

Abstract
Parkinson’s disease (PD) presents with a constellation of non-motor symptoms, notably increased anxiety, which are currently poorly treated and underrepresented in animal models of the disease. Human post-mortem studies report loss of catecholaminergic neurons in the pre-symptomatic phases of PD when anxiety symptoms emerge, and a large literature from rodent and human studies indicate that catecholamines are important mediators of anxiety via their modulatory effects on limbic regions such as the amygdala. On the basis of these observations, we hypothesized that anxiety in PD could result from an early loss of catecholaminergic inputs to the amygdala and/or other limbic structures. To interrogate this hypothesis, we bilaterally injected the neurotoxin 6-OHDA in the mouse basolateral amygdala (BL). This produced a restricted pattern of catecholaminergic (tyrosine-hydroxylase-labeled) denervation in the BL, intercalated cell masses and ventral hippocampus, but not the central amygdala or prefrontal cortex. We found that this circuit-specific lesion did not compromise performance on multiple measures of motor function (home cage, accelerating rotarod, beam balance, pole climbing), but did increase anxiety-like behavior in the elevated plus-maze and light-dark exploration tests. Fear behavior in the pavlovian cued conditioning and passive avoidance assays was, by contrast, unaffected; possibly due to preservation of catecholamine innervation of the central amygdala from the periaqueductal gray. These data provide some of the first evidence implicating loss of catecholaminergic neurotransmission in midbrain-amygdala circuits to increased anxiety-like behavior. Our findings offer an initial step towards identifying the neural substrates for pre-motor anxiety symptoms in PD.
Keywords: Anxiety, fear learning, amygdala, Parkinson disease, dopamine, noradrenaline, 6-hydroxydopamine
INTRODUCTION
According to the classical view, Parkinson’s disease (PD) is a progressive neurodegenerative motor disorder caused primarily by the loss of midbrain dopamine (DA) neurons in the substantia nigra pars compacta (SNc). However, it is increasingly appreciated that PD presents with a complex clinical phenotype comprising a constellation of non-motor symptoms, some of which predate the emergence of motor impairment (Kalia and Lang, 2015). These symptoms range from anosmia and autonomic abnormalities to disturbed sleep and depression and, of particular note, anxiety (Bower et al., 2010; Schapira et al., 2017). Anxiety symptoms affect up to sixty percent of patients with PD (Chaudhuri and Schapira, 2009; Lin et al., 2015), yet the management of these symptoms remains a significant clinical challenge because they do not respond well to existing medications (Zesiewicz et al., 2010).
In addition to the loss of DA neurons, PD involves pathophysiological alterations in a number of neurotransmitter systems. Within Braak’s scheme of the staged progression of PD pathology (Braak et al., 2004), the deterioration of noradrenergic (NA) function, including NA neuronal loss, emerges prior to the manifestation of motor symptoms. Given a large corpus of data implicating the NA system in emotional processing (McCall et al., 2017; McGaugh, 2004), NA dysfunction is one plausible mechanism contributing to the anxiety symptoms found in PD. NA neurons in the locus coeruleus (LC) and other brainstem nuclei as well as mesencephalic DAergic neurons target a number of forebrain structures known to regulate anxiety, including the medial prefrontal cortex (mPFC), ventral hippocampus (vHPC) and basolateral amygdala (BL) (Bukalo et al., 2015; Schwarz and Luo, 2015; Adhikari et al., 2015; Tovote et al., 2015). Moreover, PD patients exhibit deficits in forms of emotional recognition (Jacobs et al., 1995; Sprengelmeyer et al., 2003) and prosody (Crucian et al., 2001) that are known to be dependent upon intact amygdala function (Diederich et al., 2016).
Direct involvement of the amygdala in anxiety has been demonstrated in both human and rodent models (Büchel et al., 1999; Davidson, 2002) and, in particular, the BL-vHPC circuits have been implicated in the control of anxiety-like behavior (Tye et al., 2011; Felix-Ortiz et al., 2013). To date, the potential contribution of the amygdala and other limbic regions to anxiety in PD has received limited attention (Prediger et al., 2012; Schapira et al., 2017); reflecting the current paucity of animal models of non-motor symptoms in PD in general (Blandini and Armentero, 2012). To address this critical gap in the literature, the current study assessed the effects of a circuit-specific catecholamine ablation via infusion of a low-dose of the neurotoxin 6-hydroxydopamine (6-OHDA) directly into the BL of mice. We found that this insult produced retrograde neurodegeneration of DAergic neurons in both the SNc and ventral tegmental area (VTA), as well as selective catecholaminergic denervation of the BL, intercalated cell masses of the amygdala (ITC) and vHPC, but not the central nucleus of the amygdala (CeA) or mPFC. The neural abnormalities were associated with a significant and specific increase in anxiety-like behavior, without concomitant alterations in motor functions or conditioned fear. Together, these findings offer some of the first evidence implicating a loss of catecholaminergic neurotransmission in midbrain-amygdala circuits to increased anxiety-like behavior. Thus, the pathogenesis of anxiety symptoms in PD may result from early damage to these circuits.
MATERIALS AND METHODS
Subjects
All procedures involving animals were performed according to methods approved by the Austrian Animal Experimentation Ethics Board (license: BMWFW-66.011/0148-WF/V/3b/2014 and BMWFW-66.011/0021-WF/V/3b/2016) and NIAAA Animal Care and Use Committee and were in compliance with the European convention for the protection of vertebrate animals used for experimental and other scientific purposes (ETS number 123) and the NIH guidelines outlined in ‘Using Animals in Intramural Research’ and the local Animal Care and Use Committees. Every effort was taken to minimize animal suffering and the number of animals used. For this study, adult C57BL/6J male mice (Charles Rivers, Sulzfeld, Germany, or Jackson Laboratory, Bar Harbor, ME, USA) were used.
Intra-BL 6-OHDA administration
Anesthesia was induced with a combination of intraperitoneally injected Ketasol (80 mg/kg) and Xylazine (5 mg/kg) and maintained with 2% Sevofluran (SEVOrane). Either 6-OHDA (dissolved in 0.9% NaCI + 0.02% ascorbic acid − vehicle) or vehicle was stereotactically-injected into the BL (coordinates: DV −5.00, RC −1.30 ML ±3.35 relative to bregma). For neuropathological experiments (n=30 mice), either 0.2, 0.5, 1.0, 2.0, 4.0 μg 6-OHDA or vehicle was unilaterally infused in a volume of 0.2 μL over 10min. For behavioral experiments (n=99 mice), 1.0 μg 6-OHDA was bilaterally infused into the BL in a volume of 0.2 μL.
At the end of the respective experimental procedures, mice were deeply anesthetized with thiopental sodium (150 mg/kg, i.p.) and transcardially perfused with a fixative [4% paraformaldehyde + 15% picric acid in 0.1 M phosphate-buffer (PB), pH 7.2-7.4], to verify the site of injection and respective 6-OHDA-mediated denervation.
Tyrosine hydroxylase (TH) and norepinephrine transporter (NET) immunostaining
Coronal sections were cut (50 μm) on a Leica VT1000S vibratome (Leica Microsystems, Vienna, Austria) and immunostained against TH to quantify the extent of DA cell loss in SNc and VTA and catecholamine denervation according to previously published procedures (Dobi et al., 2013). A rabbit antibody against TH (Millipore, Temecula, CA, cat. no. AB152) was diluted 1:4,000 in 2% normal goat serum (NGS), 0.3% Triton X-100 in Tris-buffered saline (TBS; pH 7.4). After three washing steps, sections were incubated with a biotinylated goat anti-rabbit secondary antibody (1:500, Vector Laboratories, Burlingame, CA) overnight. The antigen-antibody-complex was visualized by the avidin-biotin-horseradish peroxidase procedure (Vectastain Elite ABC kit; Vector Laboratories) using 3,3’-diaminobenzidine.
In double immunofluorescence experiments, the rabbit anti-TH antibody (1:4,000, Millipore) was combined with a guinea pig antibody against the mouse NET (1:2,000, Frontiers Institute, Hokkaido, Japan, cat. no. NET-GP-Af1500), incubated for 48 hours at 6°C followed by overnight incubation with the respective secondary antibodies (anti-guinea pig Alexa Fluor™488, 1:1,000, Jackson ImmunoResearch Europe Ltd, Ely, UK; anti-rabbit Cy3, 1:500, Invitrogen, ThermoFisher Scientific, Waltham, MA). To visualize general brain morphology, sections were counterstained with a DAPI solution (2 μg/ml, Sigma, cat. no. D-9564) for 4 min. Sections were mounted onto gelatin-coated slides and coverslipped with Vectashield (Vector Laboratories). Immunofluorescence signals were examined using a Zeiss Axiolmager M1 microscope equipped with a metal-halide lamp. Images were acquired through an Orca-ER CCD-camera (Hamamatsu, Hamamatsu City, Japan) and displayed with Openlab software (version 5.5.0; Improvision, Coventry, UK).
Stereology
Stereological analyses were performed using a Nikon E-800 microscope, equipped with a Nikon DXM 1200 digital camera and the Stereoinvestigator Software (MicroBrightField Europe e.K., Magdeburg, Germany). Optical fractionator was used to define the estimated total number of cells in SNc and VTA in the lesioned and non-lesioned side. One of every four brain sections containing the VTA and SNc were used for quantification. Data are presented as percentage of neuronal loss versus the non-lesioned side.
Retrograde neuronal tracing
The retrograde tracer Cholera Toxin Subunit B (CTB) (Invitrogen) conjugated to Alexa Fluor™555 or Alexa Fluor™488 (0.5% weight/volume in phosphate-buffered saline) was unilaterally injected into BL (coordinates: DV −4.90, RC −1.40, ML ±3.30 relative to bregma; injection volume 0.3 μL) and/or vHPC (coordinates: DV −4.20, RC −3.10, ML ±3.30; injection volume 0.3 μL) using a NeuroS, 7000.5, 0.5 μL Hamilton Syringe (Reno, NV, USA) over 10 min. The needle was left in place for 5 min to ensure diffusion. CTB was injected into each region counterbalanced across mice. One week after surgery, mice were terminally anesthetized with pentobarbital (50-60 mg/kg, i.p.) and transcardially perfused with ice-cold 4% paraformaldehyde in 0.1 Μ PB. Brains were post-fixed overnight at 4°C. Coronal sections (50 μm) were cut on a vibratome (Leica VT100S) and immunostained for TH using a mouse monoclonal antibody (clone LNC1 diluted 1:1,000, Millipore-Sigma St. Louis, MO, USA, cat. no. MAB318). Sections were blocked for 2 h in a solution containing 10% Normal Goat Serum, 1% Bovine Serum Albumin, 0.3% TritonX in 0.05 M PBS and then incubated overnight at 4°C with the anti-TH primary antibody diluted in PBS containing 10% Triton X-100 and the blocking solution. The following day, sections were rinsed at least three times and incubated for 2 h at room temperature in secondary antibody solution containing Alexa 647 (1:1000, goat anti-mouse, Abcam, Cambridge, MA, USA, cat. no. AB150115) and Hoechst 33342, Trihydrochloride, Trihydrate (1:2000, Invitrogen-Life Technologies, USA cat. no. H1399,) in PBS containing 10% Triton X-100 and the blocking solution. Sections were then extensively rinsed in 0.1 M PB at room temperature, mounted and cover slipped with aqueous mounting media containing 10% 10 mM Tris-HCl (pH 8.0) and 1.42 g DABCO (Sigma-Aldrich, St. Louis, MO, USA, cat. no. D27802) and glycerol, (Sigma-Aldrich, St. Louis, MO, USA, cat. no. 5516). Correct placement of CTB infusions were verified using an Olympus BX-41 fluorescent microscope. CTB labelled cells were imaged using a Zeiss LSM 700 confocal microscope (Carl Zeiss Microscopy, Thornwood, NY, USA) under a 20× objective and tiled/stitched using filter configurations for sequential excitation/imaging via 488, 555 and 647 nm channels.
Behavioral testing
Behavioral testing began 3 weeks after intra-BL 6-OHDA infusion according to the sequences and timelines described in Fig. 3. For all motor tests, a group of non-infused, experimentally-naïve mice (n=24) was included as an additional control to test for the impact of surgery.
Fig.3. BL-targeted catecholaminergic lesion does not impair motor activity.
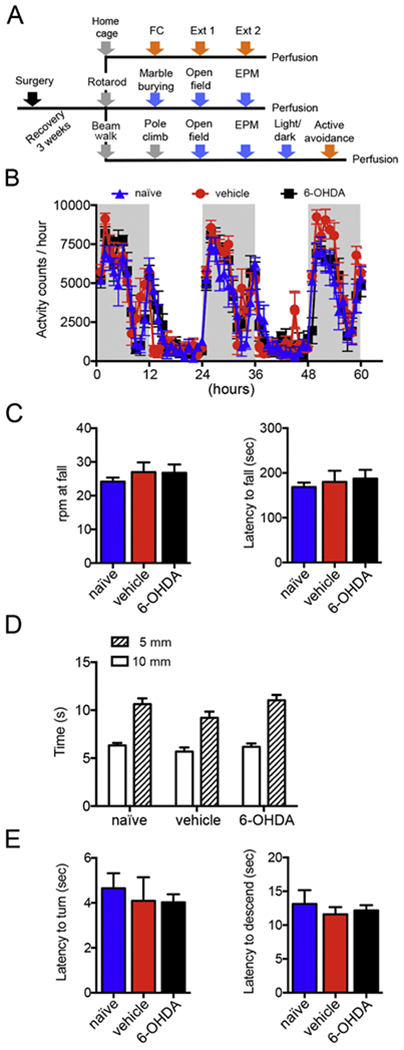
(A) Schematic diagram of the experimental design employed for behavioral assessments. Mice underwent surgery at 8 weeks of age and were allowed to recover for 3 weeks. Mice were divided in 3 different cohorts undergoing different behavioral batteries each starting with a motor test and followed by tests for anxiety-like behavior, fear-related tests or a combination of the two. The putatively most stressful test was always performed at the end. Abbreviations: EPM, elevated plus-maze; FC, cued fear conditioning; Ext, extinction training. (B) Home cage activity was measured during 3 nights and 2 days as activity counts/hour. No difference was found between naïve (n=8), vehicle- (n=6) and 6-OHDA-injected (n=8) mice. (C) In the accelerating rotarod test, the rpm at fall and latency to fall were measured for naïve (n=8), vehicle- (n=10) and 6-OHDA-injected (n=12) mice. No differences were detected among the 3 groups for the rpm at fall (p=0.70; 1-way ANOVA; 6-OHDA vs vehicle p=0.99, 6-OHDA vs naïve p=0.73, naïve vs vehicle p=0.72, Tukey multiple comparisons test) and latency to fall (p=0.70, 1-way ANOVA). (D) The time required by naïve (n=8), vehicle- (n=12) and 6-OHDA-injected (n=9) mice to traversed narrow (10 and 5mm wide) beams was comparable among groups (10mm beam: 6-OHDA vs vehicle p=0.77, 6-OHDA vs naïve p=0.97, naïve vs vehicle p=0.66; 5mm beam: 6-OHDA vs vehicle p=0.03, 6-OHDA vs naïve p=0.86, naïve vs vehicle p=0.14, 1-way ANOVA followed by Tukey multiple comparisons test). (E) In the pole climb test, naïve (n=8), vehicle- (n=10) and 6-OHDA-injected (n=8) mice showed similar latency to turn (p=0.84, 1-way ANOVA) and to descend (p=0.70, 1-way ANOVA). Data are shown as mean ± SEM.
Tests for motor function
Homecage activity:
Spontaneous locomotor activity in the home-cage was monitored over 60 hours (three dark and two light phases) using an automated infrared sensor-based system (TSE Systems, Bad Homburg, Germany).
Rotarod:
Motor coordination was assessed using the accelerating rotarod based on previously described methods (Boyce-Rustay et al., 2006a). Mice were placed on a rotating rod (4 rpm), which was gradually accelerated to a maximum of 40 rpm over 5 min. The latency to fall was used as a measure of motor coordination and dexterousness.
Beam balance:
The balance beam was used as a test of fine motor coordination based on previously described methods (Boyce-Rustay and Holmes, 2006b). Mice had to walk across two elevated beams of different width (5 and 10 mm) over a distance of 80 cm to reach a safe platform. The test was performed over 3 consecutive days: 2 days of training and 1 day of testing. Performance on the beam was quantified by measuring the time the mouse took to traverse the beam.
Pole climb:
As a test for motor strength and coordination, pole climbing was assessed, using previously described methods (Ogawa et al., 1985). Mice were placed at the top of a vertical 1 m long wooden pole facing upwards and the time to turn and descend to the base recorded.
Anxiety-related behaviors
Open field:
Mice were left to explore a 50 × 50 cm chamber for 10 min and the distance travelled as well as time spent in the aversive center (28 × 28 cm) were measured by means of a computer-assisted tracking system (VideoMot2; TSE). Illumination was set at 150 Lux.
Marble burying:
Mice were placed individually in a clean Plexiglas cage (30 × 15 × 15 cm) filled with fresh 5 cm-deep sawdust, on top of which were positioned 15 small glass marbles arranged in five evenly spaced rows of 3 marble each. Testing was conducted for 30 min at 100 Lux and the number of marbles that had been buried for at least two thirds of their height with bedding was counted.
Light-dark test:
The apparatus (TSE) consisted of a dark (<10 Lux) “safe” compartment and an illuminated aversive compartment (300 Lux). The compartments are connected by a small opening (7 × 7 cm wide) located in the center of the partition at floor level. Animals were individually placed in the apparatus facing the opening to the dark compartment and allowed to freely explore the apparatus for 10 min. A computer-assisted scanning system (VideoMot2, TSE) was used to track the behavior of each mouse.
Elevated plus-maze:
Mice were allowed to explore an elevated platform (72 cm above the floor) consisting of two opposing open (30 × 5 cm), and two opposing closed arms [30 × 5 cm] for a total of 5 min. Illumination in the open arms was 50 Lux. Mice were placed individually in the center of the platform (5 × 5 cm) facing a closed arm. The behavior of each mouse was tracked by a computer-assisted scanning system (VideoMot2, TSE). Arm entries were defined as crossing of the center point (located at approximately two thirds of the mouse body) into the arm. Percentage time spent in the open arms, arm entries and distance travelled were measured.
Conditioned freezing and avoidance
Fear conditioning and extinction:
Conditioning took place in a 17 × 17 × 25 cm chamber (Ugo Basile, Comerio, Italy) with a metal grid floor for delivering a footshock. The chamber was cleaned between subjects with 70% ethanol. Mice were placed in the chamber for a 120 sec baseline period, after which a 30 sec 80 dB white noise (75 kHz) conditioned stimulus (CS) was paired with a 0.7 mA footshock (presented during the last 2 sec of the CS) five times separated by a 120 sec interval. Fear retrieval and extinction testing occurred in a 17 × 17 × 25 cm chamber that differed from the conditioning context by having a solid Plexiglas floor and black and white stripes on the walls. The chamber was cleaned between subjects with 1% acetic acid. One day after conditioning, mice were placed in this chamber for a 120 sec baseline period, after which the CS was presented 20x with a randomized inter-CS interval ranging from 5 to 40 sec (Ext 1). This procedure was repeated the following day (Ext 2). Freezing was measured as an index of fear by ANY-maze software (Stoelting Europe, Dublin, Ireland), using a freezing minimum duration threshold of 1 sec, and manually cross-checked by an experienced experimenter.
Active avoidance:
Mice were tested using a fully automated apparatus (Ugo Basile), which consisted of a 47 × 18 × 26 cm box with a grid floor divided into two compartments. Mice were first placed into a randomly selected compartment and allowed to freely explore for a 10 min habituation session. The following day, they were again placed into a randomly selected compartment for 5 min, after which a light and 80 dB tone (7.5 kHz) compound stimulus was presented for 15 sec, co-terminating in the last 5 sec with a 0.3 mA foot shock. The compound stimulus and foot shock were presented in the same manner, irrespective of the mouse’s location, for a total of 30 trials. If the mouse made a full body transition into the other chamber during presentation of the compound stimulus, the foot shock was terminated.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Parametric statistical tests were performed on all datasets, except otherwise specified. Data were analyzed using Prism 7 software (GraphPad, La Jolla, CA, USA) using Student’s t-tests (paired or unpaired, two-sided) or analysis of variance (ANOVA) with appropriate post-hoc tests for multiple comparisons as indicated. Data were considered significant when p<0.05.
RESULTS
Intra-BL 6-OHDA causes selective limbic catecholaminergic denervation
We began by unilaterally micro-infusing the neurotoxin 6-OHDA into the BL using a range of doses, while maintaining a fixed volume of 0.2 μl. TH immunocytochemistry was used to visualize the loss of catecholaminergic fibers. A progressive loss of TH+ fibers in the amygdaloid complex was observed starting from 0.2 μg and was nearly maximal at 1 μg. At this dose, the BL, ITC and the most ventral part of the amygdalo-striatal transition zone (A-Stria) were almost completely depleted of TH+ fibers (Fig.1A). The lesion extended also to part of the piriform and perirhinal cortices, whereas the catecholaminergic innervation of the CeA, dorsal A-Stria and caudate-putamen (CPu) were largely spared (Fig. 1A). Higher doses (2 and 4 μg) of 6-OHDA produced widespread loss of TH+ fibers across these regions, as well as in the accessory basal nucleus of the amygdala and deep cortico-amygdaloid nuclei (Fig. 1A). Qualitative analysis of the rostro-caudal extent of the lesion in the BL produced by 6-OHDA at 1 μg revealed a more pronounced loss of TH+ fibers caudally (Suppl. Fig. 1A). On the basis of these observations, we extended our analysis of the 1 μg dose to other forebrain regions, finding a reduction of TH+ axons in the ventral, but not the dorsal, HPC (Suppl. Fig. 1B), and no change in density of TH+ fibers in the mPFC (Suppl. Fig. 2A). We performed double immunofluorescence staining for TH and NET in order to shed light on the DAergic versus NAergic identity of fibers in the vHPC and BL, and the denervating effects of the 1 μg 6-OHDA dose on the two catecholamines. This analysis revealed loss of both TH+ and NET+ fibers in the BL, as well as in the vHPC (Suppl. Fig. 3). However, only a small fraction of fibers was immunoreactive for TH per se in the vHPC, indicating that catecholaminergic innervation in this area was primarily NAergic and, by extension, that the BL-targeted 6-OHDA lesion caused a marked NAergic depletion in the vHPC.
Fig. 1. Intra-BL 6-OHDA causes midbrain DA neuron loss and selective limbic catecholaminergic denervation.
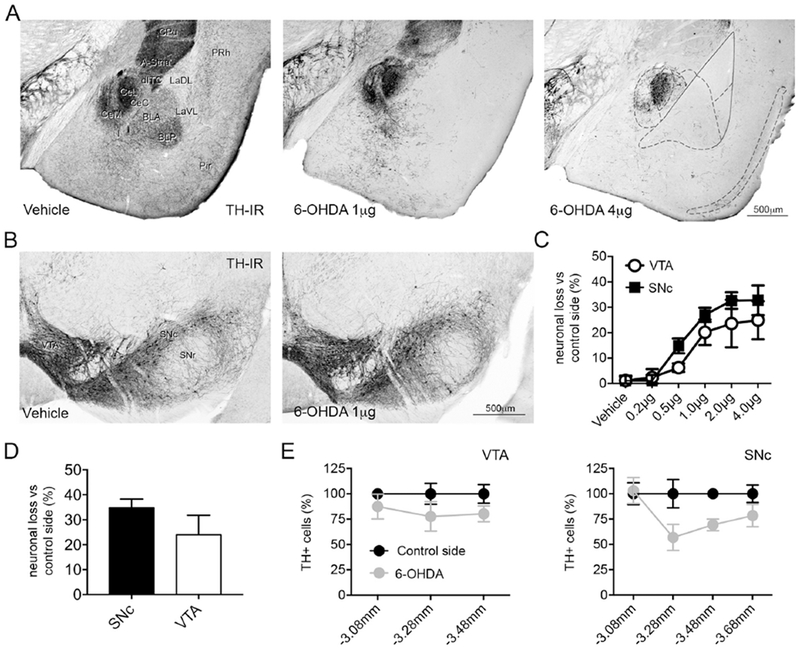
(A) Representative images of TH immunoreactivity in the amygdala and adjacent regions of C57BL/6J adult male mice injected with vehicle (0.9% NaCI + 0.02% ascorbic acid), 1 and 4 μg of 6-OHDA. Mice injected with 1 μg of 6-OHDA show a dramatic loss of TH immunolabeled fibers in the ITCs, A-Stria, BL, piriform and perirhinal cortices, whereas the CeA appears largely spared. In mice injected with 4 μg of 6-OHDA, the lesion extends to the CPu, accessory basal and CeM. (B) Representative micrographs of the VTA and SNc in vehicle and 1 μg injected animals. (C) Stereological analysis showing the loss of TH-labeled neurons in % compared to the contralateral control side. (D) Stereological analysis showing the loss of neurons, detected with Nissl staining, in % compared to the contralateral control side in mice injected with 1 μg of 6-OHDA. (E) Stereological analysis of the rostrocaudal extent of the lesion produced by 1 μg of 6-OHDA in VTA and SNc revealing a homogeneous distribution of the lesion in VTA, whereas in SNc the damage was more pronounced in the rostral part. Abbreviations: A-Stria: amygdala-striatum transition zone; BLA: basolateral amygdaloid nucleus, anterior part; BLP: basolateral amygdaloid nucleus, posterior part; CeC: central amygdaloid nucleus, capsular part; CeL: lateral part of the central amygdaloid nucleus; CeM: medial part of the central amygdaloid nucleus; CPu: caudate putamen; dITC: dorsal intercalated cell cluster; I: main intercalated cell cluster; LaVL: lateral amygdaloid nucleus, ventrolateral part; LaDL: lateral amygdaloid nucleus, dorsolateral part; Pir: piriform cortex; PRh: perirhinal cortex; SNc: substantia nigra compacta; SNr: substantia nigra reticulata; VTA: ventral tegmental area; 6-OHDA: 6-hydroxydopamine.
Intra-BL 6-OHDA causes midbrain DA neuron loss
Using a stereological approach, we found a significant, dose-dependent reduction in the number of TH+ neurons in the SNc and VTA, ipsilateral to the 6-OHDA injection side, as compared to the contralateral side (Fig. 1B-C). Of note, there was a pronounced cell loss (17% in VTA, 28% in SNc) at the 1 μg 6-OHDA dose that produced the aforementioned restricted pattern of denervation in the dorsolateral amygdala and vHPC (Fig. 1C). To exclude the possibility that this apparent loss of TH+ DAergic cells simply stemmed from a downregulation in TH expression, we counted Nissl-stained neurons in brain sections consecutive to those used for TH quantification. This confirmed that the reduction in Nissl-stained neuronal number (24% in VTA, 34% in SNc) closely corresponded to the estimates of DAergic cell loss (Fig. 1D). We also conducted an assessment of the rostro-caudal extent of the lesion-induced loss of DAergic neurons. The 1 μg 6-OHDA dose produced a homogeneous rostro-caudal decrease in TH+ cell number in VTA, whereas in SNc, the loss was more pronounced caudally (bregma levels −3,28 and −3,48) (Fig. 1E).
Next, based on our observation that catecholaminergic innervation of the CeA was largely spared by the intra-BL injection of 6-OHDA, together with prior evidence that the ventral periaqueductal gray (PAG)/dorsal raphe (DR), rather than the VTA/SNc, is the primary source of DA to the CeA (Hasue and Shammah-Lagnado, 2002; Rizvi et al., 1991), we quantified the number of TH+ neurons in the PAG, at two different bregma levels, after bilateral intra-BL infusion of 1 μg 6-OHDA. In agreement with our expectation, there was no significant reduction in the number of PAG TH+ neurons between lesioned mice and vehicle infused controls (χ2 for trend, bregma −3.28 p=0.31; bregma −3.52 p=0.16) (Suppl. Fig. 2B).
Midbrain catecholaminergic neuronal targeting of BL, vHPC and CeA
To provide an additional visualization of the topography of catecholaminergic innervation of limbic structures, we infused the retrograde tracer CTB, conjugated to Alexa-Fluor™ 488 or 555, into the BL, CeA or vHPC and immunostained midbrain neurons for TH. Following intra-BL infusion, retrogradely labeled cells were visible, though quite sparse, in both VTA and SNc. Moreover, virtually all these CTB-labeled cells were TH+ (Fig. 2A). On the one hand, the sparseness in CTB labeling could well be a function of undersampling by the tracer, as well as the small volume of CTB we infused to ensure confinement within the target region. On the other hand, other regions, such as the posterior thalamus, hippocampus and some part of the neocortex, such as the perirhinal cortex (Suppl. Fig. 4A), did exhibit dense CTB labeling in the same mouse in which there was sparse VTA/SNc labeling after BL infusion, suggesting the midbrain labeling observed may not have been a gross underestimate.
Fig. 2. CTB connectivity.

(A) Left column, schematic view of the retrograde tracer injection site (upper panel) and of the actual injection site overlaied with the atlas image (lower panel) for BL. Center, confocal tiled image of the VTA/SNc. The upper panel shows immunolabeling for TH and the lower panel CTB-488 cell body labeling. Right, magnified images from the box in center panels. Cell bodies co-labeled with TH and CTB-488 are indicated by the arrows. (B) Left, schematic view of the retrograde tracer injection site (upper panel) and of the actual injection site overlaied with the atlas image (lower panel) for vHPC. Center, confocal tiled image of the VTA/SNc showing immunolabeling for TH (upper panel) and CTB-488 cell body labeling (lower panel). Right column, magnified images from the box presented in center panels. Cell bodies labeled with CTB-488 do not show co-labeling with TH. (C) Left, schematic view of the retrograde tracer CTB-488 and CTB 555 injection sites (upper panels) and of the actual injection sites overlaied with the atlas images for BLA and vHPC (lower panels). Center, confocal tiled images of the VTA/SNc showing TH immunolabeling (upper panel), CTB-488 (middle panel) and CTB-555 cell body labeling (lower panel). Right, magnified image from the box presented in central panels. Cell bodies co-labeled with CTB-488 and TH, but not CTB-555, are marked by arrows. Scale bars: A-B center panels, 500 μm; right panels, 20 μm. C center panels, 500 μm; right panel, 50 μm.
CTB infusions into the vHPC produced a different pattern than the BL, with labeling in the paranigral and retromammillary nuclei, but not the VTA or SNc. Furthermore, only a small number of the labeled cells were positive for TH (Fig. 2B). It should be noted that for both BL and vHPC infusions, similar labeling was evident when the alternative fluorophore was infused, discounting the possibility that the labeling patterns were dependent on the fluorophore (data not shown). The same patterns of retrograde labeling in the midbrain were repeated when both CTB-488 and CTB-555 were infused into one or the other region (i.e., BL and vHPC) of the same mouse (Fig. 2E-F); here there was also no clear indication of double-labeling, suggesting minimal to no collateralization of VTA/SNc DA cells to the two forebrain structures (Fig. 2C). As a further control, we infused a cocktail of CTB-488 and CTB-555 into the same region and found, as expected, a high degree of fluorophore overlap in retrograde labeling (data not shown).
Finally, when we targeted CTB to the CeA, we detected CTB/TH+ double-positive cells in the PAG, but not the VTA/SNc, affirming the PAG as a source of DA to the CeA (Suppl. Fig. 4B-C).
BL-targeted catecholaminergic lesion does not impair motor activity
We next turned to the potential behavioral effects of the intra-BL 6-OHDA-induced catecholaminergic lesion. First, given the profound motoric effects of non-specific lesions of midbrain DA neurons, we conducted a range of assays for motor activity and coordination in mice which were bilaterally infused with 6-OHDA into the BL. Second, we tested for conditional aversive responses to a threat and anxiety-like behavior. Different cohorts of 6-OHDA-injected mice were used for these behavioral tests, in which the putatively more stressful tests were performed later in the sequence (Fig. 3A). Upon completion of the behavioral testing, injection sites and size of the lesions were examined, and mice that did not show the correct pattern were excluded from all analyses.
General locomotor activity was assessed in the home cage for 60 hours (3 nights, 2 days). 6-OHDA-lesioned mice did not differ in overall (group × time: F(2,19)= 0.95, p=0.41; interaction: F(118,1121)= 1.55, p=0.0003, 2-way ANOVA), light phase (group × time: F(2,19)= 0.98, p=0.39; interaction: F(46,437)= 1.98, p=0.0002, 2-way ANOVA) or dark phase (F(2,19)= 1.74, p=0.20; interaction: F(70,665)= 1.32, p=0.046, 2-way ANOVA) activity, as compared to vehicle-infused controls (Fig. 3B). Both groups were similar to naïve mice which had not undergone surgery (Fig. 3B), indicating there were no residual adverse effects of the surgery itself.
In the accelerating rotarod test, the latency to fall from the rod was similar among 6-OHDA-, vehicle-injected and naïve mice (p=0.70, 1-way ANOVA; 6-OHDA vs vehicle p=0.98, 6-OHDA vs naïve p=0.78, naïve vs vehicle p=0.70, Tukey multiple comparisons test) (Fig. 3C). Likewise, the time to traverse narrow beams was also no different among groups (group × time: F(2,26)= 2.11, p=0.14, 2-way ANOVA) (Fig. 3D). Finally, the latency to turn downward (F(2,23)= 0.17, p=0.84, 1-way ANOVA; 6-OHDA vs vehicle p=1.00, 6-OHDA vs naïve p=0.86, naïve vs vehicle p=0.87, Tukey multiple comparisons test) and descend to the bottom (F(2,31)=0.36, p=0.70, 1-way ANOVA; 6-OHDA vs vehicle p=0.92, 6-OHDA vs naïve p=0.84, naïve vs vehicle p=0.67, Tukey multiple comparisons test) of a wooden pole did not differ among groups (Fig. 3E).
These data show that, in contrast to the effects of more widespread ablations of midbrain DA cells, selective loss of catecholaminergic neurons innervating the dorsolateral amygdala and vHPC did not affect motor functions.
Impact of 6-OHDA bilateral microinjections in the BL on measures of fear
Given a large body of evidence suggests an important role of catecholamines in the control of conditioned fear responses (Lee et al., 2016; Pezze and Feldon, 2004), we next tested 6-OHDA-lesioned mice in cued fear conditioning (Fig. 4A). During conditioning, levels of freezing to the CS paired with a footshock increased over successive pairings (F(4,172)= 222.7, p<0.0001, 2-way ANOVA), but these levels did not differ between lesioned mice and controls (F(1,43)= 0.06, p=0.80; interaction: F(4,172)=0.95, p=0.44) (Fig. 4B). Two cohorts of animals were used for these experiments; as there was no statistical difference in freezing levels between the two cohorts (vehicle, F(1,21)=1.26, p=0.27; 6-OHDA, F(1,20)=0.77, p=0.39, 1-way ANOVA), data were pooled. Freezing levels were also similar between groups on retrieval of the CS-US association the following day (p=0.73, 1-way ANOVA), as they were over two sessions of extinction, in which the CS was unreinforced. Here, freezing levels progressively decreased across CS presentations (F(19,817)= 13.72, p<0.0001, 2-way ANOVA), but the two groups again did not differ (Ext 1, group × time freezing, F(1,43)= 1.17, p=0.69; interaction: F(19,817)=2.25, p=0.002; Ext 2, group × time freezing, F(1,43)=0.16, p=0.68, interaction: F(19,817)=2.247, p=0.0017, 2-way ANOVA) (Fig. 4C).
Fig. 4. BL-targeted catecholaminergic lesion does not impair fear behavior.
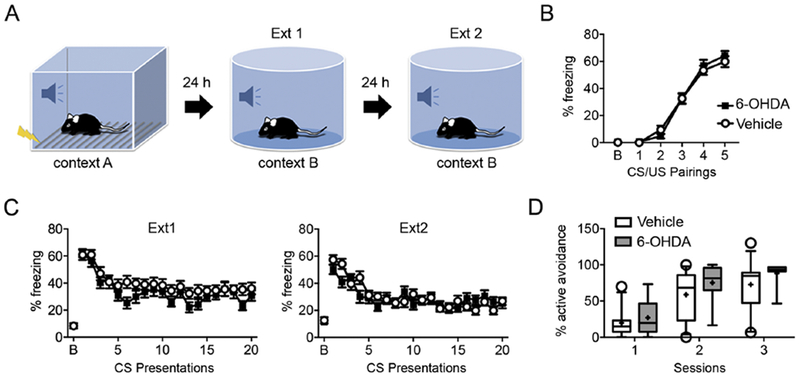
(A) Schematic diagram of the cued fear conditioning and extinction paradigms. (B) Animals received 5 CS-US pairings in context A. No difference was found in percentage of freezing between vehicle- (n=22) and 6-OHDA-injected (n=23) mice (p=0.80, 2-way ANOVA). (C) On the first day of extinction, mice were exposed in context B to 20 CS without receiving the US. Both groups of animals successfully reduced freezing to the CS, consistent with successful extinction acquisition. Mice were exposed to a second fear extinction training session composed of 20 unreinforced CS presentations 24 hours after the first fear extinction training session. Both groups of mice were equally able to extinguish the learned fear. Data are given as mean ± s.e.m. (D) Active avoidance. During 3 sessions, the percentage of successful avoidance trials was no different between vehicle- (n=12) and 6-OHDA-injected (n=8) mice. Box-whisker plots show median values and 25th/75th percentiles with 10th and 90th percentile whiskers; (+) shows the mean value. Empty circles represent values outside the10th and 90th percentiles.
Because aversive stimuli can evoke, besides freezing, other more active forms of defensive behaviors, such as escape and avoidance responses, we tested 6-OHDA-injected mice in an active avoidance paradigm. 6-OHDA-lesioned mice learned to avoid the signaled shock at a similar rate (Fig. 4D) as vehicle-injected animals (F(1,18)=1.38, p=0.26, interaction: F(2,36)=0.46, p=0.64, 2-way ANOVA).
The results of these behavioral tests indicate that two forms of conditioned fear, one based on the suppression of movement and the other on an active escape response, were unaffected by selective catecholaminergic denervation of the dorsolateral amygdala and vHPC.
BL-targeted catecholaminergic lesion increases anxiety-like behavior
In a final set of experiments, we assessed if catecholaminergic denervation of the dorsolateral amygdala and vHPC affects processing of innately aversive environments. For this purpose, we tested mice in validated rodent paradigms of anxiety-like behavior (Cryan and Holmes, 2005).
Compared to vehicle-injected controls, lesioned mice spent a similar percentage of time during the session in the aversive center of the open field (p=0.59, t=0,55, df=39, unpaired t-test) (Fig. 5A) and travelled a similar distance (6-OHDA: 4,064 ± 264, vehicle: 3,981 ± 272; p=0.83, t=0,22, df=39, unpaired t-test). Two cohorts of animals were used for these experiments; as there was no statistical difference in percentage of time spent in the aversive center of the open field between the two cohorts (vehicle: p=0.90, t=0.12, df=18; 6-OHDA: p=0.94, t=0.07, df=19, unpaired t-test), data were pooled.
Fig.5. BL-targeted catecholaminergic lesion increases anxiety-like behavior in the light-dark exploration test.
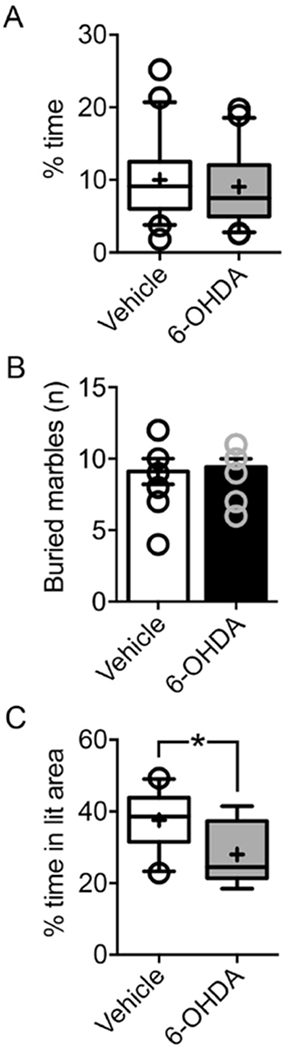
(A) Novel open field. Vehicle- (n=20) and 6-OHDA-injected (n=21) mice showed a similar percentage of time (p=0.74, unpaired t-test) in the aversive center of an open field arena. (B) Marble burying. Vehicle- (n=9) and 6-OHDA-injected (n=12) mice did not differ in the number of marbles buried (p=0.98, Mann Whitney test). Empty circles show the number of marbles buried by each animal. As different mice buried the same number of marbles, some empty circles are superimposed. (C) Light-dark exploration. 6-OHDA-injected animals (n=12) spent significantly less time (p=0.02, unpaired t-test) in the lit compartment of the light-dark exploration test, as compared to vehicle-injected controls (n=9). Box-whisker plots show median values and 25th/75th percentiles with 10th and 90th percentile whiskers; (+) shows the mean value. Empty circles represent values outside the10th and 90th percentiles. *p<0.05, unpaired t test.
In the marble burying test, the number of marbles buried over 30 min did not differ between groups (Fig. 5B, p=0.98, Mann Whitney). In contrast to the normal behavior seen in the marble burying test and open field, lesioned mice spent significantly less time (p<0.0001, t=4,76, df=39, unpaired t-test) and made fewer entries into the aversive open arms (p=0.0022, t=3.28, df=39, unpaired t-test) of the elevated plus-maze than controls (Fig. 6A-B). The total number of visits did not differ between the two groups (vehicle, 47.85 ± 4.93; 6-OHDA, 35.00 ± 4.21; p=0.054, t=1.99, df=39, unpaired t-test) as well as the ratio between open arm and total entries (p=0.224, t=1.24, df=39) (Fig.6C). Distance traveled in the open arms (p<0.0001, t=4,72, df=39, unpaired t-test) and in the maze overall (p=0.011, t=2.68, df=39, unpaired t-test) was lesser in the lesioned group (Figure 6D-E). Likewise, the ratio between open arm and total distance travelled was significantly different between the two groups (p<0.0001, t=5,31, df=39, unpaired t-test). Replicating the anxiogenic-like phenotype in the elevated plus-maze, lesioned mice spent significantly less time than controls in the aversive light compartment (p=0.0187, t=2,57, df=19, unpaired t-test) when tested for light-dark exploration (Fig. 5C).
Fig.6. BL-targeted catecholaminergic lesion increases anxiety-like behavior in the elevated plus-maze.

(A) 6-OHDA-injected animals (n=21) spent significantly less time in the open arms of the elevated plus-maze, as compared to vehicle-injected ones (n=20). Relatedly, the number of entries in the open arms were fewer in 6-OHDA- (n=21) than for vehicle-injected (n=20) mice. Two cohorts of animals were used for these experiments. Because there was no statistical difference between the two cohorts in time spent in the open arms (vehicle: p=0. 35, t=0.64, df=18; 6-OHDA: p=0.31, t=1.05, df=19; unpaired t-test), data were pooled. (B) There was a significant difference between the groups in distance travelled in open arms, as well as in total distance travelled in the maze. Box-whisker plots show median values and 25th/75th percentiles with 10th and 90th percentile whiskers; (+) shows the mean value. Empty circles represent values outside the10th and 90th percentiles. *p<0.05, **p<0.01, ***p<0.001, unpaired ttest.
The results of this set of tests indicate a significant increase in anxiety-like behavior as a result of targeted 6-OHDA lesioning to the BL and further validate a lack of locomotor impairment. The fact that an anxiety-like behavior was evident in two of three exploration-based tests could reflect a selective impairment in those tests that generate a relatively high level of anxiety-like behavior (Holmes et al., 2003).
DISCUSSION
In the current study, we used the neurotoxin 6-OHDA to produce targeted catecholaminergic denervation in the mouse dorsolateral part of the amygdala (encompassing the BL, A-Stria and ITCs) and vHPC. The major finding was that this regionally-specific lesion of mesolimbic DAergic and NAergic inputs led to increased anxiety-like behavior, without attendant alterations in either motor function or conditioned fear. These findings provide novel support for the hypothesis that loss of catecholaminergic neurotransmission in the amygdala and vHPC may account for anxiety symptoms that emerge in PD, often prior to the onset of motor dysfunction.
Intracranial infusion of 6-OHDA into different compartments of the basal ganglia has been the most extensively studied animal model of PD (Taylor et al., 2010). However, even though bilateral injections more closely approximate the pathophysiology of PD, most earlier studies have delivered 6-OHDA unilaterally, and even fewer have bilaterally infused the neurotoxin into the BL specifically. For example, one report found that unilateral application (4 μg) of 6-OHDA into the amygdala, without targeting a specific nucleus, enhanced anxiety-like behavior in female rats, but an anxiolytic-like phenotype in males (Sullivan et al., 2009). Here, by bilaterally microinfusing 6-OHDA into the BL, we were able to produce a significant and selective enhancement in anxiety-like behavior on two tests (light-dark exploration and elevated plus-maze), whereas performance on a third task (novel open field) was unaltered. The specificity of the lesion effect in two of the three tests may reflect a selective impairment in those test situations generating a relatively high level of anxiety-like behavior (Holmes et al., 2003). Of further interest was the observation that the phenotypic abnormality in the classical exploration-based assays for anxiety-like behavior did not generalize to another assay test (marble-burying) that is purported to assess compulsive-like defensive burying and is qualitatively distinct from the other tests.
While we found increased anxiety-like behavior in 6-OHDA lesioned mice, two measures of learned fear, pavlovian cued fear conditioning and active avoidance, were unaltered. On the one hand, this finding is reminiscent of an earlier report in which bilateral infusion of 3 μg of 6-OHDA into the rat BL resulted in significant amygdala DA (>60%) and NA (90%) loss, but did not affect contextual fear, though it did impair aversive conditioning to explicit cues (Selden et al., 1991). On the other hand, these findings are in apparent contradiction to numerous prior reports demonstrating a major role for both DAergic and NAergic transmission in regulating fear learning (Lee et al., 2016; Pezze et al., 2004; Sullivan et al., 1999). For example, rodent studies have repeatedly found that pharmacological blockade of DAergic signaling in the amygdala impairs the formation of pavlovian associations between cue and footshock (de la Mora et al., 2010; Lee et al., 2016). Similarly, mutant mice engineered with a selective DA deficiency (Zhou and Palmiter, 1995) were shown to be unable to learn a fear-potentiated startle response, though this could be rescued, at least in the short-term by restoration of DA in the BL (Fadok et al., 2009).
There are a number of plausible explanations for these apparent discrepancies. The first is that the effects of transient pharmacological disruption of amygdala DAergic signaling likely differ from the effects of long-term neurotransmitter depletion, including the current 6-OHDA-induced lesion. Long-lasting DA deficiency could elicit compensatory adaptations that can adequately support fear memory formation. Similarly, alterations in the NA system after lesioning may have mitigated the consequences for fear conditioning of denervating NAergic inputs to the BL and vHPC. Deficient fear learning would be expected after loss of NAergic signaling in intact animals given, for instance, the impairing effects of blocking BL β-adrenergic receptors (Bush et al., 2010) or optogenetically silencing LC NAergic fibers in the BL (Uematsu et al., 2017). On the other hand, not all studies have found fear-impairing effects of inhibiting NAergic transmission, either systemically or specifically within the BL (Bush et al., 2010; Debiec and Ledoux, 2004; Grillon et al., 2004; Lee et al., 2001; Murchison et al., 2004).
A second possibility relates to the regional specificity of experimental manipulations. In many of the previous genetic, pharmacological and neurotoxin-based models, disruption of DA neurotransmission extended to the CeA; unlike in the current study, where the catecholaminergic inputs to the CeA were spared, as evidenced by extant TH+ fibers in this nucleus. As reported in previous studies (Hasue et al., 2002; Poulin et al., 2014; Rizvi et al., 1991) and replicated here using CTB retrograde neuronal tracing, the CeA receives DAergic input from the vPAG. Moreover, in contrast to significant TH+ neuronal loss in VTA and SNc in the 6-OHDA lesioned mice, we found no reduction in TH+ cell number in vPAG. Thus, an intact vPAG→CeA DAergic circuit may mediate the main DAergic influence on fear learning. This hypothesis is strongly supported by a recent report showing that silencing vPAG DAergic neurons markedly impairs cued fear conditioning without leading to overt effects in the elevated plus-maze or light dark exploration tests (Groessl et al., 2018). Taken together our data are consistent with the defensive distance theory of threat (McNaughton and Corr, 2004; Mobbs et al., 2007), which posits that exposure to proximal danger activates a circuit comprising the vPAG, DRN and CeA to generate strong defensive reactions such as freezing and flight, whereas a more distal and ambiguous threat elicits anxiety via recruitment of an alternate network that includes the BL, vHPC and regions such as the mPFC and bed nucleus of stria terminalis.
An important avenue for future work will involve the further delineation of the relative contributions of DAergic and NAergic loss to the increased anxiety-like behavior that emerges after BL-targeted 6-OHDA lesioning (e.g. administering desipramine to prevent the uptake of 6-OHDA from NAergic terminals) (Fig. 7). Using double immunofluorescence of NET and TH, we found that hippocampal innervation by TH+ fibers corresponded for the large part to NAergic inputs from the LC, in line with other reports that the HPC receives only a very sparse DAergic innervation (Mingote et al., 2015; Verney et al., 1985). Our data also point to a clear separation between NAergic neurons targeting the dorsal and the ventral part of the HPC, with the latter component most likely giving rise to collaterals that also innervate the BL. This picture differs for midbrain DAergic neurons, which, based on our CTB retrograde tracing, send distinct projections to the BL and vHPC. Moreover, within the midbrain itself, we observed in proportion a higher loss of DA neurons in the SNc than in the VTA in the lesioned animals, consistent with the long-standing view that the SNc contains not only neurons projecting to the striatum but also to cortical and limbic areas (Bjorklund and Dunnett, 2007). Altogether, these observations point to a complex pattern of projections from midbrain DAergic and NAergic neurons to key limbic regions regulating anxiety (Poulin et al., 2014) (Fig. 7). How each of these specific circuits are affected in PD remains to be elucidated.
Fig.7. Schematic cartoon depicting catecholaminergic pathways affected by intra-BL injections of 6-OHDA.

Schematic representation of the major catecholaminergic inputs from the VTA, SNc, LC and vPAG to the amygdala, vHPC and mPFC. 6-OHDA injected into the BL causes a retrograde degeneration of the subset of neurons in the VTA, SNc and LC, but not in the vPAG, projecting to the BL, AStria and also to the vHPC (projections shown as dashed lines). Conversely, catecholaminergic neurons projecting to the dorsal HPC and mPFC were spared, suggesting minimal or no collateralization to the BL (projections shown as solid lines). The relative contribution of DAergic and NAergic loss to the increased anxiety-like behavior resulting from BL-targeted 6-OHDA lesioning remains to be determined, e.g. by administering desipramine to prevent the uptake of 6-OHDA from NAergic terminals.
Anxiety is a cardinal clinical feature in PD and can often precede the motor symptoms by several years (Schapira et al., 2017). Previous preclinical and clinical studies have suggested that reductions in DA, NA and 5-HT might underlie anxiety in PD (Bonito-Oliva et al., 2014; Remy et al., 2005; Tadaiesky et al., 2008; Taylor et al., 2009; Zhu et al., 2007), but have not identified besides a general contribution of the nigrostriatal pathway other brain circuits involved. Our study provides an important advance by implicating an impaired function of DAergic (from SNc/VTA, but not vPAG-DR) and NAergic (from LC) inputs to the dorsolateral amygdala and vHPC in the manifestation of anxiety-like behavior. We find that catecholaminergic denervation in these circuits produces a significant and selective increase in anxiety-like behavior, without discernible loss of motor function. On the basis of these findings, we hypothesize that the pathogenesis of anxiety symptoms in PD may result from an early loss of SNc and LC neurons projecting to these areas that cannot be compensated due to their limited catecholamine reserve capacity compared e.g. to the caudate-putamen. Further work is now needed to test this mechanistic hypothesis that could help to develop novel early diagnostic approaches and therapeutically-relevant manipulations that can normalize these deficits and rescue anxiety-like behavior.
Supplementary Material
Highlights.
6-OHDA infusion in the basal amygdala induces limbic-specific catecholamine loss
Loss of catecholamines in the basal amygdala elicits anxiety-like behavior
Loss of catecholamines in the basal amygdala does not impair fear learning
Basal and central amygdala receive partially independent catecholaminergic inputs
ACKNOWLEDGEMENTS
We thank Ms G. Schmid for excellent technical support.
FUNDING
We acknowledge financial support from the Austrian Science Fund (Fonds zur Förderung der Wissenschaftlichen Forschung) grant W12060-B10 and the Sonderforschungsbereich grant F44-17 to FF and F44-14 to NS, and from the NIAAA Intramural Research Program to AH, OGC and GP. The authors declare no conflict of interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare no conflict of interests. No authors have received funding from any institution, including personal relationships, interests, employment, affiliations, patents, inventions, honoraria, consultancies, royalties, stock options/ownership, or expert testimony for the last 12 months.
REFERENCES
- Bjorklund A, Dunnett SB (2007). Dopamine neuron systems in the brain: an update. Trends Neurosci 30(5): 194–202. [DOI] [PubMed] [Google Scholar]
- Blandini F, Armentero M-T (2012). Animal models of Parkinson’s disease. FEBS Journal 279(7): 1156–1166. [DOI] [PubMed] [Google Scholar]
- Bonito-Oliva A, Masini D, Fisone G (2014). A mouse model of non-motor symptoms in Parkinson’s disease: focus on pharmacological interventions targeting affective dysfunctions. Front Behav Neurosci 8: 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower JH, Grossardt BR, Maraganore DM, Ahlskog JE, Colligan RC, Geda YE et al. (2010). Anxious personality predicts an increased risk of Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society 25(13): 2105–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce-Rustay JM, Wiedholz LM, Millstein RA, Carroll J, Murphy DL, Daws LC, et al. (2006a). Ethanol-related behaviors in serotonin transporter knockout mice. Alcoholism, clinical and experimental research 30(12): 1957–1965. [DOI] [PubMed] [Google Scholar]
- Boyce-Rustay JM, Holmes A (2006b). Ethanol-related behaviors in mice lacking the NMDA receptor NR2A subunit. Psychopharmacology 187(4): 455–466. [DOI] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K (2004). Stages in the development of Parkinson’s disease-related pathology. Cell and Tissue Research 318(1): 121–134. [DOI] [PubMed] [Google Scholar]
- Buchel C, Dolan RJ, Armony JL, Friston KJ (1999). Amygdala-hippocampal involvement in human aversive trace conditioning revealed through event-related functional magnetic resonance imaging. J Neurosci 19(24): 10869–10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukalo O, Pinard CR, Silverstein S, Brehm C, Hartley ND, Whittle N, Colacicco G, Busch E, Patel S, Singewald N, Holmes A (2015) Prefrontal inputs to the amygdala instruct fear extinction memory formation. Sci Adv. July;1(6): pii: e1500251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush DE, Caparosa EM, Gekker A, Ledoux J (2010). Beta-adrenergic receptors in the lateral nucleus of the amygdala contribute to the acquisition but not the consolidation of auditory fear conditioning. Front Behav Neurosci 4: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri KR, Schapira AH (2009). Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol 8(5): 464–474. [DOI] [PubMed] [Google Scholar]
- Crucian GP, Huang L, Barrett AM, Schwartz RL, Cibula JE, Anderson JM et al. (2001). Emotional conversations in Parkinson’s disease. Neurology 56(2): 159–165. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Holmes A (2005). The ascent of mouse: advances in modelling human depression and anxiety. Nat Rev Drug Discov 4(9): 775–790. [DOI] [PubMed] [Google Scholar]
- Davidson RJ (2002). Anxiety and affective style: role of prefrontal cortex and amygdala Biol Psychiatry 51(1): 68–80. [DOI] [PubMed] [Google Scholar]
- de la Mora MP, Gallegos-Cari A, Arizmendi-Garcia Y, Marcellino D, Fuxe K (2010). Role of dopamine receptor mechanisms in the amygdaloid modulation of fear and anxiety: Structural and functional analysis. Progress in Neurobiology 90(2): 198–216. [DOI] [PubMed] [Google Scholar]
- Debiec J, Ledoux JE (2004). Disruption of reconsolidation but not consolidation of auditory fear conditioning by noradrenergic blockade in the amygdala. Neuroscience 129(2): 267–272. [DOI] [PubMed] [Google Scholar]
- Diederich NJ, Goldman JG, Stebbins GT, Goetz CG (2016). Failing as doorman and disc jockey at the same time: Amygdalar dysfunction in Parkinson’s disease. Mov Disord 31(1): 11–22. [DOI] [PubMed] [Google Scholar]
- Dobi A, Sartori SB, Busti D, Van Der Putten H, Singewald N, Shigemoto R et al. (2013). Neural substrates for the distinct effects of presynaptic group III metabotropic glutamate receptors on extinction of contextual fear conditioning in mice. Neuropharmacology 66: 274–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok JP, Dickerson TM, Palmiter RD (2009). Dopamine is necessary for cue-dependent fear conditioning. J Neurosci 29(36): 11089–11097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix-Ortiz AC, Beyeler A, Seo C, Leppla CA, Wildes CP, Tye KM (2013).BLA to vHPC inputs modulate anxiety-related behaviors. Neuron 79(4): 658–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, Baas JP, Lissek S, Smith K, Milstein J (2004). Anxious responses to predictable and unpredictable aversive events. Behav Neurosci 118(5): 916–924. [DOI] [PubMed] [Google Scholar]
- Groessl F, Munsch T, Meis S, Griessner J, Kaczanowska J, Pliota P et al. (2018). Dorsal tegmental dopamine neurons gate associative learning of fear. Nat Neurosci. doi: 10.1038/s41593-018-0174-5. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasue RH, Shammah-Lagnado SJ (2002). Origin of the dopaminergic innervation of the central extended amygdala and accumbens shell: a combined retrograde tracing and immunohistochemical study in the rat. J Comp Neurol 454(1): 15–33. [DOI] [PubMed] [Google Scholar]
- Holmes A, Kinney JW, Wrenn CC, Li Q, Yang RJ, Ma L et al. (2003). Galanin GAL-R1 receptor null mutant mice display increased anxiety-like behavior specific to the elevated plus-maze. Neuropsychopharmacology 28(6): 1031–1044. [DOI] [PubMed] [Google Scholar]
- Jacobs DH, Shuren J, Bowers D, Heilman KM (1995). Emotional facial imagery, perception, and expression in Parkinson’s disease. Neurology 45(9): 1696–1702. [DOI] [PubMed] [Google Scholar]
- Kalia LV, Lang AE (2015). Parkinson’s disease. Lancet 386(9996): 896–912. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Berger SY, Stiedl O, Spiess J, Kim JJ (2001). Post-training injections of catecholaminergic drugs do not modulate fear conditioning in rats and mice. Neurosci Lett 303(2): 123–126. [DOI] [PubMed] [Google Scholar]
- Lee JH, Lee S, Kim J-h (2016). Amygdala Circuits for Fear Memory : A Key Role for Dopamine Regulation. The Neuroscientist. [DOI] [PubMed] [Google Scholar]
- Lin CH, Lin JW, Liu YC, Chang CH, Wu RM (2015). Risk of Parkinson’s disease following anxiety disorders: a nationwide population-based cohort study. Eur J Neurol 22(9): 1280–1287. [DOI] [PubMed] [Google Scholar]
- McCall JG, Siuda ER, Bhatti DL, Lawson LA, McElligott ZA, Stuber GD et al. (2017). Locus coeruleus to basolateral amygdala noradrenergic projections promote anxiety-like behavior. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaugh JL (2004). The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci 27: 1–28. [DOI] [PubMed] [Google Scholar]
- McNaughton N, Corr PJ (2004). A two-dimensional neuropsychology of defense: fear/anxiety and defensive distance. Neurosci Biobehav Rev 28(3): 285–305. [DOI] [PubMed] [Google Scholar]
- Mingote S, Chuhma N, Kusnoor SV, Field B, Deutch AY, Rayport S (2015). Functional Connectome Analysis of Dopamine Neuron Glutamatergic Connections in Forebrain Regions. J Neurosci 35(49): 16259–16271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobbs D, Petrovic P, Marchant JL, Hassabis D, Weiskopf N, Seymour B et al. (2007). When fear is near: threat imminence elicits prefrontal-periaqueductal gray shifts in humans. Science 317(5841): 1079–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchison CF, Zhang XY, Zhang WP, Ouyang M, Lee A, Thomas SA (2004). A distinct role for norepinephrine in memory retrieval. Cell 117(1): 131–143. [DOI] [PubMed] [Google Scholar]
- Ogawa N, Hirose Y, Ohara S, Ono T, Watanabe Y (1985). A simple quantitative bradykinesia test in MPTP-treated mice. Res Commun Chem Pathol Pharmacol 50(3): 435–441. [PubMed] [Google Scholar]
- Pezze MA, Feldon J (2004). Mesolimbic dopaminergic pathways in fear conditioning. Prog Neurobiol 74(5): 301–320. [DOI] [PubMed] [Google Scholar]
- Poulin JF, Zou J, Drouin-Ouellet J, Kim KY, Cicchetti F, Awatramani RB (2014). Defining midbrain dopaminergic neuron diversity by single-cell gene expression profiling. Cell Rep 9(3): 930–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prediger RDS, Matheus FC, Schwarzbold ML, Lima MMS, Vital Ma BF (2012). Anxiety in Parkinson’s disease: A critical review of experimental and clinical studies. Neuropharmacology 62(1): 115–124. [DOI] [PubMed] [Google Scholar]
- Remy P, Doder M, Lees A, Turjanski N, Brooks D (2005). Depression in Parkinson’s disease: Loss of dopamine and noradrenaline innervation in the limbic system. Brain 128: 1314–1322. [DOI] [PubMed] [Google Scholar]
- Rizvi TA, Ennis M, Behbehani M, Shipley T (1991). Connections Between the Central Nucleus of the Amygdala and the Midbrain Periaquductal Gray Topography and Reciprocity. The Journal of Comparative Neurology 303: 121–131. [DOI] [PubMed] [Google Scholar]
- Schapira AHV, Chaudhuri KR, Jenner P (2017). Non-motor features of Parkinson disease. Nat Rev Neurosci 18(7): 435–450. [DOI] [PubMed] [Google Scholar]
- Schwarz LA, Luo L (2015). Organization of the locus coeruleus-norepinephrine system. Curr Biol 25(21): R1051–R1056. [DOI] [PubMed] [Google Scholar]
- Selden NRW, Everitt BJ, Jarrard E, Robbins TW (1991). Complementary roles for the amygdala and hippocampus in aversive conditioning to explicit and contextual cues. Neuroscience 42(2): 335–350. [DOI] [PubMed] [Google Scholar]
- Sprengelmeyer R, Young AW, Mahn K, Schroeder U, Woitalla D, Buttner T et al. (2003). Facial expression recognition in people with medicated and unmedicated Parkinson’s disease. Neuropsychologia 41(8): 1047–1057. [DOI] [PubMed] [Google Scholar]
- Sullivan GM, Coplan JD, Kent JM, Gorman JM (1999). The noradrenergic system in pathological anxiety: a focus on panic with relevance to generalized anxiety and phobias. Biol Psychiatry 46(9): 1205–1218. [DOI] [PubMed] [Google Scholar]
- Sullivan RM, Duchesne A, Hussain D, Waldron J, Laplante F (2009). Effects of unilateral amygdala dopamine depletion on behaviour in the elevated plus maze: role of sex, hemisphere and retesting. Behav Brain Res 205(1): 115–122. [DOI] [PubMed] [Google Scholar]
- Tadaiesky MT, Dombrowski Pa, Figueiredo CP, Cargnin-Ferreira E, Da Cunha C, Takahashi RN (2008). Emotional, cognitive and neurochemical alterations in a premotor stage model of Parkinson’s disease. Neuroscience 156(4): 830–840. [DOI] [PubMed] [Google Scholar]
- Taylor TN, Caudle WM, Shepherd KR, Noorian A, Jackson CR, Iuvone PM et al. (2009). Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci 29(25): 8103–8113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor TN, Greene JG, Miller GW (2010). Behavioral phenotyping of mouse models of Parkinson’s disease. Behav Brain Res 211(1): 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovote P, Fadok JP, Luthi A (2015). Neuronal circuits for fear and anxiety. Nat Rev Neurosci 16(6): 317–331. [DOI] [PubMed] [Google Scholar]
- Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C, Deisseroth K (2011). Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature 471(7338): 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uematsu A, Tan BZ, Ycu EA, Cuevas JS, Koivumaa J, Junyent F et al. (2017). Modular organization of the brainstem noradrenaline system coordinates opposing learning states. Nat Neurosci 20(11): 1602–1611. [DOI] [PubMed] [Google Scholar]
- Verney C, Baulac M, Berger B, Alvarez C, Vigny A, Helle KB (1985). Morphological evidence for a dopaminergic terminal field in the hippocampal formation of young and adult rat. Neuroscience 14(4): 1039–1052. [DOI] [PubMed] [Google Scholar]
- Zesiewicz ΤΑ, Sullivan KL, Arnulf I, Chaudhuri KR, Morgan JC, Gronseth GS et al. (2010). Practice Parameter: treatment of nonmotor symptoms of Parkinson disease: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 74(11): 924–931. [DOI] [PubMed] [Google Scholar]
- Zhou QY, Palmiter RD (1995). Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell 83(7): 1197–1209. [DOI] [PubMed] [Google Scholar]
- Zhu XR, Maskri L, Herold C, Bader V, Stichel CC, Gunturkun O et al. (2007). Non-motor behavioural impairments in parkin-deficient mice. Eur J Neurosci 26(7): 1902–1911. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.