, compared to WT hiPSCs and hiECs also showed roughly 11-fold elevated HLA II (Fig. 2d–e). B2M−/−CIITA−/− CD47 tg hiECs and hiCMs exhibited HLA I and II depletion and significant CD47 upregulation compared to their WT counterparts.
Fig. 2 |. Immune response against hiPSC derivatives.

a, WT hiPSCs first underwent B2M and CIITA gene disruption and then CD47 transgene overexpression b, Gene editing of hiPSCs was confirmed by flow cytometry (box 25th to 75th percentile with median, whiskers min–max, four independent experiments per graph, analysis of variance (ANOVA) with Bonferroni’s post-hoc test). c, WT and B2M−/−CIITA−/− CD47 tg hiPSCs were differentiated into both hiECs and hiCMs. d–e, The immune phenotype of WT and B2M−/−CIITA−/− CD47 tg hiECs (d) and hiCMs (e) is shown (box 25th to 75th percentile with median, whiskers min–max, four independent experiments per graph, two-tailed Student’s t-test). f, WT or B2M−/−CIITA−/− CD47 tg hiPSC grafts were injected into allogeneic humanized NSG-SGM3 mice. g, IFN-γ Elispots were performed after 5 days (mean ± s.d., n = 7 per group, two-tailed Student’s t-test), the background spot frequency in naïve mice is shown (mean ± s.d., four animals per group, two-tailed Student’s t-test). h, MFI of IgM binding to either hiPSC incubated with recipient serum after 5 days (mean ± s.d., five animals per group, two-tailed Student’s t-test), the background fluorescence in naïve mice is shown (mean ± s.d., three animals per group, Student’s t-test). i, IFN-γ Elispots with human NK cells were performed with B2M−/−CIITA−/− hiPSC or B2M−/−CIITA−/− CD47 tg hiPSC (box 25th to 75th percentile with median, whiskers min–max, six independent experiments, ANOVA with Bonferroni’s post-hoc test). j, WT or B2M−/−CIITA−/− CD47 tg hiEC grafts were injected into allogeneic humanized NSG-SGM3 mice. k, IFN-γ Elispots were performed after 5 days (mean ± s.d., three animals per group, two-tailed Student’s t-test), the background spot frequency in naïve mice is shown (mean ± s.d., four animals per group, two-tailed Student’s t-test). l, MFI of IgM binding to either hiEC incubated with recipient serum after 5 days (mean ± s.d., three animals per group, two-tailed Student’s t-test), the background fluorescence in naïve mice is shown (mean ± s.d., three animals per group, Student’s t-test). m, IFN-γ Elispots with human NK cells were performed with B2M−/−CIITA−/− hiECs or B2M−/−CIITA−/− CD47 tg hiECs (box 25th to 75th percentile with median, whiskers min–max, six independent experiments, ANOVA with Bonferroni’s post-hoc test). n, WT or B2M−/−CIITA−/− CD47 tg hiCM grafts were injected into allogeneic humanized NSG-SGM3 mice. o, IFN-γ Elispots were performed after 5 days (mean ± s.d., three animals per group, two-tailed Student’s t-test), the background spot frequency in naïve mice is shown (mean ± s.d., four animals per group, two-tailed Student’s t-test). p, MFI of IgM binding to either hiCM incubated with recipient serum after 5 days (mean ± s.d., three animals per group, two-tailed Student’s t-test), the background fluorescence in naïve mice is shown (mean ± s.d., three animals per group, Student’s t-test). q, IFN-γ Elispots with human NK cells were performed with B2M−/−CIITA−/− hiCMs or B2M−/−CIITA−/− CD47 tg hiCMs (box 25th to 75th percentile with median, whiskers min–max, six independent experiments, ANOVA with Bonferroni’s post-hoc test).
We next performed transplant studies in humanized CD34+ hematopoietic stem cell-engrafted NSG-SGM3 mice18, which were allogeneic to the hiPSC, hiEC and hiCM grafts. Since no syngeneic controls are available in this humanized mouse model, background measurements were collected in naïve mice. After 5 days, recipients of WT hiPSCs (Fig. 2f) showed a high splenocyte IFN-γ spot frequency (Fig. 2g) and elevated IgM levels (Fig. 2h). Recipients of B2M−/−CIITA−/− CD47 tg hiPSCs did not mount any detectable cellular IFN-γ response or antibody response. NK cell activation was assessed using in vitro incubation with human enriched CD56+ NK cells. B2M−/−CIITA−/− hiPSCs resulted in an IFN-γ release reaching roughly one-third of the spot frequency of the highly NK cell susceptible K562 line, whereas B2M−/−CIITA−/− CD47 tg hiPSCs did not provoke any measurable response (Fig. 2i). The allogeneic transplantation of WT hiECs (Fig. 2j) and WT hiCMs (Fig. 2n) resulted in strong systemic IFN-γ reactions (Fig. 2k,o) and IgM antibody increases (Fig. 2l,p) of similar intensity as WT hiPSCs, whereas hypoimmunogenic hiECs and hiCMs did not induce any cellular or humoral immune response. Moreover, in vitro, hypoimmunogenic derivatives did not trigger NK cell activation (Fig. 2m, q) or NK cell killing (Supplementary Fig. 5h). As with the miPSCs, a blocking antibody specific for human CD47 completely abolished NK cell protection of B2M−/−CIITA−/− CD47 tg hiPSCs (Supplementary Fig. 5i).
We also assessed the survival of hiPSCs, as well as their derivatives in allogeneic humanized NSG-SGM3 mice. All cell lines were transduced to express Fluc to enable tracking by BLI. There was no significant difference in HLA-A mismatch between groups (Supplementary Fig. 12j). As expected, all WT hiPSC grafts in Matrigel plugs underwent rejection (Fig. 3a) and all B2M−/−CIITA−/− CD47 tg hiPSCs formed teratomas. Similarly, WT hiECs (Fig. 3b) and WT hiCMs (Fig. 3c) were rejected, although at slightly slower rate than in the corresponding miPSC derivative experiments. This difference may be based on the reduced number, diversity and function of human immune cells in mouse recipients19, although the triple transgenic NSG-SGM3 mice specifically express human cytokines20 to minimize these limitations. All B2M−/−CIITA−/− CD47 tg hiEC and hiCM grafts showed long-term survival (50 days) and stable BLI signal levels over time. The hiECs gradually organized into structures resembling primitive vascular structures, which occasionally contained erythrocytes, and the hiCMs acquired a limited polarized architecture (Fig. 3d,e).
Fig. 3 |. Survival of hiPSCs and hiPSC derivatives in allogeneic hosts.
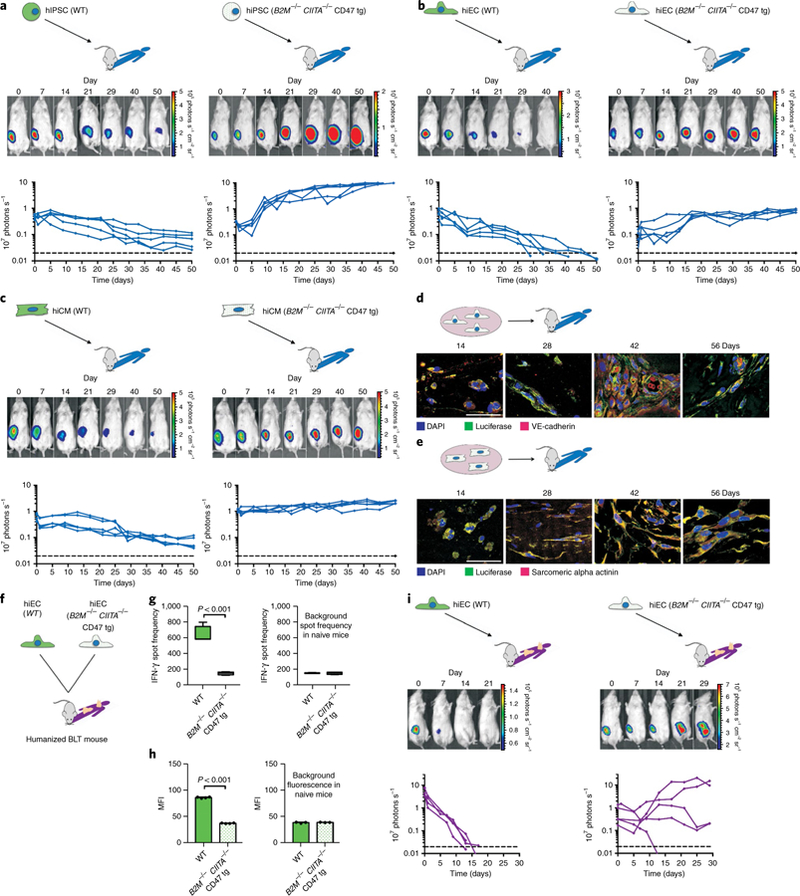
Grafts of Fluc+ WT or B2M−/−CIITA−/− CD47 tg hiPSCs, hiECs and hiCMs were transplanted into allogeneic humanized mice (NSG-SGM3 mice in a–e, BLT mice in f–i) and were longitudinally followed by BLI. One representative animal is depicted per group and the BLI values of all animals are plotted. a, BLI signals over time of WT and B2M−/−CIITA−/− CD47 tg hiPSC grafts (n = 5 per group). b, WT and B2M−/−CIITA−/− CD47 tg hiECs were transplanted as in a (n = 5 per group). c, WT and B2M−/−CIITA−/− CD47 tg hiCMs (n = 5). d, B2M−/−CIITA−/− CD47 tg hiECs started to organize into a more complex structure, which included primitive vascular structures (representative pictures of three independent experiments). Scale bar, 50 μm. e, The B2M−/−CIITA−/− CD47 tg hiCMs began to organize into a more polarized framework and maintained their sarcomeric alpha-actinin cytoskeletal structure typical of cardiomyocytes (representative pictures of three independent experiments). Scale bar, 50 μm. f, WT or B2M−/−CIITA−/− CD47 tg hiECs were transplanted into allogeneic humanized BLT mice. g, IFN-γ Elispots were performed after 5 days (box 25th to 75th percentile with median, whiskers min–max, four animals per group, two-tailed Student’s t-test), the background spot frequency in naïve mice is shown. h, MFI of IgM binding to either hiEC incubated with recipient serum after 5 days (mean ± s.d., four animals per group, two-tailed Student’s t-test), the background fluorescence in naïve mice is shown (mean ± s.d., three animals per group, two-tailed Student’s t-test). i, Grafts of Fluc+ WT or B2M−/−CIITA−/− CD47 tg hiECs were transplanted into allogeneic humanized BLT mice and were longitudinally followed by BLI. All WT hiEC grafts were rejected within roughly 14 days (four animals). Four of the five B2M−/−CIITA−/− CD47 tg hiEC grafts permanently survived, the one failure is believed not to be immune-mediated (five animals).
hiECs, the most immunogenic derivatives, were further tested in the humanized BLT mouse model. BLT humanized mice are bioengineered by implantation of human fetal liver and thymic tissue under the kidney capsule followed by intravenous transplantation with autologous CD34+ HSCs21 (Fig. 3f). This allows for T cell maturation in human thymic tissue and permits HLA restricted T cell responses. WT hiEC grafts in Matrigel plugs triggered a roughly 40% stronger IFN-γ response but 40% weaker IgM antibody response than in the previous NSG-SGM3 mice. No measurable immune activation was detected in recipients of B2M−/−CIITA−/− CD47 tg hiEC grafts (Fig. 3g,h). All WT hiEC grafts underwent rapid rejection while four out of five B2M−/−CIITA−/− CD47 tg hiEC grafts achieved survival (Fig. 3i). We assume a non-immune-related reason for the failure of the fifth graft since no immune activation could be detected in this specific recipient. We thus demonstrated that the combination of MHC class I and II depletion and CD47 overexpression renders both mouse and human stem cells, as well as their differentiated derivatives, hypoimmunogenic. In the models studied here, engineered differentiated derivatives achieved long-term survival in fully allogeneic hosts without any immunosuppression and retained basic cell-specific features after transplantation.
The initial concept of hypoimmunogenic pluripotent stem cells was based on an MHC class I knockdown and showed encouraging early results22. However, according to the ‘missing-self theory’, MHC class I-deficient mouse and human PSCs become susceptible to NK cell killing23–25. Although isolated expression of HLA-E26 or HLA-G27 in human pluripotent stem cells has been used to mitigate NK cell cytotoxicity, we observed that CD47 is a very effective non-MHC ligand to silence all innate immune responses. However, cells eluding immune monitoring may pose the long-term risks of uncontrollable malignant transformation or impaired virus clearance, although for the latter alternative mechanisms have been shown28. Inducible kill switches could enhance their overall safety.
The ability to generate substantial amounts of cardiac tissue from allogeneic iPSC-derived CMs has been well demonstrated in macaques29. However, the amounts of toxic immunosuppressive drugs required to achieve allogeneic cell survival pose a major hurdle for clinical use. Even with fully MHC class I-and II-matched allogeneic iPSC-derived CM grafts, macaque recipients required substantial and highly toxic immunosuppression to prevent cell rejection29,30. Thus, the generation of universal hypoimmunogenic iPSCs that can be differentiated into the main components of cardiac tissue and achieve long-term survival in a fully allogeneic recipient without any immunosuppression may help to develop universal cell products to treat heart failure.
Methods
Syncytiotrophoblast cells of mouse placenta.
On isolated mouse syncytiotrophoblast cells, expression of MHC class I, MHC class II and Cd47 was performed using PCR. RNA was isolated with the RNeasy Plus Mini kit (Qiagen) according to the manufacturer’s protocol. RT–PCR was performed to generate the cDNA (Applied Biosystems). The following primers were used: mouse MHC class I: 5′-AGTGGTGCTGCAGAGCATTACAA-3′, reverse: 5′-GGTGACTTCACCTTTAGATCTGGG-3′, MHC class II forward: 5′-GATGTGGAAGACCTGCG-3′, reverse: 5′-TGCATCTTCTGAGGGGTTTC-3′; mouse Cd47 forward: 5′-GGCGCAAAGCACCGAAGAAATGTT-3′, reverse: 5′-CCATGGCATCGCGCTTATCCATTT-3′. PCRs were performed on Mastercycler nexus (Eppendorf) and the amplification products were visualized by 2% agarose gel electrophoresis (Thermo Fisher).
Derivation of mouse iPSCs.
Mouse tail tip fibroblasts of mice were dissociated and isolated with collagenase type IV (Life Technologies) and maintained with Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS), 1% glutamine, 4.5 g l−1 glucose, 100 U ml−1 penicillin, and 100 μg ml−1 streptomycin (pen-strep) at 37 °C, 20% O2, and 5% CO2 in a humidified incubator. 1 × 106 mouse fibroblasts were then re-programmed using a mini-intronic plasmid carrying sequences of Oct4, Klf4, Sox2 and c-Myc as well as short hairpin RNA against p53 (10–12 μM of DNA) using the Neon Transfection system13. After transfection, fibroblasts were plated on mitomycin-inhibited CF1 mouse embryonic fibroblasts (MEF, Applied Stemcell) and kept in fibroblast media with the addition of sodium butyrate (0.2 mM) and 50 μg ml−1 ascorbic acid. When ESC-like colonies appeared, media was changed to mouse iPSC media containing DMEM + GlutaMax 31966 (Gibco) with 10% heat-inactivated fetal calf sera (FCS hi), 1% MEM-NEAA and 1% pen-strep (all Gibco). With every passage, the iPSCs were sorted for the mouse pluripotency marker SSEA-1 using antibody-coated magnetic bead based cell sorting.
Mouse iPSC culture.
After the MEF feeder cells attached and were 100% confluent, miPSCs were grown on MEF in knockout DMEM 10829 with 15% knockout Serum Replacement, 1% glutamine, 1% MEM-NEAA, 1% pen-strep (all Gibco), 0.2% beta-mercaptoethanol and 100 units LIF (both Millipore). Cells were maintained in 10 cm dishes, medium was changed daily and the cells were passaged every 2–3 days using 0.05% trypsin-EDTA (Gibco). miPSCs were cultured on gelatin (Millipore) without feeders before experiments using the standard media. Cell cultures were regularly screened for mycoplasma infections using the MycoAlert Kit (Lonza).
Pluripotency analysis by RT–PCR and immunofluorescence.
miPSC were plated in confocal dishes (MatTek) for immunofluorescence analysis 48 h after plating using the miPSC Characterization kit (Applied Stemcell). Briefly, cells were fixed, permeabilized, and stained overnight at 4 °C with the primary antibodies for Sox2, SSEA-1 and Oct4. After several washes the cells were incubated with a secondary antibody and DNA staining solution. Alkaline phosphatase activity assay was performed (Applied Stemcell). Stained cells were imaged using a fluorescent microscope.
For RT–PCR, RNA was extracted using the RNeasy Plus Mini Kit (Qiagen). Genomic DNA contamination was removed using the gDNA spin column. cDNA was generated using Applied Biosystems High-Capacity cDNA Reverse Transcription kit. Gene-specific primers of the miPSC Characterization Kit (Applied Stemcell) were used to amplify target sequences. Actin was used as housekeeping gene, which encodes a cellular cytoskeleton protein. PCR reactions were performed on Mastercycler nexus (Eppendorf) and visualized on 2% agarose gels.
Pluripotency analysis by in vivo teratoma assay.
Ten million miPSCs were injected intramuscular into the hind limb of immunodeficient SCID-beige mice and teratoma development was observed within 14 days. Teratomas were recovered and fixed in 4% paraformaldehyde in PBS, dehydrated, embedded in paraffin, and cut into sections of 5 µm thickness. For histopathology, sections were rehydrated and stained with hematoxylin and eosin (Carl Roth). Images were taken with an inverted light microscope.
Immunofluorescence staining demonstrated differentiation into ectodermal, mesodermal, and endodermal cells using antibodies against brachyury (ab20680, Abcam), cytokeratin 8 (ab 192467) and GFAP (GA5, Cell Signaling). For visualization, secondary antibodies conjugated with Alexa Fluor 555, 488 and 647 (all Invitrogen) were used, respectively. Cell nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI) and imaging was performed with a Leica SP5 laser confocal microscope (Leica).
Gene editing of mouse iPSCs.
miPSCs underwent three gene-modification steps. First, CRISPR guides targeting the coding sequence of mouse B2m gene were annealed and ligated into vectors containing the Cas9 expression cassette. Transfected miPSCs were dissociated to single cells, expanded to colonies, sequenced and tested for homogeneity. Second, these B2m−/− miPSCs were transfected with vectors containing CRISPR guides targeting Ciita. Expanded single cell colonies were sequenced and B2m−/−Ciita−/− clones were identified through the presence of aberrant sequence from the CRISPR cleavage site. Third, the Cd47 gene sequence was synthesized and the DNA was cloned into a lentiviral with blasticidin resistance. B2m−/−Ciita−/− miPSCs were transduced with the resulting lentivirus and grown in the presence of blasticidin. Antibiotic-selected pools were tested for Cd47 overexpression and B2m−/−Ciita−/− Cd47 tg miPSCs were expanded.
Generation of B2m−/− miPSCs.
CRIPSR technology was used for disruption of the B2m gene. For targeting the coding sequence of mouse B2m gene, the CRISPR sequence 5′-TTCGGCTTCCCATTCTCCGG(TGG)-3′ was annealed and ligated into the All-In-One (AIO) vectors containing the Cas9 expression cassette as per the kit’s instructions (GeneArt CRISPR Nuclease Vector Kit, Thermo Fisher). miPSC were transfected with the AIO vectors using Neon electroporation with two 1,200 V pulses of 20 ms duration. The transfected iPSC cultures were dissociated to single cells using 0.05% trypsin (Gibco) and then sorted with FACSAria cell sorter (BD Bioscience) for removing doublets and debris by selective gating on forward and side light scatter properties. Single cells were expanded to full-size colonies and tested for CRISPR editing by screening for the presence of the altered sequence from the CRISPR cleavage site. Briefly, the target sequence was amplified via PCR using AmpliTaq Gold Master Mix (Applied Biosystems) and the primers B2m gDNA forward: 5′-CTGGATCAGACATATGTGTTGGGA-3′, reverse: 5′-GCAAAGCAGTTTTAAGTCCACACAG-3′. After cleanup of the obtained PCR product (PureLink Pro 96 PCR Purification Kit, Thermo Fisher), Sanger sequencing was performed. The Ion Personal Genome Machine (PGM) Sequencing was used for the identification of the homogeneity, through sequencing of a PCR amplified 250 base pair region of the B2m gene using primers B2m gDNA PGM forward: 5′-TTTTCAAAATGTGGGTAGACTTTGG-3′ and reverse: 5′-GGATTTCAATGTGAGGCGGGT-3′. The PCR product was purified as described above and prepared using the Ion PGM Hi-Q Template Kit (Thermo Fisher). Experiments were performed on the Ion PGM System with the Ion 318 Chip Kit v.2 (Thermo Fisher).
Generation of B2m−/−Ciita−/− miPSCs.
CRIPSR technology was used for the further disruption of the Ciita gene. For targeting the coding sequence of mouse Ciita gene, the CRISPR sequence 5’-GGTCCATCTGGTCATAGAGG (CGG)-3′ was annealed and ligated into the All-In-One (AIO) vectors containing the Cas9 expression cassette as per the kit’s instructions (GeneArt CRISPR Nuclease Vector Kit, Thermo Fisher). miPSC were transfected with the AIO vectors using the same condition for B2m disruption. The transfected miPSC cultures were dissociated to single cells using 0.05% trypsin (Gibco) and then sorted with FACSAria cell sorter (BD Bioscience) for removing doublets and debris by selective gating on forward and side light scatter properties. Single cells were expanded to full-size colonies and tested for CRISPR editing by screening for the presence of an altered sequence from the CRISPR cleavage site. Briefly, the target sequence was amplified via PCR using AmpliTaq Gold Master Mix (Applied Biosystems) and the primers Ciita gDNA forward: 5′-CCCCCAGAACGATGAGCTT-3′, reverse: 5′-TGCAGAAGTCCTGAGAAGGCC-3′. After cleanup of the obtained PCR product (PureLink Pro 96 PCR Purification Kit, Thermo Fisher), Sanger sequencing was performed. Using the DNA sequence chromatogram, edited clones were then identified through the presence of altered sequence from the CRISPR cleavage site. Indel size was calculated using the TIDE tool. PCR and ICC were performed again to verify the pluripotency status of the cells.
Generation of B2m−/−Ciita−/− Cd47 tg miPSCs.
Cd47 transgene overexpression was generated using lentivirus-mediated delivery of a Cd47-expressing vector containing the antibiotic resistance cassette blasticidin. The Cd47 cDNA was synthesized and cloned into the lentiviral plasmid pLenti6/V5 (Thermo Fisher) with a blasticidin resistance. Sanger sequencing was performed to verify that no mutation had occurred. Lentivirus generation was performed with a stock titer of 1 × 107 TU per ml. The transduction was performed into 2 × 105 B2m−/−Ciita−/− miPSCs, grown on blasticidin-resistant MEF cells for 72 h with a MOI ratio of 1:10 followed by antibiotic selection with 12.5 μg ml−1 blasticidin for 7 days. Antibiotic-selected pools were tested by RT–quantitative PCR amplification of Cd47 mRNA and flow cytometry detection of Cd47 on the surface of the cells. After the confirmation of Cd47, cells were expanded and validated by running pluripotency assays.
Transduction to express firefly luciferase.
iPSCs were transduced to express Fluc. One hundred thousand miPSCs were plated in one gelatin-coated six-well plates and incubated overnight at 37 °C at 5% CO2. The next day, media was changed and one vial of Fluc lentiviral particles expressing luciferase II gene under reengineered EF1a promotor (GenTarget) was added to 1.5 ml media. After 36 h, 1 ml of cell media was added. After further 24 h, complete media change was performed. After 2 days, luciferase expression was confirmed by adding D-luciferin (Promega). Signals were quantified with IVIS 200 (Perkin Elmer) in maximum photons s−1 cm−2 sr−1
Karyotyping.
Cell collection, slide preparation and G-banded karyotyping were performed using standard cytogenetics protocols optimized for human pluripotent cells. Cells were incubated with ethidium bromide and colcemid (Gibco) and then trypsinized to detach the cells from the plate. The cells were placed in hypotonic solution (0.075 M potassium chloride, 0.559 g KCl in 100 ml water, Millipore), followed by fixation. Metaphase cell preparations were stained with Leishman’s stain. Karyotype analysis consisted of chromosomes counted in twenty cells with band-by-band analysis of eight cells.
Mice.
BALB/c (BALB/cAnNCrl, H2d), C57BL/6 (C57BL/6 J, H2b) and SCID-beige (CBySmn.CB17-Prkdcscid/J) (all 6–12 weeks) were used as recipients for different assays. The number of animals per experimental group is presented in each figure. Mice were purchased from Charles River Laboratories (Sulzfeld) and received humane care in compliance with the Guide for the Principles of Laboratory Animals. Animal experiments were approved by the Hamburg ‘Amt für Gesundheit und Verbraucherschutz’ or the University of California San Francisco (UCSF) Institutional Animal Care and Use Committee and performed according to local and EU guidelines.
Teratoma assays to study miPSC survival in vivo.
Six-to-eight-week-old syngeneic or allogeneic mice were used for transplantation of WT miPSCs or hypoimmunogenic miPSCs. Two million cells were injected in 60 µl saline into the right thigh muscle of the mice. Tumor growth was measured with a caliper every other day until day 30 and from day 30 to day 80 every tenth day. They were killed after development of tumors larger than 1.5 cm3 or following an observation period of 80 days.
Derivation and characterization of miPSC-derived endothelial cells (miECs).
miPSC were plated on gelatin in six-well plates and maintained in mouse iPSC media. After the cells reached 60% confluency, the differentiation was started and media was changed to RPMI-1640 containing 2% B-27 minus Insulin (both Gibco) and 5 μM CHIR-99021 (Selleckchem). On day 2, the media was changed to reduced media: RPMI-1640 containing 2% B-27 minus Insulin (both Gibco) and 2 μM CHIR-99021 (Selleckchem). From day 4 to day 7, cells were exposed to RPMI-1640 EC media, RPMI-1640 containing 2% B-27 minus Insulin plus 50 ng ml−1 mouse vascular endothelial growth factor (mVEGF; R&D Systems), 10 ng ml−1 mouse fibroblast growth factor basic (mFGFb; R&D Systems), 10 μM Y-27632 (Sigma-Aldrich) and 1 μM SB 431542 (Sigma-Aldrich). Endothelial cell clusters were visible from day 7 and cells were maintained in EGM-2 SingleQuots media (Lonza) plus 10% FCS hi (Gibco), 25 ng ml−1 mVEGF, 2 ng ml−1 mFGFb, 10 μM Y-27632 (Sigma-Aldrich) and 1 μM SB 431542. The differentiation process was completed after 21 days and undifferentiated cells detached during the differentiation process. For purification, cells went through magnetic-activated cell separation (MACS) purification according the manufacturer’s protocol using anti-CD15 mAb-coated magnetic microbeads (Miltenyi) for negative selection.
The highly purified miECs in the flow-through were cultured in EGM-2 SingleQuots media plus supplements and 10% FCS hi. TrypLE was used for splitting the cells 1:3 every 3–4 days. Their phenotype was confirmed by immunofluorescence for CD31 (ab28364, Abcam) and VE-Cadherin (sc-6458, Santa Cruz Biotechnology). Briefly, cells were fixed with 4% paraformaldehyde in PBS for 15 min. Cell membranes were permeabilized with Permeabilization solution (ASB-0102, Applied StemCell), followed by Blocking solution (ASB-0103, Applied StemCell) and incubation with the primary antibodies. For visualization, cells were incubated with secondary antibody conjugated with AF488 or AF555 (Invitrogen). After nuclei staining with DAPI, images were obtained and analyzed with a Leica SP5 laser confocal microscope (Leica).
Tube formation assay was performed for miEC characterization: 2.5 × 105 miECs were stained with 5 µM CFSE and 0.1 μg ml−1 Hoechst (both Thermo Fisher) for 10 min at room temperature and plated on 10 mg ml−1 undiluted Matrigel (356231, Corning) in 24-well plates. After 48 h, tube formations were visualized by immunofluorescence. PCR was performed as described above. The following primers were used: VE-Cadherin forward: 5′-GGATGCAGAGGCTCACAGAG-3′, reverse: 5′-CTGGCGGTTCACGTTGGACT-3′.
Derivation and characterization of miPSC-derived smooth muscle cells (miSMCs).
The resuspended miPSCs were cultivated on six-well, 0.1% gelatin-coated plastic petri dishes (Falcon, Becton–Dickinson) at 2 × 106 cells per well at 37 oC, 5% CO2 in 2 ml of differentiation medium with the presence of 10 μM all-trans-retinoic acid. The differentiation medium was made of DMEM, 15% FCS, 2 mM L-glutamine, 1 mM methyl thioglycolate (MTG) (Sigma-Aldrich), 1% non-essential amino acids and pen-strep. The culture was continued for 10 days with daily media changes. Starting from the 11th day, the differentiation medium was replaced by serum-free culture medium, which was composed of knock-out DMEM, 15% knock-out serum replacement, 2 mM L-glutamine, 1 mM MTG, 1% non-essential amino acids and pen-strep. The cultures were continued for another 10 days with daily change of the serum-free medium. For purification, cells were enriched according the manufacturer’s protocol using anti-CD15 mAb-coated magnetic microbeads (Miltenyi) for negative selection. The flow-through containing enriched miSMCs were cultured in RMPI-1640 Glutamax plus 20% FCS hi and 1% pen-strep (all Gibco). Their phenotype was confirmed by immunofluorescence and PCR for both Sma and Sm22.
Immunofluorescence staining was performed as described above. Primary antibodies were used against smooth muscle actin (ab21027, Abcam) and sm22 (ab14106, Abcam), followed by the corresponding secondary antibody conjugated with AF488 or AF555 (Invitrogen).
PCR was performed as described above. The following primers were used: SMA forward: 5′-CGGCTTCGCTGGTGATGAT-3′, reverse: 5′-CATTCCAACCATTACTCCCTGAT-3′; SM22 forward: 5′-AACAGCCTGTACCCTGATGG-3′, reverse: 5′-CGGTAGTGCCCATCATTCTT-3′.
Derivation and characterization of miPSC-derived cardiomyocytes (miCMs).
Before differentiation, miPSCs were passaged two times on gelatin-coated flasks to remove the feeder cells. At day 0, differentiation was started with 80,000 cells per ml in IMEM/Ham’s F12 (3/1, both Corning) +0.5% N2-Supplement, 1% B27 retinoic acid, 0.05% BSA, 1% pen-strep, 1% glutamine (Gibco), 5 mg ml−1 ascorbic acid and 40 ng ml−1 MTG (both Sigma-Aldrich) for 2 days in uncoated 10 cm plates. At day 2, cells were transferred in IMEM/Ham’s F12 (3/1, both Corning) with 0.5% N2-Supplement, 1% B27 retinoic acid, 0.05% BSA, 1% pen-strep, 1% glutamine (all Gibco), 5 mg ml−1 ascorbic acid and 40 ng ml−1 MTG (Sigma-Aldrich) for 2 days in uncoated 10 cm plates. On day 4, cells were plated in gelatin-coated six-well plates in SP34 media containing 1% glutamine, 50 μg ml−1 ascorbic acid, 5 ng ml−1 VEGF, 500 μg ml−1 hFGFb and 25 ng ml−1 hFGF10 (R&D Systems). Media was changed on day 7 to SP34 media containing 1% glutamine and 50 μg ml−1 ascorbic acid and was changed every other day. Beating of cells started around days 11–14 and demonstrated their function.
For enrichment, cells separated by MACS according the manufacturer’s protocol using anti-CD15 mAb-coated magnetic microbeads (Miltenyi) for negative selection. The flow-through containing enriched miCMs were replated and used for different assays.
Immunofluorescence staining was performed as described above to confirm their phenotype. Primary antibodies were used against α-sarcomeric actinin (EA-53, Abcam) or troponin I (ab47003, Abcam) followed by the corresponding secondary antibody conjugated with AF488 or AF555 (Invitrogen).
PCR for Gata4 forward: 5′-CTGTCATCTCACTATGGGCA-3′, reverse: 5′-CCAAGTCCGAGCAGGAATTT-3′ and Mhy6 forward: 5′-ATCATTCCCAACGAGCGAAAG-3′, reverse: 5′-AAGTCCCCATAGAGAATGCGG-3′ was performed as described above.
Flow cytometry analysis.
For the detection of MHC class I and II surface molecules on miPSCs, miECs, miSMCs and miCMs, cells were plated on gelatin-coated six-well plates in medium containing 100 ng ml−1 of IFN-γ. After collection, cells were labeled with antibodies. For MHC class I: PerCP-eFlour710-labeled anti-MHC class I antibody (clone AF6–88.5.5.3, eBioscience) or PerCP-eFlour710-labeled mouse IgG2b isotype-matched control antibody (clone eB149/10H5, eBioscience). The anti-MHC class I antibody reacts with the H-2Kb MHC class I alloantigen. For MHC class II: PerCP-eFlour710-labeled anti-MHC class II antibody (clone M5/114.15.2, eBioscience) or PerCP-eFlour710-labeled mouse IgG2a isotype-matched control antibody (clone eBM2a, eBioscience). The MHC class II antibody reacts with the mouse MHC class II, both I-A and I-E subregion-encoded glycoproteins. Cd47: Alexa Fluor 647-labeled anti-mouse Cd47 antibody (clone miap301, BD Biosciences) or Alexa Fluor 647-labeled mouse IgG2a isotype-matched control antibody (clone R35–95, BD Biosciences). The anti-Cd47 antibody specifically binds to the extracellular domain of mouse Cd47, also known as Integrin-Associated Protein. Cells were analyzed by flow cytometry (BD Bioscience) and results were expressed as fold change to isotype-matched control Ig staining.
For the assessment of purity of miPSC derivatives, antibodies against SSEA-1 (Thermo Fisher), VE-Cadherin (Sigma), SMA (Abcam) and Troponin I (Santa Cruz) were used. The miECs, miSMCs and miCMs were generated with a purity of >90%.
Elispot assays.
For uni-directional Elispot assays, recipient splenocytes were isolated from spleen 5 days after cell injection and used as responder cells. Donor cells were mitomycin-treated (50 μg ml−1 for 30 min) and used as stimulator cells. One hundred thousand stimulator cells were incubated with 1 × 106 recipient responder splenocytes for 24 h and IFN-γ and IL-4 spot frequencies were enumerated using an Elispot plate reader.
Donor-specific antibodies.
Sera from recipient mice were de-complemented by heating to 56 °C for 30 min. Equal amounts of sera and cell suspensions (5 × 106 ml) were incubated for 45 min at 4 °C. Cells were labeled with FITC-conjugated goat anti-mouse IgM (Sigma-Aldrich) and analyzed by flow cytometry (BD Bioscience).
Mouse NK cell Elispot assays in vitro.
NK cells were isolated from fresh BALB/c spleen 18 h after poly I:C injection (150 ng poly I:C in 200 μl sterile saline, intraperitoneally, Sigma-Aldrich). After red cell lysis, cells were purified by anti-CD49b mAb-coated magnetic bead-sorting and were used as responder cells. This cell population was >99% CD3-and contains NK cells (>90%) and other cells including myeloid cells (<10%). Using the Elispot principle, NK cells were co-cultured with B2m−/−Ciita−/− or B2m−/−Ciita−/− Cd47 tg miPSCs in the presence of IL-2 (1 ng ml−1, Peprotech) and their IFN-γ release was measured. YAC-1 cells (Sigma-Aldrich) served as positive control. Mitomycin-treated (50 μg ml−1 for 30 min) stimulator cells were incubated with NK cells (1:1) for 24 h and IFN-γ spot frequencies were enumerated using an Elispot plate reader.
Mouse in vivo innate cytotoxicity assay.
Five million WT miPSCs and 5 × 106 B2m−/−Ciita−/− miPSCs or 5 × 106 B2m−/−Ciita−/− Cd47 tg miPSCs were mixed and stained with 5 μM CFSE. Cells in saline with IL-2 (1 ng ml−1, Peprotech) were injected intraperitoneally into syngeneic C57BL/6 mice. After 48 h, cells were collected from the abdomen and stained with PerCP-eFlour710 labeled anti-MHC class I mAb for 45 min at 4 °C. The CFSE-positive and MHC class I-negative population was analyzed by flow cytometry (BD Bioscience) and compared between the WT and the engineered miPSC group. The assay was performed with miPSCs, miECs, miSMCs and miCMs. Some animals were pretreated with clodronate (200 μl intraperitoneally 3 days before the experiment; Liposoma) to eliminate macrophages and make the assay more specific for NK cells. Some animals were pretreated with in vivo Cd47-blocking antibody (BE0270, 100 μg intraperitoneally, 7 days and 3 days before the experiment; BioXCell) to eliminate Cd47.
NK cell stimulatory ligands.
For the detection of NK cell stimulatory ligands on miPSCs, miECs, miSMCs and miCMs, cells were blocked with mouse FcR blocking reagent (Miltenyi) according to manufacturer’s protocol. WT cells, B2m−/−Ciita−/− cells and B2m−/−Ciita−/− Cd47 tg cells were then incubated with the recombinant mouse NKp46 or NKG2D human Fc chimera protein or the recombinant control IgG1 Fc protein (R&D systems) for 45 min at 4 °C. FITC-conjugated anti-human IgG1 antibody (Invitrogen) served as secondary antibody. YAC-1 cells were used as positive control. Data analysis was carried out using flow cytometry (BD Bioscience) and FlowJo software, and results were expressed as fold change to the isotype-matched control Fc fusion protein.
Survival analysis of differentiated derivatives using BLI.
For BLI, D-luciferin firefly potassium salt (375 mg kg−1; Biosynth) was dissolved in PBS (pH 7.4) (Gibco, Invitrogen) and was injected intraperitoneally (250 μl per mouse) into anesthetized mice. Animals were imaged using the IVIS 200 system (Xenogen). Region of interest (ROI) bioluminescence was quantified in units of maximum photons per second per centimeter square per steradian (p s−1 cm−2 sr−1). The maximum signal from an ROI was measured using Living Image software (MediaCybernetics). Mice were monitored on day 0, day 1 and every other day until day 30 and every 10 days afterwards.
Matrigel plugs: cell morphology for miECs, miSMCs or miCMs.
Eight hundred thousand B2m−/−Ciita−/− Cd47 tg miECs, miSMCs or miCMs in 1:1 diluted Matrigel (Corning) were injected into allogeneic BALB/c mice. Matrigel plugs were recovered after 1, 2, 3, 4, 5, 6 and 8 weeks and fixed in 4% paraformaldehyde in PBS with 1% glutaraldehyde for 24 h. Samples were dehydrated, embedded in paraffin and cut into sections of 5 µm thickness. For histopathology, sections were stained with hematoxylin and eosin (Carl Roth) and images taken with an inverted light microscope. Origin of cells was demonstrated with immunofluorescence staining. Sections were rehydrated, and underwent antigen retrieval and blocking. Samples were incubated with antibodies against luciferase (ab21176), SMA (ab21027, Abcam), VE-Cadherin (SC-6458) or α-sarcomeric actinin (EA-53, Abcam) and a corresponding secondary antibody conjugated with AF488 or AF555 (Invitrogen). Cell nuclei were counterstained with DAPI and images taken with a Leica SP5 laser confocal microscope (Leica).
For co-staining experiments of miECs and immune cells, primary antibodies were used against VE-Cadherin (SC-6458, Sigma) and CD3 (ab16669, Abcam), followed by the corresponding secondary antibody conjugated with AF488 or AF555 (Invitrogen).
Generation of human iPSCs (hiPSCs).
The Human Episomal iPSC Line was derived from CD34+ cord blood using a three-plasmid, seven-factor (SOKMNLT; SOX2, OCT4 (POU5F1), KLF4, MYC, NANOG, LIN28 and SV40L T antigen) EBNA-based episomal system by Thermo Scientific. This cell line has been shown to be free of all reprogramming genes. These hiPSCs have a normal XX karyotype and endogenous expression of pluripotent markers including Oct4, SOX2 and NANOG (as shown by RT–PCR) and OCT4, SSEA4, TRA-1–60 and TRA-1–81 (as shown by immunofluorescence).
Gene editing of hiPSC.
hiPSC underwent two gene-modification steps. In the first step, CRISPR technology was used for a combined targeting of the coding sequence of human B2M gene with the CRISPR sequence 5′-CGTGAGTAAACCTGAATCTT-3′ and the coding sequence of human CIITA gene with the CRISPR sequence 5′-GATATTGGCATAAGCCTCCC-3′. Linearized CRISPR sequence with T7 promoter were used to synthesize gRNA as per the kit’s instructions (MEGAshortscript T7 Transcription Kit, Thermo Fisher). The obtained in vitro transcription (IVT) gRNA was then purified via the MEGAclear Transcription Clean-Up Kit. For IVT gRNA delivery, cells were electroporated with 300 ng IVT gRNA using a Neon electroporation system and the conditions 1,200 V, 30 ms, 1 pulse into hiPSC stably expressing Cas9. After electroporation, edited hiPSC were expanded for single cell seeding: hiPSC cultures were dissociated into single cells using TrypLE Express (Gibco) and stained with Alexa Fluor 488-conjugated anti-TRA-160 mAb and propidium iodide. A FACSAria II cell sorter (BD Biosciences) was used for the sorting and doublets and debris were excluded from seeding by selective gating on forward and side light scatter properties. Viable pluripotent cells were selected on the absence of propidium iodide and presence of Tra1–60 staining. Single cells were then expanded into full-size colonies, after which the colonies were tested for CRISPR editing by sequencing. CRISPR-mediated cleavage was assessed using the GeneArt Genomic Cleavage Detection Kit (Thermo Fisher) for testing of the initial edited pools. For screening of the isolated clones, genomic DNA was isolated from 1 × 106 hiPSCs and the B2M and CIITA genomic DNA regions were PCR amplified using AmpliTaq Gold 360 Master Mix and the primer sets forward: 5′-TGGGGCCAAATCATGTAGACTC-3′ and reverse: 5′-TCAGTGGGGGTGAATTCAGTGT-3′ for B2M as well as forward: 5′-CTTAACAGCGATGCTGACCCC-3′ and reverse: 5′-TGGCCTCCATCTCCCCTCTCTT-3′ for CIITA. For TIDE analysis, the obtained PCR product was cleaned up (PureLink PCR Purification Kit, Thermo Fisher) and Sanger sequencing was performed for the prediction of indel frequency. After the confirmation of B2M and CIITA disruption, cells were further characterized through karyotype analysis and the TaqMan hiPSC Scorecard Panel (Thermo Fisher). The hiPSCs were found to be pluripotent and maintained a normal (46,XX) karyotype during the genome editing process.
In the second step, the CD47 cDNA was synthesized and the DNA was cloned into a lentiviral plasmid with an EFS promotor and puromycin resistance cassette. Cells were transduced with lentiviral stocks and 8 μg ml−1 of Polybrene (Thermo Fisher). Media was changed daily after transduction. Three days after transduction, cells were expanded and selected with 0.5 μg ml−1 of puromycin. After 5 days of antibiotic selection, antibiotic-resistant colonies emerged and were further expanded to generate stable pools. The expression of CD47 transcripts was confirmed by quantitative PCR. Pluripotency assay (TaqMan hPSC Scorecard Panel, Thermo Fisher) and karyotyping was performed again to verify the pluripotent status of the cells.
Teratoma assays to study iPSC survival in vivo.
Six to eight week-old immunodeficient SCID-beige mice were used for transplantation of WT hiPSC or B2M−/−CIITA−/− CD47 tg hiPSCs. Here 1 × 107cells were resuspended in 100 µl saline solution and injected into the right thigh muscle of the mice. Teratomas were recovered, fixed in 4% paraformaldehyde, dehydrated, embedded in paraffin and cut into sections of 5 µm thickness. For histopathology, sections were rehydrated and stained with hematoxylin and eosin. Images were taken with an inverted light microscope. For immunofluorescence, slides underwent heat-induced antigen retrieval in a steamer with Dako antigen-retrieval solution (Dako), followed by antigen blocking with Image-iT FX signal enhancer solution (Invitrogen). Tissue sections were incubated with a primary antibody against brachyury (Ab20680, Abcam), followed by a goat anti-rabbit IgG secondary antibody conjugated with Alexa Fluor 555 (Invitrogen). Subsequently, sections were incubated with primary antibodies against cytokeratin 8 (EP1628Y, Abcam) and GFAP (GA5, Cell Signaling) conjugated with AF488 or AF647, respectively. DAPI was used to counterstain cell nuclei and images were acquired with a Leica SP5 laser confocal microscope (Leica).
Pluripotency analysis by RT–PCR and immunofluorescence.
hiPSCs were plated in confocal dishes (MatTek) for immunofluorescence analysis 48 h after plating using the hiPSC Characterization Kit (Applied Stem Cell). Briefly, cells were fixed, permeabilized and stained overnight at 4 °C with the primary antibodies for OCT4, SOX2, SSEA4, TRA-1–60 and TRA-1–81 (Applied Stem Cell). After several washes, the cells were incubated with a secondary antibody and DNA staining solution. Alkaline phosphatase activity assay was performed (Applied Stem Cell). Stained cells were imaged using a fluorescent microscope.
For RT–PCR, RNA was extracted using the RNeasy Plus Mini Kit (Qiagen). Genomic DNA contamination was removed using the gDNA spin column. cDNA was generated using Applied Biosystems High-Capacity cDNA Reverse Transcription Kit. Gene-specific primers of the hiPSC Characterization Kit (Applied Stem Cell) were used to amplify target sequences Actin was used as housekeeping gene. PCR reactions were performed on Mastercycler nexus (Eppendorf) and visualized on 2% agarose gels.
Humanized mice.
Humanized NSG-SGM3 mice (18–30 weeks) were purchased from Jackson Laboratories. Human CD34+ hematopoietic stem cell-engrafted NSG-SGM3 mice develop multi-lineage human immune cells, and demonstrate a functional human immune system displaying T cell-dependent immune responses with no donor cell immune reactivity towards the host. Animals were randomly assigned to experimental groups. The percentage of CD3+ cells among the human CD45+ cell population was assessed in every animal and CD3 percentages were never significantly different between WT and B2M−/−CIITA−/− CD47 tg groups (Supplementary Fig. 13a). The number of animals per experimental group is presented in each figure. All humanized NSG-SGM3 mice were HLA-A typed and the number of mismatches to the cell graft calculated (Supplementary Fig. 13b). In the Elispot assays with hiPSCs, there were 1.6 ± 0.5 and 1.7 ± 0.5 (P = 0.61), with hiCMs 1.3 ± 0.6 and 1.3 ± 0.6 (P = 1) mismatches for WT and B2M−/−CIITA−/− CD47 tg, respectively, and in the hiEC groups there were always two mismatches. The mismatches for the BLI experiments are shown in Supplementary Fig. 10j.
All BLT mice were approved by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee and were generated with the same human tissue using NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, Jackson Laboratories) and there were five out of six HLA class I and 4 out of 4 class II mismatches to the transplanted hiPSCs or derivatives. The percentage of CD3+ cells among the human CD45+ cell population was never significantly different between WT and B2M−/−CIITA−/− CD47 tg groups (Supplementary Fig. 13c). The percentage of CD3+ cells among the human CD45+ cell population was typically in the 15–65% range.
Human iPSC differentiation into hiECs.
hiPSC were plated on diluted Matrigel (356231, Corning) in six-well plates and maintained in Essential 8 Flex media (Thermo Fisher). The differentiation was started at 60% confluency and media was changed to RPMI-1640 containing 2% B-27 minus insulin (both Gibco) and 5 μM CHIR-99021 (Selleckchem). On day 2, the media was changed to reduced media: RPMI-1640 containing 2% B-27 minus insulin (Gibco) and 2 μM CHIR-99021 (Selleckchem). From day 4 to 7, cells were exposed to RPMI-1640 EC media, RPMI-1640 containing 2% B-27 minus insulin plus 50 ng ml−1 human vascular endothelial growth factor (VEGF; R&D Systems), 10 ng ml−1 human fibroblast growth factor basic (FGFb; R&D Systems), 10 μM Y-27632 (Sigma-Aldrich), and 1 μM SB 431542 (Sigma-Aldrich). Endothelial cell clusters were visible from day 7 and cells were maintained in EGM-2 SingleQuots media (Lonza) plus 10% FCS hi (Gibco), 25 ng ml−1 VEGF, 2 ng ml−1 FGFb, 10 μM Y-27632 (Sigma-Aldrich) and 1 μM SB 431542 (Sigma-Aldrich). The differentiation process was completed after 14 days und undifferentiated cells detached during the differentiation process. For purification, cells were treated with 20 μM PluriSln-1 (StemCell Technologies) for 48 h. The highly purified ECs were cultured in EGM-2 SingleQuots media (Lonza) plus supplements and 10% FCS hi (Gibco). TrypLE Express was used for passaging the cells 1:3 every 3–4 days.
Immunofluorescence staining was performed as described above to confirm their phenotype. Primary antibodies were used against CD31 (ab28364, Abcam) and VE-Cadherin (sc-6458, Santa Cruz Biotechnology), followed by the corresponding secondary antibody conjugated with AF488 or AF555 (Invitrogen). Cell nuclei were stained with DAPI. Imaging was performed using a Leica SP5 laser confocal microscope (Leica).
PCR for VE-Cadherin (forward: 5′-AAGATGCAGAGGCTCATG-3′, reverse: 5′-CATGAGCCTCTGCATCTT-3′) was performed as described above.
Human iPSC differentiation into hiCMs.
hiPSCs were plated on diluted Matrigel (356231, Corning) in six-well plates and maintained in Essential 8 Flex media (Thermo Fisher). Differentiation was started at 90% confluency, and media was changed to 5 ml of RPMI-1640 containing 2% B-27 minus Insulin (Gibco) and 6 μM CHIR-99021 (Selleckchem). After 2 days, media was changed to RPMI-1640 containing 2% B-27 minus insulin without CHIR. On day 3, 5 μl IWR1 was added to the media for two further days. At day 5, the media was changed back to RPMI-1640 containing 2% B-27 minus insulin medium and left for 48 h. At day 7, media was changed to RPMI-1640 containing B27 plus insulin (Gibco) and replaced every 3 days thereafter with the same media. Spontaneous beating of cardiomyocytes was first visible around day 10. Purification of cardiomyocytes was performed on day 10 post-differentiation. Briefly, media was changed to low glucose media and maintained for 3 days. At day 13, media was changed back to RPMI-1640 containing B27 plus insulin. This procedure was repeated on day 14.
Immunofluorescence staining was performed as described above to confirm their phenotype. Primary antibodies were used against α-sarcomeric actinin (EA-53, Abcam) and troponin I (ab47003, Abcam), followed by the corresponding secondary antibody conjugated with AF488 or AF555 (Invitrogen). Cell nuclei were stained with DAPI. Imaging was performed using a Leica SP5 laser confocal microscope (Leica).
PCR for troponin (cTNT, forward: 5′-GAGGCACCAAGTTGGGCATGAACG A-3′, reverse: 5′-GGCAGCGGAAGAGGATGCTGAA′) was performed as described above.
Flow cytometry analysis.
Human iPSCs, iCMs and iECs were plated in six-well plates in medium containing 100 ng ml−1 of IFN-γ. Cells were harvested and labeled with antibodies. APC-conjugated anti-HLA-A,B,C antibody (clone G46_2.6,BD Biosciences) or APC-conjugated IgG1 isotype-matched control antibody (clone MOPC-21,BD Biosciences). Alexa-flour647-labeled anti-HLA-DR,DP,DQ antibody (clone Tu3a, BD Biosciences) or Alexa-flour647-labeled IgG2a isotype-matched control antibody (clone G155–178, BD Biosciences). PerCP-Cy5-conjugated anti-CD47 (clone B6H12, BD Biosciences) or PerCP-Cy5-conjugated IgG1 isotype-matched control antibody (clone MOPC-21, BD Biosciences). Results were expressed as fold change to isotype-matched control Ig staining.
For the assessment of purity of hiPSC derivatives, antibodies against TRA-1–60 (Thermo Fisher), VE-Cadherin (Santa Cruz) and Troponin I (Santa Cruz) were used. The hiECs and hiCMs were generated with a purity of >95%.
Human NK cell Elispot assays.
Human NK cells were co-cultured with B2M−/−CIITA−/− or B2M−/−CIITA−/− CD47 tg hiPSC and their IFN-γ release was measured. Human NK cells were purchased from StemCell Technologies and were >99% CD3-and 95% CD56+. Flow cytometry revealed >95% NK cells and <5% other cells including myeloid cells. Donor cells were mitomycin-treated and used as stimulator cells. K562 cells (Sigma-Aldrich) served as positive control. Stimulator cells were incubated with NK cells (1:1) in RPMI-1640 containing 1% pen-strep and 1 ng ml−1 human IL-2 (Peprotech) for 24 h and IFN-γ spot frequencies were enumerated using an Elispot plate reader.
T cell Elispot using humanized mice.
For uni-directional Elispot assays, recipient splenocytes were isolated from humanized mice 5 days after cell injection and used as responder cells. Cells were incubated for 24 h in vitro with 1 μg ml−1 anti-CD3 and 1 μg ml−1 anti-CD28 before plated for Elispot Assay. Donor cells were mitomycin-treated (50 μg ml−1 for 30 min) and used as stimulator cells. One hundred thousand stimulator cells were incubated with 1 × 106 recipient responder splenocytes for 48 h and IFN-γ and IL-5 spot frequencies were enumerated using an Elispot plate reader.
DSA.
Sera from recipient mice were de-complemented by heating to 56 °C for 30 min. Equal amounts of sera and cell suspensions (5 × 106 per ml) were incubated for 45 min at 4 °C. Cells were labeled with FITC-conjugated goat anti-human IgM (BD Bioscience) and analyzed by flow cytometry (BD Bioscience).
Matrigel plugs: cell morphology.
One million B2M−/−CIITA−/− CD47 tg hiECs or hiCMs in 1:1 pro survival scaffold, consisting of 50% (vol/vol) Matrigel (Corning), 100 μM ZVAD (Millipore), 50 nM Bcl-Xl BH4 (Millipore), 200 nM cyclosporine A (Sigma-Aldrich), 100 ng ml−1 IGF-1 (Peprotech) and 50 μM Pinacidil (Sigma-Aldrich) were injected into humanized NSG-SGM3 mice. Matrigel plugs were recovered after 2, 4, 6 and 8 weeks, fixed in 4% paraformaldehyde in PBS with 1% glutaraldehyde, dehydrated and embedded in paraffin. Sections of 5 µm thickness were cut. For immunofluorescence, sections were rehydrated and underwent antigen retrieval, followed by antigen blocking. After incubation with a primary antibody against luciferase (ab21176), VE-Cadherin (SC-6458) or α-sarcomeric actinin (EA-53, Abcam), sections were incubated with a corresponding secondary antibody conjugated with AF488 or AF555 (Invitrogen). DAPI was used to counterstain cell nuclei and images were obtained with a Leica SP5 laser confocal microscope (Leica).
BLI.
For BLI, D-luciferin firefly potassium salt (375 mg kg−1) (Biosynth) dissolved in sterile PBS (pH 7.4) (Gibco, Invitrogen) was injected intraperitoneally (250 μl per mouse) into anesthetized mice. Animals were imaged using the ami HT (Spectral Instruments Imaging) ROI bioluminescence was quantified in units of maximum photons per second per centimeter square per steradian (p s−1 cm−2 sr−1). The maximum signal from an ROI was measured using Living Image software (MediaCybernetics). Humanized mice were injected with 5 × 105 or 1 × 106 cells in pro survival scaffold as described above. Mice were monitored on day 0, day 1 and every 4 days until cells were rejected or up to 50 days.
In vitro NK cell killing.
Mouse NK cells were isolated from fresh BALB/c or C57BL/6 spleens 18 h after poly I:C injection (100 μg intraperitoneally). After red cell lysis, NK cells were purified with MagniSort Mouse NK cell Enrichment Kit (Invitrogen), followed by CD49b MACS-sorting (Miltenyi). This cell population was highly selected for NK cells with a purity of >9%. Human NK cells from PBMCs were purchased from StemCell Technologies containing >99% NK cells.
NK cell killing assays were performed on the XCelligence SP platform (ACEA BioSciences). 96-well E-plates (ACEA BioSciences) were coated with collagen (Sigma-Aldrich) and 4 × 105 WT, B2m−/−Ciita−/−, or B2m−/−Ciita−/− Cd47 tg miECs or WT, B2M−/−CIITA−/− or B2M−/−CIITA−/− CD47 tg hiECs were plated in 100 μl cell-specific media containing 1 ng ml−1 mouse or human IL-2 (Peprotech). After the Cell Index value reached 0.7, NK cells were added with an effector cell / target cell (E/T) ratio of 0.5/1, 0.8/1 or 1/1. As a negative control, cell treated with 2% Triton X100 was used. Some wells were pretreated with mouse Cd47 or human CD47-blocking antibody (BioXCell) with 10 μg ml−1 media for 2 h. Data were standardized and analyzed with the RTCA software (ACEA).
NK cell stimulatory ligands.
For the detection of NKG2D, NKp80, NKp46, NKp44 and NKp30 on hiPSCs, hiECs and hiCMs, cells were plated on gelatin-coated six-well plates. K562 cells were plated in six-well plates as suspension cells. After harvesting, cells were blocked with human FcR blocking reagent (Miltenyi) according to manufacturer’s protocol. Cells were labeled with recombinant human NKG2D, Nkp80, Nkp46, Nkp44 or Nkp30 Fc chimera proteins or the recombinant control IgG1 Fc protein (all R&D Systems) for 45 min at 4 °C, followed by the secondary antibody IgG1 conjugated with FITC (Invitrogen). Data analysis was carried out by Flow Cytometry (BD Bioscience) and results were expressed as fold change to isotype control.
Statistics.
All data are expressed as mean ± s.d. or in box blot graphs showing the median and the minimum to maximum range. Intergroup differences were appropriately assessed by either an unpaired Student’s t-test or a one-way ANOVA with Bonferroni’s post-hoc test. Further information on experimental design and reagents is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
We thank C. Pahrmann for cell culture work, imaging experiments and overall assistance and L. Li for his assistance. The in vivo BLI experiments were performed at the UCSF Pre-clinical Therapeutics Core (A. Fries; with special thanks to B.C. Hann). Special thanks go to J. Wu (Stanford Cardiovascular Institute, Stanford University School of Medicine) for providing the miPSCs and the help of his laboratory with developing the protocol for hiPSC differentiation into cardiomyocytes. We thank J.-F. Garcia-Gomez (City of Hope, Duarte) for the HLA typing of humanized BLT mice. We also thank E. Maltepe and H. Pektas for providing the syncytiotrophoblast cells. D.W. was supported by the Max Kade Foundation. A.W. received funding from the National Institutes of Health (grant AI123010). J.V.G. received funding from the National Institutes of Health (AI111899 and MH108179). The cardiomyocyte research was partly made possible by a grant from the California Institute for Regenerative Medicine (Grant Number DISC1–09984). Research related to cardiomyocyte immunobiology reported in this publication was supported by National Heart, Lung, and Blood Institute of the National Institutes of Health under award number R01HL140236. L.L.L. is an American Cancer Society Professor funded by NIH AI068129 and in part by the Parker Institute for Cancer Immunotherapy. S.S. and T.D. received funding for the cardiomyocyte research from the California Institute for Regenerative Medicine (Grant Number DISC1–09984) and for the immunobiology work from the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number R01HL140236. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH, CIRM and other agencies of the State of California.
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41587-019-0016-3.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available in the paper and its Supplementary Information files.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of data availability and associated accession codes are available at https://doi.org/10.1038/s41587-019-0016-3.
References
- 1.Gyongyosi M et al. Meta-analysis of cell-based CaRdiac stUdiEs (ACCRUE) in patients with acute myocardial infarction based on individual patient data. Circ. Res 116, 1346–1360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fisher SA, Doree C, Mathur A & Martin-Rendon E Meta-analysis of cell therapy trials for patients with heart failure. Circ. Res 116, 1361–1377 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Kandala J et al. Meta-analysis of stem cell therapy in chronic ischemic cardiomyopathy. Am. J. Cardiol 112, 217–225 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Fernandez-Aviles F et al. Global position paper on cardiovascular regenerative medicine. Eur. Heart J 38, 2532–2546 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lipsitz YY, Timmins NE & Zandstra PW Quality cell therapy manufacturing by design. Nat. Biotechnol 34, 393–400 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Blair NF & Barker RA Making it personal: the prospects for autologous pluripotent stem cell-derived therapies. Regen. Med 11, 423–425 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Chakradhar S An eye to the future: researchers debate best path for stem cell-derived therapies. Nat. Med 22, 116–119 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Smith DM Assessing commercial opportunities for autologous and allogeneic cell-based products. Regen. Med 7, 721–732 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Lipsitz YY, Bedford P, Davies AH, Timmins NE & Zandstra PW Achieving efficient manufacturing and quality assurance through synthetic cell therapy design. Cell. Stem. Cell 20, 13–17 (2017). [DOI] [PubMed] [Google Scholar]
- 10.van Berlo JH & Molkentin JD An emerging consensus on cardiac regeneration. Nat. Med 20, 1386–1393 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arck PC & Hecher K Fetomaternal immune cross-talk and its consequences for maternal and offspring’s health. Nat. Med 19, 548–556 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Jaiswal S et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 138, 271–285 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diecke S et al. Novel codon-optimized mini-intronic plasmid for efficient, inexpensive, and xeno-free induction of pluripotency. Sci. Rep 5, 8081 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang CH, Fontes JD, Peterlin M & Flavell RA Class II transactivator (CIITA) is sufficient for the inducible expression of major histocompatibility complex class II genes. J. Exp. Med 180, 1367–1374 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elsner L et al. The heat shock protein HSP70 promotes mouse NK cell activity against tumors that express inducible NKG2D ligands. J. Immunol 179, 5523–5533 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Maddaluno M et al. Murine aortic smooth muscle cells acquire, though fail to present exogenous protein antigens on major histocompatibility complex class II molecules. Biomed. Res. Int 2014, 949845 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Didie M, Galla S, Muppala V, Dressel R & Zimmermann WH Immunological properties of murine parthenogenetic stem cell-derived cardiomyocytes and engineered heart muscle. Front. Immunol 8, 955 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wunderlich M et al. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia 24, 1785–1788 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shultz LD, Ishikawa F & Greiner DL Humanized mice in translational biomedical research. Nat. Rev. Immunol 7, 118–130 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Billerbeck E et al. Development of human CD4+ FoxP3+ regulatory T cells in human stem cell factor-, granulocyte-macrophage colony-stimulating factor-, and interleukin-3-expressing NOD-SCID IL2Rgamma(null) humanized mice. Blood 117, 3076–3086 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melkus MW et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat. Med 12, 1316–1322 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Deuse T et al. Human leukocyte antigen I knockdown human embryonic stem cells induce host ignorance and achieve prolonged xenogeneic survival. Circulation 124, S3–S9 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Wang D, Quan Y, Yan Q, Morales JE & Wetsel RA Targeted disruption of the beta2-microglobulin gene minimizes the immunogenicity of human embryonic stem cells. Stem Cells Transl. Med 4, 1234–1245 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dressel R et al. Pluripotent stem cells are highly susceptible targets for syngeneic, allogeneic, and xenogeneic natural killer cells. FASEB J 24, 2164–2177 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Kruse V et al. Human induced pluripotent stem cells are targets for allogeneic and autologous natural killer (NK) cells and killing is partly mediated by the activating NK Receptor DNAM-1. PLoS ONE 10, e0125544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gornalusse GG et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol 35, 765–772 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao L, Teklemariam T & Hantash BM Heterelogous expression of mutated HLA-G decreases immunogenicity of human embryonic stem cells and their epidermal derivatives. Stem Cell Res 13, 342–354 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Hou S, Doherty PC, Zijlstra M, Jaenisch R & Katz JM Delayed clearance of Sendai virus in mice lacking class I MHC-restricted CD8+ T cells. J. Immunol 149, 1319–1325 (1992). [PubMed] [Google Scholar]
- 29.Shiba Y et al. Allogeneic transplantation of iPS cell-derived cardiomyocytes regenerates primate hearts. Nature 538, 388–391 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Kawamura T et al. Cardiomyocytes derived from MHC-homozygous induced pluripotent stem cells exhibit reduced allogeneic immunogenicity in MHC-matched non-human primates. Stem Cell Rep 6, 312–320 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.