

Graphical abstract
Keywords: Virtual screening, Fragments, Drug discovery, Medicinal chemistry, Cancer
Abstract
Fascin is an actin binding and bundling protein that is not expressed in normal epithelial tissues but overexpressed in a variety of invasive epithelial tumors. It has a critical role in cancer cell metastasis by promoting cell migration and invasion. Here we report the crystal structures of fascin in complex with a series of novel and potent inhibitors. Structure-based elaboration of these compounds enabled the development of a series with nanomolar affinities for fascin, good physicochemical properties and the ability to inhibit fascin-mediated bundling of filamentous actin. These compounds provide promising starting points for fascin-targeted anti-metastatic therapies.
Introduction
Fascin 1 (hereafter termed fascin) is an evolutionarily conserved actin binding protein that cross-links filamentous actin (F-actin) into tightly packed parallel bundles driving the formation of various cell surface protrusions, such as filopodia and invadopodia, that promote cell migration and invasion.1 It’s expression is highly restricted in adult human tissues.2 Fascin is either absent or shows low expression in normal epithelial tissues but is overexpressed in a number of cancers, with expression levels correlating with overall cancer aggressiveness and predicting poor clinical outcome.3, 4 Conversely, genetic knockdown of fascin decreases tumor cell invasion both in vitro and in vivo,5, 6 suggesting that fascin may be a viable target for anticancer/antimetastatic drugs. Huang et al. have disclosed compounds which bind to fascin and inhibit actin bundling.7, 8, 9 However, in our hands a number of these compounds bound with modest (10–100 µM) affinity and suffered from poor solubility.
In order to identify novel fascin compounds, with favourable properties, we undertook a surface plasmon resonance (SPR) screen using our collection of 1050 fragments and identified 53 hits (hit rate 5%). Follow-up protein crystallography yielded a range of fragments binding to multiple sites on the protein.
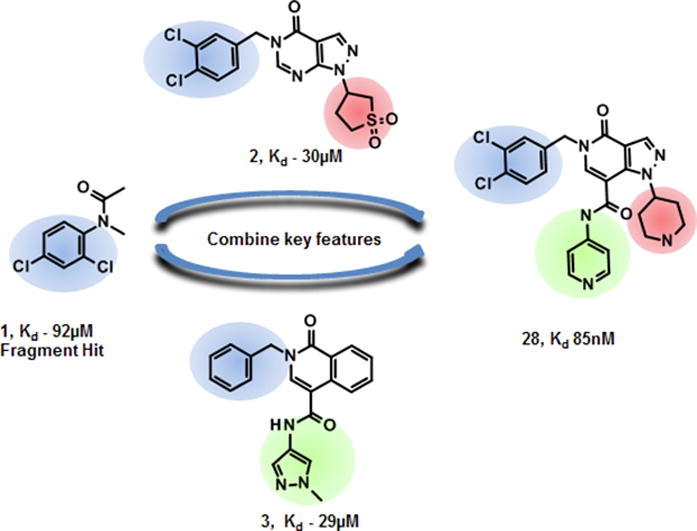
The N-phenylacetamide 1 (Fig. 1A) binds efficiently (SPR Kd = 92 μM; ligand efficiency 0.43)11 in a buried pocket between the first and second β-trefoil domain (Fig. 1B) and was therefore selected for further development. Binding of compound 1 induces a hydrophobic pocket in domain 1, mainly by displacing Trp101. In addition, domain 1 pivots by ≈15°, reorienting residues on either side of the domain interface and creating a narrow channel to the protein surface (Fig. 1C and D). The dichlorophenyl moiety of 1 fills the hydrophobic pocket, while the acetamide engages in hydrophilic interactions with domain 2, with its carbonyl accepting a hydrogen bond from the backbone of Leu214 (Fig. 1E). While the completely enclosed binding site makes compound 1 a highly efficient ligand, it hampered attempts to develop this series.
Fig. 1.

Fascin structure and fragment 1 binding. A) Chemical structure of compound 1. B) Overall structure of fascin represented as a cartoon with the four β-trefoil domains colored differently and labelled. The bound compound 1 is shown as cyan spheres (PDB id 6I0Z). C) The interface between domains 1 and 2 in apo-fascin (PDB id 3P53)10. Domains are delineated by separate surfaces colored as in B) and labelled. Residues at the interface are shown as grey sticks and labelled with single-letter amino acid codes and residue numbers. D) Compound 1 bound to fascin, representation and view as in C) with the compound shown as cyan sticks. The structures were superimposed on domain 1 only. E) Detailed view of the binding site shown as a semi-transparent surface with surrounding protein residues drawn as sticks colored and labelled. Hydrogen bonds are shown as dashed pale green lines, other conventions as for D). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The importance of this fragment binding site was highlighted when we determined the crystal structure of compound 2 (Fig. 2A) previously identified by Huang et al. to inhibit fascin-mediated actin bundling.12 To accommodate the larger compound 2, domain 1 undergoes a more dramatic conformational change, rotating 35° around an axis almost parallel to a line connecting the first two β-trefoil domains (Fig. 2B). For the compound binding site (Fig. 2C) this almost pure rotation results in a 6 Å ‘drop’ of domain 2 relative to domain 1, so that the Phe216 side chain, which in the fascin·compound 1 complex lines the channel, now extends the hydrophobic pocket (cf. Figs. 1D and 2C) where the compound’s dichlorobenzyl moiety binds. More importantly, the conformational change opens up the channel so that it can accommodate the rest of the compound. The pyrazolopyrimidinone core rests on a relatively flat surface created by the salt-bridged side chains of Glu215 and Arg217. Additionally, compound 2 accepts a hydrogen bond from Phe216 and makes a relatively weak water-bridged interaction with the side chain of Glu215 (Fig. 2D). The induced conformational changes provide 2 with access to the protein surface creating opportunities for structure-driven compound elaboration. Notably, the recently disclosed structure of fascin in complex with an unrelated small molecule bound in the same site9 shows fascin adopting a similar conformation (RMSD < 1.3 Å for 483 Cα atoms).
Fig. 2.

Fascin undergoes a significant conformational change upon binding of compounds 2 and 3. A) Chemical structures of 2 and 3. B) Apo-fascin (PDB id 3P53, red) and the fascin·2 complex (PDB id 6I10, purple) superimposed on the relatively rigid domains 2–4 (grey; RMSD = 1.1 Å for 348 Cα atoms), viewed along the domain 1 rotation axis. C) Compound 2 bound to fascin oriented as Fig. 1C/D (proteins superimposed on domain 1). D) Detail view of the fascin·2 complex. Conventions as in Fig. 1E, the red sphere represents a bound water molecule. E) Detailed view of the fascin·3 complex (PDB id 6I11). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To identify compounds which could potentially fill these additional areas a hybrid virtual screen was carried out. The first method was structure-based: 809 commercial analogues of compound 2 and ≈2.7 million lead-like molecules from our in-house virtual library were docked13 in the enclosed site. To prioritize the docked compounds, a multi-parameter optimization was performed using Pareto efficiency14 as the ranking technique with calculated free energy of binding (from docking), QED15 and interaction pattern similarity (based on compound 2 interaction fingerprints)16 as the parameters to optimize. The second method was ligand-based using shape-similarity17 to find analogues of compound 2 in our in-house library. Combining the results, 110 commercial compounds were tested by SPR at a screening concentration of 100 µM. The resulting binders were validated by co-crystallization and SPR dose-response experiments. Eighteen compounds were confirmed to bind in the same site as 2, and three yielded Kd values < 100 μM. Of these three, we decided to progress the isoquinolinone 3 (SPR Kd = 29.3 ± 5.8 µM; Fig. 2A and E), which maintains affinity similar to 2 despite substituent truncations from both the benzyl and the core ring system. The pyrazolamide moiety forms an edge-to-face aromatic interaction with the Trp101 side chain. It also moves the bridging water seen in the fascin·compound 2 complex structure deeper into the binding site. This water now hydrogen-bonds to the Thr213 side chain, the Leu214 backbone carbonyl and the ligand amide in addition to the Glu215 side chain, which ‘follows’ it by adopting a different rotamer (Fig. 2D and E). Analysis of this water with MOE 3D-RISM18 suggests it is relatively tightly bound (ΔG = −3.9 kJ/mol) and that displacing it would likely be energetically unfavorable.
In order to measure the effect of these compounds on fascin activity we developed a robust and highly reproducible in vitro F-actin bundling assay, based on a previously published method.19 Unlike compound 1, both 2 and 3 inhibit fascin in this bundling assay. Compound 3 inhibits actin bundling with an IC50 of 67.9 ± 3.5 μM, giving an approximately two-fold fall-off from SPR binding affinity. A similar fall-off is seen for other compounds in this series.
Considering that both 1 and 2 place a substituted phenyl into the hydrophobic pocket, it seemed plausible that the plain benzyl of 3 is not optimal for filling the available space. Additionally, given that this induced pocket is likely to show some plasticity, it may be possible to open it up further by extending the compound. To investigate this, we synthesized a series of benzyl derivatives of 3 (Table 1). Introduction of a para-halo substituent (compounds 4 and 5) increased binding affinity ≈14-fold compared to 3. The crystal structure of 5 bound to fascin shows a binding mode without substantial changes in either ligand or protein conformation compared to the fascin·3 complex (Fig. S1A). A single meta-fluoro substituent (6) is equipotent while m-chloro substitution (7) improved affinity, though less so than 4 or 5. This may be partly explained by the slight differences in binding mode for meta substituted compounds compared to 3, in which the benzyl moiety is rotated to line up the m-substituent with a pocket indentation (Fig. S1B). In light of this conformational divergence, it is not surprising that m,p-dihalo compounds exhibit sub-additive affinity gains, though both 8 and 9 showed improved binding over mono- or unsubstituted analogues with SPR Kd values of 1.2 and 1.5 μM respectively. The structure of the fascin·9 complex shows the compound adopts a conformation with the phenyl rotated to an intermediate position compared to the meta and para analogues (Fig. S1C). Larger meta or para substituents (e.g. 10 and 11; Fig. S1D) are tolerated by expanding the hydrophobic pocket. However, the associated conformational penalty consistently makes these compounds less potent. Introduction of an ortho substituent decreases potency both in combination with a para substituent (12) or as a single substituent (data not shown). Taken together the SAR suggests that m,p-dichloro substitution (9) is close to optimal in this series.
Table 1.
Variation of benzyl substituents.
| Compound | 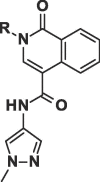 |
Kd ± SDa (µM) | IC50 ± SDb (µM) |
|---|---|---|---|
| 3 |  |
29.3 ± 5.8 | 67.9 ± 3.5 |
| 4 |  |
2.7 ± 0.1 | nd |
| 5 | 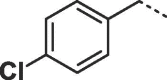 |
2.7 ± 0.1 | 4.6 ± 0.3 |
| 6 | 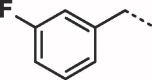 |
29.2 ± 0.9 | nd |
| 7 | 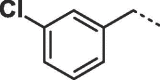 |
7.6 ± 2.5 | 11.4 ± 4.2 |
| 8 | 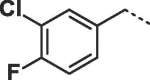 |
1.2 ± 0.1 | 2.1 ± 0.8 |
| 9 |  |
1.5 ± 0.1 | 1.3 ± 0.2 |
| 10 |  |
6.6 ± 2.0 | 8.6 ± 4.2 |
| 12 |  |
46 ± 1.7 | nd |
Kd: SPR binding affinity, values are mean ± standard deviation from n = 2–10 separate experiments.
IC50: compound concentration that reduces actin bundling by 50%, values are mean ± standard deviation.
We next investigated altering the bicyclic scaffold of 3 (Table 2), taking care to retain the pyridinone moiety, which we found to be essential for binding. Complete deletion of the outer ring (13) led to a significant loss of affinity, which can be partly recovered with the addition of a 3-amino substituent (14), which donates an additional hydrogen bond to the Phe216 backbone carbonyl. The naphthyridinones 16 and 17 maintain binding affinities similar to the parent isoquinolinone, while placement of the naphthyridinone nitrogen next to the amide (15) results in the formation of an intramolecular hydrogen bond stabilising a conformation that is not optimal for fascin binding (Fig. S2A), and thus a decrease in binding affinity. For comparison, a matched pyrazolopyridinone 19 shows 7-fold weaker binding.
Table 2.
Variation of the bicyclic scaffold.
| Compound |  |
Kd ± SDa (µM) | IC50 ± SDb (µM) |
|---|---|---|---|
| 9 | 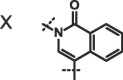 |
1.5 ± 1 | 1.3 ± 0.2 |
| 13 | 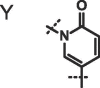 |
>100 | nd |
| 14 | 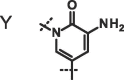 |
21 ± 4.2 | >100 |
| 15 | 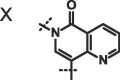 |
>100 | nd |
| 16 | 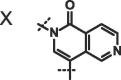 |
1.6 ± 0.1 | 5.3 ± 0.8 |
| 17 | 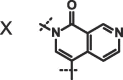 |
1.3 ± 0.1 | 3.8 ± 0.3 |
| 18 | 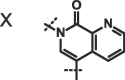 |
>100 | nd |
| 19 | 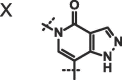 |
10.0 ± 1.4 | 10.4 ± 0.2 |
X, R = Me; Y, R = 3-pyrolidine;
Kd: SPR binding affinity, values are mean ± standard deviation from n = 2–10 separate experiments;
IC50: compound concentration that reduces actin bundling by 50%, values are mean ± standard deviation from n = 2–4 separate experiments; nd: not determined.
Considering their improved physicochemical properties and chemical tractability, we decided to progress a naphthyridinone, initially by adding substituents to replicate the space filling of the cyclic sulphone of 2. A binding mode comparison (Fig. S2B) suggested that substitution from C5 would provide the best-matched vector, though 6-substituents should be able to access the same space. Due to concerns about the potential clash between a 5-substituent and the pyrazoloamide preventing the compound from adopting the preferred fascin-binding conformation, we opted to elaborate 17 through substitution from the 6-chloro derivative (Table 3). The introduction of simple alkylamine substituents (20, 22) had no significant effect on fascin binding. The sulphonamide (21) aiming to more closely replicate the sulphone of compound 2 also failed to improve affinity. The piperazine 23 gave slightly improved fascin binding, despite ostensibly placing a partial positive charge close to the guanidine group of Arg217 (cf. Fig. 2E). The piperidine analogue (24) shows further improved fascin binding and inhibition. The crystal structure of the fascin·compound 24 complex shows the compound binding broadly as expected (Fig. S3A and B). The naphthyridinone makes an additional water-bridged interaction with the Phe216 backbone carbonyl. The piperidine orients to fill the space between Ala58 and Arg217 (Fig. S3C), but does not make any specific interactions, as its amine points towards bulk solvent. Comparing the binding modes of 24 and 9 reveals that the core ring system of 24 is rotated slightly (Fig. S3D), possibly to optimize the fit of the piperidine. This small rotation has an amplified effect on the position of the pyrazoloamide, bringing the pyrazole closer to Trp101 and Arg225 while at the same time improving the hydrogen bond from the amide to the bridging water. The displacement observed for 24 suggests that naphthyridinones with bulky 6-substituents may be sterically restricted from adopting an optimal core binding mode. In light of this we chose to revisit the pyrazolo series, where we had previously observed that the equally bulky cyclic sulphone substituent of 2 was compatible with the ‘standard’ core orientation (Fig. S2B). The truncated compound 25 showed diminished binding (Table 4), revealing that addition of either the pyrazoloamide in the 7-position (to obtain 19) or the cyclic sulphone at N1 (to obtain 2) to this compound improves fascin affinity >50-fold and 23-fold, respectively. Combining these key features into one compound gave 26, with an SPR Kd of 0.6 μM, suggesting near-additive gains in binding affinity compared to 24 and almost closing the potency gap to our best naphthyridinones.
Table 3.
6-substitution of the napthyridinone core.
| Compound | 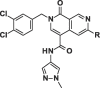 |
Kd ± SDa (µM) | IC50 ± SDb (µM) |
|---|---|---|---|
| 17 | H | 1.3 ± 0.1 | 3.8 ± 0.3 |
| 20 |  |
1.1 ± 0.2 | nd |
| 21 | 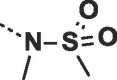 |
2.0 ± 0.1 | nd |
| 22 |  |
1.03 ± 0.03 | nd |
| 23 | 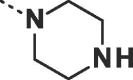 |
0.58 ± 0.02 | 0.63 ± 0.09 |
| 24 | 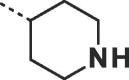 |
0.25 ± 0.1 | 0.51 ± 0.07 |
Kd: SPR binding affinity, values are mean ± standard deviation from n = 2–10 separate experiments;
IC50: compound concentration that reduces actin bundling by 50%, values are mean ± standard deviation from n = 2–4 separate experiments; nd: not determined.
Table 4.
Further substitutions of the pyrazolopyridinone scaffold.
| Compound | R1-group |
R2-group |
Kd ± SDa (µM) | IC50 ± SDb (µM) |
|---|---|---|---|---|
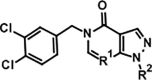 | ||||
| 25 | N | H | >500 | nd |
| 2 | N | 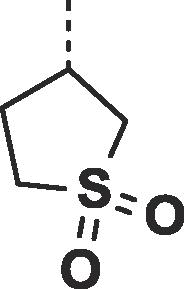 |
29.5 ± 8.3 | 67.6 ± 2.4 |
| 26 | 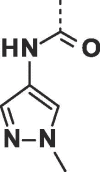 |
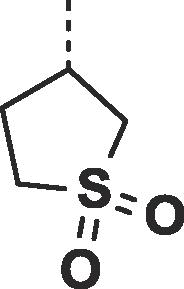 |
0.60 ± 0.05 | 0.63 ± 0.15 |
| 27 | 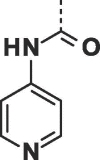 |
 |
0.27 ± 0.04 | 0.33 ± 0.06 |
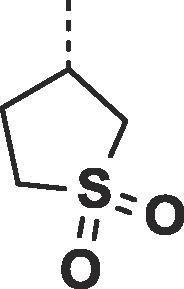
| 28 | 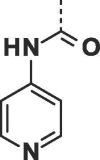 |
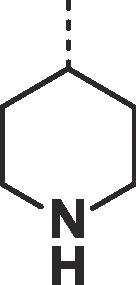 |
0.09 ± 0.02 | 0.24 ± 0.01 |
Kd: SPR binding affinity, values are mean ± standard deviation from n = 2–10 separate experiments.
IC50: compound concentration that reduces actin bundling by 50%, values are mean ± standard deviation from n = 2–4 separate experiments; nd: not determined.
We next investigated changes to the heteroaromatic amide. In line with the predicted importance of the bridging water discussed above, any changes to the amide (N-alkylation, carbonyl deletion) abolish fascin binding (not shown). On the other hand, the methylpyrazole interacts with the protein through stacking with the indole of Trp101. A number of compounds with alternative heterocycles were made to probe this interaction, focusing on five- and six-membered nitrogen-containing heterocycles. Of these the pyridin-4-yl 27 yielded the largest, albeit still modest, improvement over 26 in terms of both affinity and inhibition. QM calculations (Fig. S4) suggest that at least for (hetero)aryl derivatives improved stacking with Trp101 may be a major factor in this increase in potency.
Given the success of 24 and to alleviate concerns about chemical stability of the cyclic sulphone substituent, we opted to replace it with a 1-piperidin-4-yl.
Synthesis of this was achieved starting from the commercially available diethyl beta-ketoglutarate (29) and reacting with N,N-dimethylformamide dimethyl acetal (Scheme 1).20 Addition of tert-butyl 4-hydrazinopiperidine-1-carboxylate led to the pyrazole (31), which was further functionalised by refluxing with Bredereck’s reagent followed by the addition of dichlorobenzylamine. Partial cyclisation was observed at this stage, however, the crude product was fully cyclised with sodium ethoxide. This gave intermediate pyrazlopyridinone (32) in good yield over the three steps. Saponifaction of the ester, coupling of the 4-amino pyridyl and removal of the Boc protecting group furnished BDP-13176 (28). BDP-13176 further improved fascin affinity, giving us the best fascin binder (SPR Kd = 85 ± 0.02 nM, LE = 0.29, ITC Kd = 50 nM; Fig. 3A and B) and actin bundling inhibitor (IC50 = 240 ± 0.01 nM; Fig. 3C and D).
Scheme 1.

Reagents and conditions: (a) N, N-dimethylformamide dimethyl acetal, ethanol r.t, (75%); (b) 4 tert-butyl 4-hydrazinopiperidine-1-carboxylate, ethanol, reflux., (27%); (c-e) Bredereck’s, toluene reflux; 3,4-dichlorophenyl)methanamine, acetic acid, toluene; sodium ethoxide, ethanol (3 steps 85%) (f) Sodium hydroxide, ethanol r.t. (84%),(g) HATU, N,N-diisopropylethylamine, DMF, r.t., (83%), (h) TFA, DCM r.t. (52%).
Fig. 3.

Comparison of SPR binding affinity, ITC binding parameters and functional bundling assay potency for BDP-13176. A) Multi-cycle kinetic curve for BDP-13176 binding to immobilised (His8)2-fascin. Data were fitted using a 1:1 kinetic binding model. B) ITC titration thermogram and referenced isotherm (first point removed) of BDP-13176 (150 µM in syringe) binding to fascin (15 µM in cell). Fitting of the isotherm to a 1:1 binding model resulted in the following binding parameters: N = 1.05 ± 0.01, Kd = (45 ± 7) nM, ΔH = (−2400 ± 200) cal mol−1, ΔS = −45.9 cal mol−1 K−1. C) Inhibition of fascin bundling activity by BDP-13176. Inhibition of bundling activity can be seen as fascin and F-actin move from the pellet (P) to the supernatant (S) with increasing concentrations of BDP-13176. D) Concentration-response curve for bundling assay data shown in C).
The crystal structure of fascin in complex with BDP-13176 shows the compound adopting the expected overall binding mode (Fig. 4), but with a slight core rotation in the opposite direction from that observed for 24 (Fig. S5). The compound‘s in vitro physicochemical and DMPK properties (Table S1) are within acceptable limits with low lipophilicity (LogD7.4 = 1.8) and reasonable kinetic solubility (midpoint = 65 µM). The compound was also moderately stable in both human and mouse liver microsomes (Human Clint = 6.4, Mouse Clint = 11.8, µL/min/mg protein). However, the compound has low Caco-2 permeability (A-B Papp = 0.15x10-6 cm/s) and high efflux (efflux ratio = 37.8).
Fig. 4.

Binding of BDP-13176 to fascin. A) Crystal structure of the fascin·BDP-13176 complex (PDB id 6I18). The bridging water seen in earlier complexes is replaced by two ordered waters that together make the same contacts as the single bound water. B) Interaction plot for BDP-13176 bound to fascin.
In conclusion, fragment screening coupled with crystallography and computational approaches allowed identification of a cryptic pocket within fascin. Using compound 2 (Kd = 29.5 ± 8.3 µM)12 as a starting point combined with virtual screening and structure-based design allowed us to develop potent and functionally active fascin binders. Crystal structures reveal the compounds bind with a hydrophobic ‘hook’ in an induced pocket between the first two β-trefoil domains of fascin and from there extend towards the protein surface, effecting a substantial conformational change in domain 1. While the mechanism by which our compounds inhibit bundling remains unclear, it is worth noting that fascin’s two major proposed actin binding regions involve domain 1 and cross a domain boundary. The ligand-induced conformational change would deform both these regions and thereby disrupt actin binding (Fig. S6). We anticipate these compounds to be a useful foundation to further probe fascin’s potential role in tumor invasion and metastasis.
Acknowledgments
Acknowledgements
This research was supported by funding from Cancer Research UK (grants A17196 and A17096) and the Pancreatic Cancer Research Fund. We would like to thank Diamond Light Source for beamtime (proposals mx6683, mx8659 and mx11651) and the staff of beamlines i02, i03 and i04 for their assistance. We also thank David Sumpton and Gillian McKay for mass spectrometric analyses, and Daniel James for informatics support.
Experimental section
Experimental details can be found within the supplementary data file provided.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2019.01.035.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Li A., Dawson J.C., Forero-Vargas M. The actin-bundling protein fascin stabilizes actin in invadopodia and potentiates protrusive invasion. Curr Biol. 2010;20(4):339–345. doi: 10.1016/j.cub.2009.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hashimoto Y., Kim D.J., Adams J.C. The roles of fascins in health and disease. J Pathol. 2011;224(3):289–300. doi: 10.1002/path.2894. [DOI] [PubMed] [Google Scholar]
- 3.Tan V.Y., Lewis S.J., Adams J.C., Martin R.M. Association of fascin-1 with mortality, disease progression and metastasis in carcinomas: a systematic review and meta-analysis. BMC Med. 2013;11:52. doi: 10.1186/1741-7015-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma Y., Machesky L.M. Fascin1 in carcinomas: Its regulation and prognostic value. Int J Cancer. 2015;137(11):2534–2544. doi: 10.1002/ijc.29260. [DOI] [PubMed] [Google Scholar]
- 5.Li A., Morton J.P., Ma Y. Fascin is regulated by slug, promotes progression of pancreatic cancer in mice, and is associated with patient outcomes. Gastroenterology. 2014;146(5):1386–1396. doi: 10.1053/j.gastro.2014.01.046. e1381-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minn A.J., Gupta G.P., Siegel P.M. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436(7050):518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang F.K., Han S., Xing B. Targeted inhibition of fascin function blocks tumour invasion and metastatic colonization. Nat Commun. 2015;6:7465. doi: 10.1038/ncomms8465. [DOI] [PubMed] [Google Scholar]
- 8.Han S., Huang J., Liu B. Improving fascin inhibitors to block tumor cell migration and metastasis. Mol Oncol. 2016;10(7):966–980. doi: 10.1016/j.molonc.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang J., Dey R., Wang Y., Jakoncic J., Kurinov I., Huang X.Y. Structural Insights into the Induced-fit Inhibition of Fascin by a Small-Molecule Inhibitor. J Mol Biol. 2018;430(9):1324–1335. doi: 10.1016/j.jmb.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jansen S., Collins A., Yang C., Rebowski G., Svitkina T., Dominguez R. Mechanism of actin filament bundling by fascin. J Biol Chem. 2011;286(34):30087–30096. doi: 10.1074/jbc.M111.251439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hopkins A.L., Groom C.R., Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today. 2004;9(10):430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 12.Huang X-Y, Shue, CY. Methods for Inhibiting Fascin. WO/2014/031732; 2014.
- 13.Friesner R.A., Murphy R.B., Repasky M.P. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 14.Deb K., Agrawal S., Pratap A., Meyarivan T. Springer Berlin Heidelberg; Berlin, Heidelberg: 2000. A Fast Elitist Non-dominated Sorting Genetic Algorithm for Multi-objective Optimization: NSGA-II; pp. 849–858. [Google Scholar]
- 15.Bickerton G.R., Paolini G.V., Besnard J., Muresan S., Hopkins A.L. Quantifying the chemical beauty of drugs. Nat Chem. 2012;4(2):90–98. doi: 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng Z., Chuaqui C., Singh J. Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein-ligand binding interactions. J Med Chem. 2004;47(2):337–344. doi: 10.1021/jm030331x. [DOI] [PubMed] [Google Scholar]
- 17.Sastry G.M., Dixon S.L., Sherman W. Rapid shape-based ligand alignment and virtual screening method based on atom/feature-pair similarities and volume overlap scoring. J Chem Inf Model. 2011;51(10):2455–2466. doi: 10.1021/ci2002704. [DOI] [PubMed] [Google Scholar]
- 18.Truchon J.F., Pettitt B.M., Labute P. A cavity corrected 3D-RISM functional for accurate solvation free energies. J Chem Theory Comput. 2014;10(3):934–941. doi: 10.1021/ct4009359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamashiro-Matsumura S., Matsumura F. Purification and characterization of an F-actin-bundling 55-kilodalton protein from HeLa cells. J Biol Chem. 1985;260(8):5087–5097. [PubMed] [Google Scholar]
- 20.Grošelj U., Pušavec E., Golobič A., Dahmann G., Stanovnik B., Svete J. Synthesis of 1,5-disubstituted-4-oxo-4,5-dihydro-1H-pyrazolo[4,3-c]pyridine-7-carboxamides. Tetrahedron. 2015;71(1):109–123. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.