

Summary
DNA methylation is essential for gene regulation, imprinting and silencing of transposable elements (TEs). Although bursts of transposable elements are common in many plant lineages, how plant DNA methylation is related to transposon bursts remains unclear. Here we explore the landscape of DNA methylation of tea, a species thought to have experienced a recent transposon burst event. This species possesses more transposable elements than any other sequenced asterids (potato, tomato, coffee, pepper and tobacco). The overall average DNA methylation levels were found to differ among the tea, potato and tomato genomes, and methylation at CHG sequence sites was found to be significantly higher in tea than that in potato or tomato. Moreover, the abundant TEs resulting from burst events not only resulted in tea developing a very large genome size, but also affected many genes involved in importantly biological processes, including caffeine, theanine and flavonoid metabolic pathway genes. In addition, recently transposed TEs were more heavily methylated than ancient ones, implying that DNA methylation is proportionate to the degree of TE silencing, especially on recent active ones. Taken together, our results show that DNA methylation regulates transposon silencing and may play a role in genome size expansion.
Keywords: DNA methylation, tea methylome, genome expansion, TE burst
Introduction
DNA methylation is a stable epigenetic mark that is widespread among eukaryotic species. Extensive studies have shown that DNA methylation plays an important role in regulation gene expression and transposon silencing (Finnegan et al., 2000; Gehring and Henikoff, 2007). Unlike in animals, where methylation often occurs exclusively on CG sequences, in plants, methylation is common in three different contexts: CG, CHG (where H can be A, T or C) and CHH (Cokus et al., 2008; Feng et al., 2010; Lister et al., 2008). The maintenance of CG context methylation is functionalized by methyltransferase 1 (MET1), which is recruited to hemi‐methylated CG sites during DNA replication (Bostick et al., 2007). In contrast, CHG methylation is maintained by plant‐specific chromomethylase 3 (CMT3); this methylation is strongly associated with dimethylation of lysine 9 on histone 3 (H3k9me2), and CMT3 acts together with histone methyltransferase and SUVH5/6 to form a self‐reinforcing loop (Jackson et al., 2002; Lindroth et al., 2001). CHH methylation is established and maintained by CMT2, and like CHG methylation, it is associated with methylation in H3K9me2 regions (Gouil and Baulcombe, 2016). De novo establishment of DNA methylation in all three contexts is mediated by the RNA‐directed DNA methylation (RdDM) pathway guided by small RNAs and domains rearranged methyltransferase 2 (DRM2) (Cao and Jacobsen, 2002). Recent studies have showed that RdDM and CMT2 often complement each other. Moreover, CMT2 also plays a role in CHG methylation (Law and Jacobsen, 2010; Stroud et al., 2013).
Current knowledge of DNA methylation in plants is primarily based on studies of Arabidopsis thaliana, which has a compact genome and is relatively tolerant to significant reductions in DNA methylation. However, most plant genomes are much larger and possess an abundance of transposons (Dodsworth et al., 2015). Genome‐wide DNA methylation patterns may vary considerably, even among closely related plant species (Kim et al., 2015; Wang et al., 2018). Advances in DNA methylation sequencing have permitted the construction of the methylomes of many plant species, often profiled at single‐base resolution (Niederhuth et al., 2016). Comparative methylome studies have shown that genome‐wide average DNA methylation levels are positively correlated with genome size and TE contents (Takuno et al., 2016; Wang et al., 2018); thus, the larger the genome, the higher the DNA methylation level of the DNA.
Tea is one of the world's most important beverage crop, and provides a wealth of health benefits. Analysis of tea genome has revealed that this species experienced a burst event of transposable elements in its evolutionary past, and now possesses one of the largest genomes of all sequenced asteroid species (Wei et al., 2018; Xia et al., 2017). The availability of the tea genome allows us to explore the DNA methylation landscape at the whole genome level and to determine the relationship between DNA methylation and the abundance of transposons in the tea genome. To better understand the role of DNA methylation in the TE‐abundant tea genome, we present a single‐base resolution DNA methylation landscape of the tea by whole genome bisulphite sequencing. We found that the levels and patterns of DNA methylation are different from those found in other asterids, such as potato and tomato, and that CHG methylation level is significantly higher in the tea genome relative to other plant species. This fact is likely due to the presence of abundant repetitive DNA elements and the large overall size of the genome. In addition, average DNA methylation levels of TEs are reduced as the evolutionary ages increased, suggesting DNA methylation roles on silencing of recent active transposons.
Results
DNA methylation pathway‐related genes in the tea genome
Genetic studies of the model plant Arabidopsis thaliana have defined many of the key components involved in the DNA methylation pathways, including MET1, DRM2, CMT2, and CMT3 (Law and Jacobsen, 2010; Matzke et al., 2015). To comprehensively assess the function of these genes in tea, we searched the tea genome for homologs of A. thaliana methylation pathway‐related genes by using BLASTP and the HMM algorithm. We found that most of the DNA methylation pathway‐related genes found in A. thaliana are conserved in tea, including MET1, CMT2, CMT3 and DRM2 (Table 1). In addition, at least one copy of homologs of genes involved in the RNA‐directed DNA methylation pathway (RdDM), including SHH1, NRPD1, NRPE1, DMS3 and DCL2, were also found in the tea genome. Furthermore, RNA‐seq data showed that those genes were expressed (Table 1). Taken together, our data suggest that the DNA methylation pathway is functional and conserved in tea.
Table 1.
Putative DNA methylation pathway genes in tea tree
| Tea tree (Camellia sinensis) | ||||
|---|---|---|---|---|
| Name (Arabidopsis) | Length (a.a)* | Orthologs | Expression level (FPKM) | |
| MET1 | VIM1,2,3,4,5,6 | 645 | CSA002777.1 | 2.55959 |
| MET1,2a,2b,3 | 1534 | CSA018787.1 | 0.987532 | |
| CMT3 | SUVH4 | 624 | CSA002812.1 | 11.2352 |
| CMT2 | 1295 | CSA032127.1 | 2.48205 | |
| CMT3 | 839 | CSA030966.1 | 3.81276 | |
| Pol IV recruit | CLSY1/CLSY2 | 1256 | CSA023919.1 | 11.5655 |
| SHH1/SHH2 | 258 | CSA015026.1 | 2.85557 | |
| Pol IV | NRPD1 | 1453 | CSA001027.1 | 4.89804 |
| Pol IV+V | NRPD2/NRPE2 | 1172 | CSA013061.1 | 3.41522 |
| Pol IV+V | NRPD4/NRPE4 | 205 | CSA033129.1 | 5.28803 |
| Pol V | NRPE1 | 1976 | CSA002792.1 | 8.60674 |
| Pol V | NRPE5 | 222 | CSA008802.1 | 23.2597 |
| Pol V | NRPE9B | 114 | CSA022741.1 | 9.89088 |
| Pol V recruit | DRD1 | 888 | CSA033157.1 | 6.42526 |
| DMS3 | 420 | CSA003130.1 | 3.2686 | |
| RDM1 | 163 | CSA033069.1 | 5.54529 | |
| SUVH2/9 | 650 | CSA033444.1 | 15.037 | |
| RdDM | RDR2 | 1133 | CSA003297.1 | 5.3044 |
| DCL1 | 1910 | CSA034540.1 | 1.34299 | |
| DCL2 | 1388 | CSA001781.1 | 3.48661 | |
| DCL3 | 1580 | CSA030036.1 | 10.0673 | |
| DCL4 | 1702 | CSA011120.1 | 1.0754 | |
| HEN1 | 942 | CSA016903.1 | 1.51594 | |
| AGO4 | 924 | CSA016856.1 | 57.3644 | |
| KTF1 | 1493 | CSA021730.1 | 11.9791 | |
| IDN2 | 647 | CSA020735.1 | 8.02238 | |
| SUVR2 | 740 | CSA021101.1 | 2.91624 | |
| DMS4 | 346 | CSA023931.1 | 10.5941 | |
| UBP26 | 1067 | CSA033342.1 | 20.4559 | |
| DRM2 | 626 | CSA001531.1 | 13.0907 | |
| LDL1 | 844 | CSA034653.1 | 4.18459 | |
| LDL2 | 746 | CSA000647.1 | 1.91191 | |
| JMJ14 | 954 | CSA018589.1 | 3.90207 | |
| Others | HDA6 | 471 | CSA007759.1 | 12.977 |
| RDR6 | 1196 | CSA020358.1 | 11.412 | |
| MORC6 | 663 | CSA024443.1 | 4.76682 | |
| DDM1 | 764 | CSA030020.1 | 2.27703 | |
For each gene family, the length of protein indicates the longest protein in this gene family, and only one ortholog was identified in Tea tree genome.
Genome‐wide profile of DNA methylation in tea
To explore single‐base resolution of DNA methylation in tea, we performed whole genome bisulphite sequencing (BS‐seq) of young leaf tissue. We used two independent biological replicates and generated 1.3 billion reads in total. BS‐seq reads were mapped to the Camellia sinensis var. assamica genome (Xia et al., 2017), and methyl‐cytosines were identified by BSMAP (Xi and Li, 2009). More than 70% of the total reads were uniquely mapped, yielding an average ~25 coverage sequencing depth of each replicate (Table S1). Meanwhile, approximately 80% of the total cytosines were covered by at least 1 read (Figure S1). A pairwise comparison of the methylation levels of 2 kb bins across the genome revealed that two biological replicates were well correlated in all three‐sequence contexts, indicating high reproducibility and adequate coverage of our replicates (Figure S2).
The genome‐wide landscape of DNA methylation of the first ten scaffolds are shown in Figure 1a. Consistent with Figure S2, we found that the DNA methylation patterns of both replicates were like one another along the entire length of the scaffolds, and that TE density was correlated with DNA methylation level. The genome‐wide average methylation of the CG, CHG and CHH contexts were 82%, 70% and 10%, respectively; these levels were much higher than those found in potato at CG and CHG sites (Figure 1b). We also observed that the position‐wise frequency of CG and CHG methylation showed bimodal distribution; this pattern was seen in both potato and tomato (Figure S3A). Moreover, highly methylated CHG sites (i.e. those with methylation levels >75%) were found in greater abundance than in potato and tomato. However, the proportion of methyl‐cytosines of the different contexts were similar among the three asterids genomes, and very different from the proportions found in other plant lineages, implying that the proportion of methyl‐cytosines is lineage‐specific (Figure S3B). The DNA methylation of symmetric sites (CG and CHG) was also found to be highly correlated highly in Arabidopsis, but the weak correlation of the CG and CHG sites has also been observed in a closely related relative, Arabis alpina (Willing et al., 2015). In tea, we also observed a weaker correlation of DNA methylation between two strands at CG and CHG sites than was found in potato and tomato (Figure S4). This suggests that the maintenance of both CG and CHG methylation is relatively weak. In addition, we found that CHG methylation was significantly higher than in both potato and tomato, and was comparable to other plants such as maize and Norway spruce (Ausin et al., 2016; Niederhuth et al., 2016). Recent studies have confirmed that the proportion of the genome composed of transposable elements and methylation levels of symmetrical contexts are both positively correlated with genome size (Takuno et al., 2016; Wang et al., 2018), partly explaining the high CHG methylation levels found in the tea genome.
Figure 1.
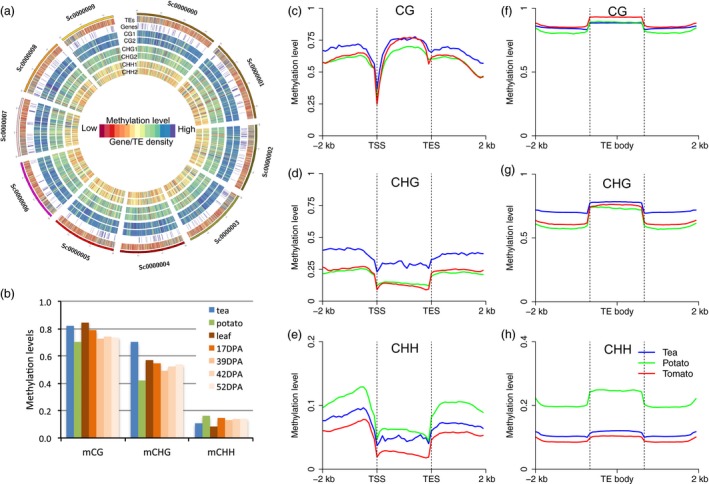
The DNA methylome landscape of tea tree. (a) The landscape of DNA methylation in the first 10 chromosomes of tea. From outer to inner: TE density, gene density, and methylation of CG rep1, CG rep2, CHG rep1, CHG rep2, CHH rep1 and CHG rep2. Blue indicates high methylation levels and high gene/TE density. Red indicates low methylation and low gene/TE density. (b) DNA methylation comparison between tea and other asterids, including potato and tomato (leaf, and fruit ripening stages: 17 days post anthesis (DPA), 39DPA, 42 DPA and 52DPA). (c–e). DNA methylation patterns of gene body and flanking regions in the CG, CHG and CHH sequence contexts. (f–h) DNA methylation patterns of transposon and flanking regions in the CG, CHG and CHH sequence contexts. In all cases, −2 kb indicates the 2000 bp upstream from transcriptional start sites (TSS), and 2 kb indicates the 2000 bp downstream from transcriptional end sites (TES). Upstream regions, gene bodies and downstream regions were divided into 20 proportionally sized bins to draw the metaplot.
Since genome‐wide average DNA methylation was very different among tea and other two asterids examined (potato and tomato), we compared the DNA methylation levels of genic and TE regions in tea, potato and tomato, and to investigate the relative patterns of DNA methylation in gene and TE regions in tea compared to potato and tomato. We found that both gene and TE methylation patterns were similar to patterns found in potato and tomato (Figure 1c–h), as well as in other plants (Feng et al., 2010; Niederhuth et al., 2016; Wang et al., 2015, 2018). As expected, we found that CHG methylation was higher in both gene body and TE regions compared to those in potato and tomato (Figure 1d,g), which is consistent with genome‐wide average DNA methylation comparisons (Figure 1b). In addition, we found that CHH methylation was similar in tea and tomato in both gene and TE regions, but was much higher in potato, which confirms the findings of a previous study (Figure 1e,h; Wang et al., 2018). Taken together, CHG methylation was the most different methylation comparing to other asterids plants, and might be related to abundant transposable elements contained in the tea genome.
Association between DNA methylation and gene transcription
To assess the relationship between DNA methylation and gene transcription, we first grouped genes into five groups according to their expression levels (Figure 2a). A positive correlation was observed between gene body CG methylation and gene expression levels (Figure 2b,c), but not between CHG and CHH methylation and expression (Figure S5). In addition, consistent with previous studies in other plant species (Li et al., 2012; Wang et al., 2015; Zemach et al., 2010; Zhang et al., 2006), moderately highly expressed genes were the most heavily methylated in the CG sequence context (i.e. the third and fourth groups of genes in this study), and not the group of genes showing the lowest expression (i.e. the first group of genes) or the group showing the highest expression (i.e. the fifth group of genes) (Figure 2c). Unexpectedly, we observed that promoter and downstream CG methylation in the fifth group of genes was hypo‐methylated relative to the methylation levels of the other groups of genes. In fact, the other four groups of genes showed no differences in both promoter and downstream CG methylation, implying that methylation of flanking and gene regions act together to regulate gene expression. Unlike CG methylation, a negative relationship was found between non‐CG methylation and gene expression, and the lowest expressed genes possessed the highest methylation levels in gene body regions (Figure 2d,e).
Figure 2.
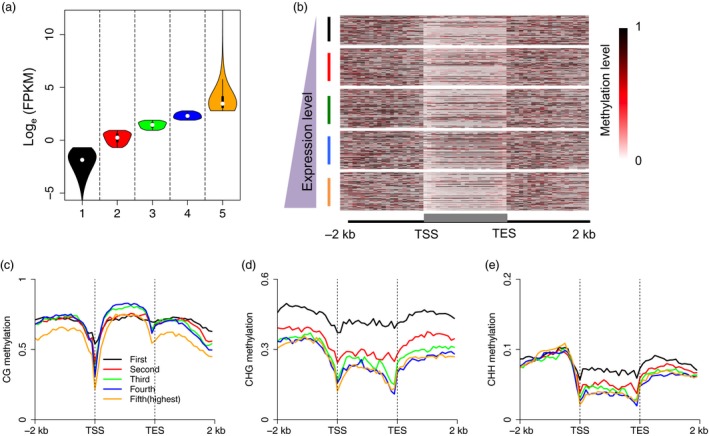
Association between DNA methylation and gene expression. (a) Genes were divided into five groups according to their expression levels. (b) CG methylation levels across gene body and flanking regions of the five groups of genes. (c–e) Association between gene expression and DNA methylation in the CG, CHG and CHH sequence contexts. Genes with FPKM value <0.5 were sorted into the first group, and all other genes were proportionally divided into the other four groups by their gene expression level. This meant that the fifth group of genes contained the highest expressed genes. Each upstream 2 kb, gene body, and downstream 2 kb region was divided proportionally into 20 bins, and average methylation levels were estimated for each bin.
To further examine the relationship between DNA methylation and gene expression in different sequence contexts (CG, CHG and CHH) and in different genomic regions (i.e. in promoters, gene regions, downstream regions, see the Methods section for further explanation), we compared the methylation levels of lowly‐ and highly expressed genes in the CG, CHG and CHH sequence contexts in different genomic regions. Our results showed that lowly expressed genes were methylated to a significantly higher degree in the CG and CHG contexts of promoter regions (Mann–Whitney test, P‐value <0.001), but not in the CHH context in these regions (Figure 3a–c). In addition, the DNA methylation levels of gene body and downstream regions were also higher in lowly expressed genes compared to highly expressed genes. Conversely, we also classified genes into highly‐ and lowly methylated genes, and assessed whether these genes differed in expression level. We also included the methylation levels of different genomic regions in this analysis. Consistent with our results described above, we found that highly methylated genes were always expressed at significantly lower levels than genes that were lowly methylated. This was true for all genic regions in the CG and CHG sequence contexts (Mann–Whitney test, P‐value <0.001), but not for highly methylated promoter regions in the CHH sequence context (Figure 3d–f). Taken together, our data suggest that the relationship between DNA methylation and gene expression is complicated, and is also affected by sequence context and genomic region.
Figure 3.
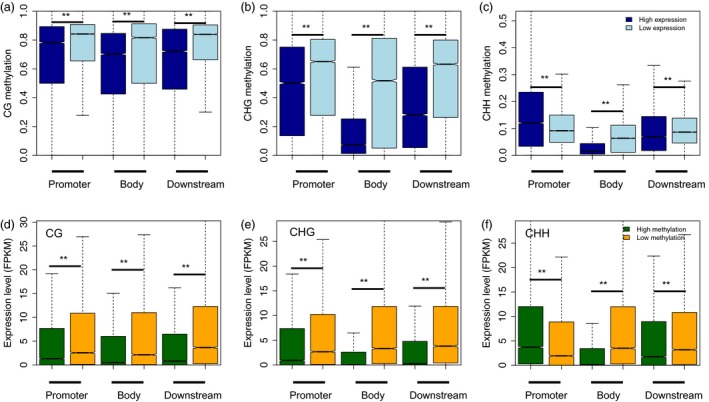
Association between gene expression and methylation at different genic regions. Comparison of methylation between lowly and highly expressed genes for the CG (a), CHG (b) and CHH (c) sequence contexts for each genic regions (i.e. promoter (upstream 2 kb), gene body and downstream 2 kb). Also shown are: a comparison of the expression levels between lowly‐ and highly methylated genes for the CG (d), CHG (e) and CHH (f) sequence contexts of each genic regions. Highly‐ and lowly expressed genes were defined as the one‐third of genes with the highest and lowest expression levels, respectively. Highly‐ and lowly methylated genes were defined as the one‐third of genes with the highest and lowest methylation levels with respect to each genic region, respectively. P‐values were calculated by Mann–Whitney tests, and double stars indicate a P‐value <0.001.
Intragenic TEs induced increased DNA methylation of orthologous genes in the tea genome
The striking differences of DNA methylation levels between tea and other two asterids studied here, potato and tomato, are probably due to difference in TE contents in their respective genomes. We also found that, relative to potato and tomato, in tea both gene and TE regions were hypermethylated in the CHG sequence context (Figure 1d,g). To assess the effect of TE abundance on genome structure and DNA methylation, we compared the genomes of tea, potato and tomato and explored the collinear regions in these three genomes. We found that when comparing homologous regions of the tea genome always harboured more TE insertions than the other two species, resulting in tea genome reaching a larger size. Figure 4a shows collinear regions in tea, potato and tomato: in total, we found 128 kb homologous regions in tea, but only 83 kb, and 78 kb homologous regions in the genomes of potato and tomato, respectively. Detailed examination revealed 180 TE insertions in the homologous regions of the tea genome, but only 19 and 56 TE insertions in the genome of potato and tomato, respectively. To investigate the effect of TE insertions in orthologous genes of collinear regions among these three plants, we compared the DNA methylation levels of orthologous genes in tea, potato and tomato in the three sequence contexts. As expected, higher DNA methylation levels were observed in tea than in potato and tomato, and this was especially true in the CHG sequence context (Mann–Whitney test, P‐value <0.01) (Figure 4b,c). This finding was consistent with the genome‐wide DNA methylation and metaplot analyses of gene and TE regions, in which CHG methylation was also found to be much higher in tea than in potato or tomato (Figure 1b,d,g). We know that DNA methylation could spread from TE boundaries to closed gene regions, and that this generally results in increased DNA methylation levels of protein‐coding genes near to TE regions (Gent et al., 2013).
Figure 4.
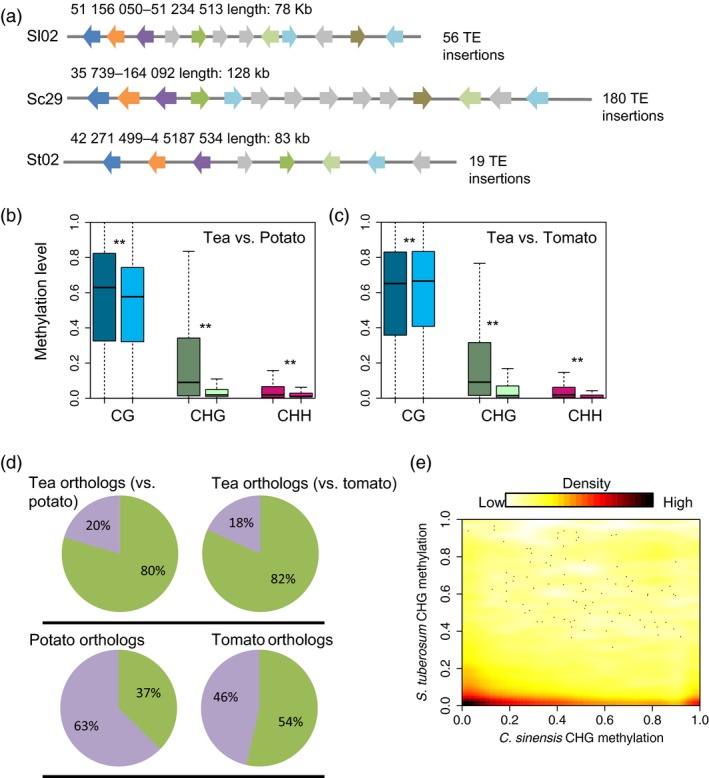
Comparisons of orthologous gene pairs in tea, potato and tomato. (a) A comparison of collinear regions of tea, tomato and potato. Orthologous gene pairs are indicated by the same colours, and the grey colour indicates non‐homologues genes. DNA methylation comparisons of orthologous gene pairs between tea and potato (b), and tea and tomato (c) are shown for all three sequence contexts. Double stars indicate Mann–Whitney test P‐value <0.001. Dark colours indicate the CG (blue), CHG (green) and CHH (red) sequence contexts in tea, and light colours indicate the same in potato and tomato. (d) Pie charts illustrating the percentage of TE‐associated (green) and non‐TE associated (purple) orthologous genes in tea and potato, tea and tomato, and their orthologous copies in potato and tomato, respectively. TE‐associated genes are defined as genes that overlap with TEs. (e) Distribution of CHG methylation levels of 20 990 orthologous gene pairs between tea and potato.
Next, we compared the proportions of TE‐associated genes in tea versus potato or tomato separately. We defined TE‐associated genes as those protein‐coding genes that overlap with TEs in their flanking 2 kb regions. Strikingly, we found more TE‐associated genes in tea than in potato or tomato (80% vs 37% in tea and potato, 82% vs 54% in tea and tomato) (Figure 4d). Interestingly, we found that many genes encoding secondary metabolites were enriched in TE‐associated genes. These genes included genes involved in caffeine metabolism (e.g. IMPDH, TCS, SAMS and AMPDA), flavonoid metabolism (e.g. 4CL, PAL, C4H, F3'H and FLS) and theanine metabolism (e.g. TS).
Recent studies have verified that the DNA methylation levels of homologous genes in different plants are often very similar to one another. This is especially true of CG and CHG methylation (Niederhuth et al., 2016; Takuno and Gaut, 2013; Wang et al., 2018), However, we did not observe this phenomenon in our comparison of tea with potato and tomato. On the contrary, we found that no correlation between tea and potato orthologs with respect to CHG methylation levels (Figure 4e). This finding was also true of comparisons between tea and tomato in all sequence contexts (Figure S6). In summary, our data imply that high levels of TE insertions can not only result in genome size enlargement, but also result in higher levels of DNA methylation than in closely related plants with low levels of TE insertions.
Reduced DNA methylation levels of TEs along evolutionary ages
Recent studies have shown that recently duplicated genes were hypermethylated relative to older ones, and that duplicates show decreases in DNA methylation as evolutionary time passes (Keller and Yi, 2014). To explore the relationship between DNA methylation and TE insertion time, Kimura distances (Kimura, 1980) were calculated for all TE copies found in each family to estimate the age of TE insertion. Like the Ks value (synonymous substitution divergence), which estimates the age of duplicated genes, Kimura distance estimates are based on TE similarity: the presence of copies of TEs that are similar (i.e. a low Kimura distance) indicates recent insertion, while the presence of unique or divergent copies of TEs (i.e. a high Kimura distance) indicates that these TEs originate from relatively ancient transposition events. Consistent with a recent study regarding TE expansion in the tea genome (Xia et al., 2017), we observed a TE burst event around 20 Kimura distance (Figure S7). Like duplicated genes in human genome, we found a negative correlation between the age of TE insertion and DNA methylation level in all three sequence contexts (Figure 5a–c). Due to a TE burst event in the past (Kimura distance ~20), we found many TE copies with a Kimura distance of approximately 20. Among the three sequence contexts, the CHH methylation levels of different TEs showed greater reductions through evolutionary time (Figure 5d) than the CG and CHG methylation levels (Figure S8A,B). This implies that the RdDM pathway plays an important role in silencing recently active TEs but gradually loses its effectiveness in silencing more ancient ones.
Figure 5.
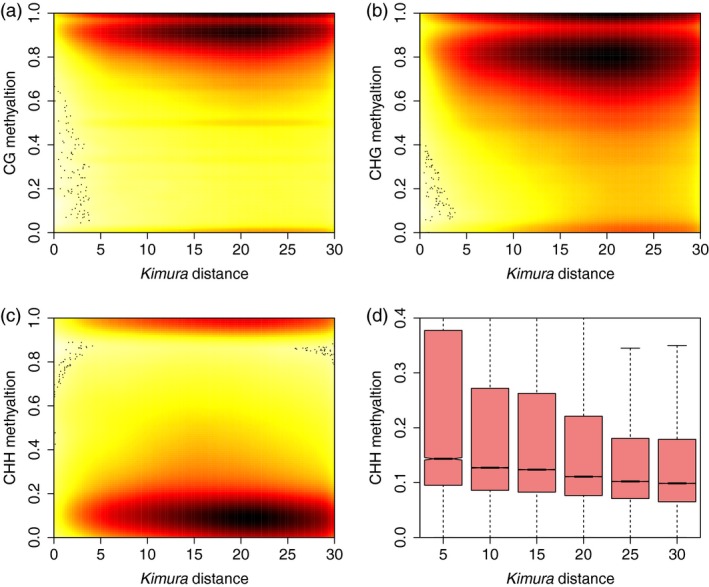
Association between the age of transposons and DNA methylation. A scatterplot showing the negative correlation between Kimura distance and CG (a), CHG (b) and CHH (c) DNA methylation. Kimura distance was used as a proxy for transposon age. (d) Boxplot showing the reduced CHH methylation of transposons as the Kimura distance increased.
Discussion
In this study, we provide the first high‐resolution genome‐wide DNA methylation map of the tea genome. Tea has numerous healthy and medicinal benefits for humans due to its abundance of secondary metabolites (Wei et al., 2018; Xia et al., 2017). Given the important role that DNA methylation plays in the control of gene expression and transposon silencing, our data serve as an important resource for the scientific and agronomic community.
In present study, we first identified DNA methylation related genes and found most of them have at least one copy in tea tree genome, and RNA‐seq data showed relatively high expression level, which is consistent to our RT‐PRC results (Figure S9 and Table S2). These data suggest DNA methylation pathway is conserved and functional in tea tree genome. Comparative analysis of the methylomes of tea, potato and tomato revealed that a considerable proportion of the variation of DNA methylation occurs between species. The greatest difference in DNA methylation between these species was observed in the CHG sequence context, one hypothesis is that tea may have a more robust maintenance methylation mechanism for CHG sites than potato and tomato. But weak DNA methylation correlations between the two strands of DNA were found at symmetrical CG and CHG sites in the tea genome, implying that the methylation maintenance pathway at CG and CHG sites are not as robust in tea as they are in potato and tomato. Tea also has higher TE contents in general than in potato and tomato, and recent studies have shown that both CG and CHG methylation levels are significantly positively correlated with TE content and genome size (Niederhuth et al., 2016; Takuno et al., 2016; Wang et al., 2018). Thus, the other hypothesis is that the superabundance of TEs presented in the tea genome has resulted in higher CHG methylation than is present in potato or tomato. We also observed that CHG methylation levels in both gene and TE regions were much higher in tea than in potato and tomato, which also suggests an effect of genic TE insertions generated from a TE burst event. Furthermore, we also observed that many genes encoding secondary metabolism were TE‐associated genes, such as caffeine, theanine and flavonoid metabolic pathway genes. This implies that epigenetic modifications such as DNA methylation may affect tea flavour by regulating secondary metabolic pathways.
Examination of the DNA methylation of TEs from different evolutionary periods revealed that recent active TEs are heavily methylated, while methylation is reduced in older TEs. This indicates that in tea the average DNA methylation levels of TEs are negatively correlated with evolutionary time, as measured by Kimura distance. This finding is consistent with observations of duplicated genes in the human genome (Keller and Yi, 2014). Taken together, this study demonstrates that heavy CHG methylation is present in the tea genome (along with abundant TE contents), and that the variation in DNA methylation levels occurs through evolutionary time. Recently, non‐canonical DNA modifications have been identified and characterized in many eukaryotes (Greer et al., 2015; Liang et al., 2018; Zhou et al., 2018), such as N6‐methyladenine (also referred as 6 mA). In plants, 6 mA was also identified in Arabidopsis and rice, and both plants showed that 6 mA was associated with gene expression, suggesting its important roles on plant development. In rice, 6 mA also was correlated with 5 mC at CG sites in gene body regions, and is complementary at CHH site of transposon regions (Liang et al., 2018; Zhou et al., 2018). Therefore, future studies on DNA methylation should combine with more methylation modifications to comprehensively study their regulation on gene expression. Taken together, this study may help us to understand the extent of DNA methylation variation observed among plants, and tea in particular.
Experimental procedures
Kimura distance calculation and estimation of TE burst event
Kimura distance between genome copies and a consensus sequence from a TE library were determined by using the RepeatMasker pipeline. The rates of transitions and transversions were calculated on alignments and transformed to Kimura distance (Kimura, 1980) using the following equation: K = −1/2 ln(1−2p−q)−1/4 ln(1−2q). Here, q is the proportion of sites with transversions and p is the proportion of sites with transitions.
BS‐seq data analysis
For tomato BS‐seq data (AC cultivar), we used public data (SRA accession SRR046092) from leaf and fruit of different ripening stages, for example, 17 DPA (immature), 39 DPA (mature green), 42 DPA (breaker) and 52 DPA (red ripe) (Zhong et al., 2013). For potato BS‐seq data (Atlantic cultivar), we downloaded data from GEO database (accession GSE86983; Wang et al., 2018). We first mapped BS‐seq reads to reference genome of tea, potato and tomato using BSMAP v2.90 (Xi and Li, 2009). Only uniquely mapped reads were kept for further analysis. Mapped reads were permitted four mismatches per 100‐bp length. Methylation levels were calculated as weighted methylation level (Schultz et al., 2012). Promoter methylation levels were calculated as the average methylation levels of the DNA sequence starting at the transcription start site and counting upstream for 2000 bp. Downstream methylation levels were calculated as the average methylation level of the DNA sequence starting at the transcription end site and counting downstream for 2000 bp.
RNA‐seq data analysis
For RNA‐seq data, we first mapped clean reads to the tea reference genome by tophat v2.1.0, (Trapnell et al., 2009). We kept only uniquely mapped alignments. Expression values were quantified by Cufflinks with FPKM (fragments per kilobase per million mapped reads) with default parameters (Trapnell et al., 2012).
Real‐time PCR validation
Total RNA for real‐time PCR analysis was extracted from tea leaf that was the same stage for RNA‐seq following the cetyltrimethylammonium bromide (CTAB) protocol. RNA quality and quantity were each assessed, respectively, by Agilent 2100 BioAnalyzer (Agilent Technologies, Santa Clara, CA) and NanoDrop 2000 (Thermo Scientific, Waltham, MA). cDNA was synthesized from the total RNA using SuperScript II Reverse Transcriptase system (Invitrogen, Carlsbad, CA) with oligo(dT) following the manufacturer's instructions. The primers (Table 1) for real‐time PCR were designed using NCBI Primer‐BLAST tools according to cDNA sequences. Reactions were carried out on StepOnePlus Real‐Time PCR System (Applied Biosystems, Foster City, CA) using three‐step cycling conditions of 95 °C for 1 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 30 s and 72 °C for 30 s. After the amplification steps, the melting curve was determined for each primer pair at a final stage of 15 s at 95 °C, 15 s at 60 °C and 15 s at 95 °C to verify the presence of only one specific product. Relative quantification was performed with the 2‐ΔΔCT method. Teaβ‐actin gene (Forward primer: 5′‐GCCATCTTTGATTGGAATGG‐3′, Rverse primer: 5′‐GGTGCCACAACCTTGATCTT‐3′) was used as reference for messenger RNA (mRNA) expression. The Pearson correlation analysis of RNA‐seq and qRT‐PCR was performed by R software (www.r-project.org).
Homologous gene and collinear regions identification
Homologous genes of tea tree genome were identified by MCScanX (Wang et al., 2012), which is an algorithm that can simultaneously scan multiple genomes to identify homologous chromosomal regions and subsequently align these regions by using genes. Collinear regions among tea, potato and tomato were also identified by MCScanX with default parameters, and orthologous genes pairs were extracted from collinear regions.
Correlation analysis of methylation data
Correlation between biological replicates of BS‐seq was calculated as methylation levels of total Cs in 2000 bp regions. First, tea tree genome was splited into 2000 bin bins, and methylation levels were calculated as the average #C/(#C+#T) for all cytosines in each bin, #C is the number of C reads and #T is the number of T reads. Then, Pearson correlation coefficients were calculated between the two biological replicates.
Correlation analysis between orthologous gene pairs of tea and tomato/potato was performed by corr.test() of R project. DNA methylation level was first calculated of each gene in tea, tomato, and tomato of CG, CHG and CHH sequence contexts. Then, smooth scatter plots were conducted by smoothScatter() of R project (www.r-project.org).
For correlation between DNA methylation and kimura distance, the DNA methylation level of each TE was calculated by custom Perl script, and kimura distance were extracted from RepeatMasker pipeline.
Metaplot analysis
For metaplot analysis, each gene body region was proportionally divided into 20 bins. Both upstream and downstream 2000 bp regions were also divided into 20 bins (i.e. the length of each bin is 100 bp), separately. Then, the average level of DNA methylation of each bin was calculated for all genes and plotted by R script.
Accession numbers
The data reported in this paper were deposited in the Gene Expression Omnibus (GEO) database (accession number GSE119992).
Author contributions
H.W. and H.Y. designed the research; H.W., L.W., Y.S. and X.C. performed the research and analysed the data; S.J. and X.C. collected the data; Q.Z. and C.Y. provided the tools; H.W. wrote the paper.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Supporting information
Figure S1 BS‐seq coverage shown as the proportion of cytosines that were covered by at least ‘X’ reads.
Figure S2 Correlation analysis between replicates in the CG, CHG and CHH sequence contexts.
Figure S3 DNA methylation pattern comparison between tea and other plants.
Figure S4 DNA methylation correlations between Watson and Crick strand in CG and CHG symmetrical sequence contexts in the tea, potato and tomato genomes.
Figure S5 DNA methylation levels of gene body and flanking regions for five group genes.
Figure S6 Distribution of DNA methylation levels of orthologous gene pairs between tea and tomato/potato.
Figure S7 Distribution of Kimura distances of different types of transposons.
Figure S8 Reduced DNA methylation levels of transposons over evolutionary time.
Figure S9 Expression correlation between RT‐PCR and RNA‐seq of 37 DNA methylation pathway related genes.
Table S1 Summary of BS‐seq reads mapping.
Table S2 The qRT‐PCR primers and 2−ΔΔCT value.
Acknowledgements
We thank laboratory members and colleagues for comments and discussions. This work was supported by the funds from National Natural Science Foundation of China Grant 31501031, the Program for Excellent Youth Talents and the Program for New Century Excellent Talents in Fujian Province University, the Pre‐eminent Youth Fund and Distinguished Young Scholars in Fujian Province (to H.W.). This work was also supported by the Youth Backbone Teachers Plant of XYNU (2016GGJS‐13) and Nanhu Scholars Program for Young Scholars of XYNU.
Contributor Information
Hongyu Yuan, Email: yhongyu92@163.com.
Haifeng Wang, Email: haifengwang@fafu.edu.cn.
References
- Ausin, I. , Feng, S. , Yu, C. , Liu, W. , Kuo, H.Y. , Jacobsen, E.L. , Zhai, J. et al (2016) DNA methylome of the 20‐gigabase Norway spruce genome. Proc. Natl Acad. Sci. USA, 113, E8106–E8113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick, M. , Kim, J.K. , Esteve, P.O. , Clark, A. , Pradhan, S. and Jacobsen, S.E. (2007) UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science, 317, 1760–1764. [DOI] [PubMed] [Google Scholar]
- Cao, X. and Jacobsen, S.E. (2002) Locus‐specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc. Natl Acad. Sci. USA, 99(Suppl 4), 16491–16498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus, S.J. , Feng, S. , Zhang, X. , Chen, Z. , Merriman, B. , Haudenschild, C.D. , Pradhan, S. et al (2008) Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature, 452, 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodsworth, S. , Leitch, A.R. and Leitch, I.J. (2015) Genome size diversity in angiosperms and its influence on gene space. Curr. Opin. Genet. Dev. 35, 73–78. [DOI] [PubMed] [Google Scholar]
- Feng, S. , Cokus, S.J. , Zhang, X. , Chen, P.Y. , Bostick, M. , Goll, M.G. , Hetzel, J. et al (2010) Conservation and divergence of methylation patterning in plants and animals. Proc. Natl Acad. Sci. USA, 107, 8689–8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan, E.J. , Peacock, W.J. and Dennis, E.S. (2000) DNA methylation, a key regulator of plant development and other processes. Curr. Opin. Genet. Dev. 10, 217–223. [DOI] [PubMed] [Google Scholar]
- Gehring, M. and Henikoff, S. (2007) DNA methylation dynamics in plant genomes. Biochim. Biophys. Acta, 1769, 276–286. [DOI] [PubMed] [Google Scholar]
- Gent, J.I. , Ellis, N.A. , Guo, L. , Harkess, A.E. , Yao, Y. , Zhang, X. and Dawe, R.K. (2013) CHH islands: de novo DNA methylation in near‐gene chromatin regulation in maize. Genome Res. 23, 628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouil, Q. and Baulcombe, D.C. (2016) DNA methylation signatures of the plant chromomethyltransferases. PLoS Genet. 12, e1006526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer, E.L. , Blanco, M.A. , Gu, L. , Sendinc, E. , Liu, J. , Aristizábal‐Corrales, D. , Hsu, C.H. et al (2015) DNA methylation on N6‐Adenine in C. elegans. Cell, 161, 868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, J.P. , Lindroth, A.M. , Cao, X. and Jacobsen, S.E. (2002) Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature, 416, 556–560. [DOI] [PubMed] [Google Scholar]
- Keller, T.E. and Yi, S.V. (2014) DNA methylation and evolution of duplicate genes. Proc. Natl Acad. Sci. USA, 111, 5932–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K.D. , El Baidouri, M. , Abernathy, B. , Iwata‐Otsubo, A. , Chavarro, C. , Gonzales, M. , Libault, M. et al (2015) A comparative epigenomic analysis of polyploidy‐derived genes in soybean and common bean. Plant Physiol. 168, 1433–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M. (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. [DOI] [PubMed] [Google Scholar]
- Law, J.A. and Jacobsen, S.E. (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Zhu, J. , Hu, F. , Ge, S. , Ye, M. , Xiang, H. , Zhang, G. et al (2012) Single‐base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 13, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, Z. , Shen, L. , Cui, X. , Bao, S. , Geng, Y. , Yu, G. , Liang, F. et al (2018) DNA N(6)‐Adenine methylation in Arabidopsis thaliana . Dev. Cell, 45, 406–416.e3. [DOI] [PubMed] [Google Scholar]
- Lindroth, A.M. , Cao, X. , Jackson, J.P. , Zilberman, D. , McCallum, C.M. , Henikoff, S. and Jacobsen, S.E. (2001) Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science, 292, 2077–2080. [DOI] [PubMed] [Google Scholar]
- Lister, R. , O'Malley, R.C. , Tonti‐Filippini, J. , Gregory, B.D. , Berry, C.C. , Harvey Millar, A. and Ecker, J.R. (2008) Highly integrated single‐base resolution maps of the epigenome in Arabidopsis. Cell, 133, 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzke, M.A. , Kanno, T. and Matzke, A.J. (2015) RNA‐directed DNA methylation: the evolution of a complex epigenetic pathway in flowering plants. Annu. Rev. Plant Biol. 66, 243–267. [DOI] [PubMed] [Google Scholar]
- Niederhuth, C.E. , Bewick, A.J. , Ji, L. , Alabady, M.S. , Kim, K.D. , Li, Q. , Rohr, N.A. et al (2016) Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 17, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, M.D. , Schmitz, R.J. and Ecker, J.R. (2012) ‘Leveling’ the playing field for analyses of single‐base resolution DNA methylomes. Trends Genet. 28, 583–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud, H. , Greenberg, M.V. , Feng, S. , Bernatavichute, Y.V. and Jacobsen, S.E. (2013) Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell, 152, 352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuno, S. and Gaut, B.S. (2013) Gene body methylation is conserved between plant orthologs and is of evolutionary consequence. Proc. Natl Acad. Sci. USA, 110, 1797–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuno, S. , Ran, J.H. and Gaut, B.S. (2016) Evolutionary patterns of genic DNA methylation vary across land plants. Nat. Plants, 2, 15222. [DOI] [PubMed] [Google Scholar]
- Trapnell, C. , Pachter, L. and Salzberg, S.L. (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Roberts, A. , Goff, L. , Pertea, G. , Kim, D. , Kelley, D.R. , Pimentel, H. et al (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Tang, H. , Debarry, J.D. , Tan, X. , Li, J. , Wang, X. , Lee, T.H. et al (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , Beyene, G. , Zhai, J. , Feng, S. , Fahlgren, N. , Taylor, N.J. , Bart, R. et al (2015) CG gene body DNA methylation changes and evolution of duplicated genes in cassava. Proc. Natl Acad. Sci. USA, 112, 13729–13734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Xie, J. , Hu, J. , Lan, B. , You, C. , Li, F. , Wang, Z. et al (2018) Comparative epigenomics reveals evolution of duplicated genes in potato and tomato. Plant J. 93, 460–471. [DOI] [PubMed] [Google Scholar]
- Wei, C. , Yang, H. , Wang, S. , Zhao, J. , Liu, C. , Gao, L. , Xia, E. et al (2018) Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl Acad. Sci. USA, 115, E4151–E4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willing, E.M. , Rawat, V. , Mandakova, T. , Maumus, F. , James, G.V. , Nordström, K.J. , Becker, C. et al (2015) Genome expansion of Arabis alpina linked with retrotransposition and reduced symmetric DNA methylation. Nat. Plants, 1, 14023. [DOI] [PubMed] [Google Scholar]
- Xi, Y. and Li, W. (2009) BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics, 10, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, E.H. , Zhang, H.B. , Sheng, J. , Li, K. , Zhang, Q.J. , Kim, C. and Zhang, Y. (2017) The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol. Plant, 10, 866–877. [DOI] [PubMed] [Google Scholar]
- Zemach, A. , Kim, M.Y. , Silva, P. , Rodrigues, J.A. , Dotson, B. , Brooks, M.D. and Zilberman, D. (2010) Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl Acad. Sci. USA, 107, 18729–18734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Yazaki, J. , Sundaresan, A. , Cokus, S. , Chan, S.W. , Chen, H. , Henderson, I.R. et al (2006) Genome‐wide high‐resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell, 126, 1189–1201. [DOI] [PubMed] [Google Scholar]
- Zhong, S. , Fei, Z. , Chen, Y.R. , Zheng, Y. , Huang, M. , Vrebalov, J. , McQuinn, R. et al (2013) Single‐base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 31, 154–159. [DOI] [PubMed] [Google Scholar]
- Zhou, C. , Wang, C. , Liu, H. , Zhou, Q. , Liu, Q. , Guo, Y. , Peng, T. et al (2018) Identification and analysis of adenine N(6)‐methylation sites in the rice genome. Nat. Plants, 4, 554–563. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 BS‐seq coverage shown as the proportion of cytosines that were covered by at least ‘X’ reads.
Figure S2 Correlation analysis between replicates in the CG, CHG and CHH sequence contexts.
Figure S3 DNA methylation pattern comparison between tea and other plants.
Figure S4 DNA methylation correlations between Watson and Crick strand in CG and CHG symmetrical sequence contexts in the tea, potato and tomato genomes.
Figure S5 DNA methylation levels of gene body and flanking regions for five group genes.
Figure S6 Distribution of DNA methylation levels of orthologous gene pairs between tea and tomato/potato.
Figure S7 Distribution of Kimura distances of different types of transposons.
Figure S8 Reduced DNA methylation levels of transposons over evolutionary time.
Figure S9 Expression correlation between RT‐PCR and RNA‐seq of 37 DNA methylation pathway related genes.
Table S1 Summary of BS‐seq reads mapping.
Table S2 The qRT‐PCR primers and 2−ΔΔCT value.