

Abstract
Motivation
High throughput (HT) screens in the omics field are typically analyzed by automated pipelines that generate static visualizations and comprehensive spreadsheet data for scientists. However, exploratory and hypothesis driven data analysis are key aspects of the understanding of biological systems, both generating extensive need for customized and dynamic visualization.
Results
Here we describe WIlsON, an interactive workbench for analysis and visualization of multi-omics data. It is primarily intended to empower screening platforms to offer access to pre-calculated HT screen results to the non-computational scientist. Facilitated by an open file format, WIlsON supports all types of omics screens, serves results via a web-based dashboard, and enables end users to perform analyses and generate publication-ready plots.
Availability and implementation
We implemented WIlsON in R with a focus on extensibility using the modular Shiny and Plotly frameworks. A demo of the interactive workbench without limitations may be accessed at http://loosolab.mpi-bn.mpg.de. A standalone Docker container as well as the source code of WIlsON are freely available from our Docker hub https://hub.docker. com/r/loosolab/wilson, CRAN https://cran.r-project.org/web/packages/wilson/, and GitHub repository https://github.molgen.mpg.de/loosolab/wilson-apps, respectively.
1 Introduction
High-throughput (HT) screens of complex biological systems conducted on the genome, proteome or metabolome level generate a massive amount of data. These screens are frequently supplied by technical platforms/facilities of research institutions and clinics in order to focus the necessary technical expertise and to provide access and assistance to end users such as biologists or clinicians. Here we refer to such data producing HT screens as omics. Primary HT results are typically generated by automated software pipelines, and provide relevant features such as genes, probes, or proteins, summarized in extensive spreadsheet tables. In addition, graphical representations automatically generated from these pipelines are often provided as static file formats such as PDF, where interactive adaptation is limited. As interactive exploration and visualization of HT data is a key aspect of the analysis and understanding of the biological systems under investigation such presentations are of limited value for end users without programming skills. Therefore, individual exploration steps are often performed by computational scientist at service platforms, generating additional workload. Interactive tools such as START (Nelson et al., 2017), shinyNGS (https://github.com/pinin4fjords/shinyngs), ExpressionDB (Hughes et al., 2017) and MicroScope (https://www.biorxiv.org/content/early/2016/07/04/034694) have recently become available and address this problem by empowering the end user to generate plots based on high throughput experiments. However, these tools come with limitations regarding input formats, online filtering, adjustment of plotting parameters, or the ability to easily access variable datasets without adapting the source code. Furthermore, these tools are typically not agnostic to the source of the underlying data and focused on RNAseq data. Here we describe the WIlsON_App, an interactive web-based workbench for visual data exploration of multi-omics datasets, based on an identically named R package. It relies on a flexible spreadsheet format suitable for the output of virtually all types of HT screens and experimental settings, and provides access via a convenient, web based dashboard for data exploration. All layers of data (annotations/individual sample/condition/pairwise contrast) are accessible, enabling the user to generate standard plots (such as volcano or MA plots) as well as all comparisons of interest. WIlsON and WIlsON_App are intended to be used by data generating service platforms (e.g. sequencing facilities/array facilities/bioinformatical groups) as a powerful tool to provide an interactive result viewer which imposes a low technical burden on the end user. Nevertheless, WIlsON can be used on local machines utilizing R-studio, making it very attractive for individual end users as well.
2 Results
WIlsON_App is based on a hierarchical structure of R Shiny modules provided as an R package, that constitute a flexible and performant foundation, even when handling gene/protein tables exceeding 100 000 features. Based on the free R Shiny framework (https://shiny.rstudio.com), the infrastructure can be effectively hosted on modest server hardware. The R package and container build recipes are intended to be used to automatically provide interactive data access to individual datasets for the respective end users (Fig. 1).
Fig. 1.
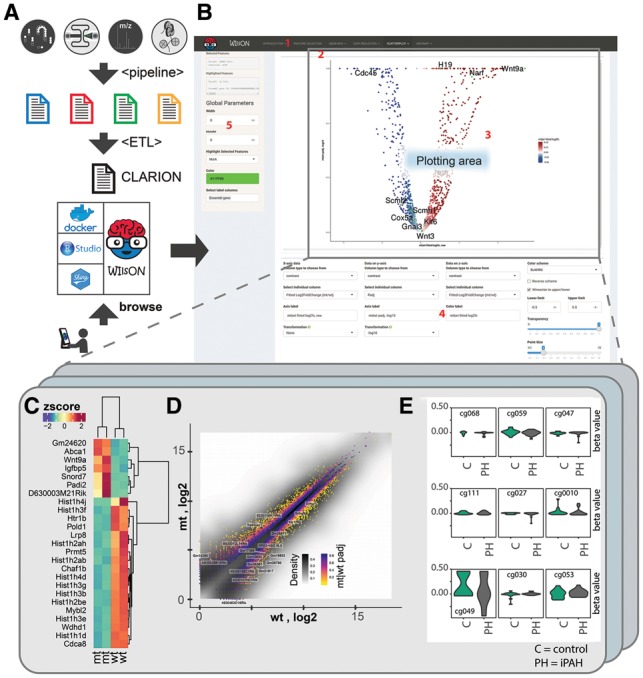
(A) The WIlsON workflow starting from the top: a screening platform generates raw data that is analyzed by a platform-specific software pipeline, providing a platform-specific result format (blue to yellow spreadsheets). An ETL (Extract, Transform, Load) process extracts relevant data generating a CLARION file, that is loaded into the WIlsON_App (containerized infrastructure-> Docker; local-> Rstudio; Client/Server -> Shiny). The end user can access the data with a web browser. (B) Screenshot of the WIlsON_App: the dashboard is divided into subsections as indicated, including a main selection panel (1), allowing data filtering and a plotting module selection. Plotting module specific submenus give access to plotting subtypes (i.e. static and interactive variants) (2); a general plotting area for all plots (3); a plot type specific parameter section (4); and a global parameter section and logging module (5). (C) Heatmap based on PRMT5 (Zhang et al., 2015) dataset: expression data from individual samples for both conditions (wt/mt) were selected, filtered for the top 25 genes considering the adjusted P-value denoting significant differential expression, and a row-wise z-score transformation was applied. Clustering was performed to rows and columns, and a ‘spectral’ color palette was selected. By choosing the static heatmap module, all labels were automatically scaled to be readable. (D) Scatterplot from PRMT5 (Zhang et al., 2015) dataset: all genes were selected and illustrated by choosing mean wt expression values for x axis and mean mutant signal for y axis. Both axes were selected to be log2 transformed. A third dimension was added via color coding based on the adjusted P-value using color palette ‘magma’. For a second data layer, all lncRNA were selected and 25 of these were picked for labeling using the gene symbol. (E) Violin plots from iPAH (Hautefort et al., 2017) dataset: all sites were filtered for nine methylation sites at chromosome 1 with proximity to gene MXRA8. Beta values for controls and all iPAH patients were chosen for grouping
2.1 Design and overview of the WIlsON_App
WIlsON_App is based on a modular design to ensure reusable source code and extensibility, as well as future support. Modules implementing basic functionality (e.g. a statistical transformation or selection of a color palette) may thus be included into other modules to enhance their functionality. Besides a variety of basic functionality, higher level modules encompass the calculation and generation of box plots, bar plots, line plots, violin plots, heatmaps, scatter plots and dimension reductions (PCA, global correlation heatmaps). Most of these support interactive (scroll, zoom, mouse over popup), as well as static modes. Global modules for data labeling and changes on image dimensions are provided as well. While WIlsON is intended to simplify plot generation by providing a graphical user interface, hiding complex plotting scripts in R, log files keep record of all analysis steps. Furthermore, Rdata objects can be downloaded for manual reproduction and manipulation of the visualizations by skilled R users. These Rdata files also serve to encapsulate input and plot functions for long term storage. All interactively generated plots can be subjected to an export function for the generation of high resolution PDF and PNG files for later use. WIlsON is intended to present results from HT screens to the end user, either via a centralized R Shiny server, via applications such as RStudio, or packaged inside a virtualized Docker container for offline usage (Fig. 1A).
2.2 CLARION: a flexible spreadsheet file format
WIlsON relies on a simple tab delimited text format called CLARION, developed to be used with WIlsON. It minimally includes a set of features (e.g. genes, proteins, metabolites, or sequence probes) with an assigned set of numerical values (e.g. counts, scores, fold changes, P-values) and is therefore suitable for the output of virtually all types of HT screens. Information on contained data types (e.g. unique IDs, intensities, calculated contrasts, textual annotations, categories and multiple-value-categories) is described in a metadata header section that is interpreted by the workbench modules to enable selection, filtering and evaluation of numeric expressions. This format keeps data import very flexible, since all -omics HT screens analyses can produce such tables with little modification. For popular HT formats [e.g. MaxQuant based proteomics output (Cox and Mann, 2008)] we included data import functionality in our R package.
2.3 Visual data exploration workflow
WIlsON interprets data columns according to the data type definition (annotation, sample, condition, contrast) in the CLARION file format. Initially, data is parsed within sub modules based on the respective datatype. Additional levels, (e.g. fold change and P-value) within upper level entities (e.g. contrast) are grouped and presented for data transformation and exploration (e.g. zscore on P-value on the x axis of a scatter plot). Typically, a meaningful visualization (e.g. scatterplot, boxplot, or heatmap) is supposed to be created from a set of known features (e.g. genes: hypothesis driven) or from a set of features resulting from an unbiased screening perspective (exploratory/data mining). Often, the data is visualized sequentially as part of an iterative analysis process. WIlsON supports this scenario by providing functionality for: i) the selection, sorting and highlighting of entities of interest by filtering on all provided columns; ii) many different visualization types including dimension reduction methods; and iii) the functionality of a second feature layer (e.g. selection of ‘special’ genes which are plotted in a different way than the rest of the genes considering color or labeling) (Fig. 1B).
2.4 Import of common omics file formats
As proof of principle, we used tables adapted from the widely used tools DESeq2 (Love et al., 2014) taken from a published RNAseq experiment of PRMT5 mutant and wild type samples (Zhang et al., 2015), ADMIRE output (Preussner et al., 2015) based on methylation arrays of patients with pulmonary arterial hypertension (Hautefort et al., 2017), and Maxquant (Tyanova et al., 2016) proteomics analyses of patients with ovarian cancer (Worzfeld et al., 2018). The workbench was used to generate plots exemplified in Fig. 1C, D and E).
3 Conclusion
Technical service groups such as sequencing or proteomics platforms face a growing burden of individual requests for custom visualization that defy automation. The WIlsON app is intended to present such platforms, as well as end users with limited scripting experience, the means to satisfy these requirements. WIlsON includes the infrastructure needed for usage of containers in a multi user environment where access has to be restricted. Furthermore, the WIlsON container is an optimal tool to seal data, software and dependencies in an audit-proof manner for offline usage, load efficient deployment and data storage.
Acknowledgements
We thank Peter Hofmann for technical support.
Funding
This work was supported by the Max-Planck Society (MPG), the Excellence Cluster Cardio-Pulmonary System (ECCPS, Bioinformatics Platform) and from the Deutsche Forschungsgemeinschaft (KLIFO309, Epigenetics Unit).
Conflict of Interest: none declared.
References
- Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol., 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Hautefort A. et al. (2017) Pulmonary endothelial cell DNA methylation signature in pulmonary arterial hypertension. Oncotarget, 8, 52995–53016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes L.D. et al. (2017) ExpressionDB: an open source platform for distributing genome-scale datasets. PLoS One, 12, e0187457.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I. et al. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson J.W. et al. (2017) The START App: a web-based RNAseq analysis and visualization resource. Bioinformatics, 33, 447–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preussner J. et al. (2015) ADMIRE: analysis and visualization of differential methylation in genomic regions using the Infinium HumanMethylation450 Assay. Epigenet. Chromatin., 8, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S. et al. (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc., 11, 2301–2319. [DOI] [PubMed] [Google Scholar]
- Worzfeld T. et al. (2018) Proteotranscriptomics Reveal Signaling Networks in the Ovarian Cancer Microenvironment. Mol Cell Proteomics, 17, 270–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T. et al. (2015) Prmt5 is a regulator of muscle stem cell expansion in adult mice. Nat. Commun., 6, 7140. [DOI] [PMC free article] [PubMed] [Google Scholar]