

Abstract
We describe the use of comparative structural analysis and structure-guided molecular design to develop potent and selective inhibitors (10d and (S)-17b of matrix metalloproteinase 13 (MMP-13). We applied a three-step process, starting with a comparative analysis of the X-ray crystallographic structure of compound 5 in complex with MMP-13 with published structures of known MMP-13·inhibitor complexes followed by molecular design and synthesis of potent but nonselective zinc-chelating MMP inhibitors (e.g., 10a and 10b). After demonstrating that the pharmacophores of the chelating inhibitors (S)-10a, (R)-10a, and 10b were binding within the MMP-13 active site, the Zn2+ chelating unit was replaced with nonchelating polar residues that bridged over the Zn2+ binding site and reached into a solvent accessible area. After two rounds of structural optimization, these design approaches led to small molecule MMP-13 inhibitors 10d and (S)-17b, which bind within the substrate-binding site of MMP-13 and surround the catalytically active Zn2+ ion without chelating to the metal. These compounds exhibit at least 500-fold selectivity versus other MMPs.
1. Introduction
MMP-13 is known to be mainly responsible for the cleavage of type II collagen in osteoarthritis (OA).1,2 The expression of MMP-13 is highly upregulated (>40-fold) in the cartilage of OA patients but is hardly detectable in healthy individuals.3 Transgenic animal models indicate that overexpression of MMP-13 induces joint abnormalities characteristic of human OA.4 Recent reports demonstrate that MMP-13 activity is involved in inflammatory bowel diseases as well as melanoma cell invasion and breast cancer metastasis, which make MMP-13 an even more interesting therapeutic target.5–7
The 24-membered MMP family is highly conserved, with sequence similarity between 56 and 64% in their active domains.8 The common structural element in the MMP active site is a Zn2+ ion coordinated by a tris(histidine) motif.9 The first MMP inhibitors, discovered in the 1990s, were not selective for any particular MMP because of their zinc-chelating functional units.10,11 Several of these compounds entered clinical trials, but all were withdrawn due to the occurrence of musculoskeletal toxicities evoked by unselective binding within the MMP family.12–14 More recently, a selective Zn-binding inhibitor containing a 1,2,4-triazole ring as the metal coordinating group showed promising results in the inhibition of collagen release from cartilage in vitro.15 A more detailed analysis of the MMP active site led to the discovery of six subsites (S1–S3 and S1′–S3′) surrounding the catalytic Zn2+ ion.16 Of these, the S1′ subsite is surrounded by a specificity loop (Ω-loop), which encloses the so-called S1′* specificity pocket and varies in the length and amino acid sequence for different MMP isoforms.16 Targeting Lys140, which is unique at the bottom of the S1′* subsite of MMP-13 vs other MMP isozymes, has provided the basis for the development of highly selective MMP-13 inhibitors. Consequently, various agents possessing a benzoic acid unit, which can form a salt bridge interaction with Lys140, have emerged as highly specific MMP-13 inhibitors (1–4, Figure 1).17–19 However, no MMP-13 inhibitor has yet received FDA approval. Some of the most promising recent selective MMP-13 inhibitors had poor solubility, permeability, biodistribution, metabolic stability, and/or bioavailability, and thus the search for new MMP-13 inhibitors continues.20
Figure 1.

Highly selective MMP-13 inhibitors 1–4.
Structure-guided drug design has been increasingly utilized in modern drug discovery and provides many opportunities for the rational development of drug candidates. Indeed, the rapidly expanding number of protein X-ray structures constitutes a significant resource of structural information useful for structure-guided drug design and has greatly facilitated the drug discovery processes.21–23 In this article, we report the use of comparative structural analysis and molecular design algorithms to design potent and selective MMP-13 inhibitors. Our molecular design approach was validated by the results of biochemical assays and a series of X-ray cocrystal structures of MMP-13 complexed with the designed inhibitors, as reported herein.
2. Results
MMP-13 in Complex with 5 (Figure 2A–C)24 and Design of Zn-Chelating Agents
Figure 2.

Comparative structural analysis and design of Zn-chelating inhibitors. (A) X-ray crystallographic structure of MMP13·5 complex (PDB 4L19). Hydrogen bond interactions of 5 with the amide backbone units of Thr245 and Thr247 are represented in black dashed lines. Leu239, Phe252, and Pro255 form hydrophobic contacts with 5. (B) The 4-methylphenyl ring of 5 is oriented toward the Zn binding site and the MMP-13 S1 subsite. (C) The cyclopentyl ring of 5 occupies the S1′ subsite of MMP-13. (D) Superimposition of X-ray cocrystal structures of MMP-13·5 and MMP-13·6 (PDB 1ZTQ) complexes. (E) Docking model of the designed inhibitor (S)-10a in the MMP-13 active site (ligand–protein interactions are shown in black dashed lines). (F) Superimposition of the X-ray crystallographic (green) and model (blue) structures of MMP-13·(S)-10a complex. The X-ray crystallographic structure of MMP-13·(S)-10a was obtained at 1.60 Å (authors will release the atomic coordinates and experimental data upon article publication; PDB 5UWK). The docking structure was superimposed with the X-ray crystallographic structure by forming Cα atom pairs of MMP-13 using Schrödinger Maestro suites. The two ligand structures matched with 1.4 Å RMSD. Pymol was used to generate figures.
We performed a comparative structural analysis of our previously published MMP-13·524 complex with multiple MMP-13·inhibitor crystallographic structures currently available in the Protein Data Bank (PDB).25 On the basis of this analysis, we found that the benzofuran ring of the Zn-chelating MMP-13 inhibitor 626 substantially overlaps with the cyclopentapyrimidinone unit (core scaffold) of 5 (Figure 2D). Although these two fragments do not have any common structural features, they share the same MMP-13 binding site. On the basis of this observation, inhibitor (S)-10a was designed and its binding pose in the active site of MMP-13 was predicted via Glide docking27,28 (Figure 2E). Compound (S)-10a fits perfectly into the MMP-13 active site with the carboxylic acid unit chelating the active site Zn2+ ion and the hydrogen atom and oxygen atom of the sulfonamide unit forming hydrogen bond interactions with the amide backbone of Ala186 and Leu185, respectively. Furthermore, the pyrimidinone −NH and carbonyl groups form hydrogen bonding interactions with three residues in the Ω-loop (Thr245, Tyr246, and Thr247).
As a proof of concept, compounds (S)-10a, (R)-10a, and achiral 10b were synthesized by using the synthetic route outlined in Scheme 1. Treatment of 4-(bromomethyl)biphenyl (7) with the thioxopyrimidinone fragment 829 in DMF in the presence of triethylamine provided 9 in high yield. Chlorosulfonation of 9 followed by treatment with either l- or d- valine or glycine as the nucleophile gave the chelating MMP-13 inhibitors (S)-10a, (R)-10a, and 10b, respectively.
Scheme 1. Synthesis of Compounds 10a–f.

All three compounds displayed significant inhibition potency toward MMP-13 (IC50 values of 2.2, 7.0, and 1.6 nM, respectively) with inhibition constants (Ki) of 2.3, 1.6, and 1.8 nM, respectively (Table 1). This constituted an almost 1000-fold improvement of inhibition potency compared to the starting inhibitor, 5, and was achieved in only two rounds of SAR optimization. We subsequently validated the binding mode of these inhibitors by determining the X-ray crystallographic structure of MMP-13 in complex with (S)-10a (PDB 5UWK), which was obtained at 1.60 Å. The X-ray structure established that 10a–b are indeed Zn-chelating agents and validates our computational design approach for the development of potent MMP-13 inhibitors. In addition, the docking structure of MMP-13·10a was superimposed on the experimentally determined X-ray structure by forming Cα atom pairs of MMP-13 using the Schrödinger Maestro suites. As shown in Figure 2F, the theoretical and experimental structures are in excellent agreement (1.4 Å RMSD). This result validates the reliability of the docking studies that we performed in the course of this work.
Table 1. IC50 and Ki Values and Selectivity Data for 5 and 10a–f.
| IC50(nM)a | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| R-NH2 | Inhibitor Type |
Ki (nM) MMP-13 |
MMP-13 | MMP-1 | MMP-2 | MMP-8 | MMP-9 | MMP-14 | |
| 5 | – | 800 | 2400 | -b | -b | -b | -b | -b | |
| (S)-10a | 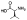 |
Zn2+ chelator | 2.3 | 2.2±0.8 | -c | -c | -c | -c | -c |
| (R)-10a | 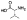 |
Zn2+ chelator | 1.6 | 7.0±1.2 | -c | -c | -c | -c | -c |
| 10b | 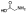 |
Zn2+ chelator | 1.8 | 1.6 | -c | -c | -c | -c | -c |
| 10c | 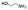 |
non-chelator | 9.2±2.0 | 12.0±2.2 | 4000 | >5000 | >5000 | >10000 | >10000 |
| 10d | 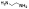 |
non-chelator | 13.5±37 | 3.4±0.4 | >5000 | 730 | 600 | >10000 | >10000 |
| 10e | 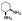 |
non-chelator | - | 17.9±1.2 | >10000 | 2700 | 3500 | >10000 | >10000 |
| 10f | 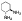 |
non-chelator | - | 16.9±1.0 | >10000 | >10000 | >10000 | >10000 | >10000 |
The IC50 values were determined by using fluorescence resonance energy transfer triple-helical peptides (fTHP) as substrates in the enzyme assay.24,30,31
Compound 5 was tested against MMP-1, -2, -8, -9, and -14 at a single concentration and these data are reported in ref 28 (Roth et al.).
Because (S)-10a, (R)-10a, and 10b are expected to be Zn-chelating agents, these were only tested at a single concentration; these data are reported in Supporting Information, Figure S1. Determination of inhibition constants and modalities were conducted by incubating the range of fTHP-15 substrate concentrations (2–25 μM) with 4 nM MMP-13 at room temperature in the presence of varying concentrations of inhibitors (0.5–50 nM). Experiments were performed 1–3 times in duplicates or triplicates.
To assess the selectivity among the MMP family we tested all compounds discussed in this manuscript for their inhibition of MMP-1, -2, -8, -9, and -14, which are the close relatives of MMP-13 with sequence homologies higher than 60% and which are also capable of cleaving different types of collagen.8,32–34 Triple-helical peptides (THPs) containing a fluorophore and a quencher within the same peptide chain were used as enzyme substrates, whereby fluorescence resonance energy transfer (FRET) measurements assessed enzymatic conversion.31 Because of the conformational features of THPs, the interaction with MMP subsites is more precise than in the case of single-stranded substrates.30,35
It is known that MMP inhibitors that chelate to the Zn2+ ion are often promiscuous,36 as indeed proved to be the case for 10a,b (Supporting Information, Figure S1). Nevertheless, we intentionally included the carboxylic acid unit in our initial inhibitor design, with the intent that it function as an anchor to place the inhibitor in a predictable position in the MMP-13 active site. Subsequently, we explored ways to convert the chelating carboxylate of 10a,b into a non-Zn binding unit via molecular design. Therefore, the amino acid units in 10a,b were replaced by amines, such as in 10c and 10d. The binding poses of 10c and 10d were also predicted via Glide docking, which suggested that these compounds would not be Zn2+ chelating inhibitors (Figure 3A). Both compounds were synthesized following the sequence as described in Scheme 1 and tested for potency and selectivity for inhibition of MMP-13 vs other members of the MMP family.
Figure 3.

Binding poses of 10d in the active site of MMP-13. (A) Docking model of 10d in the active site of MMP-13. (B) X-ray crystallographic structure of 10d in complex with MMP-13 at 3.20 Å resolution (authors will release the atomic coordinates and experimental data upon article publication; PDB 5UWN). The hydrogen bond interactions between the amide backbone units of Gly183, Leu185, and Ala186 with the sulfonamide and amine moieties of the inhibitors are presented in black dashed lines. (C) X-ray crystallographic structure of MMP-13·10d complex. Flexible specificity loops are red, the ethylenediamine units are marked with a blue circle, and the hydrophobic surfaces near the ethylenediamine unit are displayed in yellow. (D) (E) Docking models of inhibitors 10e (D) and 10f (E) in the MMP-13 active site. Hydrophobic areas of the MMP-13 binding site are yellow surfaces.
Compounds 10c and 10d proved to be potent MMP-13 inhibitors, with 12.0 and 3.4 nM IC50s, respectively (Table 1). The selectivity of both compounds toward other MMP isozymes was significantly improved compared to the Zn-chelating inhibitors 10a,b (Table 1 and Supporting Information, Figure S2). The inhibition potency of 10c toward MMP-1, -2, -8, -9, and -14 was ca. 400-fold weaker compared to MMP-13. Compound 10d inhibits MMP-2 and MMP-8 with IC50 values of 730 and 600 nM, respectively, but does not inhibit MMP-1, -9, and -14 at the highest concentration tested (10 μM). We also determined the X-ray crystallographic structure of MMP-13 with bound 10d, which demonstrated that the predicted (docked) binding mode of 10d reasonably matched the inhibitor binding mode the experimental structure except for the orientation of the ethylenediamine unit (Figure 3B,C). The X-ray crystallographic structure of the MMP-13·10d complex includes five different protein chains, which have very similar conformations apart from the specificity loop and the ethylene diamine unit of 10d (Figure 3C). The ethylenediamine units adopt gauche conformations in all five structures, with the terminal primary amine in each case projecting into a solvent accessible area. The solvent exposure and poorly defined electron density observed for the terminal primary amines suggest that multiple conformations are available to the protein-bound ethylenediamine unit of 10d.
In view of the apparent gauche conformation of the ethylene diamine unit of 10d in the MMP-13-bound structures, a 1,2-diaminocyclohexane ring was introduced in compounds 10e and 10f to engage hydrophobic contacts with the protein surface composed of Leu184, Pro242, and Ile243 (yellow surface in Figure 3C). The docking models of 10e and 10f are shown in Figure 3D,E. Two amine units (terminal −NH3+ and −NH of sulfonamide) of 10e are predicted to form a bifurcated salt bridge with Glu223 in the MMP-13 active site. The terminal primary amine of 10f should be able to interact with the amide backbone of Pro242 via a hydrogen bond interaction. Furthermore, the cyclohexane unit of 10f is anticipated to form hydrophobic contacts with Leu184 and Leu185.
Compounds 10e and 10f are marginally weaker MMP-13 inhibitors (IC50 = 18 and 17 nM, respectively) compared to 10d but also exhibit high selectivity against other MMP isozymes (MMP-1, -2, -8, -9, and -14), as shown by the data in Table 1. Inhibitor 10f has IC50 > 10000 nM against all five of these other MMP’s, while 10e exhibits weak inhibition of MMP-2 and MMP-8 with IC50 values of 2.7 and 3.5 μM, respectively.
Application of the Design Method to Non-Zn Chelating Agents
These results validated our use of comparative structural analysis and molecular design for the development of potent and selective MMP-13 inhibitors. As a next step, we extended this method to other known MMP-13-inhibitor crystallographic structures in an attempt to further validate this approach and to design an additional series of potent and highly selective MMP-13 inhibitors.
We intended to keep the core of 5 as part of a second-generation set of inhibitors and to replace the phenylsulfonamide moiety in 10a. To accomplish this, the structure of MMP-13 complexed with 1137 was superimposed with our MMP-13·5 crystallographic structure. The benzofuran unit of 11 and the cyclopentapyrimidinon scaffold of 5 occupy the same space (S1′ site) within the MMP-13 active site without having common pharmacophores (Figure 4A). Thus, (S)-17a was designed based on docking experiments (Figure 4B) and subsequently synthesized (Scheme 2).
Figure 4.

Comparative structural analysis and design of non-Zn chelating MMP-13 inhibitors. (A) Superimposition of MMP-13·5 (green) structure with MMP-13·11 complex (yellow, PDB 3I7I). (B) Predicted binding pose of the designed non-Zn chelating inhibitor (S)-17a. (C) X-ray cocrystal structure of MMP-13·(S)-17a complex. (D) X-ray cocrystal structure of MMP-13·(R)-17a complex. (E) Superimposition of X-ray crystallographic structures of MMP-13 in complex with (S)-17a (green) and (R)-17a (cyan).
Scheme 2. Synthesis of MMP-13 Inhibitors 17a–c.

The Suzuki coupling reaction38 of bromofuran 12 and arylboronic acids 13 and 14 yielded the expected biaryl fragments, which were subsequently converted into the benzylic bromide intermediates 15. After alkylation of 15 with the thioxopyrimidinone fragment 8, syntheses of 17a–c were completed following ester hydrolysis and amide formation.
The inhibition potency of (S)-17a (IC50 = 9.4 nM) vs MMP-13 was nearly 300-fold improved compared to 5. However, (S)-17a proved to be a moderately active inhibitor of MMP-2 and MMP-8 when tested at 200 nM in a single-dose assay (Supporting Information, Figure S3).
Replacement of the l-valine unit (in (S)-17a) with the unnatural amino acid d-valine (to give (R)-17a) resulted in a ca. 40-fold loss of inhibition activity vs MMP-13 (Table 2). The X-ray crystallographic structures of MMP-13 in complex with both enantiomers of 17a were obtained. The MMP-13·(R)-17a structure shows that the (R)-valine unit is located in the substrate binding site and forms hydrophobic contacts with Pro242 and Ile243 (Figure 4E, red dashed circle). However, the terminal N-methylamide unit of (S)-17a, which is 40-fold more potent, forms hydrogen bond interactions with the backbone amide groups of Gly183 and Tyr244 (Figure 4C,E, black dashed lines), while the terminal N-methylamide unit of (R)-17a is oriented toward Leu185 (Figure 4D,E, dark-blue stick). We surmised from these observations that hydrophobic contacts with Ile243 in the substrate binding site are critical for selective ligand binding to MMP-13 compared to other members of the MMP family, more so than the hydrogen bond interaction with the backbone amide groups of Gly183 and Tyr244 (as seen in the (S)-17a structure). To enhance isozyme specificity while retaining high MMP-13 activity, the ortho-position of the phenyl ring of 17a was substituted with a fluorine atom, and the natural amino acid l-valine was replaced with d-valine as well as an unnatural amino acid containing a cyclohexyl ring (hexahydro-Phe, as in 17c). Compound (S)-17b, with an ortho-fluorophenyl ring, exhibited improved selectivity for MMP-13 compared to (S)-17a while retaining its inhibition potency (Table 2). The Ki of (S)-17b is 1.7 nM, and it is comparable to its IC50 (2.7 nM). The introduction of the unnatural amino acid d-valine ((R)-17b) resulted in a drop of activity by almost 100-fold compared to (S)-17b. Furthermore, (S)-17c was a low nanomolar MMP-13 inhibitor (IC50 = 6.3 nM) and had an excellent selectivity profile, with >1000-fold selectivity when tested against the MMP isozymes. Again, enantiomeric (R)-17c lost activity toward MMP-13 (IC50 = 159 nM) but still exhibited a very clean selectivity profile within the collagenases MMP-1, -2, -8, -9, and -14.
Table 2. IC50 Values and Selectivity Data for 17a–c.
| IC50(nM)a | ||||||||
|---|---|---|---|---|---|---|---|---|
| X | R | MMP-13 | MMP-1 | MMP-2 | MMP-8 | MMP-9 | MMP-14 | |
| (S)-17a | H | 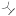 |
9.4±1.7 | -b | -b | -b | -b | -b |
| (R)-17a | H | 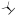 |
356±95 | -c | -c | -c | -c | -c |
| (S)-17bd | F | 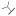 |
2.7±0.6 | >5000 | >5000 | >5000 | >5000 | >5000 |
| (R)-17b | F | 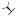 |
257±47 | >5000 | 3100 | >5000 | >5000 | >5000 |
| (S)-17c | F | 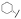 |
6.3±1.5 | >5000 | >5000 | >5000 | >5000 | >5000 |
| (R)-17c | F | 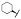 |
159±60 | >5000 | >5000 | >5000 | >5000 | >5000 |
Inhibitor (S)-17a was tested against MMP-1, -2, -8, -9, and -14 at a single concentration and data are reported in Supporting Information, Figure S3.
Not tested due to decrease in potency compared to (S)-17a.
Ki of (S)-17b is 1.7 ± 0.2 nM.
Inhibition of Type II Collagen Cleavage
The potential of inhibitors 10c–f and 17a–c for modifying the degradation of articular cartilage by MMP-13 was evaluated in an in vitro type II collagen cleavage assay.39 These compounds exhibited >90% inhibition of collagenolysis at 20 μM, while 5 is nearly inactive at this concentration (Figure 5). Further dose–response type II collagen cleavage assay revealed that the highly selective MMP-13 inhibitors 10d, (S)-17b, and (S)-17c possess low nM inhibition potency (IC50 = 8.3, 8.1, and 7.9 nM, respectively) against the collagen cleavage activity of MMP-13.
Figure 5.

Inhibition of collagen cleavage activity of MMP13. Inhibition was determined as a percent of intact type II collagen remaining after 24 h of incubation at 37 °C. Collagen oligomers are observed at >250 kDa. “Empty” refers to type II collagen and MMP-13 in the absence of any inhibitor.
Specificity Profiling of 10d and (S)-17b
The protease selectivity of highly potent MMP-13 inhibitors (10d and (S)-17b) was evaluated by using a profiling assay against 25 proteases (Table 3). As expected, 10d and (S)-17b exhibited high inhibition potency vs MMP-13 (97% at 1 μM) in this profiling assay but were substantially if not entirely inactive vs most other proteases tested. Interestingly, 10d is a modestly active inhibitor of MMP-12 (40% inhibition at 1 μM), while (S)-17b is moderately active against MMP-3 and MMP-12, with 63% and 81% inhibition, respectively, at 1 μM. Subsequently, the inhibition IC50 values for 10d and (S)-17b vs MMP-3 and MMP-12 were determined in a 10-point dilution assays using FRET single-stranded peptide substrates.40 These determinations established that the IC50 values for 10d and (S)-17b as inhibitors of MMP-12 are 470 and 1800 nM, respectively (e.g., 140- and 700-fold less active than their activity as MMP-13 inhibitors). Because of high structural similarity between (S)-17b and (S)-17c, these two inhibitors possess similar inhibition potency toward MMP-13, selectivity against other MMP isozymes, and inhibition of type II collagen cleavage as shown in Table 2 and Figure 5. As such, it is also expected that (S)-17c should display similar selectivity profiling as compared to (S)-17b.
Table 3. Protease Selectivity Profiling for 10d and (S)-17b.
| % inhibitiona |
% inhibitiona |
||||
|---|---|---|---|---|---|
| enzyme | 10d | (S)-17b | enzyme | 10d | (S)-17b |
| ACE | 7 | 0 | cathepsin-S | 12 | 2 |
| ACE2 | 1 | 0 | factor-XA | 3 | 0 |
| ADAM10 | 0 | 0 | furin | 0 | 0 |
| BACE-1 | 1 | 0 | IDE | 0 | 0 |
| caspase-1 | 1 | 0 | MMP-3 (stromelysin) | 13 | 63 (IC50 = 4.4 μM)b |
| caspase-2 | 0 | 0 | MMP-7 (matrilysin) | 7 | 4 |
| caspase-3 | 0 | 0 | MMP-12 | 40 (IC50 = 467 nM)b | 81 (IC50 = 1.8 μM)b |
| caspase-5 | 0 | 0 | MMP-13 | 97 | 97 |
| caspase-6 | 0 | 0 | neprilysin | 3 | −6 |
| caspase-7 | 1 | 8 | TACE | 4 | −3 |
| cathepsin-D | 3 | 0 | thrombin | −3 | −8 |
| cathepsin-K | 0 | 2 | uPA | 3 | 1 |
| cathepsin-L | 9 | 7 | |||
Percent inhibition was determined by using single-stranded peptide substrates at an inhibitor concentration of 1 μM. Assays were performed in duplicates, % inhibition was determined, and average values are present.
Additional specificity assays against MMP-3 and MMP-12 were performed to determine the IC50 values.
3. Discussion
The traditional strategy for the development of MMP-13 inhibitors has targeted the catalytically active Zn2+ ion. However, it is now well established that specificity and toxicity issues arise due to Zn2+ chelation, and so efforts are now focusing on development of isoform specific inhibitors by other means. Recently, Lys140 located in the S1′* site of MMP-13 has been targeted for the development of selective MMP-13 inhibitors.17 Most of these specific inhibitors possess a benzoic acid unit (Figure 1) that can form a salt bridge interaction with Lys140.18,19 However, the benzoic acid moiety is potentially vulnerable to phase II drug metabolism such as glucuronidation, leading to potential limitations in in vivo efficacy studies.41,42 Hence, the design of new types of MMP-13 specific inhibitors that do not target either the S1′ subsite only or the Zn2+ ion is an important objective.
We demonstrated here that highly specific MMP-13 inhibitors (e.g., 10d and (S)-17b) can be designed by taking advantage of substrate–inhibitor interactions close to the Zn2+ ion in the substrate-binding site. These inhibitors bind within the substrate binding site and bridge from the S1 subsite over the Zn2+ ion toward a solvent accessible area without chelating to the metal.
The strategy that we utilized in the design of these new, potent, and selective inhibitors (e.g., 10d and (S)- 17b) is highly efficient; only two rounds of analogue design and synthesis were required to progress from the starting point 5 to the final inhibitors 10d and (S)-17b.
Key to the success of this effort were systemic analyses of structural features of known MMP-13 inhibitors, design of new inhibitor candidates by using molecular modeling, and X-ray crystallographic validation of binding poses of new generations of inhibitors.
Detailed structural analyses of enzyme–inhibitor interactions in close proximity to the catalytically active Zn2+ ion provided a clear basis for the structure-based design and development of highly potent and specific MMP-13 inhibitors. While the binding pose of the Zn-chelating inhibitor (S)-10a was precisely predicted by the molecular docking study, the orientation of the ethylene amine moiety in 10d was not correctly predicted in the modeling effort. This discrepancy mainly resulted from the overemphasis of scoring functions influenced by the ligand–enzyme hydrogen bonding interaction (Figure 3B,C). Interestingly, all five chains in the X-ray cocrystal structure of MMP-13·10d have very similar binding poses without any specific interaction of the terminal amine with the target enzyme. Presumably, the hydrophobic contacts between the ethylene unit and Leu184 and Pro242, which were not recognized by the docking analysis, make important contributions to the binding of these MMP-13 inhibitors. The significance of hydrophobic contacts in this region is also highlighted by the X-ray cocrystal structures of MMP-13 in complex with the two enantiomers (S)-17a or (R)-17a (Figure 4C,D). The binding pose of (S)-17a was predicted in the docking model, and we assumed its enantiomer (R)-17a possesses the same hydrogen bond network with the isopropyl unit pointing toward the opposite direction. However, the X-ray cocrystal structure of (R)-17a shows that the terminal N-methyl amide unit is twisted to enable the isopropyl units to form hydrophobic contacts with Ile243 and Pro242. Because the N-methyl amide unit of (R)-17a is not involved in any direct interaction with the target enzyme (or hydrophobic–π interactions with Leu185), the hydrophobic contacts of the isopropyl group with Ile243 seemed to be an important contributor for inhibitor binding. This conclusion was instrumental in the decision to synthesize (S)-17b.
4. Conclusion
We have developed a very useful strategy for the design of highly potent and selective MMP-13 inhibitors. The process started with a comparative analysis of the X-ray crystallographic structure of compound 5 in complex with MMP-13 with published structures of known MMP-13·inhibitor complexes to design, and then to synthesize potent, but nonselective zinc-chelating MMP inhibitors. After demonstrating that the pharmacophores of our inhibitors were properly binding in the MMP-13 active site, the Zn2+ chelating unit was removed and replaced with polar residues that bridged over the Zn2+ ion and reached into a solvent accessible area. This approach led to the development of highly potent and isoform-selective MMP-13 inhibitors 10d and (S)-17b. To our knowledge, this strategy has not been previously used in the development of MMP inhibitors. This approach, namely designing metal-chelating inhibitors and subsequently removing the chelating moiety to improve selectivity, could become a useful method for the structure-based design of inhibitors of other metal-containing enzymes, including other MMP isozymes.
Assessment of this new set of selective MMP-13 inhibitors in in vitro and in vivo pharmacokinetic studies and ultimately in in vivo disease models are in progress and will be reported in due course.
Supplementary Material
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b00514.
Experimental procedures for MMP-13 assays, X-ray structure determination and analysis, synthesis of inhibitors, and docking studies(PDF)
Molecular formula strings (CSV)
Docking model MMP-13-10e (PDB)
Docking model MMP-13-10f (PDB)
Acknowledgments
This research was generously supported by NIH R01 grant AR063795 (G.B.F., P.J.H., and W.R.R.). P.J.H. was additionally funded by the Welch Foundation AQ-1399. R.F. was supported by a Schroedinger Postdoctoral Fellowship, financed by the Austrian Science Fund (FWF). Support for the X-ray Crystallography Core Laboratory by the UTHSCSA Office of the Vice President for Research and the Cancer Therapy & Research Center (NIH P30 CA054174) is gratefully acknowledged. The X-ray structure data presented in this work were obtained at the Northeastern Collaborative Access Team beamlines, funded by NIH (P41 GM103403). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357.
Abbreviations Used
- MMP
matrix metalloproteinase
- RMSD
root-mean-square deviation
- THP
triple helical peptide
- FAM-fTHP
fluorescein amidite–fluorescence resonance energy transfer triple-helical peptide
- OA
osteoarthritis
- PDB
Protein Data Bank
- ACE
angiotensin-converting enzyme
- ADAM
a disintegrin and metalloproteinase
- BACE
β-secretase
- IDE
insulin degrading enzyme
- TACE
tumor necrosis factor-α converting enzyme
- uPA
urokinase-type plasminogen activator
Footnotes
Accession Codes
PDB ID codes for MMP-13·(S)-10a, MMP-13·(S)-17a, MMP-13·(R)-17a, and MMP-13·10d complexes are 5UWK, 5UWL, 5UWM, and 5UWN, respectively. Authors will release the atomic coordinates and experimental data upon article publication.
ORCID
William R. Roush: 0000-0001-9785-5897
Author Contributions
J.Y.C. and R.F. contributed equally. J.Y.C and R.F. performed design, synthesis, and structure analysis. A.M.K and L.S. performed the enzyme assays. A.T., X.C, and P.J.H. obtained the X-ray crystallographic structures. G.B.F. and W.R.R. designed the overall study. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- (1).Takaishi H, Kimura T, Dalal S, Okada Y, D’Armiento J. Joint diseases and matrix metalloproteinases: A role for MMP-13. Curr Pharm Biotechnol. 2008;9:47–54. doi: 10.2174/138920108783497659. [DOI] [PubMed] [Google Scholar]
- (2).Neuhold LA, Killar L, Zhao WG, Sung MLA, Warner L, Kulik J, Turner J, Wu W, Billinghurst C, Meijers T, Poole AR, et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J Clin Invest. 2001;107:35–44. doi: 10.1172/JCI10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bau B, Gebhard PM, Haag J, Knorr T, Bartnik E, Aigner T. Relative messenger RNA expression profiling of collagenases and aggrecanases in human articular chondrocytes in vivo and in vitro. Arthritis Rheum. 2002;46:2648–2657. doi: 10.1002/art.10531. [DOI] [PubMed] [Google Scholar]
- (4).Baragi VM, Becher G, Bendele AM, Biesinger R, Bluhm H, Boer J, Deng H, Dodd R, Essers M, Feuerstein T, Gallagher BM, et al. A new class of potent matrix metalloproteinase 13 inhibitors for potential treatment of osteoarthritis: Evidence of histologic and clinical efficacy without musculoskeletal toxicity in rat models. Arthritis Rheum. 2009;60:2008–2018. doi: 10.1002/art.24629. [DOI] [PubMed] [Google Scholar]
- (5).Morrison C, Mancini S, Cipollone J, Kappelhoff R, Roskelley C, Overall C. Microarray and proteomic analysis of breast cancer cell and osteoblast co-cultures: role of osteoblast matrix metalloproteinase (MMP)-13 in bone metastasis. J Biol Chem. 2011;286:34271–34285. doi: 10.1074/jbc.M111.222513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Vandenbroucke RE, Dejonckheere E, Van Hauwermeiren F, Lodens S, De Rycke R, Van Wonterghem E, Staes A, Gevaert K, López-Otin C, Libert C. Matrix metalloproteinase 13 modulates intestinal epithelial barrier integrity in inflammatory diseases by activating TNF. EMBO Mol Med. 2013;5:1000–1016. doi: 10.1002/emmm.201202100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zigrino P, Kuhn I, Bauerle T, Zamek J, Fox JW, Neumann S, Licht A, Schorpp-Kistner M, Angel P, Mauch C. Stromal expression of MMP-13 is required for melanoma invasion and metastasis. J Invest Dermatol. 2009;129:2686–2693. doi: 10.1038/jid.2009.130. [DOI] [PubMed] [Google Scholar]
- (8).Lukacova V, Zhang Y, Mackov M, Baricic P, Raha S, Calvo JA, Balaz S. Similarity of binding sites of human matrix metalloproteinases. J Biol Chem. 2004;279:14194–14200. doi: 10.1074/jbc.M313474200. [DOI] [PubMed] [Google Scholar]
- (9).Lovejoy B, Welch AR, Carr S, Luong C, Broka C, Hendricks RT, Campbell JA, Walker KAM, Martin R, Van Wart H, Browner MF. Crystal structures of MMP-1 and −13 reveal the structural basis for selectivity of collagenase inhibitors. Nat Struct Biol. 1999;6:217–221. doi: 10.1038/6657. [DOI] [PubMed] [Google Scholar]
- (10).Rothenberg ML, Nelson AR, Hande KR. New drugs on the horizon: matrix metalloproteinase inhibitors. Stem Cells. 1999;17:237–240. doi: 10.1002/stem.170237. [DOI] [PubMed] [Google Scholar]
- (11).Blagg JA, Noe MC, Wolf-Gouveia LA, Reiter LA, Laird ER, Chang S-PP, Danley DE, Downs JT, Elliott NC, Eskra JD, Griffiths RJ, et al. Potent pyrimidinetrione-based inhibitors of MMP-13 with enhanced selectivity over MMP-14. Bioorg Med Chem Lett. 2005;15:1807–1810. doi: 10.1016/j.bmcl.2005.02.038. [DOI] [PubMed] [Google Scholar]
- (12).Mengshol JA, Mix KS, Brinckerhoff CE. Matrix metalloproteinases as therapeutic targets in arthritic diseases: bull’s-eye or missing the mark? Arthritis Rheum. 2002;46:13–20. doi: 10.1002/1529-0131(200201)46:1<13::aid-art497>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- (13).Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci, Landmark Ed. 2006;11:529–543. doi: 10.2741/1817. [DOI] [PubMed] [Google Scholar]
- (14).Fingleton B. MMPs as therapeutic targets—still a viable option? Semin Cell Dev Biol. 2008;19:61–68. doi: 10.1016/j.semcdb.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nara H, Kaieda A, Sato K, Naito T, Mototani H, Oki H, Yamamoto Y, Kuno H, Santou T, Kanzaki N, Terauchi J, et al. Discovery of novel, highly potent, and selective matrix metalloproteinase (MMP)-13 inhibitors with a 1,2,4-triazol-3-yl moiety as a zinc binding group using a structure-based design approach. J Med Chem. 2017;60:608–626. doi: 10.1021/acs.jmedchem.6b01007. [DOI] [PubMed] [Google Scholar]
- (16).Dormán G, Cseh S, Hajdú I, Barna L, Kónya D, Kupai K, Kovács L, Ferdinandy P. Matrix metalloproteinase inhibitors. Drugs. 2010;70:949–964. doi: 10.2165/11318390-000000000-00000. [DOI] [PubMed] [Google Scholar]
- (17).Nara H, Sato K, Naito T, Mototani H, Oki H, Yamamoto Y, Kuno H, Santou T, Kanzaki N, Terauchi J, Uchikawa O, et al. Discovery of novel, highly potent, and selective quinazoline-2-carboxamide-based matrix metalloproteinase (MMP)-13 inhibitors without a zinc binding group using a structure-based design approach. J Med Chem. 2014;57:8886–8902. doi: 10.1021/jm500981k. [DOI] [PubMed] [Google Scholar]
- (18).Gege C, Bao B, Bluhm H, Boer J, Gallagher BM, Korniski B, Powers TS, Steeneck C, Taveras AG, Baragi VM. Discovery and evaluation of a non-Zn chelating, selective matrix metalloproteinase 13 (MMP-13) inhibitor for potential intra-articular treatment of osteoarthritis. J Med Chem. 2012;55:709–716. doi: 10.1021/jm201152u. [DOI] [PubMed] [Google Scholar]
- (19).Johnson AR, Pavlovsky AG, Ortwine DF, Prior F, Man CF, Bornemeier DA, Banotai CA, Mueller WT, Mcconnell P, Yan C, Baragi V, et al. Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without joint fibroplasia side effects. J Biol Chem. 2007;282:27781–27791. doi: 10.1074/jbc.M703286200. [DOI] [PubMed] [Google Scholar]
- (20).Nara H, Sato K, Kaieda A, Oki H, Kuno H, Santou T, Kanzaki N, Terauchi J, Uchikawa O, Kori M. Design, synthesis, and biological activity of novel, potent, and highly selective fused pyrimidine-2-carboxamide-4-one-based matrix metalloproteinase (MMP)-13 zinc-binding inhibitors. Bioorg Med Chem. 2016;24:6149–6165. doi: 10.1016/j.bmc.2016.09.009. [DOI] [PubMed] [Google Scholar]
- (21).Thiel KA. Structure-aided drug design’s next generation. Nat Biotechnol. 2004;22:513–519. doi: 10.1038/nbt0504-513. [DOI] [PubMed] [Google Scholar]
- (22).Sliwoski G, Kothiwale S, Meiler J, Lowe EW. Computational methods in drug discovery. Pharmacol Rev. 2014;66:334–395. doi: 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kuntz ID. Structure-based strategies for drug design and discovery. Science. 1992;257:1078–1082. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- (24).Spicer TP, Jiang JW, Taylor AB, Choi JY, Hart PJ, Roush WR, Fields GB, Hodder PS, Minond D. Characterization of selective exosite-binding inhibitors of matrix metalloproteinase 13 that prevent articular cartilage degradation in vitro. J Med Chem. 2014;57:9598–9611. doi: 10.1021/jm501284e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wu J, Rush TS, III, Hotchandani R, Du X, Geck M, Collins E, Xu Z-B, Skotnicki J, Levin JI, Lovering FE. Identification of potent and selective MMP-13 inhibitors. Bioorg Med Chem Lett. 2005;15:4105–4109. doi: 10.1016/j.bmcl.2005.06.019. [DOI] [PubMed] [Google Scholar]
- (27).Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- (28).Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide:- A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- (29).Roth J, Minond D, Darout E, Liu Q, Lauer J, Hodder P, Fields GB, Roush WR. Identification of novel, exosite-binding matrix metalloproteinase-13 inhibitor scaffolds. Bioorg Med Chem Lett. 2011;21:7180–7184. doi: 10.1016/j.bmcl.2011.09.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lauer-Fields JL, Fields GB. Triple-helical peptide analysis of collagenolytic protease activity. Biol Chem. 2002;383:1095–1105. doi: 10.1515/BC.2002.118. [DOI] [PubMed] [Google Scholar]
- (31).Minond D, Lauer-Fields JL, Cudic M, Overall CM, Pei DQ, Brew K, Visse R, Nagase H, Fields GB. The roles of substrate thermal stability and P-2 and P-1 ′ subsite identity on matrix metalloproteinase triple-helical peptidase activity and collagen specificity. J Biol Chem. 2006;281:38302–38313. doi: 10.1074/jbc.M606004200. [DOI] [PubMed] [Google Scholar]
- (32).Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- (33).Bigg HF, Rowan AD, Barker MD, Cawston TE. Activity of matrix metalloproteinase-9 against native collagen types I and III. FEBS J. 2007;274:1246–1255. doi: 10.1111/j.1742-4658.2007.05669.x. [DOI] [PubMed] [Google Scholar]
- (34).Fields GB. Interstitial collagen catabolism. J Biol Chem. 2013;288:8785–8793. doi: 10.1074/jbc.R113.451211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lauer-Fields JL, Broder T, Sritharan T, Chung L, Nagase H, Fields GB. Kinetic analysis of matrix metalloproteinase activity using fluorogenic triple-helical substrates. Biochemistry. 2001;40:5795–5803. doi: 10.1021/bi0101190. [DOI] [PubMed] [Google Scholar]
- (36).Overall CM, Kleifeld O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br J Cancer. 2006;94:941–946. doi: 10.1038/sj.bjc.6603043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Heim-Riether A, Taylor SJ, Liang S, Gao DA, Xiong Z, August EM, Collins BK, Farmer BT, II, Haverty K, Hill-Drzewi M, Junker H-D, et al. Improving potency and selectivity of a new class of non-Zn-chelating MMP-13 inhibitors. Bioorg Med Chem Lett. 2009;19:5321–5324. doi: 10.1016/j.bmcl.2009.07.151. [DOI] [PubMed] [Google Scholar]
- (38).Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem Rev. 1995;95:2457–2483. [Google Scholar]
- (39).Chung L, Dinakarpandian D, Yoshida N, Lauer-Fields JL, Fields GB, Visse R, Nagase H. Collagenase unwinds triple-helical collagen prior to peptide bond hydrolysis. EMBO J. 2004;23:3020–3030. doi: 10.1038/sj.emboj.7600318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Neumann U, Kubota H, Frei K, Ganu V, Leppert D. Characterization of Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2, a fluorogenic substrate with increased specificity constants for collagenases and tumor necrosis factor converting enzyme. Anal Biochem. 2004;328:166–173. doi: 10.1016/j.ab.2003.12.035. [DOI] [PubMed] [Google Scholar]
- (41).Shipkova M, Armstrong VW, Oellerich M, Wieland E. Acyl glucuronide drug metabolites: toxicological and analytical implications. Ther Drug Monit. 2003;25:1–16. doi: 10.1097/00007691-200302000-00001. [DOI] [PubMed] [Google Scholar]
- (42).Sawamura R, Okudaira N, Watanabe K, Murai T, Kobayashi Y, Tachibana M, Ohnuki T, Masuda K, Honma H, Kurihara A, Okazaki O. Predictability of idiosyncratic drug toxicity risk for carboxylic acid-containing drugs based on the chemical stability of acyl glucuronide. Drug Metab Dispos. 2010;38:1857–1864. doi: 10.1124/dmd.110.034173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.