

Abstract
The potential to develop materials with antibody-like molecular recognition properties has helped sustain interest in protein-imprinted polymers over the past several decades. Unfortunately, despite persistent research, the field of noncovalent protein imprinting has seen limited success in terms of achieving materials with high selectivity and high affinity. In this Perspective, important yet sometimes overlooked aspects of the imprinting and binding processes are reviewed to help understand why there has been limited success. In particular, the imprinting and binding processes are viewed through the scope of free radical polymerization and hydrogel swelling theories to underscore the complexity of the synthesis and behavior of protein-imprinted polymers. Additionally, we review the metrics of success commonly used in protein imprinting literature (i.e., adsorption capacity, imprinting factor, and selectivity factor) and consider the relevance of each to the characterization of an imprinted polymer’s recognition characteristics. Throughout, common shortcomings are highlighted, and experiments that could help verify or disprove the efficacy of noncovalent protein imprinting are discussed.
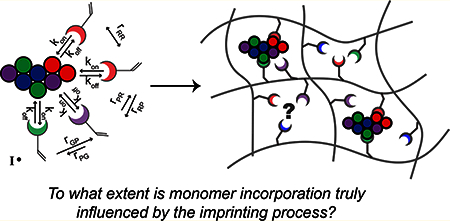
Graphical Abstract:
■. INTRODUCTION
Molecular imprinting is the most important polymer chemistry approach for designing and producing synthetic receptors. Although the term “molecular imprinting” was not coined until the 1970s, the idea of including a template molecule during synthesis to drive self-assembly and generate materials with specificity for the template stems back to a study by Dickey in 1949.1 Inspired by Pauling’s proposition that an antibody’s selectivity was achieved by self-assembling around its antigen, Dickey demonstrated improved affinity of silica gels for specific dye molecules when the gels were prepared in the presence of template dyes.
Since Dickey’s seminal paper, interest in the idea of generating molecularly imprinted polymers (MIPs) grew. In the production of these “artificial antibodies”, monomers with functional groups capable of forming favorable interactions with a molecule of interest, called the template, are used as building blocks instead of amino acids. In the production of antibodies, cells incorporate amino acids in an exact order based on the genetic code. In the production of MIPs, on the other hand, scientists rely on self-assembly of the functional monomers and template to try to influence monomer incorporation. The intention is that, after initiating polymerization, the functional groups will be locked into a well-defined pattern that is complementary to the template.
Originally this preassembly was achieved using a combination of reversible-covalent and noncovalent interactions,2 but the technique became more adaptable when systems relying entirely on noncovalent interactions were introduced.3,4 Crosslinking molecules are typically included at high percentages to minimize the mobility of the polymer chains and ultimately generate cavities that correspond to template size.3
Much of the early work in the molecular imprinting field was aimed at separation of small molecules (e.g., for making chromatography resins), which is a challenging feat in its own right.5 In many cases, high selectivity for the template was achieved,4,6 while in other cases, it was found that selectivity was template dependent. For example, Shea et al. showed that, using the same imprinting strategy, selectivity depended not only on separation distance of functional groups in the imprinted cavity but also on template identity.7 For one template, they achieved selectivity factors (i.e., ratio of template bound to nontemplate bound) as high as 3.8, while for another template tested, the selectivity factors were less than 1 (~0.6), suggesting that the polymer had an inherent selectivity for the nonimprinted molecule rather than the template.7 In another study, the high cross-reactivity of amino acid-imprinted materials brought the authors to the conclusion that “...interactions involved in binding to MIPs are more complex than generally envisaged.”8
Despite the challenges and cross-reactivity observed in many cases, the success stories of small molecule imprinting motivated researchers to push forward to more complex templates, including peptides,9 proteins,10 viruses,11,12 and whole cells.13,14 There is no doubt that synthetic materials capable of protein recognition are alluring as low-cost platforms for applications in drug delivery,15 sensing,16 and tissue engineering.17 However, molecular imprinting of protein templates poses additional challenges stemming from the (1) large size, (2) chemical and structural complexity, and (3) environmental instability of proteins.
First, the large size of proteins leads to significant diffusion limitations. Specifically, the diffusion coefficient of a template inside a polymer network relative to that in water decreases greatly when the template size approaches the network mesh size (i.e., as the restriction parameter (template radius/network mesh size) approaches 1). As a result, proteins are unable to diffuse into, or out of, many of the imprinted cavities within a bulk MIP. To overcome this diffusion limitation, methods for producing MIPs with smaller dimensions (i.e., crushing films into microparticles to expose binding sites, nanoscale imprinting, and surface imprinting) were established (Figure 1).19 Nanoscale and surface imprinting strategies are particularly advantageous for protein imprinting because they require less protein template and thus reduce cost relative to that of macroscale film synthesis.
Figure 1.

Evolution of molecular imprinting. Molecular imprinting was first introduced for small molecule templates, and imprinting was done on the bulk scale. As the complexity of the templates evolved to larger templates such as proteins and viruses, methods for reducing the dimensionality of the polymers to improve binding site accessibility (i.e., crushing films into microparticles, nanoscale imprinting, or imprinting on the surface of substrates) emerged to help overcome diffusion limitations.
A few noteworthy strategies have been developed in response to the increased complexity of macromolecular templates such as using aptamers as macromonomers20,21 or similarly (meth)-acrylate containing assistant recognition polymer chains (ARPCs) that assemble with the protein before initiating polymerization.22,23 However, in most reports, the usual imprinting strategy (i.e., using small, commercially available monomers that form noncovalent interactions with the protein) is still used. As for the environmental instability, the conditions necessary for proteins to be in their native form (i.e., aqueous buffer, temperature around 37 °C or lower, no surfactants) are the opposite of the conditions that would be ideal for MIPs: a catch-22.
Nonetheless, there are many studies reporting high selectivity for macromolecules as a result of imprinting.24–26 The question, then, is whether or not the imprinting process is truly responsible for improved selectivity, despite all of the obstacles.
■. DISCUSSION
1. Understanding the Imprinting Process.
Protein MIPs are made by including protein molecules in the prepolymer mixture to try to influence monomer incorporation. To help researchers unfamiliar with the field understand the imprinting process, a schematic very similar to that shown in Figure 2 is commonly included in literature reports. The first frame of these prototypical schematics shows static interactions between a template protein and functional monomers. Then, the second frame shows the formed polymer where monomers were locked into place based on how they had preassembled with the protein template. Lastly, it shows an empty cavity with a size that remains unchanged after protein extraction. This schematic is at best oversimplified and at worst misleading. A more accurate but still simplified depiction of the imprinting process is shown in Figure 3.
Figure 2.

Traditional schematic of the molecular imprinting process. The first frame shows static interactions between monomers and a template protein. After initiation, the second frame shows a pore of the polymer network, where the monomers were locked in their preassembled order. The third frame shows the pore after the template has been extracted with the same rigid structure as before the template was removed.
Figure 3.

Schematic depiction of the dynamic nature of noncovalent protein imprinting. (a) Interactions between monomer and template protein are highly dynamic. The residence time (i.e., time that the monomer is bound to the protein) depends on the off rate (koff) and is dependent on the strength of the noncovalent interactions between the monomer and protein. The on rate (kon) is a function of diffusion as well as long-range electrostatic interactions. (b) Upon initiation, the “pre-assembled” system may look different than just moments before initiation. The ability of monomers to polymerize together is determined by both localization and reactivity ratios of the monomers. Long and short arrows depict high and low preference, respectively, for reacting with the nearby monomer. (c) If two monomers are in proximity to the growing radical oligomer, the monomer with which the radical reacts will be influenced by the reactivity ratios. (d) By the time the next propagation step is occurring, the monomers or growing oligomers may diffuse away from the protein template, although higher molecular weight species will have a slower koff (i.e., a longer residence time) due to the multivalent nature of the interaction. (e) After the polymerization is complete, some of the pores in the hydrogel are imprinted while others are not, leading to nonspecific binding sites. (f) After the template is extracted, the MIP is likely to swell or collapse depending on the solvent, environmental conditions (e.g., pH, ionic strength), and amount of template removed.
First, the more accurate MIP schematic (Figure 3) shows that the interactions between monomers and protein molecules are highly dynamic, particularly in aqueous buffers where water and salt molecules compete with monomers for interactions with the protein. The dynamic interactions can be described by the association (kon) and dissociation (koff) rate constants of the monomer—template complex (Figure 3a). The association rate constant is primarily dependent on the diffusion coefficients of the template and monomer, although long-range electrostatic interactions can enhance kon via charge steering.27,28 Although kon can vary depending on the small molecule—protein pair, it is typically diffusion limited, and thus, the upper limit and common values are on the order of 108–109 M−1s−1.29 The dissociation rate constant, on the other hand, primarily depends on the number and strength of noncovalent interactions between the monomer and template. For example, higher molecular weight compounds have more functional groups that can interact with the protein, so they typically have lower koff and, in turn, longer residence times (tR = 1/koff).28
Thermodynamically, this makes sense. Imagine two monomers, one which can form only one hydrogen bond with the template and another which can form two. Knowing that the Gibbs free energy (ΔG) is related to the dissociation constant (KD = koff/kon = e ΔG/RT) and assuming an average ΔG for a hydrogen bond (−2.2 kcal/mol)30 and equivalent kon for both monomers, KD and koff for the doubly hydrogen bonding monomer will be 40 times lower and will stay bound to the protein 40 times longer than the singly hydrogen bonding monomer. The actual strength of a hydrogen bond will vary depending on the acidity of the hydrogen bond donor and basicity of hydrogen bond acceptor, orientation of the hydrogen bonding pair, and reaction conditions (e.g., pH, temperature, and ionic strength).31 Nevertheless, hydrogen bonds break and reform on fast time scales, from picoseconds to tens of nanoseconds, depending on the strength of the hydrogen bonds.32,33 Thus, for imprinting systems that rely solely on hydrogen bonding for preassembly, monomer- template interactions are forming and breaking in fractions of a second. Thus, hour-long preassembly steps are not only unnecessary because of the rapid reversal of monomer- template interactions but potentially detrimental due to monomer-induced protein instability.34
Similar to increasing the number of noncovalent interactions, increasing the strength of the noncovalent interaction can increase residence time. For example, Coulombic interactions are typically stronger than hydrogen bonds in low ionic strength solutions and thus are commonly exploited in the synthesis of protein-imprinted polymers. Unfortunately, the trade-off when using ionizable monomers is increased crossreactivity.35 Additionally, when ionizable monomers are used in the synthesis of protein-imprinted polymers, it is important to take the ionic strength of the polymerization buffer into consideration, as the ionic strength will affect the formation of Coulombic interactions. Specifically, the length over which an electrostatic effect persists in an electrolyte solution (i.e., the Debye length, λD) is inversely proportional to the square root of the ionic strength (I) as shown in eq 1:
| (1) |
where εo is the dielectric constant of the solution, εr is the permittivity of free space, kB is the Boltzmann constant, T is the temperature (K), Na is Avogadro’s number, and e is the charge of an electron. Clearly, as ionic strength is increased, the attraction of two oppositely charged species (e.g., an anionic amino acid residue and cationic monomer) will be diminished.
Going back to the traditional MIP schematic (Figure 2), in the second frame, where the monomers are shown to have polymerized with adjacent monomers, important aspects of free radical polymerization are disregarded. First, the rate of polymerization depends on both the initiator and monomer concentrations and thus will change as the reaction proceeds.36 Specifically, as the initiator and monomer are consumed to form growing polymer chains that serve as macroradicals, initiation efficiency decreases as a result of diffusional limitations of the macroradicals.37 Overall, polymerization is fastest initially before large oligomers form, during the time when monomer—template interactions will be most rapidly forming and breaking. While studies have shown that inclusion of templates affects polymerization kinetics, the rate enhancement can be detrimental if a low-affinity monomer happens to be the first monomer to reach the template near the growing radical chain, resulting in unfavorable monomer placement.38,39
Furthermore, the dependence of polymerization on monomer reactivity ratios is not evident in the second frame of Figure 2. In free radical polymerization, incorporation of comonomers will follow a statistical distribution that depends on the feed ratio and reactivity ratios of the monomers (Figure 3b). A reactivity ratio describes the preference of a radical to react with a monomer of the same identity versus a monomer with a different identity. Factors affecting monomer reactivity include steric effects, resonance stabilization of the radical site, and polarity of the double bond. Reactivity ratios also depend on the reaction conditions (i.e., bulk vs solution, solvent, temperature, and pH). In extreme cases, it is possible that a radical strongly prefers to react with either the comonomer (reactivity ratio →0) or a monomer of its own kind (reactivity ratio → infinity). In an ideal copolymerization, the incorporation of monomers is completely random, meaning that the radical has an equal preference for reacting with all monomers present. However, most copolymerizations are not perfectly random.36 Thus, even if two monomers are close to one another due to association with a protein template, unfavorable reactivity ratios may impede the desired or expected incorporation (Figures 3c and d).
Lastly, the traditional MIP scheme (Figure 2) shows only a single cavity in the network when in reality much of the crosslinked polymer will not be imprinted. In other words, even though monomers may be incorporated near the protein in a somewhat altered manner, random polymerization of monomers not associated with protein molecules will lead to nonspecific binding sites (Figure 3e). While the imprinted cavities may be reminiscent of the “hot spots” that are known to be important for protein—protein binding,40,41 the extent to which monomer distribution was altered by the imprinting process is unknown. Instead, the presence of the protein during polymerization may change the morphological properties (e.g., increased porosity where template was successfully extracted, increased stiffness due to residual protein serving as “crosslinks”)42,43 or chemical properties (e.g., functional groups from residual protein)35 of the polymer in ways not explained by the traditional concept of molecular imprinting. Overall, the standard view of how imprinting influences the protein binding properties of cross-linked polymers is inaccurate or at least incomplete.
3. Understanding the Binding Process from the Context of Hydrogel Swelling Theory.
In the prototypical imprinting scheme (Figure 2), the recognition cavity is shown to be the same size before extracting the protein as it is during rebinding. For small molecules, this is reasonably realistic, as a very high percent of cross-linking monomer is used to help achieve size exclusion and maintain rigidity. However, for proteins, less cross-linker is included to enable proteins to diffuse into and out of the network. Instead of being viewed as rigid materials, protein-imprinted polymers should be regarded as hydrogels, which are well-known to exhibit environmentally responsive swelling behavior (Figure 3f). The amount that a hydrogel swells depends on the functional groups present in the hydrogel and the density and distribution of covalent crosslinks or other tie-points (e.g., chain entanglements, hydrogen bonds, electrostatic interactions). For hydrogels that do not contain any ionizable functional groups, the swelling behavior can be described by the Peppas—Merrill equation.44 From this equation, the equilibrium volume swelling ratio and mesh size can be calculated.45,46
For nonionic hydrogels, the degree of swelling and mesh size depends primarily on molecular weight between cross-links. However, for hydrogels containing ionizable monomers, which are very common in protein imprinting literature, the swelling is also dependent on external factors, specifically pH and ionic strength as described by the modified Brannon—Peppas eq (Figure 4).47
Figure 4.

Swelling behavior of ionizable hydrogels. Equilibrium volume swelling ratio (Q) calculated as a function of pH or ionic strength using the modified Brannon—Peppas equation. (a) Q for anionic hydrogels increases as the hydrogels transition above the pKa of the acidic group, while Q for cationic hydrogels increases as they transition below the pKa of the basic group. (b) Q decreases rapidly with increasing ionic strength. This swelling behavior is important to keep in mind during template extraction and rebinding steps.
Mesh size is an important value to consider for imprinting studies, as it affects the diffusion of a solute (e.g., the template) through a hydrogel network.48,49 As described above, if the mesh size is smaller than or similar to the diameter of the template, which is the case for protein templates, diffusion of the template into the network will be greatly hindered. Unlike binding to an antibody, where the binding sites are freely exposed in solution, many of the binding sites in bulk-imprinted polymers are not accessible to protein template. As mentioned in the Introduction, MIP films can be crushed into microparticles to expose otherwise inaccessible binding sites. Alternatively, MIPs can be synthesized on a surface and/or on the nanoscale to achieve more complete protein extraction and faster protein binding.19 Even though the most accessible binding sites reside on or very near the surface of the MIP, it is beneficial to allow the gel to swell during the extraction step such that protein diffusion is less hindered and the amount of template extracted can be maximized. This can be achieved by increasing the pH (for anionic hydrogels) or decreasing the pH (for cationic hydrogels). Ideally, the change in pH for protein extraction should also eliminate charge—charge interactions between the protein and polymer, as favorable interactions will further impede the diffusion of the protein out of the network.
When doing rebinding, it is a trade-off between having an open mesh (to facilitate protein diffusion) and having the polymer in the same conditions as those where the imprinting was performed (such that the recognition cavity’s mesh size more closely matches the size of the protein). Even if the same buffer is used for both imprinting and rebinding, it is likely that the mesh size will be different for two reasons. First, functional groups that once interacted with the protein template can now interact with one another (e.g., monomers with opposite charge), decreasing the mesh size. For large templates, it is less likely that functional groups will be able to interact with one another, but it is a conceivable problem, especially for smaller proteins or peptides. Second, removing the proteins which once served as tie points will increase the mesh size. The main point of this discussion is to emphasize how the complexity of hydrogel swelling can cause the behavior of MIPs to deviate from traditional descriptions of these materials.
4. Metrics for the Success of Imprinting.
The primary goal of protein imprinting is to develop polymers capable of selective recognition of a target protein. The most commonly reported values for quantifying the success of imprinting are the adsorption capacity (Q), imprinting factor (IF), and selectivity factor (α). We will walk through each of these to better explain what information is contained within these values. We also suggest other values that should be reported and experiments that can and should be performed when developing new protein-imprinted polymers. In all cases, it is crucial to repeat synthesis and binding studies to demonstrate reproducibility and be able to make substantial claims about the effect(s) of imprinting. Unfortunately, much of the current data in protein imprinting literature are presented without statistics. Repeating synthesis of free radical polymerization is particularly important due to the inherently uncontrolled nature of this polymerization technique. Future researchers are urged to demonstrate reproducibility and provide appropriate statistical analysis to make their conclusions more convincing.
Although the reason has not been definitively proved, synthesizing cross-linked polymers in the presence of protein molecules does improve adsorption capacity in many cases.50 Q is a measure of the mass of protein that can be absorbed per mass of polymer and is calculated by eq 2:
| (2) |
where Co and Ce are the initial and equilibrium concentration of the protein in solution (mg/mL), respectively, V is the rebinding volume (mL), and m is the mass of particles used (mg). A high adsorption capacity is beneficial for many applications, but what information about the molecular recognition behavior of the polymers does it provide? Adsorption capacity can provide a measure of relative affinity in certain situations. Specifically, if MIPs prepared the same way are tested against multiple different proteins and the concentration of MIPs is kept constant (i.e., the number of binding sites is constant), the adsorption capacity is a reasonable measure of relative protein affinity, as fractional occupancy is related to equilibrium protein concentration, Ce, and the dissociation constant, KD, by eq 3:
| (3) |
However, if two polymers are morphologically different (e.g., a nonimprinted polymer (NIP) and the corresponding MIP), the number of binding sites per mass of particle will be different; thus, differences in adsorption capacity should not be used to make conclusions about relative affinity. Instead, KD should be measured from available techniques such as surface plasmon resonance, quartz crystal microbalance, or isothermal titration calorimetry. At the very least, data should be fit to an isotherm such as the Langmuir, Freundlich, or Langmuir— Freundlich isotherms, from which KD can be approximated.51 Affinity is an important measure because it provides information on how much protein will bind to the polymer at a given concentration. For example, in the development of biosensors, affinity is important to know to determine the limit of detection. For example, if an MIP has a KD of 1 μM, 50% of the binding sites will be filled when the protein concentration is 1 , μM. At concentrations below this, most sites will remain empty, and it is unlikely that a detectable signal will be produced.
A value very much related to Q is the imprinting factor (IF), which is the adsorption capacity of the MIP divided by that of a NIP made from the same constituent monomers (eq 4):
| (4) |
The IF is often used to make conclusions about how much imprinting improved the affinity of a polymer for the template, but this generalization cannot be made. As discussed above, two morphologically different polymers will have a different number of binding sites, and thus IF is not a fair way to compare affinity. Instead, IF simply provides a measure of how much more protein binds to MIPs than NIPs. Reported values for IF are almost always greater than 1, as imprinting likely increases the porosity of the material due to the presence of protein molecules during polymerization serving as porogens. To demonstrate increased porosity in protein imprinted materials, the MIP and NIP should be compared by techniques such as electron microscopy, atomic force microscopy, and/or mercury porosimetry. Another good control would be to compare the target-imprinted polymer to a different protein-imprinted polymer that is likely to have similar morphological properties to the target MIP.52
As MIPs are often claimed to be “plastic antibodies”, a critical property to characterize is their selectivity.9,53 Unfortunately, much of the data reported in literature does not support that MIPs are selective. The selectivity factor is calculated by eq 5:
| (5) |
and can be used to compare relative affinity of the same polymer for the two proteins, although it is still advisable to obtain KD values. The selectivity factor can be reported for experiments that were done noncompetitively (i.e., proteins incubated individually with the polymer) or competitively (i.e., two or more proteins simultaneously incubated with the polymer). Noncompetitive binding experiments provide information on what is driving protein binding. For example, if the polymer preferentially binds proteins with similar isoelectric points, it is likely electrostatic attraction driving the binding. If only proteins below the molecular weight of the template bind, it is likely size exclusion. For most MIPs, particularly those containing ionizable functional groups, crossreactivity is inevitable. This is useful for some applications (e.g., differential sensing)54 but prohibitive for other applications (e.g., targeted drug delivery).
Competitive studies are more representative of the complex mixtures that MIPs would actually be used in (e.g., bodily fluids, wastewater, and separations). In some literature reports, competitive binding experiments are not even performed. When these experiments are performed, they often have some flaws in the experimental design or interpretation of results. First, it is imperative to perform the competitive studies with both MIPs and NIPs made from the same monomers. Then, if the conclusions from these studies are along the lines of “imprinting resulted in improved selectivity” or “selectivity was achieved by molecular imprinting”, then the NIP must not show similar selectivity. In cases where the NIP exhibits similar selectivity, the commentary should be supported by the data, focusing on the improved adsorption capacity and inherent selectivity of the starting polymer. It is indeed advantageous if the polymer formulation chosen is selective for the desired template, but it cannot be claimed that imprinting was responsible for the observed selectivity.
Several reports have demonstrated that NIPs are good predictors of the selectivity that will be observed for corresponding MIPs, enabling rational design.55 Thus, we believe this rational design strategy is an imperative first step before attempting molecular imprinting. Time and money will be saved if a polymer that is selective for the target protein is identified or designed before imprinting. If it is desirable to have increased adsorption capacity, then molecular imprinting can be implemented.
5. Rational Design and Choosing Appropriate Controls.
There are many resources available for obtaining more detailed information on proteins that can help rationally design recognitive polymers. First, reviewing published literature on what drives protein—protein binding is a crucial step.40,41,56 Then, with the target protein in mind, the primary sequence should be analyzed to determine the distribution of amino acids. The primary sequence as well as crystal structure (i.e., secondary and tertiary structure) of many proteins can be obtained from the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do). Additionally, PyMOL (https://www.pymol.org/) is a useful software for visualizing proteins. One of the particularly useful features of PyMOL is its ability to quickly calculate vacuum electrostatics of protein surfaces (i.e., a qualitative view of the distribution of charges on the protein surface), which can help guide monomer selection.
A more quantitative tool is PDBePISA (http://www.ebi.ac.uk/pdbe/pisa/), a free online tool from which information such as the solvent accessible surface area (SASA) of proteins57 can be found. SASA is an important protein property for designing MIPs because, as the name implies, it gives information on the relative accessibility (specifically for a water molecule) of the amino acids in a protein. For example, for a protein with an exceptionally high percentage of hydrophobic SASA, hydrophobic monomers should be considered in the polymer design. SASA can also provide information on which charged residues are the most solvent exposed versus ones that are buried.
All of these computational resources can also be used to appropriately choose nontemplate proteins for investigating the selectivity of a protein-imprinted polymer. While choosing a protein with similar pi and/or molecular weight (MW) to that of the template is a good first step, these two metrics alone do not provide a complete picture of the differences between two proteins. A good example of this is lysozyme, one of the three most commonly imprinted proteins, and cytochrome c, lysozyme’s most common competitor. Lysozyme and cytochrome c have similar pi and MW; however, the majority of lysozyme’s cationic residues are arginine, while the majority of cytochrome c’s are lysine. Arginine is known to be enriched in protein—protein interfaces and can form several more non- covalent interactions than lysine. Considering the earlier discussion of koff and the importance of multivalent interactions, polymers containing hydrogen bond acceptors and anionic functional groups will undoubtedly show preference for lysozyme over cytochrome c.50
In another example, many MIPs are developed using boronic acid containing monomers in an effort to achieve selective recognition of glycoproteins. In one case, horse radish peroxidase (HRP) was imprinted and then selectivity was probed by comparing its binding to other nonglycoproteins and one other glycoprotein, ovalbumin.58 However, what was not mentioned was that HRP is over 20% glycosylation by weight,59 while ovalbumin is only 3% glycosylation by weight.60 It is not surprising, then, that a polymer made to bind the diols of a glycoprotein would preferentially bind the more heavily glycosylated protein. At first look, another study showed promise as they imprinted several different glycoproteins using boronic acid monomers and demonstrated selectivity for even the less glycosylated proteins. However, the polymerizations for each template were carried out for different amounts of time, affecting polymer thickness. Thus, the effects of polymerization time and imprinting on the observed selectivity cannot be decoupled.61
In general, control proteins for competitive binding assays should be more carefully selected. If a protein with truly similar properties can be used, that is ideal, but, at the very least, differences in the properties of the proteins used in the selectivity studies should be mentioned in the discussion. Alternatively, biological tools have been established for making protein mutations (e.g., site-directed mutagenesis).62 Mutants of the original template could be very useful in demonstrating the importance of specific amino acids in the imprinting and recognition processes.
6. Additional Shortcomings in Protein Imprinting.
Beyond the challenges and shortcomings described above, there are several system-specific challenges. For example, the cost of many proteins that would be interesting templates are prohibitively expensive, which is why the majority of literature reports are on the same few low-cost proteins.10 Relatively high protein concentrations are typically used in the synthesis of MIPs, diminishing the low-cost advantage of imprinting when expensive proteins are used as templates.
Additionally, there are added challenges associated with surface imprinted nanomaterials, a popular class of MIPs for overcoming the diffusion limitation associated with proteins. While nanoscale polymers are unquestionably advantageous for protein binding, the methods available for making nanoscale polymers often require conditions that are likely to denature the protein. For example, microemulsion polymerizations require surfactants, and precipitation polymerizations often necessitate the use of high temperatures. While there are a few examples of using emulsion polymerizations for making nanoscale protein-imprinted polymers,63,64 the design of these emulsions requires special considerations, as some surfactants or solvents used in these systems are more likely than others to denature proteins.65
Other researchers have reported achieving thin, uniform nanoscale MIP layers on the surface of nanomaterials using nonemulsion strategies.24,66 While this is an ideal method, better material characterization is necessary. In particular, it would be beneficial for the field of protein imprinting if high quality transmission electron microscopy (TEM) images were presented to clearly show that uniform, nanoscale shells were indeed achieved. Ultrathin polymer layers can be hard to visualize by TEM because under- or overfocusing can generate light or dark Fresnel fringes, respectively, that could be mistaken as thin imprinted layers.67 Additionally, the method used for staining the particles should be explicitly stated because, without staining, polymer shells do not have sufficient electron density for observable contrast by TEM. In general, better material characterization would make positive imprinting results more convincing.
■. CONCLUSIONS AND OUTLOOK
Several of the early developers and leaders in the molecular imprinting field have acknowledged the persistent challenges of macromolecule imprinting. For example, in his most recent review, Wulff said, “Imprinting of high molecular weight biopolymers is still problematic...it is still unclear whether a really high selectivity for very similar proteins can be achieved.”68 Additionally, Shea and his team suggested that, “The synthesis of NIPs with intrinsically high affinity and selectivity to a target toxin without molecular imprinting or affinity purification would streamline the process of antidote development” and, indeed, their team was able to do just that.69 Likewise, it is our opinion that if imprinting does not afford substantially improved affinity and selectivity, then it is not worth the increased time and cost associated with this step. For those who want to continue pursuing noncovalent protein imprinting, rational design of the polymer, careful selection of competitive proteins, and thorough discussion of the benefits of imprinting based on data and not preconceived notions are necessary to revitalize this field.
■ ACKNOWLEDGMENTS
The authors would like to acknowledge a grant from the National Institutes of Health (R01-EB022025).
Biographies
Heidi R. Culver received a B.S. in Materials Science and Engineering from The Johns Hopkins University and a Ph.D. in Biomedical Engineering from The University of Texas at Austin. She was the recipient of an NSF Graduate Research Fellowship, a Donald D. Harrington Dissertation Fellowship, a Thrust 2000-O.L. Chenoweth Endowed Graduate Fellowship from the Cockrell School of Engineering, and a Scholar Award from the Philanthropic Educational Organization. Her dissertation research focused on the development of nanoscale hydrogels with molecular recognition characteristics for use as protein receptors in biosensing applications.
Nicholas A. Peppas is a professor of Biomedical Engineering, Chemistry, Surgery and Perioperative Care, and Pharmacy and the director of the Institute for Biomaterials, Drug Delivery, and Regenerative Medicine at The University of Texas at Austin. He is a member of the National Academy of Engineering, the National Academy of Medicine, the American Academy of Arts and Sciences, the National Academy of Inventors, the Academy of Athens, the National Academy of Pharmacy of France, and the Academy of Pharmacy of Spain. He received his D. Eng. from the National Technical University of Athens, Greece and his Sc.D. from MIT, both in Chemical Engineering.
Footnotes
Notes
The authors declare no competing financial interest.
■ REFERENCES
- (1).Dickey FH The Preparation of Specific Adsorbents. Proc. Natl. Acad. Sci. U. S. A 1949, 35, 227–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wulff G; Sarhan A; Zabrocki K Enzyme-Analogue Built Polymers and Their Use for the Resolution of Racemates. Tetrahedron Lett. 1973, 14, 4329–4332. [Google Scholar]
- (3).Arshady R; Mosbach K Synthesis of Substrate-Selective Polymers by Host-Guest Polymerization. Makromol. Chem. 1981, 182, 687–692. [Google Scholar]
- (4).Sellergren B Moleclar Imprinting by Noncovalent Interactions: Tailor-Made Chiral Stationary Phases of High Selectivity and Sample Load Capacity. Chirality 1989, 1, 63–68. [Google Scholar]
- (5).Kempe M; Mosbach K Molecular Imprinting Used for Chiral Separations. J. Chromatogr. A 1995, 694, 3–13. [Google Scholar]
- (6).Mayes AG; Andersson LI; Mosbach K Sugar Binding Polymers Showing High Anomeric and Epimeric Discrimination Obtained by Noncovalent Molecular Imprinting. Anal. Biochem. 1994, 222, 483–488. [DOI] [PubMed] [Google Scholar]
- (7).Shea KJ; Dougherty TK Molecular Recognition on Synthetic Amorphous Surfaces. The Influence of Functional Group Positioning on the Effectiveness of Molecular Recognition. J. Am. Chem. Soc 1986, 108, 1091–1093. [Google Scholar]
- (8).Allender CJ; Brain KR; Heard CM Binding Cross-Reactivity of Boc-Phenylalanine Enantiomers on Molecularly Imprinted Polymers. Chirality 1997, 9, 233–237. [Google Scholar]
- (9).Hoshino Y; Kodama T; Okahata Y; Shea KJ Peptide Imprinted Polymer Nanoparticles: A Plastic Antibody. J. Am. Chem. Soc 2008, 130, 15242–15243. [DOI] [PubMed] [Google Scholar]
- (10).Kryscio DR; Peppas NA Critical Review and Perspective of Macromolecularly Imprinted Polymers. Acta Biomater. 2012, 8, 461–473. [DOI] [PubMed] [Google Scholar]
- (11).Cumbo A; Lorber B; Corvini PF-X; Meier W; Shahgaldian P A Synthetic Nanomaterial for Virus Recognition Produced by Surface Imprinting. Nat. Commun 2013, 4, 1503. [DOI] [PubMed] [Google Scholar]
- (12).Wangchareansak T; Thitithanyanont A; Chuakheaw D; Gleeson MP; Lieberzeit PA; Sangma C A Novel Approach to Identify Molecular Binding to the Influenza Virus H5N1: Screening Using Molecularly Imprinted Polymers (MIPs). Med Chem Comm 2014, 5, 617–621. [Google Scholar]
- (13).Hayden O; Dickert FL Selective Microorganism Detection with Cell Surface Imprinted Polymers. Adv. Mater 2001, 13, 1480–1483. [Google Scholar]
- (14).Ren K; Banaei N; Zare RN Sorting Inactivated Cells Using Cell-Imprinted Polymer Thin Films. ACS Nano 2013, 7, 6031–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hilt JZ; Byrne ME Configurational Biomimesis in Drug Delivery: Molecular Imprinting of Biologically Significant Molecules. Adv. Drug Delivery Rev 2004, 56, 1599–1620. [DOI] [PubMed] [Google Scholar]
- (16).Whitcombe MJ; Chianella I; Larcombe L; Piletsky SA; Noble J; Porter R; Horgan A The Rational Development of Molecularly Imprinted Polymer-Based Sensors for Protein Detection. Chem. Soc. Rev 2011, 40, 1547–1571. [DOI] [PubMed] [Google Scholar]
- (17).Neves MI; Wechsler ME; Gomes ME; Reis RL; Granja PL; Peppas NA Molecularly Imprinted Intelligent Scaffolds for Tissue Engineering Applications. Tissue Eng., Part B 2017, 23, 27–43. [DOI] [PubMed] [Google Scholar]
- (18).Spizzirri UG; Peppas NA Structural Analysis and Diffusional Behavior of Molecularly Imprinted Polymer Networks for Cholesterol Recognition. Chem. Mater 2005, 17, 6719–6727. [Google Scholar]
- (19).Gao D; Zhang Z; Wu M; Xie C; Guan G; Wang D A Surface Functional Monomer-Directing Strategy for Highly Dense Imprinting of TNT at Surface of Silica Nanoparticles. J. Am. Chem. Soc 2007, 129, 7859–7866. [DOI] [PubMed] [Google Scholar]
- (20).Bai W; Gariano NA; Spivak DA Macromolecular Amplification of Binding Response in Superaptamer Hydrogels. J. Am. Chem. Soc 2013, 135, 6977–6984. [DOI] [PubMed] [Google Scholar]
- (21).Bai W; Spivak DA A Double-Imprinted Diffraction-Grating Sensor Based on a Virus-Responsive Super-Aptamer Hydrogel Derived from an Impure Extract. Angew. Chem., Int. Ed 2014, 53, 2095–2098. [DOI] [PubMed] [Google Scholar]
- (22).Guo M-J; Zhao Z; Fan Y-G; Wang C-H; Shi L-Q; Xia JJ; Long Y; Mi H-F Protein-Imprinted Polymer with Immobilized Assistant Recognition Polymer Chains. Biomaterials 2006, 27, 4381–4387. [DOI] [PubMed] [Google Scholar]
- (23).Liu D; Yang Q; Jin S; Song Y; Gao J; Wang Y; Mi H Core-shell Molecularly Imprinted Polymer Nanoparticles with Assistant Recognition Polymer Chains for Effective Recognition and Enrichment of Natural Low-Abundance Protein. Acta Biomater. 2014, 10, 769–775. [DOI] [PubMed] [Google Scholar]
- (24).He H; Fu G; Wang Y; Chai Z; Jiang Y; Chen Z Imprinting of Protein over Silica Nanoparticles via Surface Graft Copolymerization Using Low Monomer Concentration. Biosens. Bioelectron 2010, 26, 760–765. [DOI] [PubMed] [Google Scholar]
- (25).Lin Z; Xia Z; Zheng J; Zheng D; Zhang L; Yang H; Chen G Synthesis of Uniformly Sized Molecularly Imprinted Polymer- Coated Silica Nanoparticles for Selective Recognition and Enrichment of Lysozyme. J. Mater. Chem 2012, 22, 17914. [Google Scholar]
- (26).Tan L; Kang C; Xu S; Tang Y Selective Room Temperature Phosphorescence Sensing of Target Protein Using Mn-Doped ZnS QDs-Embedded Molecularly Imprinted Polymer. Biosens. Bioelectron. 2013, 48, 216–223. [DOI] [PubMed] [Google Scholar]
- (27).Gabdoulline RR; Wade RC Protein-Protein Association: Investigation of Factors Influencing Association Rates by Brownian Dynamics Simulations. J. Mol. Biol 2001, 306, 1139–1155. [DOI] [PubMed] [Google Scholar]
- (28).Pan AC; Borhani DW; Dror RO; Shaw DE Molecular Determinants of Drug-receptor Binding Kinetics. Drug Discovery Today 2013, 18, 667–673. [DOI] [PubMed] [Google Scholar]
- (29).Copeland RA; Pompliano DL; Meek TD Drug-target Residence Time and Its Implications for Lead Optimization. Nat. Rev. Drug Discovery 2006, 5, 730–739. [DOI] [PubMed] [Google Scholar]
- (30).Myers JK; Pace CN Hydrogen Bonding Stabilizes Globular Proteins. Biophys. J 1996, 71, 2033–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Abraham MH; Grellier PL; Kamlet MJ; Doherty RM; Taft RW; Abboud J-LM The Use of Scales of Hydrogen-Bond Acidity and Basicity in Organic Chemistry. Rev. Port Quim 1989, 31, 85–92. [Google Scholar]
- (32).Kooijman EE; Tieleman DP; Testerink C; Munnik T; Rijkers DTS; Burger KNJ; de Kruijff B An Electrostatic/ Hydrogen Bond Switch as the Basis for the Specific Interaction of Phosphatidic Acid with Proteins. J. Biol. Chem 2007, 282, 1135611364. [DOI] [PubMed] [Google Scholar]
- (33).Martiniano HFMC; Galamba N Insights on Hydrogen- Bond Lifetimes in Liquid and Supercooled Water. J. Phys. Chem. B 2013, 117, 16188–16195. [DOI] [PubMed] [Google Scholar]
- (34).Kryscio DR; Fleming MQ; Peppas NA Protein Conformational Studies for Macromolecularly Imprinted Polymers. Macromol. Biosci 2012, 12, 1137–1144. [DOI] [PubMed] [Google Scholar]
- (35).Hjerten S; Liao J-L; Nakazato K; Wang Y; Zamaratskaia G; Zhang H-X Gels Mimicking Antibodies in Their Selective Recognition of Proteins. Chromatographia 1997, 44, 227–234. [Google Scholar]
- (36).Painter PC; Coleman MM Fundamentals of Polymer Science: An Introductory Text, 2nd ed.; Technomic Pub. Co: Lancaster, PA, 1997. [Google Scholar]
- (37).Achilias DS; Kiparissides C Development of a General Mathematical Framework for Modeling Diffusion-Controlled Free- Radical Polymerization Reactions. Macromolecules 1992, 25, 37393750. [Google Scholar]
- (38).Oral E; Peppas NA Dynamic Studies of Molecular Imprinting Polymerizations. Polymer 2004, 45, 6163–6173. [Google Scholar]
- (39).Tan YY; van Ekenstein ORA A Generalized Kinetic Model for Radical-Initiated Template Polymerizations in Dilute Template Systems. Macromolecules 1991, 24, 1641–1647. [Google Scholar]
- (40).Bogan AA; Thorn KS Anatomy of Hot Spots in Protein Interfaces. J. Mol. Biol 1998, 280, 1–9. [DOI] [PubMed] [Google Scholar]
- (41).Keskin O; Ma B; Nussinov R Hot Regions in Protein–Protein Interactions: The Organization and Contribution of Structurally Conserved Hot Spot Residues. J. Mol. Biol 2005, 345, 1281–1294. [DOI] [PubMed] [Google Scholar]
- (42).Bossi A; Bonini F; Turner APF; Piletsky SA Molecularly Imprinted Polymers for the Recognition of Proteins: The State of the Art. Biosens. Bioelectron 2007, 22, 1131–1137. [DOI] [PubMed] [Google Scholar]
- (43).Nematollahzadeh A; Sun W; Aureliano CSA; Lutkemeyer D; Stute J; Abdekhodaie MJ; Shojaei A; Sellergren B High- Capacity Hierarchically Imprinted Polymer Beads for Protein Recognition and Capture. Angew. Chem 2011, 123, 515–518. [DOI] [PubMed] [Google Scholar]
- (44).Peppas NA; Merrill EW Crosslinked Poly (Vinyl Alcohol) Hydrogels as Swollen Elastic Networks. J. Appl. Polym. Sci 1977, 21, 1763–1770. [Google Scholar]
- (45).Peppas NA; Huang Y; Torres-Lugo M; Ward JH; Zhang J Physicochemical Foundations and Structural Design of Hydrogels in Medicine and Biology. Annu. Rev. Biomed. Eng 2000, 2, 9–29. [DOI] [PubMed] [Google Scholar]
- (46).Bell CL; Peppas NA Water, Solute and Protein Diffusion in Physiologically Responsive Hydrogels of Poly(methacrylic Acid-GEthylene Glycol). Biomaterials 1996, 17, 1203–1218. [DOI] [PubMed] [Google Scholar]
- (47).Sen M; Guven O Prediction of Swelling Behaviour of Hydrogels Containing Diprotic Acid Moieties. Polymer 1998, 39, 1165–1172. [Google Scholar]
- (48).Am Ende MT; Peppas NA Transport of Ionizable Drugs and Proteins in Crosslinked Poly (Acrylic Acid) and Poly (Acrylic Acid-Co-2-Hydroxyethyl Methacrylate) Hydrogels. II. Diffusion and Release Studies. J. Controlled Release 1997, 48, 47–56. [Google Scholar]
- (49).Peppas NA; Wright SL Drug Diffusion and Binding in Ionizable Interpenetrating Networks from Poly (Vinyl Alcohol) and Poly (Acrylic Acid). Eur. J. Pharm. Biopharm 1998, 46, 15–29. [DOI] [PubMed] [Google Scholar]
- (50).Culver HR; Steichen SD; Peppas NA A Closer Look at the Impact of Molecular Imprinting on Adsorption Capacity and Selectivity for Protein Templates. Biomacromolecules 2016, 17, 4045–4053. [DOI] [PubMed] [Google Scholar]
- (51).Umpleby R; Baxter S; Rampey A; Rushton G; Chen Y; Shimizu K Characterization of the Heterogeneous Binding Site Affinity Distributions in Molecularly Imprinted Polymers. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci 2004, 804, 141–149. [DOI] [PubMed] [Google Scholar]
- (52).Clegg JR; Zhong JX; Irani AS; Gu J; Spencer DS; Peppas NA Characterization of Protein Interactions with Molecularly Imprinted Hydrogels That Possess Engineered Affinity for High Isoelectric Point Biomarkers. J. Biomed. Mater. Res., Part A 2017, 105, 1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Poma A; Guerreiro A; Whitcombe MJ; Piletska EV; Turner APF; Piletsky SA Solid-Phase Synthesis of Molecularly Imprinted Polymer Nanoparticles with a Reusable Template-”Plastic Antibodies. Adv. Funct. Mater 2013, 23, 2821–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Greene NT; Shimizu KD Colorimetric Molecularly Imprinted Polymer Sensor Array Using Dye Displacement. J. Am. Chem. Soc 2005, 127, 5695–5700. [DOI] [PubMed] [Google Scholar]
- (55).Baggiani C; Giovannoli C; Anfossi L; Passini C; Baravalle P; Giraudi G A Connection between the Binding Properties of Imprinted and Nonimprinted Polymers: A Change of Perspective in Molecular Imprinting. J. Am. Chem. Soc 2012, 134, 1513–1518. [DOI] [PubMed] [Google Scholar]
- (56).Sheinerman FB; Norel R; Honig B Electrostatic Aspects of Protein—protein Interactions. Curr. Opin. Struct. Biol 2000, 10, 153–159. [DOI] [PubMed] [Google Scholar]
- (57).Richmond TJ Solvent Accessible Surface Area and Excluded Volume in Proteins: Analytical Equations for Overlapping Spheres and Implications for the Hydrophobic Effect. J. Mol. Biol 1984, 178, 63–89. [DOI] [PubMed] [Google Scholar]
- (58).Wei J; Ni Y; Zhang W; Zhang Z; Zhang J Detection of Glycoprotein through Fluorescent Boronic Acid-Based Molecularly Imprinted Polymer. Anal. Chim. Acta 2017, 960, 110–116. [DOI] [PubMed] [Google Scholar]
- (59).Yang BY; Gray JS; Montgomery R The Glycans of Horseradish Peroxidase. Carbohydr. Res. 1996, 287, 203–212. [DOI] [PubMed] [Google Scholar]
- (60).Nisbet AD; Saundry RH; Moir AJ; Fothergill LA; Fothergill JE The Complete Amino-Acid Sequence of Hen Ovalbumin. Eur. J. Biochem 1981, 115, 335–345. [DOI] [PubMed] [Google Scholar]
- (61).Wang S; Ye J; Bie Z; Liu Z Affinity-Tunable Specific Recognition of Glycoproteins via Boronate Affinity-Based Controllable Oriented Surface Imprinting. Chem. Sci 2014, 5, 1135. [Google Scholar]
- (62).Ho SN; Hunt HD; Horton RM; Pullen JK; Pease LR Site-Directed Mutagenesis by Overlap Extension Using Polymerase Chain Reaction. Gene 1989, 77, 51–59. [DOI] [PubMed] [Google Scholar]
- (63).Tan CJ; Tong YW The Effect of Protein Structural Conformation on Nanoparticle Molecular Imprinting of Ribonuclease A Using Miniemulsion Polymerization. Langmuir 2007, 23, 2722–2730. [DOI] [PubMed] [Google Scholar]
- (64).Tan CJ; Wangrangsimakul S; Bai R; Tong YW Defining the Interactions between Proteins and Surfactants for Nanoparticle Surface Imprinting through Miniemulsion Polymerization. Chem. Mater 2008, 20, 118–127. [Google Scholar]
- (65).Moore PN; Puvvada S; Blankschtein D Role of the Surfactant Polar Head Structure in Protein—Surfactant Complexation: Zein Protein Solubilization by SDS and by SDS/C 12 E N Surfactant Solutions. Langmuir 2003, 19, 1009–1016. [Google Scholar]
- (66).Fu G; He H; Chai Z; Chen H; Kong J; Wang Y; Jiang Y Enhanced Lysozyme Imprinting Over Nanoparticles Functionalized with Carboxyl Groups for Noncovalent Template Sorption. Anal. Chem 2011, 83, 1431–1436. [DOI] [PubMed] [Google Scholar]
- (67).Williams DB; Carter CB Transmission Electron Microscopy: A Textbook for Materials Science, 2nd ed.; Springer: New York, 2008. [Google Scholar]
- (68).Wulff G Fourty Years of Molecular Imprinting in Synthetic Polymers: Origin, Features and Perspectives. Microchim. Acta 2013, 180, 1359–1370. [Google Scholar]
- (69).Hoshino Y; Koide H; Furuya K; Haberaecker WW; Lee S-H; Kodama T; Kanazawa H; Oku N; Shea KJ The Rational Design of a Synthetic Polymer Nanoparticle That Neutralizes a Toxic Peptide in Vivo. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]