

ABSTRACT
Bone marrow provides a unique microenvironment favoring the colonization and outgrowth of metastatic tumor cells. Despite the high incidence of bone metastasis in breast and prostate cancer patients, many of the molecular mechanisms controlling metastatic progression remain unclear. Several gene signatures associated with bone metastasis have been reported, but no metastasis‐specific gene alterations have been identified. Therefore, there has been considerable interest in understanding how the bone microenvironment impacts the behavior of disseminated tumor cells (DTCs) prior to and following colonization of the bone. Substantial evidence indicates that disruption of normal bone homeostasis by tumor‐derived factors establishes a premetastatic niche within the bone that favors DTC colonization. Following dissemination, bone resident cells and the surrounding stroma provide critical signals that support tumor cell colonization, survival, and eventual outgrowth. Clinical data suggest that patients can harbor DTCs for years to decades prior to developing overt bone metastases, suggesting a period of tumor dormancy occurs in the bone marrow. Several dormancy‐promoting factors have been recently identified; however, critical questions surrounding the molecular triggers and timing of tumor cell emergence from dormancy remain. Here, we review how metastatic tumor cells co‐opt the bone marrow microenvironment for metastatic progression and discuss emerging insights into how to more effectively target DTCs and prevent metastasis. © 2018 The Authors. JBMR Plus published by Wiley Periodicals, Inc. on behalf of American Society for Bone and Mineral Research
Keywords: BONE METASTASIS, PREMETASTATIC NICHE, HOMING, COLONIZATION, DORMANCY
Introduction
The mechanisms that regulate tumor cell dissemination from a primary tumor and the establishment of a metastasis are complex and poorly understood. Metastasis is the leading cause of cancer‐related deaths, but disseminated tumor cells (DTCs) encounter multiple challenges, making metastatic progression a highly inefficient process. Initially, DTCs must survive in the circulation before homing to and colonizing a foreign microenvironment in a distant organ. Upon arrival at the metastatic site, DTCs enter a dormant state for some period, often months to decades, before eventually becoming reactivated and developing into an overt metastasis. An extensive body of clinical and experimental research supports Stephen Paget's “seed and soil” hypothesis1 that proposed tumor cells preferentially metastasize to particular secondary sites. This nonrandom tumor cell distribution, referred to as metastatic organotropism, is likely facilitated in part by blood flow dynamics, but most prominently by the fertile “soil” at distant sites.2
Metastasis to the bone occurs in approximately 60% of patients with metastatic breast or prostate cancer and to a lesser extent in other cancers, including lung and melanoma.3 The bone microenvironment provides a uniquely fertile soil for the homing of DTCs for several reasons. First, the bone marrow houses numerous cell types implicated in metastatic progression, including hematopoietic and mesenchymal stem cells, endothelial cells, osteoblasts, and osteoclasts, and the bone matrix itself provides a rich source of growth factors and cytokines.4 Second, the bone marrow is the primary site for hematopoietic stem cell (HSC) maintenance and contains two specialized niches: the endosteal “osteoblastic” niche and the perivascular niche.5 These niches are established and maintained by systemic signals and HSC interaction with resident cells, including osteoblasts and endothelial cells. Finally, the vasculature of the bone marrow results in varying oxygen levels ranging from <1% to 6% throughout the bone marrow, making the bone a particularly hypoxic tissue.6 Thus, the bone marrow provides an ideal microenvironment for metastasis and ample opportunities for DTCs to co‐opt these physiological niches to promote their own survival and outgrowth.
Genetic Drivers of Bone Metastasis
Metastatic progression has traditionally been thought of as a late event that occurs following numerous genetic or epigenetic aberrations; however, recent literature suggests that dissemination can occur early in tumor progression.7, 8 Currently, two fundamental models of metastatic progression exist: linear progression and parallel progression. The linear progression model implies that metastatic founder cells evolve with the primary tumor, arising late in tumor progression, followed by delayed dissemination and adaptation to the microenvironment at the distant metastatic site. In contrast, parallel progression suggests early dissemination and acquisition of additional mutations in the metastatic tumor cells that are not detected in the primary tumor.
Advancement of single‐cell genomic analyses that are able to identify rare clonal populations and evaluate genetic alterations in DTCs has allowed for investigation into the biological significance of these progression models. The competing views and the evidence for each model in various tumor types have recently been reviewed.9 In the context of bone metastasis, early evidence of parallel progression came from analysis of DTCs in the bone marrow of breast cancer patients with and without clinically detectable metastasis.10, 11 Patients without metastasis harbored DTCs with less genetic heterogeneity compared to the primary tumor or DTCs isolated from M1 patients.10, 11 These findings suggest that metastatic cells in the bone follow the parallel progression model, acquiring additional genetic abnormalities after dissemination to a distant site. Similar results have been reported for patients with prostate cancer.12, 13 In support of these clinical data, studies using murine models of breast cancer demonstrated that invasive subpopulations disseminate from very early lesions to distant organs and eventually initiate overt metastasis.14, 15 Despite these corroborative findings, it is possible these genomic analyses failed to capture every unique subclone within the primary tumor and DTC populations. Thus, the presence of a rare subclone within the primary tumor that is able to give rise to DTCs and metastasis cannot be excluded.
Although these comparative genomic studies have illuminated the timing of tumor cell dissemination to distant metastatic sites, they have not provided considerable insight into the specific mechanisms controlling the ability of tumor cells to disseminate and home to a distant site. Thus, gene expression studies comparing primary human tumors and respective bone metastases have been performed to identify metastasis‐promoting genes that are associated with bone metastasis and poor outcome. Massague and colleagues reported a bone metastasis 102 gene signature,16 which included genes involved in bone marrow homing (CXCR4), extracellular matrix alteration (MMP1, ADAMTS1, proteoglycan‐1), angiogenesis (FGF5, CTGF), and osteoclastogenesis (IL11). Several of these genes were shown to cooperate to promote bone colonization and tumor‐induced osteolysis in vivo, and likely cooperate with other unidentified genes to promote this phenotype. Subsequently, several other bone metastasis gene signatures, including signatures driven by Src‐dependent17 or Irf7‐regulated genes,18 have been described. Of important note, very little overlap occurs between the reported gene signatures, which may be because of tumor heterogeneity or differences in tumor source (e.g., analysis of primary tumors versus metastatic tumors to predict bone metastasis). Thus, the clinical significance and applicability of these gene signatures remains unclear.
To date, no metastasis‐specific mutations have been identified, implying that numerous genes become altered and act cooperatively to drive metastatic progression.19 These global gene expression changes are proposed to be a result of alterations to the epigenetic landscape, including DNA methylation and histone acetylation modifications.20, 21 Among the most frequently mutated genes in human cancers are epigenetic modifying enzymes,21 which are likely responsible for the increased DNA and histone methylation observed in tumors that efficiently metastasize to bone, brain, lung, and liver.22, 23 Presumably, these global epigenetic changes would result in abnormal gene expression and generation of additional mutations to promote a prometastatic phenotype. For example, DNA and histone methylation changes allow for the accessibility of VHL‐HIF target genes, namely CYTIP and CXCR4, to promote bone and lung metastasis in clear cell renal carcinoma.24
Premetastatic Niches
Accumulating evidence suggests that several types of premetastatic niches (PMNs) exist to support the homing, survival, and colonization of metastatic tumor cells.4 The PMN is established by systemic signals secreted from the primary tumor that alter the extracellular matrix and recruit supportive stromal cells to create a conducive environment in the secondary site. The importance of tumor‐derived factors in establishing the PMN through recruitment of bone‐marrow‐derived cells to the secondary site has been extensively investigated.4 However, because these cells normally reside in the bone marrow, the mechanisms controlling PMN formation in the bone remain less clear. Nonetheless, disruption of normal bone homeostasis appears to be a driving mechanism in PMN establishment in the bone (Fig. 1 A). For example, hypoxic breast cancer cells in the primary tumor secrete the collagen‐crosslinking enzyme lysyl oxidase (LOX), which acts directly on osteoblasts and osteoclasts in the bone marrow to favor bone resorption and promote colonization of DTCs.25 Additional secreted factors, including tumor‐derived CCL2,25, 26 interleukin‐6 (IL‐6),27, 28, 29 the Notch ligand, Jagged1 (JAG1),30 and the Wnt inhibitor, DKK1,31 can enhance osteoclast differentiation and activity to promote skeletal metastasis. Interestingly, though CCL2 also promotes lung metastasis by recruiting macrophages to the metastatic site,25 tumor‐secreted DKK1 prevents lung metastasis by inhibiting stromal cell recruitment.31 These data suggest that there may be site‐specific effects of these tumor‐derived factors that require further investigation.
Figure 1.
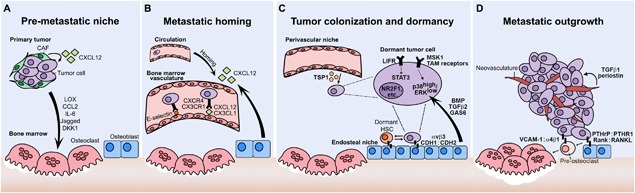
Metastatic progression in the bone. (A) Tumor‐derived factors promote the formation of a premetastatic niche in the bone prior to tumor cell dissemination. Factors such as lysyl oxidase (LOX) and C‐C motif chemokine ligand 2 (CCL2) disrupt normal bone homeostasis thereby favoring tumor cell colonization. (B) Disseminated tumor cells (DTCs) enter the circulation and can eventually home to the bone via microenvironmental signals including CXCL12:CXCR4 and E‐selectin. (C) Following extravasation, interaction with resident bone cells and signaling molecules such as leukemia inhibitory factor receptor (LIFR), p38, and thrombospondin‐1 (TSP1) maintain tumor cells in a dormant state. (D) Emergence of DTCs from dormancy results in the outgrowth into overt metastases. Tumor cell proliferation and osteolytic bone destruction is mediated by parathyroid hormone‐related protein (PTHrP), receptor activator of NFκB ligand (RANKL), and vascular cell adhesion molecule‐1 (VCAM‐1). The growth of neovasculature within the metastasis produces transforming growth factor beta 1 (TGFβ1) and periostin to further promote the proliferation of metastatic tumor cells. CAF = cancer‐associated fibroblast; CXCL12 = CXC Chemokine Ligand 12; LOX = lysyl oxidase; CCL2 = C‐C Motif Chemokine Ligand; IL‐6 = Interleukin‐6; DKK1 = Dickkopf WNT Signaling Pathway Inhibitor 1; CXCR4 = CXC Motif Chemokine Receptor 4; CX3CL = CX3C motif Chemokine Ligand; CX3CR = CX3C Motif Chemokine Receptor; TSP1 = Thrombospondin‐1; LIFR = Leukemia Inhibitory Factor Receptor; STAT3 = Signal transducer and activator of transcription 3; MSK1 = Ribosomal Protein S6 Kinase A5; NR2F1= Nuclear Receptor Subfamily 2 Group F Member 1; TAM receptors = TYRO3 Protein Tyrosine Kinase, AXL Receptor Tyrosine Kinase, MER Proto‐Oncogene Tyrosine Kinase; p38 = Mitogen Activated Protein Kinase 14; ERK = Extracellular‐signal Regulated Kinase; BMP = Bone Morphogenetic Protein; TGFb = Transforming Growth Factor Beta; GAS6 = Growth Arrest Specific 6; CDH1 = E‐cadherin; CDH2 = N‐cadherin; PTHrP = Parathyroid Hormone related Protein; PTHR1= Parathyroid Hormone Receptor 1; RANK = TNF Superfamily Member 11; VCAM1 = Vascular Cell Adhesion Molecule 1.
Extracellular vesicles (EVs) are a heterogeneous collection of membrane‐encapsulated particles classified by size and origin as exosomes or microvesicles that are secreted by cells. Tumor‐derived EVs have gained considerable interest in the metastasis field because of their ability to educate cells at distant sites.32 Although several studies have provided insight into the role of EVs in establishing the PMN in the liver33 and lung,34, 35, 36 limited evidence has been reported for their role in the bone. Using organotropic sublines of human MDA‐MB‐231 breast cancer cells, secreted EVs were shown to distribute to their preferential site following injection and establish premetastatic niches in an integrin‐dependent manner in vivo.37 Of particular interest, priming of naïve mice with lung‐tropic EVs enhanced lung colonization of bone‐tropic breast cancer cells.37 Exosomes secreted from highly metastatic melanomas contain MET‐related signaling proteins that can reprogram bone marrow cells toward a provascular and prometastatic phenotype, creating a PMN.38 Further, exosomes derived from a weakly metastatic melanoma line reduced metastatic burden in the bone.38 Numerous in vitro studies have also described the prometastatic effects of tumor‐derived exosomes on normal bone cells.39, 40, 41, 42 Thus, these data suggest that tumor‐derived EVs may contain vastly different cargos depending on the metastatic potential of the primary tumor and that further understanding of these differences may prove a viable approach to inhibit metastasis.
Metastatic Homing
The CXCL12:CXCR4 axis is one of the most well‐described and prominent mechanisms favoring tumor cell homing and colonization of the bone (Fig. 1 B). Bone marrow stromal cells and osteoblasts normally express high levels of CXCL12 (also known as SDF‐1) to regulate the homing of HSCs to the bone marrow.43 Overexpression of its cognate receptor, CXCR4, by many cancer types,44 including breast and prostate, facilitates the priming of tumor cells by CXCL12‐secreting cancer‐associated fibroblasts to colonize and survive in the CXCL12‐rich bone microenvironment.45 This signaling cascade is further propagated by recruitment of CXCR6+ mesenchymal stem cells (MSCs) to the primary tumor in response to tumor‐secreted CXCL16.46 Activation of the CXCL16:CXCR6 pathway converts MSCs into cancer‐associated fibroblasts, which subsequently secrete high levels of CXCL12.46
Endothelial cells are the initial cell type encountered by tumor cells after homing to the bone microenvironment (Fig. 1 B). Therefore, understanding the mechanisms controlling tumor cell adhesion to the endothelium is critical. Following intravasation, CXCL12:CXCR4 also serves as a chemoattractant to the bone47 and facilitates tumor cell binding to marrow endothelial cells.48, 49 Cell adhesion molecules and integrins have been heavily implicated in regulating tumor cell colonization of the bone.50, 51 Loss of E‐selectin ligand, β1 integrin, and Rac1 disrupts the ability of prostate tumor cells to adhere to and breach E‐selectin (also known as CD62E or ELAM‐1) positive bone marrow endothelial cells, resulting in decreased metastasis incidence.52 Similarly, CX3CL1:CX3CR153, 54 and ANXA2: ANXA2R55 promote the adhesion of breast and prostate tumor cells to bone marrow endothelial cells.
Pre‐Existing Niches and Bone Colonization
Pre‐existing niches within the secondary site, especially those involved in maintaining adult stem cell populations, are often exploited by metastatic tumor cells as receptive microenvironments. The endosteal and perivascular niches are the two specialized compartments critical for HSC maintenance and self‐renewal in the bone marrow.5 Bone‐lining osteoblasts are the key component of the endosteal niche necessary for HSC maintenance, whereas endothelial and mesenchymal cells regulate this process in the perivascular niche.5 Direct competition of HSCs with tumor cells for occupancy of the endosteal niche has been demonstrated in murine models of prostate cancer.56 This competition is facilitated by the direct interaction of tumor cells with osteoblasts and induction of HSC maturation by tumor cells, resulting in HSC egression from the niche.56 Notably, manipulation of the niche size resulted in a concomitant change in the number of DTCs. Specifically, osteoblast ablation led to decreased colonization by prostate cancer cells.56 Substantial evidence indicates that interaction of metastatic tumor cells with bone resident cells facilitates their successful colonization of the bone marrow. For example, bone colonization is mediated by the formation of heterotopic adherens junctions between E‐cadherin (CDH1) positive breast cancer cells and N‐cadherin (CDH2) positive osteoblasts.57 Additionally, tumor cell αvβ3 integrin expression is a critical mediator of tumor cell adhesion to bone matrix proteins and bone resident cells such as osteoblasts and osteoclasts through vitronectin and osteopontin.58 These interactions have been shown to be necessary for successful colonization of breast and prostate cancer cells and the enhancement of tumor‐induced osteolysis.59, 60, 61 Recent clinical and experimental evidence in breast cancer implicates RUNX2 as a transcriptional driver of αv (ITGA5) expression to promote CTC colonization of the bone.62
Using murine models and organotypic cultures, Ghajar and colleagues demonstrated that breast cancer cells preferentially localize to the perivascular niche, where they are maintained in a nonproliferative state through interactions with endothelial‐derived thrombospondin‐1 (TSP1).63 This preferential homing of breast cancer cells was recently observed using real‐time in vivo microscopy in which DTCs home to E‐selectin‐ and CXCL12‐rich perivascular regions.64 Similarly, disseminated melanoma cancer cells interact with MSCs through CD146 (also known as melanoma cell adhesion molecule [MCAM]) and CXCR4 to facilitate their colonization.65 Although the perivascular niche also contains resident stem cells, direct competition of tumor cells for niche occupancy has not been reported.
Tumor Dormancy
The physiological role of the stem cell niche is to provide survival, quiescence, and self‐renewal signals from the microenvironment. Thus, tumor cells preferentially localize to these niches within the bone marrow to promote their own survival and dormancy (Fig. 1 C). Increasing clinical evidence suggests that even patients without detectable metastasis harbor reservoirs of dormant tumor cells in the bone marrow. Breast cancer patients without nodal involvement have an approximate 20% risk of developing bone metastases 5 to 20 years after primary diagnosis.66 Accordingly, nonproliferating DTCs have been detected in the circulation,67, 68 as well as in the bone upon autopsy69, 70 in approximately 70% of breast or prostate cancer patients.69 Intriguingly, the presence of DTCs in the bone marrow of patients is not only predictive of metastasis to the bone, but also to the lungs, liver, and brain.71 This predictive capability also applies to cancer types that rarely metastasize to the bone. For example, despite the low incidence of bone metastasis, DTCs are detected in patients with colorectal and gastric cancer, suggesting that these cells very rarely escape dormancy.67 Combined, these studies suggest that dormant DTCs may lie in the bone marrow for an extended period, putting cancer survivors at significant risk of developing bone metastases should these DTCs become reactivated. Despite the recent advances in our understanding of tumor dormancy, many of the complex molecular mechanisms remain unclear.
Microenvironmental factors known to regulate HSC quiescence include bone morphogenetic proteins (BMPs),72 TGFβ2,73, 74 and growth arrest‐specific protein 6 (GAS6),75, 76 which were among the first factors identified to induce dormancy of prostate cancer cells and head and neck squamous cell carcinoma. These secreted factors as well as other molecular signals, including retinoic acid77 and urokinase plasminogen activator receptor (uPAR),78, 79 have been shown to alter the ratio of ERK and p38 MAPK signaling, which has become one of the most well‐established mechanisms for inducing tumor cell dormancy.74 Specifically, preferential activation of p38 MAPK over ERK signaling (p38high/ ERKlow) results in the induction of DTC dormancy. TGFβ2, which is proposed to activate the p38 MAPK pathway in bone‐disseminated tumor cells, has been shown to be more abundant in the bone marrow compared to other organs (liver, spleen, lung), suggesting potential organ‐specific mechanisms of tumor dormancy.80 Additionally, a downstream mediator of p38 MAPK signaling, mitogen‐ and stress‐activated kinase 1 (MSK1), was recently identified as an important regulator of metastatic dormancy in breast cancer cells.81 Clinical data also implicate p38 MAPK/ERK in bone metastasis because a p38‐regulated dormancy gene signature was associated with increased time to metastasis in breast cancer patients.82, 83
The Tyro3, Axl, and MERTK (TAM) receptor tyrosine kinases compete for the GAS6 ligand secreted by osteoblasts.75, 76 Xenograft models of prostate cancer revealed that GAS6‐mediated Axl signaling induces dormancy, whereas GAS6‐activated Tyro3 promotes escape into a proliferative state. Recent evidence indicates that GAS6:Axl signaling is critical for TGFβ2‐mediated dormancy.84 Finally, MERTK was recently shown to promote dormancy escape in prostate cancer cells through multiple transcriptional and epigenetic mechanisms.85 Combined, these data suggest that the ratio of the TAM receptors on DTCs may be one mechanism controlling the fate of DTCs in the bone marrow.
The tumor suppressor leukemia inhibitory factor (LIF) receptor (LIFR) was also recently identified as a mediator of tumor dormancy in breast cancer cells.86, 87, 88 Loss of LIFR and downstream STAT3 signaling in DTCs resulted in dormancy escape and enhanced osteolytic bone destruction in vivo.86 Activation of LIFR:STAT3 is mediated by several IL‐6 family cytokines including oncostatin M (OSM) and LIF, which have been previously implicated in regulating metastasis to the bone, lung, and liver.89, 90, 91 Thus far, because of the complexity of LIFR signaling and abundance of ligands in the bone marrow, the specific factor(s) responsible for the prodormancy effects of LIFR signaling has not yet been identified. Of particular interest are the findings that LIFR expression is downregulated by hypoxia,86, 92 suggesting that oxygen gradients in the bone marrow may regulate the emergence of tumor cells from a dormant state.
Reversible epigenetic modifications are known to regulate stem cell plasticity, suggesting that these mechanisms are also likely to be involved in tumor dormancy. Indeed, several genes belonging to the aforementioned p38‐regulated gene signature,82, 83 including NR2F1, TGFB2, and DNMT1, are known epigenetic modulators of stem cell quiescence and have been identified as key regulators of tumor dormancy. Further investigation using experimental head and neck squamous cell carcinoma models revealed that NR2F1 drives global chromatin changes primarily to promote the survival of DTCs and, to a lesser extent, their dormancy in the bone marrow.77 In contrast to the bone, NR2F1 predominantly drives DTC dormancy in the lung and spleen through SOX9 and RARβ.77 Examination of stemness in prostate cancer DTCs revealed that traditional stem cell markers (e.g., CD44 and CD133) were not enriched in quiescent DTCs in the bone marrow, but these cells were far more tumorigenic than their proliferative counterparts. Interestingly, these cells also expressed higher levels of CXCR4, suggesting that the quiescent cells may be more adept at bone marrow homing.93
Metastatic Outgrowth
DTCs can persist in a dormant state for years to decades before becoming reactivated and developing into overt metastases. Although our understanding of metastatic outgrowth remains incomplete, many mechanisms regulating the switch of dormant tumor cells into proliferative metastases have been identified (Fig. 1 D). Disruption of bone homeostasis is one of the primary switches that causes tumor cells to exit a dormant state. The “vicious cycle” of osteolytic bone metastasis is the most well‐defined mechanism that disrupts bone homeostasis and is observed in numerous cancer types including breast, lung, and multiple myeloma.94 The vicious cycle is initiated by the secretion of molecules by tumor cells, including parathyroid hormone‐related protein (PTHrP) and IL‐11, which stimulate RANKL‐mediated differentiation and activation of osteoclasts.94 Osteoclasts resorb the surrounding bone matrix, releasing stored mitogenic factors, namely TGFβ, that subsequently fuel cancer cell proliferation and the feed‐forward cycle by stimulating PTHrP95 and its upstream regulator GLI2.96, 97 The role for TGFβ signaling through the TGFβ type I receptor in propagating the vicious cycle is well‐established95, 98, 99, 100 and is in contrast with the proposed role for TGFβ2 induction of tumor cell dormancy in the bone marrow. This suggests an important temporal role for TGFβ signaling that extends beyond its dual role at the primary and metastatic sites. In contrast to breast cancer, prostate cancer cells predominantly form osteoblastic lesions as a result of excessive induction of osteoblast differentiation and proliferation.101 The positive‐feedback loop for osteoblastic metastases is initiated by the secretion of osteoblast‐activating factors such as bone morphogenic proteins (BMPs) and epidermal growth factor (EGF) from tumor cells, which in turn results in the production of osteoblast‐derived factors including IL‐6 and monocyte chemotactic protein‐1 (MCP1) that promote tumor cell proliferation.101 It is worth noting that antiresorptive therapies have been effective in reducing bone pain in prostate cancer patients,102, 103 suggesting that a resorptive phase precedes the formation of osteoblastic lesions.
Although it remains largely unclear whether PTHrP is expressed in breast cancer cells prior to dissemination or turned on following extravasation into the bone marrow, there have been several studies investigating how the rigidity of the bone microenvironment impacts breast cancer cell expression of PTHrP. Bone metastatic breast and lung cancer cells grown on increasingly rigid substrates exhibited similar increases in PTHrP and GLI2, which was mediated by Rho‐associated kinase (ROCK) activation of TGFβ signaling,104 as well as integrin β3.105 Interestingly, MCF7 cells, which home to bone, but do not induce much bone destruction,86, 106 do not increase PTHrP levels in response to increasing rigidity.104 These data suggest that cells primed for the bone are more responsive to bone matrix rigidity.
Because of the initiating role of PTHrP in osteolytic bone destruction, numerous studies have investigated its role in bone colonization. Inhibition of PTHrP shortly after tumor inoculation107 or 2 weeks after inoculation100 effectively reduces tumor‐induced osteolysis. Overexpression of PTHrP in otherwise dormant human MCF7 breast cancer cells results in aggressive colonization and osteolysis of the bone through enhanced osteoclastogenesis.108 PTHrP overexpression in breast cancer cells also reduces pro‐dormancy gene expression, suggesting that PTHrP may play a role in tumor cell exit from dormancy.86, 109 Further, the enabling of dormant tumor cells to aggressively colonize the bone following PTHrP overexpression appears to be independent of parathyroid hormone receptor type I (PTH1R) and cAMP‐mediated signaling and may rely on the calcium signaling pathway.109
Aberrant expression of vascular cell adhesion molecule 1 (VCAM‐1) on breast cancer cells has been shown to recruit osteoclast progenitors expressing the cognate receptor integrin α4β1, thus enhancing local osteoclast activity.110 Pharmacological targeting of VCAM‐1 or integrin α4 effectively reduced progression from dormancy into overt metastasis.110 Clinical data suggest Src activation is associated with bone metastasis.17 Although Src has no effect on the homing of breast cancer cells to the bone, its activation results in enhanced metastatic outgrowth in the bone.17 Similar to the primary tumor, the vasculature is known to play an important role in metastasis.4 In contrast to the suppressive cues of mature vessels, the sprouting neovasculature promotes progression of metastatic outgrowth by secreting TGF‐β1 and periostin.63 These findings identified an unexpected source of these tumor‐promoting factors, suggesting that vascular homeostasis is critical for initiating emergence from dormancy.
Recent work by Lawson and colleagues provides a novel look at the vicious cycle and multiple myeloma dormancy using longitudinal intravital imaging through an optical window in the mouse tibia.111 Using this model, myeloma cells colonized the endosteal niche and their dormancy status was determined by the balance between prodormancy signals from osteoblasts and proproliferative signals from osteoclasts in the endosteal niche.111 Intriguingly, when proliferative tumor cells were isolated and subsequently reintroduced into mice, a small portion localized to the endosteal niche and did not divide.111 Thus, regardless of their prior proliferative capacity, re‐engagement of tumor cells with the endosteal niche was able to induce dormancy.
A critical question arises from the findings presented above: What initiates the eventual switch from a dormant to proliferative state? Given the evidence supporting early dissemination of tumor cells, it is possible that primary tumor‐derived factors are responsible for altering DTCs or the bone metastatic niche prior to detection of the primary tumor. Age‐related changes have also been postulated to trigger the emergence of tumor cells from dormancy. With increasing age, more cells enter an irreversible senescent state, which is associated with a senescence‐associated secretory phenotype (SASP).112 This SASP results in the elevated secretion of cytokines, chemokines, and growth factors that may establish a tumor‐promoting microenvironment that can act on surrounding dormant tumor cells. Indeed, senescent osteoblasts enhanced breast cancer bone colonization through IL‐6‐mediated osteoclastogenesis.27 Additionally, systemic sex steroid levels may also play a role in this process. These molecules can regulate bone homeostasis by inducing osteoclast apoptosis and promoting osteoblast proliferation.113, 114, 115 Reduced hormone levels can also result in enhanced bone resorption, which leads to accelerated metastasis formation in hormone‐responsive tumors, including breast and prostate.116, 117
Bone Metastasis Therapies
Over the past decade, significant progress has been made in elucidating the molecular mechanisms that control metastatic niches, tumor dormancy, and the emergence of clinically detectable bone metastases. Preferential metastasis to the bone marrow occurs for many tumor types, with breast and prostate being the most notable.69 Bone‐modifying agents that target resorption, including bisphosphonates and denosumab, are commonly used to effectively manage bone metastasis‐related morbidities including pain and hypercalcemia.117, 118, 119 However, because these therapies target osteolysis and not tumor cells themselves, mortality rates in patients with bone metastasis has not significantly improved. Conventional therapies have limited success in preventing or treating bone metastasis in part because of the complex nature of the bone microenvironment, tumor heterogeneity, and therapeutic resistance of DTCs. Until recently, the persistence of dormant tumor cells in secondary sites and their resistance to therapeutics that preferentially target proliferating cells was not appreciated. Thus, there is significant need to identify novel therapeutic strategies to prevent the colonization and outgrowth of DTCs in the bone.
A complicating factor of bone metastasis is that the kinetics of tumor cell dissemination and metastatic outgrowth remain unclear, and the emergence of DTCs from a dormant state may not be a synchronized event.111 Temporally or spatially regulated factors may promote dormancy escape in a subset of DTC clones and maintain dormancy of others. These possibilities complicate the development of therapies targeting dissemination, colonization, and metastatic outgrowth. Therapeutic strategies designed to induce DTC dormancy and/or prevent reactivation or those that promote proliferation and mobilization of bone DTCs into the circulation have been proposed.120 Both strategies have advantages and disadvantages, highlighting the need for more investigation into their effectiveness in preclinical animal models.
Preventing tumor cell dissemination and colonization by disrupting the factors shown in Table 1 may prove to be a promising strategy to prevent metastasis. In support of this notion, disruption of the CX3CR1 or CXCR4 pathway in murine models of breast and prostate cancer, respectively, reduces metastatic incidence and tumor burden in the bone.45, 53, 121, 122 Similarly, substantial preclinical evidence suggests that pharmacological targeting of αvβ3 integrin effectively prevents metastatic colonization of the bone.123, 124, 125 Clinical trials using neutralizing antibodies and small molecule inhibitors against αvβ3 have shown promise; however, these have not specifically focused on bone metastasis.126, 127 Microenvironmental factors such as TGFβ and VEGF have also been identified as potential therapeutic targets to prevent bone metastasis.128, 129 Therapeutic approaches targeting tumor cell intrinsic mechanisms have also been proposed for bone metastases. A combination of Src and ERK inhibition has been shown to effectively reduce breast cancer metastasis to the lungs.130 Given the known roles of Src17 and ERK73, 74, 79 signaling in promoting dormancy escape of tumor cells in the bone, this combination treatment may be an effective treatment to maintain tumor cells in a dormant state and prevent recurrence. Given the newly identified role of epigenetics in regulating tumor dormancy, epigenetic‐modulating therapies may also represent promising therapeutic options to induce dormancy in DTCs. LIFR and other prodormancy genes are upregulated in breast cancer cells following HDAC inhibitor treatment,86 indicating this may be a mechanism to induce a chronic state of dormancy; however, it has also been suggested that HDAC induction of LIFR may contribute to therapeutic resistance by tumor cells, suggesting the dormant cells may be more difficult to target long‐term.131 A combination treatment of demethylating agents and all‐trans retinoic acid (ATRA) induced dormancy in a p38‐dependent manner in a murine model of head and neck squamous cell carcinoma,77 suggesting that the combination of epigenetic‐modulating drugs may be a promising therapeutic avenue.
Table 1.
Factors Controlling Metastatic Progression in the Bone
| Function | Protein Interaction | Reference(s) | |
|---|---|---|---|
| Pre‐metastatic Niches | Disrupt bone homeostasis to promote colonization | LOX | 25 |
| CCL2 | 25, 26 | ||
| IL‐6 | 27‐29 | ||
| JAG1 | 30 | ||
| DKK1 | 31 | ||
| Promote colonization | Extracellular vesicles (MET proteins) | 32‐38 | |
| Metastatic Homing | Tumor‐derived chemoattractants for bone marrow derived cells | CXCL16: CXCR6 | 46 |
| Bone‐derived chemoattractants for tumor cells | CXCL12: CXCR4 | 43‐45 | |
| Promote endothelial cell adhesion | CXCL12: CXCR4 | 47‐49 | |
| CD62E | 52 | ||
| CX3CL1: CX3CR1 | 53, 54 | ||
| ANXA2: ANXA2R | 55 | ||
| Pre‐exisiting Niches | Interaction with bone marrow cells | Competition of tumor cells with HSCs for niche occupancy | 56 |
| CDH1 (tumor cells) and CDH2 (osteoblasts) | 57 | ||
| Integral αvβ3 | 58‐62 | ||
| CD62E | 64 | ||
| CXCL12: CXCR4 | 64 | ||
| CD146, CXCR4 | 65 | ||
| Tumor dormancy | Regulate tumor dormancy | TSP1 | 63 |
| BMP7 | 72 | ||
| TGFβ2 | 73, 74 | ||
| GAS6 | 75, 76 | ||
| ATRA | 77 | ||
| uPAR | 78, 79 | ||
| p38high/ERKlow signaling | 74, 8O, 82, 83 | ||
| MSK1 | 81 | ||
| Gas6: Tyro3, Axl, MERTK | 75, 76 | ||
| LIFR, PTHrP | 86‐88, 109 | ||
| Epigenetic modifiers (NR2F1, TGFβ2, DXMT1) | 77, 82, 83 | ||
| Metastatic outgrowth | Enhance tumor cell proliferation | PTHrP, RANKL, TGFβ | 94, 95‐100 |
| VCAM‐1 | 110 | ||
| SRC | 17 | ||
| TGF‐βl, periostin (endothelial‐derived) | 63 |
LOX = lysyl oxidase; CCL2 = C‐C motif chemokine ligand 2; IL6 = interleukin‐6; JAG1 = jagged1; DKK1 = Dickkopf WNT signaling pathway inhibitor 1; CXCL = CXC chemokine ligand; CXCR = CXC motif chemokine receptor; CD62E = E‐selectin; CX3CL = CX3C motif chemokine ligand; CX3CR = CX3C motif chemokine receptor; ANXA2 = annexin II; ANXA2R = annexin A2 receptor; CDH1 = E‐cadherin; CDH2 = N‐cadherin; CD146 = melanoma cell adhesion molecule; TSP1 = thrombospondin‐1; BMP7 = bone morphogenetic protein 7; TGFβ = transforming growth factor beta; GAS6 = growth arrest specific 6; ATRA = all‐trans retinoic acid; uPAR = urokinase plasminogen activator receptor; p38 = mitogen‐activated protein kinase 14; ERK = extracellular‐signal regulated kinase; MSK1 = ribosomal protein S6 kinase A5; Tyro3 = TYRO3 protein tyrosine kinase; Axl = AXL receptor tyrosine kinase; MERTK = MER proto‐oncogene tyrosine kinase; LIFR = leukemia inhibitory factor receptor; NR2F1 = nuclear receptor subfamily 2 group F member 1; DNMT1 = DNA methyltransferase 1; PTHrP = parathyroid hormone‐related protein; RANKL = TNF superfamily member 11; VCAM1 = vascular cell adhesion molecule 1; SRC = SRC proto‐oncogene.
Conclusions
Although we have uncovered many of the factors that regulate tumor cell homing and colonization of the bone marrow, many challenges still remain. The gaps in knowledge regarding the formation of a premetastatic niche in the bone marrow will likely take some time to elucidate; however, much progress has been made in identifying the factors that mediate tumor cell homing to the bone marrow and the molecules that govern metastatic outgrowth of DTCs in the bone marrow. The tumor dormancy field is still emerging, but has already produced a number of potential therapeutic targets that may be applicable to bone metastases. The field has continued to evolve, producing innovative ways to approach the therapeutic targeting of DTCs, and this is likely to expand as we learn more about the bone‐specific mechanisms that promote tumor homing and dissemination. The cellular processes and molecular mechanisms that trigger the metastatic cascade to the bone marrow and allow dormant tumor cells to transition into a proliferative state still require further investigation. Elucidating these mechanisms will identify the most effective ways to prevent bone metastasis and tumor recurrence in bone.
Disclosures
The authors have no conflicts of interest to declare.
Acknowledgments
RWJ and MES are supported by NIH award R00CA194198 (RWJ) and DoD Breakthrough Award W81XWH‐17‐BCRP (RWJ).
References
- 1. Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889. 133: 571–73. doi: 10.1016/S0140-6736(00)49915-0 [DOI] [PubMed] [Google Scholar]
- 2. Smith HA, Kang, Y. Determinants of organotropic metastasis. Ann Rev Cancer Biol. 2017; 1:403–23. doi: 10.1146/annurev-cancerbio-041916-064715 [DOI] [Google Scholar]
- 3. Macedo, F , Ladeira K, Pinho F, et al. Bone metastases: an overview. Oncol Rev. 2017; 11: 321. doi: 10.4081/oncol.2017.321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peinado H, Zhang H, Matei IR, et al. Pre‐metastatic niches: organ‐specific homes for metastases. Nat Rev Cancer. 2017; 17: 302–17. doi: 10.1038/nrc.2017.6 [DOI] [PubMed] [Google Scholar]
- 5. Crane GM, Jeffery E, Morrison SJ. Adult haematopoietic stem cell niches. Nat Rev Immunol. 2017; 17: 573–90. doi: 10.1038/nri.2017.53 [DOI] [PubMed] [Google Scholar]
- 6. Johnson RW, Sowder ME, Giaccia AJ. Hypoxia and bone metastatic disease. Curr Osteoporos Rep. 2017; 15: 231–38. doi: 10.1007/s11914-017-0378-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Husemann Y, Geigl JB, Schubert F, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008; 13: 58–68. doi: 10.1016/j.ccr.2007.12.003 [DOI] [PubMed] [Google Scholar]
- 8. Sanger N, Effenberger KE, Riethdorf S, et al. Disseminated tumor cells in the bone marrow of patients with ductal carcinoma in situ. Int J Cancer. 2011; 129: 2522–26. doi: 10.1002/ijc.25895 [DOI] [PubMed] [Google Scholar]
- 9. Naxerova K, Jain RK. Using tumour phylogenetics to identify the roots of metastasis in humans. Nat Rev Clin Oncol. 2015; 12: 258–72. doi: 10.1038/nrclinonc.2014.238 [DOI] [PubMed] [Google Scholar]
- 10. Schmidt‐Kittler O, Ragg T, Daskalakis A, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S.A. 2003; 100: 7737–42. doi: 10.1073/pnas.1331931100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klein CA, Blankenstein TJ, Schmidt‐Kittler O, et al. Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer. Lancet. 2002; 360: 683–89. doi: 10.1016/s0140-6736(02)09838-0 [DOI] [PubMed] [Google Scholar]
- 12. Weckermann D, Polzer B, Ragg T, et al. Perioperative activation of disseminated tumor cells in bone marrow of patients with prostate cancer. J Clin Oncol. 2009;27:1549–56. doi: 10.1200/JCO. 2008.17.0563 [DOI] [PubMed] [Google Scholar]
- 13. Gundem G, Van Loo P, Kremeyer B, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015; 520: 353–57. doi: 10.1038/nature14347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hosseini H, Obradović MM, Hoffmann M, et al. Early dissemination seeds metastasis in breast cancer. Nature. 2016. doi: 10.1038/nature20785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harper KL, Sosa MS, Entenberg D, et al. Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature. 2016. doi: 10.1038/nature20609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kang Y, Siegel PM, Shu W, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003; 3: 537–49. doi: 10.1016/s1535-6108(03)00132-6 [DOI] [PubMed] [Google Scholar]
- 17. Zhang XH, Wang Q, Gerald W, et al. Latent bone metastasis in breast cancer tied to Src‐dependent survival signals. Cancer Cell. 2009; 16: 67–78. doi: 10.1016/j.ccr.2009.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bidwell BN, Slaney CY, Withana NP, et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med. 2012; 18: 1224–31. doi: 10.1038/nm.2830 [DOI] [PubMed] [Google Scholar]
- 19. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013; 339: 1546–58. doi: 10.1126/science.1235122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baxter E, Windloch K, Gannon F, Lee JS. Epigenetic regulation in cancer progression. Cell Biosci. 2014; 4: 45. doi: 10.1186/2045-3701-4-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012; 22: 9–20. doi: 10.1016/j.ccr.2012.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mehrotra J, Vali M, McVeigh M, et al. Very high frequency of hypermethylated genes in breast cancer metastasis to the bone, brain, and lung. Clin Cancer Res. 2004; 10: 3104–9. doi: 10.1158/1078-0432.ccr-03-0118 [DOI] [PubMed] [Google Scholar]
- 23. Shi X, Tasdogan A, Huang F, Hu Z, Morrison SJ, DeBerardinis RJ. The abundance of metabolites related to protein methylation correlates with the metastatic capacity of human melanoma xenografts. Sci Adv. 2017; 3 (11): eaao5268. doi: 10.1126/sciadv.aao5268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vanharanta S, Shu W, Brenet F, et al. Epigenetic expansion of VHL‐HIF signal output drives multiorgan metastasis in renal cancer. Nat Med. 2013; 19: 50–6. doi: 10.1038/nm.3029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu X, Kang Y. Chemokine (C‐C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J Biol Chem. 2009; 284: 29087–96, doi: 10.1074/jbc.M109.035899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li X, Loberg R, Liao J, et al. A destructive cascade mediated by CCL2 facilitates prostate cancer growth in bone. Cancer Res. 2009; 69: 1685–92. doi: 10.1158/0008-5472.CAN-08-2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo X, Fu Y, Loza AJ, et al. Stromal‐initiated changes in the bone promote metastatic niche development. Cell Rep. 2016; 14: 82–92. doi: 10.1016/j.celrep.2015.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zheng Y, Basel D, Chow SO, et al. Targeting IL‐6 and RANKL signaling inhibits prostate cancer growth in bone. Clin Exp Metastasis. 2014; 31: 921–33. doi: 10.1007/s10585-014-9680-3 [DOI] [PubMed] [Google Scholar]
- 29. Zheng Y, Chow SO, Boernert K, et al. Direct crosstalk between cancer and osteoblast lineage cells fuels metastatic growth in bone via auto‐amplification of IL‐6 and RANKL signaling pathways. J Bone Miner Res. 2014; 29: 1938–49. doi: 10.1002/jbmr.2231 [DOI] [PubMed] [Google Scholar]
- 30. Sethi N, Dai X, Winter CG, Kang Y. Tumor‐derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell. 2011; 19: 192–205. doi: 10.1016/j.ccr.2010.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhuang X, Zhang H, Li X, et al. Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nat Cell Biol. 2017;19:1274–85. doi: 10.1038/ncb3613 [DOI] [PubMed] [Google Scholar]
- 32. Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in cancer: cell‐to‐cell mediators of metastasis. Cancer Cell. 2016; 30: 836–48. doi: 10.1016/j.ccell.2016.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Costa‐Silva B, Aiello NM, Ocean AJ, et al. Pancreatic cancer exosomes initiate pre‐metastatic niche formation in the liver. Nat Cell Biol. 2015; 17: 816–26. doi: 10.1038/ncb3169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Y, Gu Y, Han Y, et al. Tumor exosomal RNAs promote lung pre‐metastatic niche formation by activating alveolar epithelial TLR3 to recruit neutrophils. Cancer Cell. 2016; 30: 243–56. doi: 10.1016/j.ccell.2016.06.021 [DOI] [PubMed] [Google Scholar]
- 35. Jung T, Castellana D, Klingbeil P, et al. CD44v6 dependence of premetastatic niche preparation by exosomes. Neoplasia. 2009; 11: 1093–105. doi: 10.1593/neo.09822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grange C, Tapparo M, Collino F, et al. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011; 71: 5346–56. doi: 10.1158/0008-5472.CAN-11-0241 [DOI] [PubMed] [Google Scholar]
- 37. Hoshino A, Costa‐Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015; 527: 329–35. doi: 10.1038/nature15756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Peinado H, Alečković M, Lavotshkin S, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro‐metastatic phenotype through MET. Nat Med. 2012; 18: 883–91. doi: 10.1038/nm.2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rossi M, Battafarano G, D'Agostini M, Del Fattore A. The role of extracellular vesicles in bone metastasis. Int J Mol Sci. 2018; 19 (4): pii: E1136. doi: 10.3390/ijms19041136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karlsson T, Lundholm M, Widmark A, Persson E. Tumor cell‐derived exosomes from the prostate cancer cell line TRAMP‐C1 impair osteoclast formation and differentiation. PLoS ONE. 2016;11:e0166284. doi: 10.1371/journal.pone.0166284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taverna S, Pucci M, Giallombardo M, et al. Amphiregulin contained in NSCLC‐exosomes induces osteoclast differentiation through the activation of EGFR pathway. Sci Rep. 2017; 7 (1): 3170. doi: 10.1038/s41598-017-03460-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Raimondi, L , De Luca A, Amodio N, et al. Involvement of multiple myeloma cell‐derived exosomes in osteoclast differentiation. Oncotarget. 2015;6:13772–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jung Y, Wang J, Schneider A, et al. Regulation of SDF‐1 (CXCL12) production by osteoblasts; a possible mechanism for stem cell homing. Bone. 2006; 38: 497–508. doi: 10.1016/j.bone.2005.10.003 [DOI] [PubMed] [Google Scholar]
- 44. Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res. 2014;124:31–82. doi: 10.1016/B978-0-12-411638-2.00002-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang XH, Jin X, Malladi S, et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. 2013; 154: 1060–73. doi: 10.1016/j.cell.2013.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jung Y, Kim JK, Shiozawa Y, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun. 2013; 4:1795. doi: 10.1038/ncomms2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang J, Loberg R, Taichman RS. The pivotal role of CXCL12 (SDF‐1)/CXCR4 axis in bone metastasis. Cancer Metastasis Rev. 2006; 25: 573–87. doi: 10.1007/s10555-006-9019-x [DOI] [PubMed] [Google Scholar]
- 48. Kukreja P, Abdel‐Mageed AB, Mondal D, Liu K, Agrawal KC. Up‐regulation of CXCR4 expression in PC‐3 cells by stromal‐derived factor‐1alpha (CXCL12) increases endothelial adhesion and transendothelial migration: role of MEK/ERK signaling pathway‐dependent NF‐kappaB activation. Cancer Res. 2005;65:9891–8. doi: 10.1158/0008-5472.CAN-05-1293 [DOI] [PubMed] [Google Scholar]
- 49. Engl T, et al. CXCR4 chemokine receptor mediates prostate tumor cell adhesion through alpha5 and beta3 integrins. Neoplasia. 2006; 8: 290–301. doi: 10.1593/neo.05694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Graham N, Qian B. ‐Z. Mesenchymal stromal cells: emerging roles in bone metastasis. Int J Molec Sci. 2018; 19: 1121. doi: 10.3390/ijms19041121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nature Revs Cancer. 2018;18:533–48. doi: 10.1038/s41568-018-0038-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barthel SR, Hays DL, Yazawa EM, et al. Definition of molecular determinants of prostate cancer cell bone extravasation. Cancer Res. 2013; 73: 942–52. doi: 10.1158/0008-5472.CAN-12-3264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shen F, Zhang Y, Jernigan DL, et al. Novel small‐molecule CX3CR1 antagonist impairs metastatic seeding and colonization of breast cancer cells. Mol Cancer Res. 2016;14:518–27. doi: 10.1158/1541-7786.MCR-16-0013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jamieson WL, Shimizu S, D'Ambrosio JA, Meucci O, Fatatis A. CX3CR1 is expressed by prostate epithelial cells and androgens regulate the levels of CX3CL1/fractalkine in the bone marrow: potential role in prostate cancer bone tropism. Cancer Res. 2008; 68: 1715–22. doi: 10.1158/0008-5472.CAN-07-1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shiozawa Y, Havens AM, Jung Y, et al. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J Cell Biochem. 2008;105:370–80. doi: 10.1002/jcb.21835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shiozawa Y, Pedersen EA, Havens AM, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011; 121: 1298–1312. doi: 10.1172/JCI43414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang H, Yu C, Gao X, et al. The osteogenic niche promotes early‐stage bone colonization of disseminated breast cancer cells. Cancer Cell. 2015; 27: 193–210. doi: 10.1016/j.ccell.2014.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schneider JG, Amend SR, Weilbaecher KN. Integrins and bone metastasis: integrating tumor cell and stromal cell interactions. Bone. 2011;48:54–65. doi: 10.1016/j.bone.2010.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kwakwa KA, Sterling JA. Integrin alphavbeta3 signaling in tumor‐induced bone disease. Cancers (Basel). 2017; 9 (7): pii: E84. doi: 10.3390/cancers9070084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pécheur I, Peyruchaud O, Serre CM, et al. Integrin αvβ3 expression confers on tumor cells a greater propensity to metastasize to bone. FASEB J. 2002; 16: 1266–8. doi: 10.1096/fj.01-0911fje [DOI] [PubMed] [Google Scholar]
- 61. Sloan EK, Pouliot N, Stanley KL, et al. Tumor‐specific expression of αvβ3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res. 2006; 8 (2): R20. doi: 10.1186/bcr1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li XQ, Lu JT, Tan CC, Wang QS, Feng YM. RUNX2 promotes breast cancer bone metastasis by increasing integrin alpha5‐mediated colonization. Cancer Lett. 2016; 380: 78–86. doi: 10.1016/j.canlet.2016.06.007 [DOI] [PubMed] [Google Scholar]
- 63. Ghajar CM, Peinado H, Mori H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013; 15: 807–17. doi: 10.1038/ncb2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Price TT, Burness ML, Sivan A, et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci Transl Med. 2016; 8 (340): 340ra73. doi: 10.1126/scitranslmed.aad4059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Correa D, Somoza RA, Lin P, Schiemann WP, Caplan AI. Mesenchymal stem cells regulate melanoma cancer cells extravasation to bone and liver at their perivascular niche. Int J Cancer. 2016; 138: 417–27. doi: 10.1002/ijc.29709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pan H, Gray R, Braybrooke J, et al. 20‐year risks of breast‐cancer recurrence after stopping endocrine therapy at 5 years. N Engl J Med. 2017; 377: 1836–46. doi: 10.1056/NEJMoa1701830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004; 4: 448–56. doi: 10.1038/nrc1370 [DOI] [PubMed] [Google Scholar]
- 68. Morgan TM, Lange PH, Porter MP, et al. Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clin Cancer Res. 2009; 15: 677–83. doi: 10.1158/1078-0432.ccr-08-1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Johnson RW, Schipani E, Giaccia AJ. HIF targets in bone remodeling and metastatic disease. Pharmacol Ther. 2015; 150, 169–77. doi: 10.1016/j.pharmthera.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Coleman, R. E. Metastatic bone disease: clinical features, pathophysiology and treatment strategies. Cancer Treat Rev. 2001; 27: 165–76. doi: 10.1053/ctrv.2000.0210 [DOI] [PubMed] [Google Scholar]
- 71. Braun S, Vogl FD, Naume B, et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N Engl J Med. 2005; 353; 793–802. doi: 10.1056/NEJMoa050434 [DOI] [PubMed] [Google Scholar]
- 72. Kobayashi A, Okuda H, Xing F, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem‐like cells in bone. J Exp Med. 2011; 208: 2641–55. doi: 10.1084/jem.20110840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bragado P, Estrada Y, Parikh F, et al. TGF‐beta2 dictates disseminated tumour cell fate in target organs through TGF‐beta‐RIII and p38alpha/beta signalling. Nat Cell Biol. 2013; 15: 1351–61. doi: 10.1038/ncb2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yu‐Lee LY, Yu G, Lee YC, et al. Osteoblast‐secreted factors mediate dormancy of metastatic prostate cancer in the bone via activation of the TGFbetaRIII‐p38MAPK‐pS249/T252RB pathway. Cancer Res. 2018; 78: 2911–24. doi: 10.1158/0008-5472.CAN-17-1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Taichman RS, Patel LR, Bedenis R, et al. GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS One. 2013; 8: e61873. doi: 10.1371/journal.pone.0061873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shiozawa Y, Pedersen EA, Patel LR, et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia. 2010; 12: 116–27. doi: 10.1593/neo.91384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sosa MS, Parikh F, Maia AG, et al. NR2F1 controls tumour cell dormancy via SOX9‐ and RARbeta‐driven quiescence programmes. Nat Commun. 2015; 6: 6170. doi: 10.1038/ncomms7170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Xue A, Xue M, Jackson C, Smith RC. Suppression of urokinase plasminogen activator receptor inhibits proliferation and migration of pancreatic adenocarcinoma cells via regulation of ERK/p38 signaling. Int J Biochem Cell Biol. 2009; 41: 1731–38. doi: 10.1016/j.biocel.2009.03.004 [DOI] [PubMed] [Google Scholar]
- 79. Aguirre‐Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERKMAPK to p38MAPK activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell. 2001; 12: 863–79. doi: 10.1091/mbc.12.4.863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sosa MS, Bragado P, Aguirre‐Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nature Rev Cancer. 2014; 14: 611–22. doi: 10.1038/nrc3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gawrzak S, Rinaldi L, Gregorio S, et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER+ breast cancer. Nat Cell Biol. 2018; 20: 211–21. doi: 10.1038/s41556-017-0021-z [DOI] [PubMed] [Google Scholar]
- 82. Adam AP, George A, Schewe D, et al. Computational identification of a p38SAPK‐regulated transcription factor network required for tumor cell quiescence. Cancer Res. 2009; 69: 5664–72. doi: 10.1158/0008-5472.CAN-08-3820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kim RS, Avivar‐Valderas A, Estrada Y, et al. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS One. 2012; 7: e35569. doi: 10.1371/journal.pone.0035569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yumoto K, Eber MR, Wang J, et al. Axl is required for TGF‐beta2‐induced dormancy of prostate cancer cells in the bone marrow. Sci Rep. 2016; 6: 36520. doi: 10.1038/srep36520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cackowski FC, et al. Mer tyrosine kinase regulates disseminated prostate cancer cellular dormancy. J Cell Biochem. 2017; 118: 891–902. doi: 10.1002/jcb.25768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Johnson RW, Finger EC, Olcina MM, et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat Cell Biol. 2016; 18: 1078–89. doi: 10.1038/ncb3408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chen D, Sun Y, Wei Y, et al. LIFR is a breast cancer metastasis suppressor upstream of the Hippo‐YAP pathway and a prognostic marker. Nat Med. 2012; 18: 1511–7. doi: 10.1038/nm.2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Iorns E, Ward TM, Dean S, et al. Whole genome in vivo RNAi screening identifies the leukemia inhibitory factor receptor as a novel breast tumor suppressor. Breast Cancer Res Treat. 2012; 135: 79–91. doi: 10.1007/s10549-012-2068-7 [DOI] [PubMed] [Google Scholar]
- 89. Bolin C, Tawara K, Sutherland C, et al. Oncostatin m promotes mammary tumor metastasis to bone and osteolytic bone degradation. Genes Cancer. 2012; 3: 117–30. doi: 10.1177/1947601912458284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wysoczynski M, Miekus K, Jankowski K, et al. Leukemia inhibitory factor: a newly identified metastatic factor in rhabdomyosarcomas. Cancer Res. 2007; 67: 2131–40. doi: 10.1158/0008-5472.can-06-1021 [DOI] [PubMed] [Google Scholar]
- 91. Maruta S, Takiguchi S, Ueyama M, et al. A role for leukemia inhibitory factor in melanoma‐induced bone metastasis. Clin Exp Metastasis. 2008; 26: 133. doi: 10.1007/s10585-008-9223-x [DOI] [PubMed] [Google Scholar]
- 92. Jeong CH, Lee HJ, Cha JH, et al. Hypoxia‐inducible factor‐1 alpha inhibits self‐renewal of mouse embryonic stem cells in vitro via negative regulation of the leukemia inhibitory factor‐STAT3 pathway. J Biol Chem. 2007; 282: 13672–79. doi: 10.1074/jbc.M700534200 [DOI] [PubMed] [Google Scholar]
- 93. Wang N, Docherty F, Brown HK, et al. Mitotic quiescence, but not unique “stemness, ” marks the phenotype of bone metastasis‐initiating cells in prostate cancer. FASEB J. 2015; 29, 3141–50. doi: 10.1096/fj.14-266379 [DOI] [PubMed] [Google Scholar]
- 94. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002; 2: 584, doi: 10.1038/nrc867 [DOI] [PubMed] [Google Scholar]
- 95. Kakonen SM, Selander KS, Chirgwin JM, et al. Transforming growth factor‐beta stimulates parathyroid hormone‐related protein and osteolytic metastases via Smad and mitogen‐activated protein kinase signaling pathways. J Biol Chem. 2002; 277: 24571–8. doi: 10.1074/jbc.M202561200 [DOI] [PubMed] [Google Scholar]
- 96. Sterling JA, Oyajobi BO, Grubbs B, et al. The hedgehog signaling molecule Gli2 induces parathyroid hormone‐related peptide expression and osteolysis in metastatic human breast cancer cells. Cancer Res. 2006; 66: 7548–53. doi: 10.1158/0008-5472.can-06-0452 [DOI] [PubMed] [Google Scholar]
- 97. Johnson RW, Nguyen MP, Padalecki SS, et al. TGF‐beta promotion of Gli2‐induced expression of parathyroid hormone‐related protein, an important osteolytic factor in bone metastasis, is independent of canonical hedgehog signaling. Cancer Res. 2011; 71: 822–31. doi: 10.1158/0008-5472.can-10-2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Javelaud D, Mohammad KS, McKenna CR, et al. Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res. 2007; 67: 2317–24. doi: 10.1158/0008-5472.can-06-3950 [DOI] [PubMed] [Google Scholar]
- 99. Mohammad KS, Javelaud D, Fournier PG, et al. TGF‐β‐RI kinase inhibitor SD‐208 reduces the development and progression of melanoma bone metastases. Cancer Res. 2011; 71: 175–84. doi: 10.1158/0008-5472.CAN-10-2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Biswas S, Nyman JS, Alvarez J, et al. Anti‐transforming growth factor ß antibody treatment rescues bone loss and prevents breast cancer metastasis to bone. PLoS ONE. 2011; 6: e27090. doi: 10.1371/journal.pone.0027090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ottewell PD. The role of osteoblasts in bone metastasis. J Bone Oncol. 2016; 5: 124–27. doi: 10.1016/j.jbo.2016.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ernst DS, Tannock IF, Winquist EW, et al. Randomized, double‐blind, controlled trial of mitoxantrone/prednisone and clodronate versus mitoxantrone/prednisone and placebo in patients with hormone‐refractory prostate cancer and pain. J Clini Oncol. 2003;21:3335–42. doi: 10.1200/jco.2003.03.042 [DOI] [PubMed] [Google Scholar]
- 103. Saad F, Gleason DM, Murray R, et al. Long‐term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone‐refractory prostate cancer. J Natl Cancer Inst. 2004; 96: 879–82. doi: 10.1093/jnci/djh141 [DOI] [PubMed] [Google Scholar]
- 104. Ruppender NS, Merkel AR, Martin TJ, Mundy GR, Sterling JA, Guelcher SA. Matrix rigidity induces osteolytic gene expression of metastatic breast cancer cells. PLoS One. 2010; 5: e15451. doi: 10.1371/journal.pone.0015451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Page JM, Merkel AR, Ruppender NS, et al. Matrix rigidity regulates the transition of tumor cells to a bone‐destructive phenotype through integrin β3 and TGF‐β receptor type II. Biomaterials. 2015; 64: 33–44. doi: 10.1016/j.biomaterials.2015.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Thomas RJ, Guise TA, Yin JJ, et al. Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology. 1999; 140: 4451–8. doi: 10.1210/endo.140.10.7037 [DOI] [PubMed] [Google Scholar]
- 107. Guise TA, Yin JJ, Taylor SD, et al. Evidence for a causal role of parathyroid hormone‐related protein in the pathogenesis of human breast cancer‐mediated osteolysis. J Clin Invest. 1996; 98: 1544–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Thomas RJ, Guise TA, Yin JJ, et al. Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology. 1999; 140: 4451–8. doi: 10.1210/endo.140.10.7037 [DOI] [PubMed] [Google Scholar]
- 109. Johnson RW, Sun Y, Ho PWM, et al. Parathyroid hormone‐related protein negatively regulates tumor cell dormancy genes in a PTHR1/cyclic AMP‐independent manner. Front Endocrinol (Lausanne). 2018;9:241. doi: 10.3389/fendo.2018.00241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lu X, Mu E, Wei Y, et al. VCAM‐1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1‐positive osteoclast progenitors. Cancer Cell. 2011; 20: 701–14. doi: 10.1016/j.ccr.2011.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Lawson MA, McDonald MM, Kovacic N, et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun. 2015; 6: 8983. doi: 10.1038/ncomms9983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Campisi J. Aging, cellular senescence, and cancer. Ann Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Clarke BL, Khosla S. Androgens and bone. Steroids. 2009; 74: 296–305. doi: 10.1016/j.steroids.2008.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Smith EP, Specker B, Korach KS. Recent experimental and clinical findings in the skeleton associated with loss of estrogen hormone or estrogen receptor activity. J Steroid Biochem Mol Biol. 2010; 118: 264–72. doi: 10.1016/j.jsbmb.2009.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Nakamura T, Imai Y, Matsumoto T, et al. Estrogen prevents bone loss via estrogen receptor α and induction of Fas ligand in osteoclasts. Cell. 2007; 130: 811–23. doi: 10.1016/j.cell.2007.07.025 [DOI] [PubMed] [Google Scholar]
- 116. Ottewell PD, Wang N, Brown HK, et al. OPG‐Fc inhibits ovariectomy‐induced growth of disseminated breast cancer cells in bone. Int J Cancer. 2015; 137: 968–77. doi: 10.1002/ijc.29439 [DOI] [PubMed] [Google Scholar]
- 117. Morrissey C, Roudier MP, Dowell A, et al. Effects of androgen deprivation therapy and bisphosphonate treatment on bone in patients with metastatic castration‐resistant prostate cancer: Results from the University of Washington Rapid Autopsy Series. J Bone Miner Res. 2013; 28: 333–40. doi: 10.1002/jbmr.1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Smith MR, Coleman RE, Klotz L, et al. Denosumab for the prevention of skeletal complications in metastatic castration‐resistant prostate cancer: comparison of skeletal‐related events and symptomatic skeletal events. Ann Oncol. 2015; 26: 368–74. doi: 10.1093/annonc/mdu519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Fizazi K, Carducci M, Smith M, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration‐resistant prostate cancer: a randomised, double‐blind study. Lancet. 2011; 377: 813–22. doi: 10.1016/S0140-6736(10)62344-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ghajar CM. Metastasis prevention by targeting the dormant niche. Nat Rev Cancer. 2015; 15: 238–47. doi: 10.1038/nrc3910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Sun YX, Schneider A, Jung Y, et al. Skeletal localization and neutralization of the SDF‐1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J Bone Miner Res. 2005;20:318–29. doi: 10.1359/JBMR. 041109 [DOI] [PubMed] [Google Scholar]
- 122. Conley‐LaComb MK, Semaan L, Singareddy R, et al. Pharmacological targeting of CXCL12/CXCR4 signaling in prostate cancer bone metastasis. Mol Cancer. 2016; 15: 68. doi: 10.1186/s12943-016-0552-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhao Y, Bachelier R, Treilleux I, et al. Tumor alphavbeta3 integrin is a therapeutic target for breast cancer bone metastases. Cancer Res. 2007; 67: 5821–30. doi: 10.1158/0008-5472.CAN-06-4499 [DOI] [PubMed] [Google Scholar]
- 124. Khalili P, Arakelian A, Chen G, et al. A non‐RGD‐based integrin binding peptide (ATN‐161) blocks breast cancer growth and metastasis in vivo. Mol Cancer Ther. 2006;5:2271–80. doi: 10.1158/1535-7163.MCT-06-0100 [DOI] [PubMed] [Google Scholar]
- 125. Brooks P, Clark R, Cheresh D. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994; 264: 569–71. doi: 10.1126/science.7512751 [DOI] [PubMed] [Google Scholar]
- 126. Cirkel GA, Kerklaan BM, Vanhoutte F, et al. A dose escalating phase I study of GLPG0187, a broad spectrum integrin receptor antagonist, in adult patients with progressive high‐grade glioma and other advanced solid malignancies. Invest New Drugs. 2016;34:184–92. doi: 10.1007/s10637-015-0320-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hersey P, Sosman J, O'Day S, et al. A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin αvβ3, ± dacarbazine in patients with stage IV metastatic melanoma. Cancer. 2010; 116: 1526–34. doi: 10.1002/cncr.24821 [DOI] [PubMed] [Google Scholar]
- 128. Bierie B, Moses HL. TGFβ: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506. doi: 10.1038/nrc1926 [DOI] [PubMed] [Google Scholar]
- 129. Raymaekers K, Stegen S, van Gastel N, Carmeliet G. The vasculature: a vessel for bone metastasis. BoneKEy Rep. 2015; 4: 742. doi: 10.1038/bonekey.2015.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. El Touny LH, Vieira A, Mendoza A, Khanna C, Hoenerhoff MJ, Green JE. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J Clin Invest. 2014;124:156–68. doi: 10.1172/JCI70259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Zeng H, Qu J, Jin N, et al. Feedback activation of leukemia inhibitory factor receptor limits response to histone deacetylase inhibitors in breast cancer. Cancer Cell. 2016;30:459–73. doi: 10.1016/j.ccell.2016.08.001 [DOI] [PubMed] [Google Scholar]