

Abstract
Chronic sustained inflammation underlies many pathological conditions including neurodegenerative diseases. Divalent manganese (Mn) exposure can stimulate neurotoxicity by increasing inflammation. In this study, we examined whether Mn activates the NLRP3 inflammasome to prime neuroinflammation. Exposing LPS-primed microglial cells to Mn significantly augmented NLRP3, caspase-1 cleavage, and IL-1β maturation. Mn exposure in mouse models also augmented the abundance of NLRP3 inflammasome signaling components, including cleaved caspase-1, IL-1β and NLRP3. Furthermore, Mn impaired mitochondrial ATP generation, basal respiratory rate, and spare capacity in microglia cells. These data suggest that Mn-induced mitochondrial defects drove the inflammasome signal amplification. We found that Mn induced cell-to-cell transfer of ASC in exosomes. Furthermore, primed microglial cells exposed to exosomes from Mn-treated animals released more IL-1β than cells exposed to exosomes from control treated animals. We also observed that welders exposed to Mn fumes have plasma exosomes that contained more ASC than those from a matched control group. Collectively, these results demonstrate that the divalent metal manganese acts as a key amplifier of NLRP3 inflammasome signaling and exosomal ASC release.
One-sentence summary:
Exosomes containing the adaptor ASC spread NLRP3 inflammasome activation between cells after manganese exposure.
Editor’s summary:
Exosomes transfer inflammasome activation
Chronic occupational exposure to manganese (Mn) is associated with the risk of developing Parkinson’s disease. Sarkar et al. found that exposing primed microglial cells or mice to the transition metal Mn increased NLRP3 inflammasome expression and activation. Microglial cells exposed to Mn exhibited mitochondrial dysfunction and released exosomes containing the inflammasome adaptor ASC. The effects of Mn on inflammasome activation were sensitive to reduced endocytosis and transferable between cells exposed to purified exosomes from ASC-sufficient cells. Similarly, serum exosomes from welders contained more ASC and stimulated stronger IL-1β expression than did those from control donors, suggesting that occupational Mn exposure may increase systemic inflammasome activation due to exosome-mediated transfer of ASC.
Introduction
Hyperactivation of immune cells, which leads to the production of a plethora of proinflammatory factors, is a key pathophysiological hallmark of multiple disorders including neurodegenerative disorders, autoimmune diseases, and inflammatory bowel syndrome (IBS) (1–3). Though inflammation exacerbates multiple pathological conditions, the signaling cascade underlying this hyperactivated state is unclear. The etiology of neurodegenerative disorders including Alzheimer’s disease (AD) and Parkinson’s disease (PD) is multifactorial. Microglial cells as well as various inflammatory factors, including tumor necrosis factor-α (TNF-α) and IL-1β, are increased in PD patients (1, 4, 5). Blocking microglial activation is neuroprotective in animal models of PD (5). Furthermore, use of anti-inflammatory agents is protective in cell culture and animal models of PD (6–10). Use of anti-inflammatory drugs reduces the incidence of PD (11). Though the effects of chronic inflammation are well documented in neurodegenerative disorders like PD, the mechanism underlying this sustained immune response remains unclear (12).
Inflammasome activation is a key determinant of chronic inflammation in a variety of inflammatory cells (13) and pathological conditions. A multimeric protein complex, inflammasomes that contain NLRP3 (nucleotide binding and oligomerization domain-like receptor family pyrin domain containing 3), NLRC4 (NLR family CARD domain-containing protein 4), and AIM2 (absent in melanoma 2) among others, are involved in neurodegenerative disorders like PD, AD, and traumatic brain injury (TBI) (14). The NLRP3 inflammasome oligomeric complex is comprised of an adapter protein ASC (apoptosis-associated speck-like protein containing a CARD), Caspase 1, and NLRP3. Multiple external signals, including misfolded β-amyloid (15) and αSynPFF (16), activate the NLRP3 inflammasome complex. NLRP3 inflammasome signaling requires two signals for its activation. Lipopolysaccharide (LPS), TNF-α, or any signal capable of activating NFκB can act as signal 1 (or a priming signal) to increase the expression of NLRP3 and pro-IL-1β. A second activation signal, such as mitochondrial dysfunction and lysosomal damage, that is known as signal 2 triggers the assembly and activation of the NLRP3 inflammasome complex. The adapter molecule ASC links the NLRP3 protein to the recruited caspase-1, which promotes caspase-1 dimerization, autocatalytic processing, and the generation of active caspase-1. Active caspase-1 processes pro-IL-1β into its mature IL-1β form that stimulates inflammation (17, 18). NLRP3 inflammasome propagation is modulated by a cell-to-cell transmission of the ASC adaptor protein by an unknown mechanism (19). However, the exact mechanisms of inflammasome activation and propagation remain elusive.
Chronic exposure to transition metals like manganese (Mn), iron (Fe), copper (Cu), and zinc (Zn) from occupational or environmental sources (e.g., various food products and well-water) is linked to various neurodegenerative disorders such as AD and PD. Chronic exposure to Mn in occupational settings is observed in miners (20, 21) and welders (22). Occupational exposure to Mn increases the risk of developing PD in welder populations (23). Mn toxicity can also manifest as a disease called manganism, which shares similar symptoms as PD (24, 25). Mn accumulates mainly in the globus pallidus as well as in the striatum leading to Parkinsonian neurological deficits (26).
Mn toxicity is well documented in neurodegeneration and glial dysfunction (26–31). More specifically, Mn induces TNF-α and inducible nitrogen synthase (iNOS) in astrocytes (32, 33), which causes mitochondrial dysfunction by accumulating in mitochondria (34). Additionally, iNOS induces neurotoxicity in a rat model of Mn neurotoxicity by modulating microgliosis (35). Mn exposure induces classical activation (M1 activation) in microglial cells (36) as well as astrocytes(31) in the central nervous system, leading to the production of pro-inflammatory cytokines. A plethora of pro-inflammatory factors causes neuronal damage (37). Furthermore, Mn enhances LPS-induced production of pro-inflammatory factors in microglial cells (29). Mn-induced microglial activation also activates astrocytes, leading to enhanced production of pro-inflammatory factors (38). However, the molecular signaling mechanism of Mn-induced microglial activation is still not well understood. Microglia express multiple functional inflammasomes, including NLRP3, NLRC4, and AIM2 inflammasomes (13). However, the effect of Mn on microglial inflammasome activation and propagation remains ill-defined.
In this study, we characterized the role of Mn in inflammasome activation in microglial cells. We found that Mn acted as signal 2 to activate the NLRP3 inflammasome in microglial cells, which stimulated mitochondrial dysfunction in microglial cells. Mn exposure reduced the abundance of mitochondrial fusion protein 2 (also Mitofusin 2 or Mfn2) and vacuolar protein sorting-associated protein 35 (VPS35), a retromer complex protein which promotes Mfn2 ubiquitination. Uptake of ASC-containing exosomes played a role in Mn-induced inflammasome propagation. Finally, welder serum exosomes had a higher ASC load compared to age-matched controls, and welder serum had increased amounts of serum pro-inflammatory cytokines.
Results
Mn induces NLRP3 inflammasome activation in microglial cells in vitro and in vivo
The NLRP3 inflammasome can be activated by various external signals including, ATP, pathogen-activated molecular patterns (PAMP’s), and danger-associated molecular patterns (DAMP’s) (15). Because the role of environmental PD neurotoxicants, like Mn, in inducing inflammasome activation has yet to be described, we investigated the ability of Mn to stimulate the processing and release of IL-1β and caspase-1 cleavage from LPS-primed microglial cells. Microglial cells were primed with LPS for 3 h, after which the cells were treated with Mn for a further 24 h. We found no detectable mature IL-1β protein in unstimulated, only LPS-primed , or only Mn-treated cells (Fig. 1A). However, mature IL-1β was only detected in LPS-primed, Mn-treated microglial cells (Fig. 1A). Similarly, a multiplexed Luminex assay confirmed that LPS-primed microglial cells released IL-1β after Mn exposure in a dose-dependent manner (Fig. 1B). Howwver, Mn treatment alone had no effect on IL-1β processing and release. Furthermore, Western blot analysis revealed that Mn stimulated the cleavage of caspase-1 in LPS-primed cells when compared to either unstimulated or LPS-primed cells (Fig. 1A). These results confirmed that Mn exposure served as a signal 2 to augment microglial inflammasome signaling (39).
Fig. 1. Manganese treatment induced NLRP3 inflammasome activation in microglial cells in vitro.
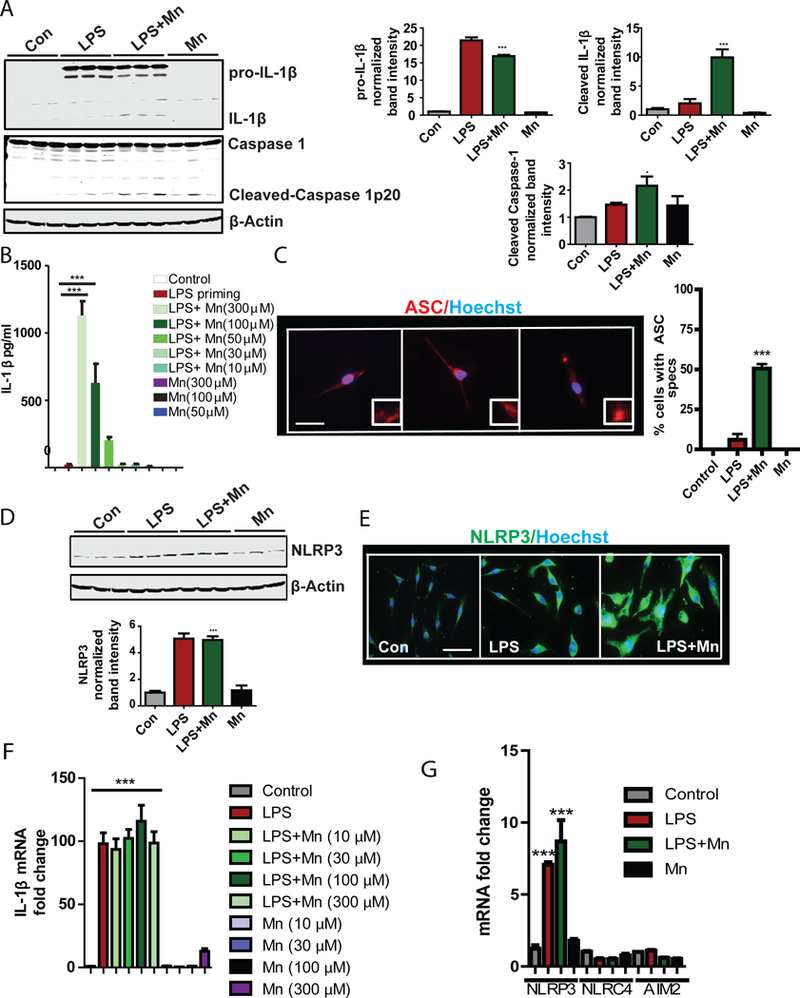
(A) Western blot analysis of Caspase 1 cleavage and IL-1β maturation in lysates from a LPS-primed microglial cell line treated with Mn, as indicated. Blots (left) are representative of 4 independent experiments. Normalized band intensity data (right) are means ± SEM from all experiments. (B) Luminex analysis of IL-1β production by a microglial cell line primed with LPS and treated for 24 h with Mn at the indicated dose. Data are means ± SEM pooled from 8 independent experiments. (C) Immunofluorescence microscopy analysis of ASC spec formation in a microglial cell line primed with LPS and treated as indicated. Images (left) are representative of 3 independent experiments. Quantified data (right) are means ± SEM from all experiments. Scale bar, 20 μm. (D and F) Western blot (D) and Immunofluorescence microscopy (E) analyses of NLRP3 abundance in a microglial cell line primed with LPS and treated as indicated. Blots and images are representative of 3 independent experiments. Normalized band intensity data (D, lower) are means ± SEM from all experiments. Scale bar, 20 μm. (F and G) qRT-PCR analysis of IL-1β (F) and NLRP3, NLRC4, and AIM2 (G) mRNA expression following treatment of an LPS-primed microglial cell line. Data are means ± SEM pooled from 3 independent experiments. *P<0.05, **P <0.01, ***P<0.001 by ANOVA with Tukey post analysis.
ASC, a component of inflammasome activation, can form speck-like structures that propagate inflammasome activation from cell-to-cell (19). By immunofluorescence analysis we found that ASC was evenly distributed throughout unstimulated or LPS-primed cells. In contrast, the LPS-primed microglial cells exposed to Mn formed intense ASC specks, which are indicative of inflammasome activation (Fig. 1C). When we tested by Western blot which inflammasome was activated by Mn, we found that NLRP3 abundance was significantly increased in LPS-primed and Mn-treated, LPS-primed microglial cells (Fig. 1D). Immunocytochemical (ICC) analysis further confirmed NLRP3 abundance was increased (Fig. 1E) by LPS priming of microglial cells. Similarly, qRT-PCR analysis showed LPS increased mRNA expression of pro-IL-1β in microglial cells, and treatment with various doses of Mn did not further augment expression (Fig. 1F). Furthermore, by qRT-PCR analysis we found that LPS priming and LPS+Mn increased NLRP3 mRNA expression, but not expression of Absent In Melanoma 2 (AIM2) and NLR Family CARD Domain Containing 4 (NLRC4) (Fig. 1G). Mn-treated unprimed cells did not increase expression of pro-IL-1β or NLRP3, indicating that Mn exposure had no effect on the priming step of the NLRP3 inflammsome. To further verify the specificity of Mn in metal-induced inflammasome activation, primary microglial cells were treated with Mn, copper (Cu), and zinc (Zn). Because Zn can be toxic to microglial cells, a non-toxic dose of Zn was used (fig. S1A). Cu was used at the same concentration as Mn. Our Luminex analysis revealed that Mn increased IL-1β release in LPS-primed cells, whereas Cu and Zn did not (fig. S1B). In contrast, LPS-treated cells stimulated with Mn, Cu or Zn released comparable amounts of TNF-α (fig. S1C). These data validated that inflammasome activation was not a property of all divalent transition metals. Moreover, Mn exposure increased LPS-induced Nos2 mRNA expression (fig. S2A) and protein abundance (fig. S2B and Fig. S2C), as well as nitrite release (fig. S2D). All these findings together showed that Mn activated the NLRP3 inflammasome in primed microglial cells.
The etiology of PD is complex and multifactorial and gene-environment interaction is likely involved in PD pathogenesis (40–42). Aggregated αSyn is an integral component of PD-related Lewy bodies and Lewy neurites, which can induce NLRP3 inflammasome activation (16). We hypothesized that Mn may augment αSynPFF-induced microglial NRLP3 inflammasome activation. Co-treatment of Mn with αSynPFF increased the abundance of αSynPFF-induced NLRP3 and iNOS proteins (Fig. 2A). Luminex assay revealed that Mn further increased the αSynPFF-induced release of IL-1β (Fig. 2B) but not IL-6 (fig. S3A) or TNF-α (fig. S3B). These findings indicate that Mn increased αSynPFF-induced inflammasome activation in microglial cells.
Fig. 2. Manganese exposure induced NLRP3 inflammasome activation in microglial cells in vivo.
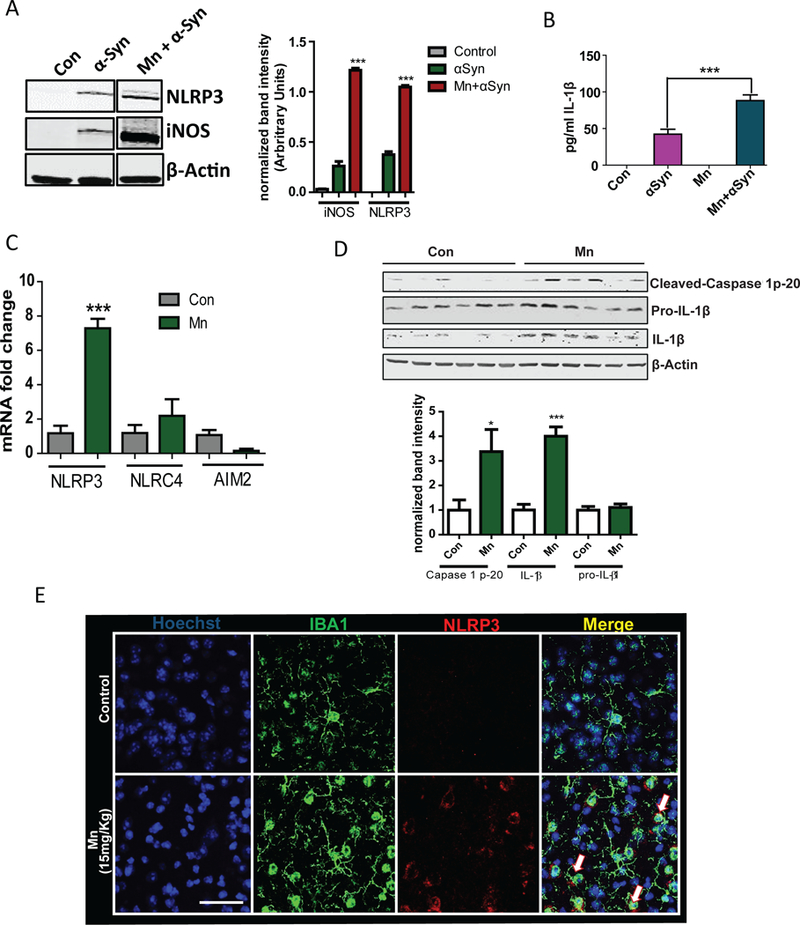
(A) Western Blot analysis of the NLRP3 and iNOS expression in wild-type microglial cells treated with Mn and αSynAgg as indicated. Blots (left) are representative of 3 independent experiments. Normalized band intensity data (right) are means ± SEM from all experiments. (B) Luminex analysis of IL-1β production by wild-type microglial cells treated with Mn and αSynAgg as indicated. Data are means ± SEM pooled from 4 independent experiments. (C) qRT-PCR analysis of NLRP3, NLRC4, and AIM2 mRNA expression in the striata of C57BL mice exposed to Mn for 30 days. Data are means ± SEM pooled from 5 mice from 3 experiments. (D) Western blot analysis of Caspase 1 cleavage and IL-1β maturation in lysates from striatum samples from mice treated as indicated. Blots (upper) are representative of 6 mice from 3 experiments. Normalized band intensity data (lower) are means ± SEM from all experiments. (E) Immunofluorescence microscopy analysis of NLRP3 abundance in IBA1-positive microglial cells in the striatal region of mice treated as indicated. Images are representative of 3 mice from 3 experiments. Scale bar, 15 μm. *P<0.05 and ***P<0.001 by ANOVA with Tukey post analysis.
We verified that Mn can promote NLRP3 inflammasome-mediated inflammation in an animal model of manganese neurotoxicity. Mn exposure induces a neurotoxic effect (43) and production of factors like TNF-α from neurons (44), which can potentially act as signal 1. These stress signals can activate microglial cells. Hence, in an in vivo scenario, Mn may act as both signal 1 and signal 2. When C57BL mice were orally exposed to Mn for 30 days, we found that Mn specifically induced NLRP3 mRNA expression, but not AIM2 and NLRC4, as revealed by qRT-PCR (Fig. 2C). Western blot analysis showed that Mn significantly increased caspase-1 cleavage and mature IL-1β (Fig. 2D), although we did not observe increased abundance of pro-IL-1β. The Mn-induced increase in cleaved caspase-1 was strongly correlated with an increase in the amount of released IL-1β (fig. S4). IHC studies further revealed that this NLRP3 increase occurred in IBA1 (microglia marker)-positive cells (Fig. 2E). Collectively, all these findings showed that Mn activated the NLRP3 inflammasome in microglial cells both in vitro and in vivo. This NLRP3 signaling cascade could be the putative mechanism by which chronic inflammation is sustained in Mn-induced Parkinsonism.
Mn leads to mitochondrial dysfunction in primed microglial cells
Although the mechanisms of inflammasome activation are currently unclear, mitochondrial dysfunction and mitochondrial superoxide generation may stimulate inflammasome activation (17, 18, 45–47). Because Mn severely impairs mitochondria under stress conditions (48–51), we tested the effects of Mn on mitochondrial bioenergetics of LPS-primed microglia. Primary microglial cells were primed with LPS for 3 h followed by 24 h of treatment with Mn before analysis of oxygen consumption and extracallular acidificaiton on a Seahorse Bioscience extracellular flux analyzer. Exposing LPS-primed microglia to Mn significantly impaired mitochondrial function (Fig. 3A), as indicated by the decreased mitochondrial ATP production (Fig. 3B), basal respiration rate (Fig. 3C), maximal respiration (Fig. 3D), and spare respiratory capacity (Fig. 3E) of the cells after exposure to Mn. Mn exposure in unprimed microglial cells showed no significant changes in mitochondrial dynamics. This is consistent with no increase in oxidative stress in N9 microglia after a high-concentration Mn exposure (29). Exposure of LPS-primed primary microglia to Mn significantly decreased the stressed oxygen consumption rate (OCR) without changing the stressed extracellular acidification rate (ECAR), as measured by the cell energy phenotype test. These data suggest that Mn exposure reduced the mitochondrial metabolism rate without modulating non-mitochondrial function (fig. S5).
Fig. 3. Mn treatment induced mitochondrial damage by modulating Mfn2 and Mul1 expression in microglial cells.
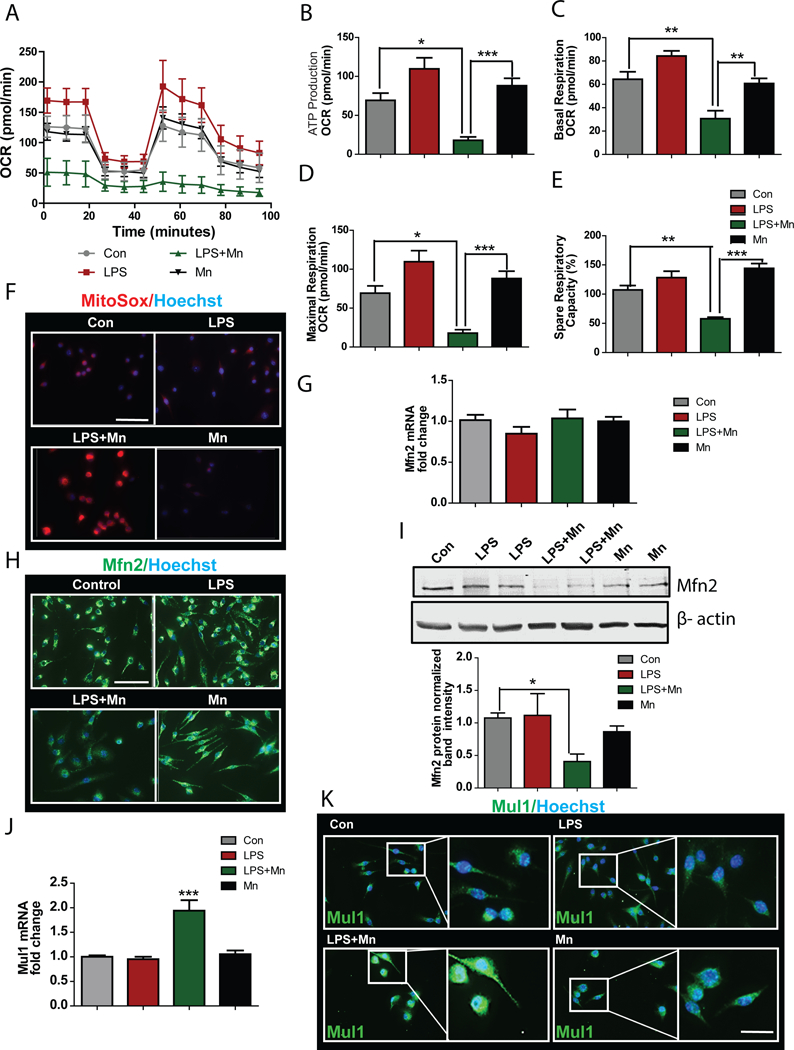
(A to E) Seahorse analysis of the oxygen consumption rate in Mn-exposed LPS-primed primary microglial cells. Data were analyzed to extrapolate mitochondrial ATP production (B), basal respiration rate (C), maximal respiration (D), and spare respiratory capacity (E). Data are means ± SEM pooled from 5 independent experiments (F) Immunofluorescence microscopy analysis of mitochondrial superoxide generation from microglial cells primed with LPS and exposed to Mn. Images are representative of 3 independent experiments. Scale bar, 100 μm. (G) qRT-PCR analysis of Mfn2 mRNA expression in Mn exposed microglial cells. Data are means ± SEM pooled from 3 independent experiments. (H and I) Immunofluorescence microscopy (H) and western blot analysis (I) of Mfn2 abundance in microglial cells primed with LPS and exposed to Mn. Images and blots are representative of 4 experiments. Normalized band intensity data (I, lower) are means ± SEM from all experiments. Scale bar, 100 μm. (J) qRT-PCR analysis of Mul1 mRNA expression in Mn-exposed microglial cells. Data are means ± SEM pooled from 3 independent experiments. (K) Immunofluorescence microscopy analysis measuring Mul1 protein on Mn exposure. Images are representative of 3 independent experiments. Scale bar, 20 μm. *P<0.05 and ***P<0.001 by ANOVA with Tukey post analysis.
Mitochondrial dysfunction can lead to superoxide generation. When we assayed ROS activity by MitoSox staining, we found that LPS-primed microglial cells treated with Mn for 24 h generated significant amounts of superoxide (Fig. 3F), as expected. These data futher confirmed the effects of Mn treatment on mitochondrial dysfunction in LPS-primed microglia. Because LPS-primed cells and unprimed microglial cells exposed to Mn both exhibited minimal or no changes in mitochondrial superoxide production (Fig. 3F), we hypothesized that LPS may increase Mn transporters in microglia. Indeed, qRT-PCR analysis revealed that LPS significantly increased the Mn importer SLC11A2 (fig. S6A), but Mn exposure alone stimulated expression of the Mn exporter SLC30A10 (fig. S6B) without changing importer expression. Mn uptake was increased in LPS-primed, Mn-treated microglial cells when compared to unprimed Mn-treated cells, as determined by ICP-MS analysis (fig. S6C). These data suggest that LPS priming increased the uptake of Mn and Mn transporter expression. Further studies in transporter knockdown cells are required to understand the mechanism of Mn uptake in LPS-treated microglia.
Mfn2 protein promotes mitochondrial quality control by eliminating damaged mitochondria by mitochondrial fusion (52–54). Although our qRT-PCR analysis of LPS-primed microglial cells showed no significant change in Mfn2 expression (Fig. 3G), our immunofluorescence (Fig. 3H) and Western blot (Fig. 3I) analyses showed that Mfn2 abundance was decreased in primed microglial cells treated with Mn. These data suggest that the Mn-induced mitochondrial dysfunction in primed microglia may be due to Mfn2 degradation. One candidate for ubiquitinating Mfn2 is mitochondrial ubiquitin ligase 1(Mul1). In fact, the mRNA expression of Mul1 in Mn-treated, LPS-primed cells doubled when compared to other groups (Fig. 3J). Furthermore, the increased expression of Mul1 was Mn dose-dependent (fig. S7). ICC analysis also indicated an increase in the Mul1 protein abundance in Mn-treated, LPS-primed cells (Fig. 3K). Collectively, these data suggest that Mn-induced inflammasome activation in microglia was related to mitochondrial dysfunction and that may be promoted by Mul1-mediated degradation of Mfn2.
Retromer complex proteins are involved in Mn-induced mitochondrial dysfunction in LPS-primed microglia
Mutating or reducing the abundance of the retromer complex protein VPS35 (Park 17) induces mitochondrial dysfunction by increasing Mul1 in dopaminergic neurons (55). Because we observed that Mn exposure increased the amount of Mul1 expressed in LPS-primed microglia, we further investigated whether this increased Mul1 might be due to decreased expression of VPS35. We treated an LPS-primed microglial cell line with Mn for 24 h in preparation for qRT-PCR, ICC, and immunoblot analyses. Our qRT-PCR analysis revealed that Mn exposure reduced VPS35 mRNA expression in LPS-primed microglial cells when compared to other groups (Fig. 4A). Expression of another retromer complex protein, VPS29, was also decreased in primed microglial cells treated with Mn (Fig. 4B). Furthermore, ICC (Fig. 4C) and Western blot (Fig. 4D) analyses confirmed that VPS35 is diminished in Mn-exposed, LPS-primed microglial cells. We further validated our cell culture findings in an animal model of Mn toxicity. Mice were similary treated with Mn as described above. Striatal VPS35 mRNA expression decreased in Mn-gavaged mice (Fig. 4E).
Fig. 4. VPS35, a retromer complex protein, can modulate Mfn2 degradation during Mn-induced inflammasome activation.
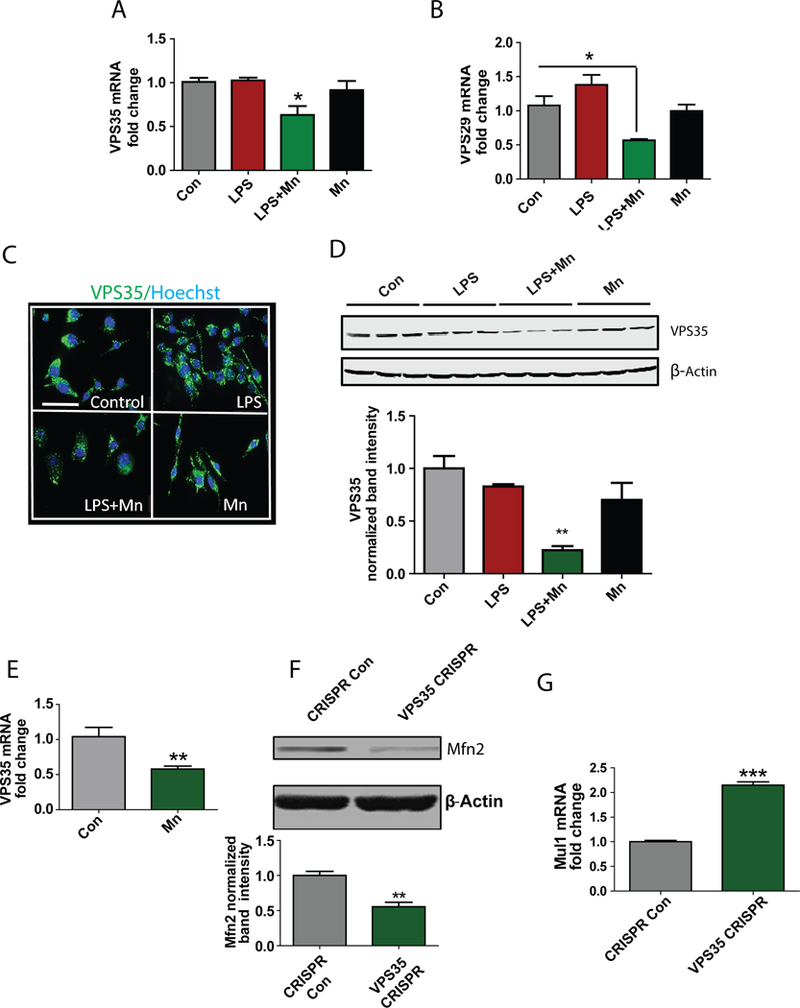
(A and B) qRT-PCR analysis of the retromer complex components VPS35 and VPS29 mRNA expression. Data are means ± SEM pooled from 3 independent experiments. (C and D) Immunofluorescence microscopy (C) and western blot (D) analysis of VPS35 abundance in microglial cells exposed to Mn. Images and blots are representative of 3 independent experiments. Normalized band intensity data (D, lower) are means ± SEM from all experiments. Scale bar, 20 μm. (E) qRT-PCR analysis of VPS35 mRNA expression in the striatum of mice exposed to Mn. Data are means ± SEM pooled from 6 mice from 3 independent experiments. (F) Western blot analysis of Mfn2 abundance in lysates from WT and VPS35 CRISPR-cas9 KD microglial cells. Blots are representative of 4 independent experiments. Normalized band intensity data (D, lower) are means ± SEM from all experiments. (G) qRT-PCR analysis of Mul1 mRNA expression in VPS35 KD microglial cells compared to wild-type cells. Data are means ± SEM pooled from 3 independent experiments. *P<0.05, **P <0.01, ***P<0.001 by ANOVA with Tukey post analysis.
To further elucidate the role of VPS35 in maintaining microglial mitochondria, we developed a stable VPS35 knockdown (KD) microglial cell line using the CRISPR-Cas9 system (fig. S8A). Our Western blot analysis showed VPS35 KD cells have less Mfn2 (Fig. 4F). Mn treatment enhanced the loss of Mfn2 in VPS35 KD cells (fig. S8B). Moreover, qRT-PCR revealed that Mul1 mRNA was significantly elevated in the VPS35 KD cells (Fig. 4G). These data collectively suggest that VPS35 is upstream of Mfn2 and the Mul1 pathway and played a role in maintaining mitochondrial integrity.
Exosomes mediate Mn-induced cell-to-cell transmission of the inflammasome component ASC in LPS-primed microglia
A major inflammasome adaptor component, ASC propagates inflammasome activation through cell-to-cell transmission (19). We hypothesized that ASC may migrate from one cell to another through exosomes and that Mn may promote inflammasome propagation by increasing ASC loading in exosomes. To determine whether exosomal uptake plays a role in inflammasome propagation, we knocked down endocytosis mediators, caveolin and clathrin, that are responsible for distinct pathways of exosomal uptake in microglial cells using the CRISPR-Cas9 system. Control, Cav1-KD, and Cltc-KD microglial cells were treated with Mn for 24 h following LPS priming. Reduced caveolin expression inhibited Mn-stimulated expression of pro-IL-1β (Fig. 5A) and NLRP3 (Fig. 5B) mRNA in LPS-primed cells. The expression of nos2 mRNA was reduced when both caveolin and clathrin abundance was limited (fig. S9A). Furthermore, Luminex assay revealed that Mn-exposed, LPS-primed Cav1-KD and Cltc-KD cells produced significantly less mature IL-1β (Fig. 5C), without affecting TNF-α abundance (fig. S9B). These data suggest that endocytosis played a role in inflammasome activation and that limiting exosomal uptake reduced Mn-induced inflammasome propagation.
Fig. 5. Exosomal release of the inflammasome component ASC propagates Mn-induced inflammasome activation.
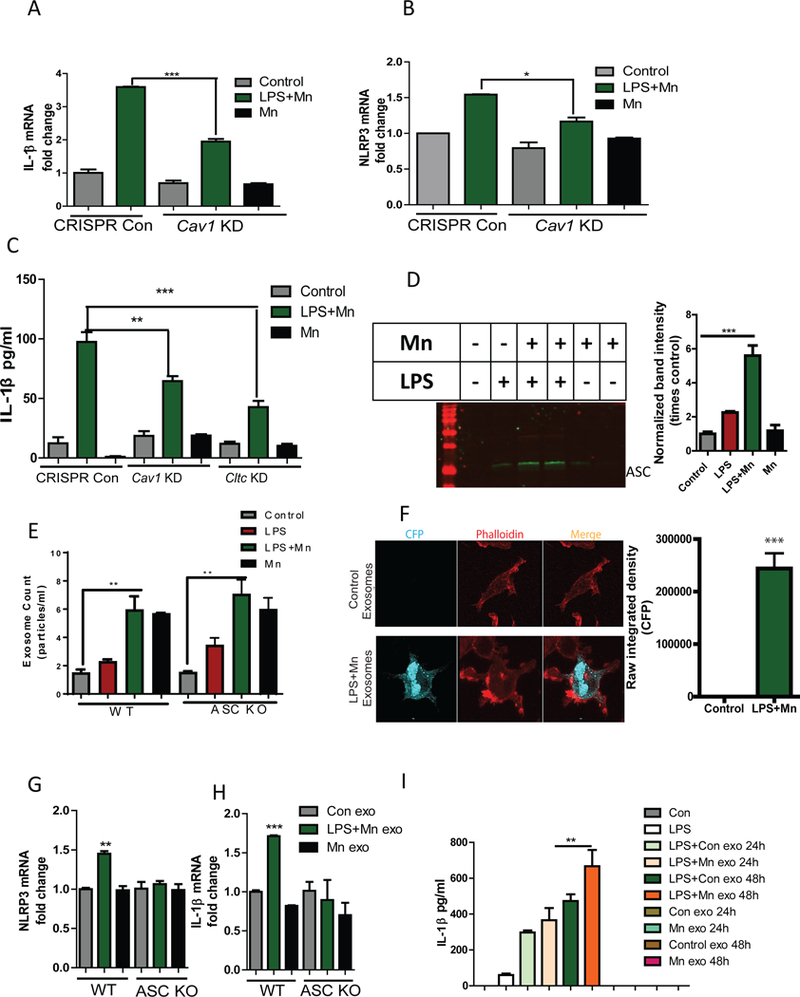
(A and B) qRT-PCR analysis of pro-IL-1β (A) and NLRP3 (B) mRNA expression in WT and Cav1-KD microglial cells after Mn-treatment. Data are means ± SEM pooled from 3 independent experiments. (C) Luminex analysis of IL-1β secreted by WT and Cav1-KD or Cltc-KD microglial cells after Mn-treatment. Data are mean ± SEM pooled from 8 independent experiments. (D) Western blot analysis of ASC abundance in lysates of exosomes isolated from Mn-treated microglial cells treated as indicated. The blot (left) is representative of 3 independent experiments. Normalized band intensity data (right) are means ± SEM from all experiments. (E) NanoSight analysis of the number of exosomes released from wild-type and ASC KO microglial cells. Data are mean ± SEM from 3 independent experiments. (F) Immunofluorescence microscopy analysis of CFP expression in wild-type microglial cells after treatment with exosomes isolated from cells overexpressing ASC-CFP. Images (left) are representative of 3 independent experiments. Quantified data (right) are means ± SEM from all experiments. Scale bar, 20 μm. (G and H) qRT-PCR analysis of NLRP3 (G) and pro-IL-1β (H) mRNA expression in microglial cells after treatment with exosomes isolated from wild-type and ASC KO microglial cells. Data are means ± SEM pooled from 3 independent experiments. (I) Luminex analysis of IL-1β secreted by microglial cells treated with exosomes isolated from the serum of Mn-exposed animals. Data are means ± SEM pooled from 8 independent experiments. *P<0.05, **P <0.01, ***P<0.001 by ANOVA with Tukey post analysis.
To further identify the importance of exosome cargo for inflammasome propagation, exosomes were isolated from treatment media of microglial cells treated with Mn for 24 h. Immunoblot analysis showed that Mn exposure enhanced exosomal release of ASC from LPS-primed microglial cells (Fig. 5D), but exposure to LPS or Mn alone had only minimal effects. Furthermore, Mn treatment significantly enhanced the number of exosomes released from primed microglial cells (Fig. 5E) without altering exosome size (fig. S10). When we exposed ASC-KO cells to Mn, we found no decrease in exosome number (Fig. 5E). These data indicated that the increase in exosome number was independent of inflammasome activation. To test whether exosome transfer ASC between cells, exosomes were collected from the medium of ASC-CFP-overexpressing microglial cells untreated or treated with Mn for 24 h after LPS-priming. When wild-type microglial cells were exposed to an equal number of exosomes for 24 h, confocal microscopy revealed that exosomes from Mn-treated, LPS-primed cells transferred ASC-CFP, whereas exosomes from untreated cells did not (Fig. 5F). These data confirmed that ASC migrated from cell-to-cell through exosomes and that Mn increased the exosomal ASC content. When we exposed cells to exosomes from LPS-primed Mn-treated microglial cells, qRT-PCR analysis revealed significantly increased NLRP3 (Fig. 5G) and pro-IL-1β (Fig. 5H) mRNA expression relative to exposure to exosomes from untreated microglial cells. This increase in NLRP3 and pro-IL-1β mRNA was not seen when wild-type microglial cells were treated with exosomes isolated from Mn-treated, LPS-primed ASC KO cell line (Fig. 5G-H), further validating that exosomal transfer of ASC is involved in propagation of inflammasome activation. Also, exosomes isolated from serum of Mn-gavaged animals significantly elevated the release of IL-1β when administered to LPS-primed primary microglial cells for 24 and 48 h (Fig. 5I), however no significant change in IL-6 or TNF-α release was observed (fig. S11, A-B). Collectively, these findings suggest that Mn induced cell-to-cell transmission of ASC through exosomes, further propagating inflammasome activation.
Serum exosomes and serum from welder population have higher load of ASC and higher pro-inflammatory cytokines compared to age-matched controls
Welders exposed to Mn exhibit a dose-dependent progression in Parkinsonian syndrome (56). To demonstrate the human relevance of our findings from cell culture and animal models of Mn exposure, we utilized serum exosomes collected from welders. When we counted serum exosomes and normalized our Western blot analysis to the number of exosomes isolated from an equal volume of serum, we found that welders’ serum exosomes had significantly more ASC than age-matched controls (Fig. 6B). These data further corroborate our in vitro and in vivo data, suggesting that Mn exposure increases exosomal release of ASC. We analyzed the amount of Mn in whole blood from control and welder samples, but we did not observe any significant differences (fig. S12). Most of the welders used in the study were exposed to low amounts of Mn, and their blood Mn concentration measured at a single time point may not be an accurate measure of toxicity for low-grade exposures. A better measure of chronic weak Mn exposure can be achieved by taking MRI-T1 weighted brain scans (57).
Fig. 6. Serum exosomes and serum from welder population have higher load of ASC and higher pro-inflammatory cytokines, respectively, when compared to age-matched controls.
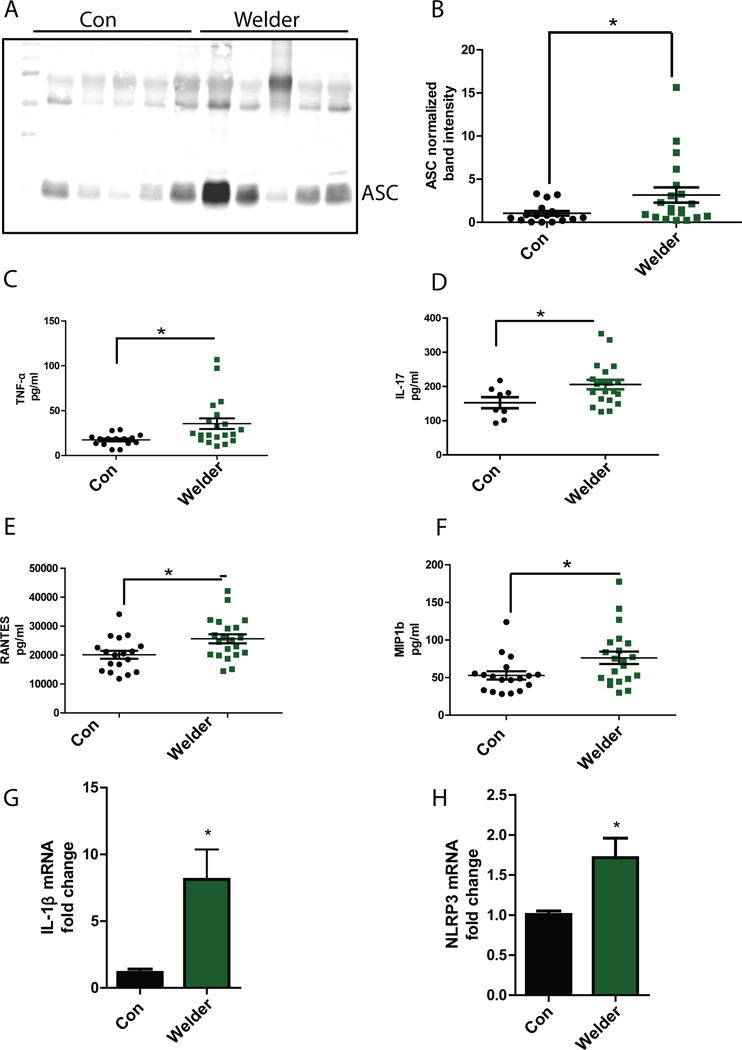
(A and B) Western blot analysis of exosomal ASC load in welder population and age-matched controls. Blot (A) is representative of 17 controls and 20 welder samples. Normalized band intensity data (B) are means ± SEM from all experiments. (C to F) Luminex analysis of TNF-α (C), IL-17 (D), Rantes (E), and MIP-1b (F) abundance in control and welder serum. Data are means ± SEM from 17 Control and 20 welder samples. (G and H) qRT-PCR analysis of pro-IL-1β (G) and NLRP3 (H) mRNA expression in microglial cells treated with exosomes isolated from welders or age-matched controls. Data are means ± SEM from 5 welders and 4 age-matched controls. *P<0.05 by Student-t test.
We also analyzed welder serum for cytokine and chemokine profiling (Fig. 5C-F and Supplementary Fig. 12). We found that the serum pro-inflammatory cytokines TNF-α (Fig. 6C) and IL-17(Fig. 6D), and the chemokines RANTES (Fig. 6E) and MIP1b (Fig. 6F) were significantly increased in welders when compared to age-matched controls. However, the small amount of IL-1β (fig. S13A) detected in the serum of welders and control subjects may be attributed to the short half-life of IL-1β (58). To verify whether the exosomes isolated from welder serum can induce NLRP3 expression, we treated microglial cells with serum exosomes from 4 welders and 5 age-matched controls for 24 h. Our qRT-PCR analysis revealed that the cells treated with welder exosomes significantly increased expression of pro-IL-1β (Fig. 6G) and NLRP3 (Fig. 6H) when compared to exposure to serum exosomes from healthy donors. Together, these data suggested that exosomal ASC was increased in human populations occupationally exposed to Mn and that environmental exposure to Mn induced an inflammatory response. Moreover, exosomes from welders stimulated NLRP3 inflammasome signaling.
Discussion
A diverse variety of signals including ATP (59), various crystals (60, 61), mitochondrial inhibitors (17, 62), and misfolded proteins (15, 16) stimulate NLRP3 inflammasome activation. Inflammasome activation is linked to various neurodegenerative disorders including PD, AD, and TBI (14). In the present study, we showed that the divalent metal Mn promoted NLRP3 inflammasome activation in microglial cells in vitro and in vivo. Furthermore, this inflammasome activation in microglial cells was associated with mitochondrial dysfunction and correlated with Mfn2 degradation. We also showed that Mn-stimulated exosomes that contained the inflammasome component ASC propagated NLRP3 inflammasome activation. Finally, we demonstrated that occupational exposure to Mn in welders increased serum exosomal ASC load and circulating pro-inflammatory cytokine abundance when compared to age-matched controls.
The link between Mn and neurodegenerative and neuroimmune processes is becoming clearer. Chronic Mn exposure damages the nigrostriatal region (38, 43, 63–67) and induces microglial activation in the SNpc region in a primate model (68) and astroglial inflammation (38). Mn exposure also promotes the pro-inflammatory cytokine produced by glial cells (69–71) and increases the NFκB and p-38 MAPK pathway signaling in glial cells (37, 70). Although the mechanism underlying sustained and chronic inflammation in Mn-induced neurotoxicity is not well understood, inflammasome activation promotes inflammatory responses in various disease models (13). Exposure to silica and asbestos also stimulates NLRP3 inflammasome activation in various cell types including lymphocytes and macrophages, among others (61). Furthermore, NLRP3 inflammasome activation is linked to Parkinsonian syndrome (16, 72). Here, using cell culture and animal models of Mn toxicity, we showed that Mn exposure activated the NLRP3 inflammasome specifically in microglial cells.
Though a variety of stimuli has been shown to act as signal 2 in activating the NLRP3 inflammasome cascade, the exact mechanism underlying its assembly is not well understood. Various factors, including mitochondrial dysfunction (17), mitochondrial superoxide generation (18), and lysosomal dysfunction (73), can promote NLRP3 inflammasome assembly and activation (44). Mn causes mitochondrial dysfunction by accumulating in mitochondria (26, 34, 51, 74) in neurons and astrocytes. In this study, we showed that Mn exposure lead to mitochondrial dysfunction and mitochondrial superoxide generation in microglial cells. Mfn2 plays an important role in mitochondrial fusion and is one of the targets of the PD-associated gene PARKIN (75, 76). Manipulating Mfn2 expression alters mitochondrial dynamics (54, 55, 77), and Mfn2 plays a role in Mn-induced neurotoxicity in neuronal cells (78). In this study, we demonstrated that Mn exposure lead to decreased expression of Mfn2 in microglial cells, which was accompanied by mitochondrial dysfunction and inflammasome activation.
Most studies with VPS35 focus on neurons, but largely ignore the role of this retromer complex protein in microglia. VPS35 mutation or reduced expression can increase mitochondrial dysfunction either by promoting Mfn2 degradation in dopaminergic neurons (55) or by recycling the dynamin-like protein 1 (DLP1) complex (79). Here we showed that Mn exposure reduced VPS35 in LPS-primed microglial cells and that VPS35 increased Mfn2 degradation that correlated with Mul1 expression. Although exactly how Mn treatment decreases VPS35 abundance is not clear, another retromer complex protein VPS29 contains Mn binding sites and VPS35 wraps around these metal binding sites (80, 81). Thus Mn-induced reduction in retromer complex proteins may be due to excessive binding of Mn to these metal binding sites, but more studies are required to confirm the exact mechanism. Additionally, VPS35 modulates the cellular biogenesis and release of exosomes (82, 83). Retromer pathway dysfunction also promotes amyloid precursor protein processing (84).
Release of exosomes, the so-called “garbage bag” of cells, can promote the progression of neurodegenerative disorders including AD and PD. Exosomal export of aggregated α-Syn to other cells (85, 86) acts as a key signal between cells. Intercellular communication through exosome cargo can also modulate neuroinflammation (87). NLRP3 inflammasome propagation has become an active area of research. A novel pyroptotic-phagocytic cascade has been shown to spread ASC and the NLRP3 inflammasome assembly between cells, in a “prionoid” fashion, thus propagating the inflammasome (19). In this study, we demonstrated that ASC loaded in exosomes can stimulate inflammasome activation in neighboring cells. Mn exposure induced the exosomal release of ASC from LPS-primed microglial cells, whereas blocking exosomal uptake reduced IL-1β release (Fig 5). This finding may have broad implications as exosomes can cross the blood-brain-barrier and can communicate to various organs.
Mn can induce Parkinsonism in humans (20, 56, 63, 64, 88). Due to their exposure to welding-derived metal nanoparticles, Swedish welders have an increased inflammatory profile (89). Our data suggest a probable mechanism for inflammation stemming from occupational exposure to Mn. Though we were not able to detect significant amounts of IL-1β in welder serum, we did show an increased ASC load in welder serum exosomes. The lack of elevated IL-1β may be attributed to its short half-life (Fig. 6) (58, 90). Moreover, we also showed that exosomes isolated from welder serum stimulated the expression of NLRP3 and pro-IL1β, further validating the role of exosomes in modulating inflammasome propagation. Our findings provide some insight into the mechanism of communication between peripheral inflammation and CNS inflammation in neurodegenerative disorders, including AD and Parkinsonian syndrome. Future studies should explore the role of exosomal biogenesis during inflammation in different disease models including Parkinson’s disease. Overall, our study describes how Mn dysregulates VPS35-Mul1-Mfn signaling to activate and propagate the inflammasome (Fig. 7).
Fig. 7. Mn may induce mitochondrial dysfunction leading to inflammasome activation by degradation of Mfn2.
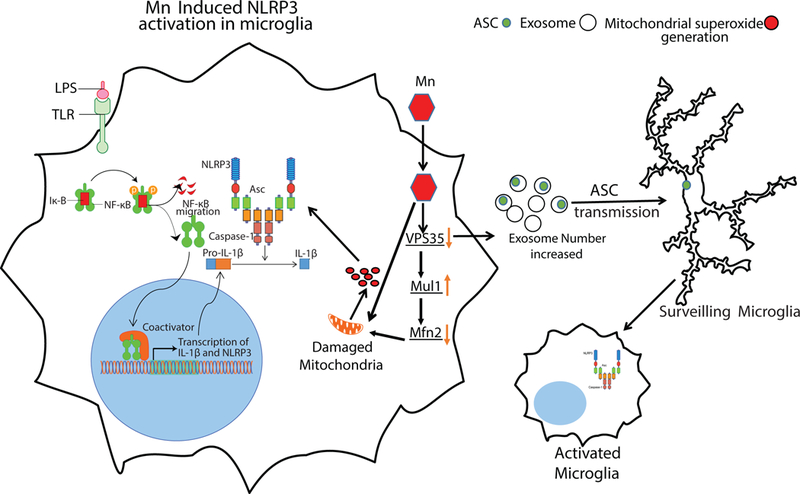
Mn may also stimulate the release of exosomal ASC, thereby leading to NLRP3 inflammasome propagation.
Materials and Methods
Cell culture, Primary culture and Treatments
Primary microglial cells were isolated using a magnetic bead separation technique described in our previous publications (91, 92). Primary microglial cells were cultured in DMEM-F12, 10% FBS, 1% sodium pyruvate, 1% glutamine, 1% penicillin-streptavidin, and 1% non-essential amino acids. The wild-type microglial cell line and ASC-CFP microglial cell line were kind donations from Dr. D.T. Golenbock (University of Massachusetts Medical School, Worcester, MA). ASC KO macrophage cell line were kind donations from Dr. K. Fitzgerald (University of Massachusetts). The wild-type microglial cell line was characterized by Halle et al. (15), and our group (93), and cultured in DMEM medium, 10% FBS, 1% glutamine, and 1% penicillin-streptavidin. Treatments were done in 2% FBS-containing medium. For LPS-priming treatments, cells were treated with LPS (100 ng/mL) for 3 h. Next, the cells were triple-washed with full serum medium to remove any excess LPS, and then treated with 100 μM Mn for 6–24 h unless otherwise stated. For exosome treatment, cells were treated with an equal number of exosomes from welders and age-matched controls for 24 h following which qRT-pCR analysis was performed.
CRISPR-Cas9 knockdown (KD) cell generation
CRISPR guide RNA was obtained for VPS35, Cav1 and Cltc from Sigma and transfected using protocols from our previous publications (9, 94). Following transfection, cells were incubated for 48 h before treatment. The lentivirus-based CRISPR/Cas9 KO plasmid against gene Vps35, with target site CAAGTCATTTCCTCAATCCAGG, in a U6gRNA-Cas9–2A-RFP vector, was purchased from Sigma-Aldrich. The lentivirus-based CRISPR/Cas9 plasmids, pLV-U6g-EPCG-Cav1 and pLV-U6g-EPCG-Cltc with the Cav1 and Cltc gRNA target sequences GTTGAGATGCTTGGGGTCGCGG and TACTGAAGCCAATGTTTGCTGG, respectively, were purchased from Sigma-Aldrich. To make the lentivirus, the lenti-CRISPR/Cas9 Vps35 KO plasmid and nontarget control plasmid were transfected into 293FT cells using the Mission Lentiviral Packaging Mix from Sigma-Aldrich according to manufacturer’s instructions. The lentivirus was harvested 48 h post-transfection and added to the microglial cell line at an MOI of 100 to knockdown VPS35, Caveolin-1, and Clathrin expression.
Animal study
Eight-week-old male C57BL/6Ncrl mice, obtained from Charles River, were housed under standard conditions of constant temperature (22 ± 1°C), humidity (relative, 30%), and a 12 h light/dark cycle. After acclimating for 3 days, mice were gavaged with 15 mg/kg body weight Mn for 30 days, and then sacrificed. This Mn dose regimen was chosen based on previous studies in humans and animals (95–97). Use of the animals and protocol procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Iowa State University (Ames, IA, USA).
Western blot
Western Blot analyses were performed following our previous publications (94, 98). Briefly, cells or tissues were lysed using modified RIPA buffer. Proteins were normalized using Bradford assay before loading on SDS-acrylamide gels. For separation of proteins, 20–40 μg of protein was loaded in each well of 10–15% acrylamide gels. Acrylamide gels were run at 110 V for 2 h at 4°C. Following separation, proteins were transferred on a nitrocellulose membrane at 27 V for 18 h at 4°C. After transfer, the membranes were blocked with LI-COR blocking buffer for 45 min and incubated in primary antibodies following manufacturer’s protocol. Following primary antibody incubation, membranes were washed with PBS-TWEEN 20 (0.05%) for 1 h and incubated in LI-COR IR secondary antibodies for 1 h at room temperature, washed with PBS-TWEEN for 1 h, and imaged using an Odyssey scanner. The primary antibodies used are as follows: anti-caspase-1 (Adipogen, 1:1000) (AB_2490248), anti-NLRP3 (Adipogen, 1:1000), anti-iNOS (Santa Cruz, 1:1000), anti-IL-1β (R&D systems, 1:500), anti-Mfn2 (Cell Signaling, 1:1000), anti-VPS35 (Santa Cruz, 1:500), and anti-ASC (Adipogen, 1:1000) (AB_2490440). The secondary antibodies used are as follows: IR-800 conjugated goat anti-mouse IgG (LI-COR, 1:20000) and IR-700 conjugated goat anti-rabbit IgG (LI-COR 1:20000). For loading control, β-actin (Sigma, 1:10000) antibody was used.
Human Samples
In central Pennsylvania, USA, and local communities, eighty-one subjects were recruited. Controls are those who have no history of welding, whereas welders were defined as persons who have welded at any point in their lifetime. All subjects were male. Written informed consent was obtained in accordance with guidelines approved by the Penn State Hershey Internal Review Board. Controls were age-matched volunteers from the same regional community with various occupations. Prior to sending the serum samples to Iowa State, samples were blinded. This serum was used to isolate exosomes as described. Out of the exosome samples, 39 samples were chosen randomly for further Western blot, qRT-PCR and Luminex analysis.
qRT-PCR
RNA was isolated from tissues and cells according to a previous publication (99, 100). In short, cells or tissues were lysed in 1 mL of TRIzol reagent and incubated for 5 min. After incubation, 0.2 mL of chloroform was added to each tube, incubated for 2 min and centrifuged at 12,000 x g for 15 min at 4°C. After centrifugation, the top clear layer containing RNA was transferred to a fresh tube containing 0.7 mL of isopropanol, incubated for 15 min and centrifuged at 12,000 x g for 10 min at 4°C to precipitate the RNA. After precipitation, the supernatant was discarded, the pellet was washed with 75% ethanol, air-dried, and dissolved in water. A NanoDrop was used to quantify the RNA and 1 μg of RNA was utilized for converting into cDNA. For cDNA synthesis, the high capacity cDNA synthesis kit from Applied Biosystems (Cat#4368814) was used according to manufacturer’s protocol. Quantitative SYBR green PCR assay was performed using qRT2PCR SYBR Green Mastermix (Agilent) and pre-validated primers. The following validated primers from Qiagen were used: pro-IL-1β (QT01048355), NLRP3 (QT00122458), NLRC4 (QT00264670), AIM2 (QT00266819 ), Nos2 (QT00100275), Mfn2 (QT00134295 ), Mul1 (QT00132734), SLC30A10 (QT01199009), SLC11A2 (QT01047368), VPS35 (QT00160258), VPS29 (QT00137228), and 18S (QT02448075). The fold change in gene expression was determined by ΔΔCt method where Ct is the threshold value. 18S was used as the housekeeping gene.
Immunohistochemistry and Immunocytochemistry
Immunohistochemistry (IHC) was performed on striatal sections as described in our previous publications (6, 8, 101). Striatal sections (30 μm) were used for IHC. Citrate buffer was used to perform antigen retrieval (10 mM sodium citrate, pH 8.5). Following antigen retrieval, sections were washed with PBS, blocked with blocking buffer (2% BSA, 0.5% Triton 100X, and 0.05% TWEEN 20), and incubated in primary antibodies overnight at 4°C. Next, sections were washed with PBS, incubated in secondary antibodies for 1 h and stained with the nuclear dye Hoechst. Finally, sections were mounted on pre-coated slides and dried overnight before visualizing them under a microscope. Confocal imaging was performed on these sections at the Iowa State University Microscopy Facility using a Leica DMEIR2 confocal microscope with 63X oil objective. For z-stacking, each section consisted of 10–15, 0.5-μm slices.
For immunocytochemical (ICC) studies on microglial cells and microglial primary culture, 4% paraformaldehyde (PFA) was used to fix the cells. Next, fixed cells were washed with PBS, blocked using blocking buffer, and incubated in primary antibodies following manufacturer’s protocol. Following primary antibody incubation, cells were washed with PBS, incubated in secondary antibody and mounted on slides using Fluoromount aqueous mounting medium (Sigma). Samples were visualized using an inverted fluorescence microscope (Nikon TE-2000U). The following primary antibodies were used: IBA1 (Wako, 1:1000) (AB_2314667), IBA1 (Abcam, 1:500) (AB_870576), Mul1 (Abcam, 1:500) (AB_1860863), NLRP3 (Adipogen, 1:500) (AB_2490202), ASC (Adipogen, 1:500), Nos2 (Santa Cruz, 1:500) (AB_2152867), VPS35 (Santa Cruz, 1:500) (AB_2215220), and Mfn2 (Cell Signaling, 1:500). Alexa dye-conjugated secondary antibodies were used for ICC and IHC experiments.
Multiplex cytokine assay
Multiple cytokine assay was performed according to our previous publication (94). Briefly, 40 μL of treatment media was incubated overnight with 40 μL of primary antibodies conjugated with magnetic beads. Following incubation with primary antibodies, samples were washed, incubated with a biotinylated antibody and biotin/streptavidin, and then read in a Bio-Plex plate reader. Standards were obtained from PeproTech. For welder serum cytokine analysis, Bio-Plex Pro Human Cytokine 27-plex Assay (M500KCAF0Y, Bio-Rad) was performed according to manufacturer’s protocol.
MTS cell viability assay
Cells were plated in 96-well tissue plates. Following treatment, 10 μL of MTS reagent (Promega) was added and incubated at 37°C for 1.5 h. After incubation, a plate reader was used to quantify the absorbance at 490 nm.
Griess assay
Griess assay was performed as previously described in our publication (94). 50,000–100,000 microglial cells were plated in 96-well plates and treated in 2% FBS containing medium. After treatment, 50 μL of medium was collected, and incubated with 50 μL of Griess reagent for 10 min. A plate reader was used to read the absorbance at 540 nm. Sodium nitrite solution was used for making standard curve.
Exosome isolation
Wild type microglial cell line-produced exosomes were isolated by filtering the cell-culture supernatant through 0.22-μm filters, followed by high-speed ultracentrifugation. Briefly, cells were grown in T-175 cell culture flasks and treated with 100 μM Mn. For each exosome sample, conditioned media from two T175 flasks were pooled with an original seeding density of 8×106 cells per flask. Post treatment, the media was collected and centrifuged at 300 x g for 10 min, and 10,000 x g for 15 min successively to get rid of any cell debris before passing it through a 0.22-μm syringe filter. The filtrate was then centrifuged at 100,000 x g for 90 min using a Beckman Optima L-100 XP ultracentrifuge. The pellet containing exosomes was washed once with cold PBS and centrifuged again at 100,000 x g for 90 min using a Beckman optima MAX ultracentrifuge.
Inductively coupled plasma mass spectroscopy (ICP-MS)
Microglial cell line samples were carefully scraped and counted prior to sample preparation. Samples were analyzed for different metals including, Cd, Ca, Cr, Co, Cu, Fe, Mg, Mn, Mo, P, K, Se, Na, and Zn using ICP-MS (Analytik Jena Inc. Woburn, MA, USA) in CRI mode with hydrogen as the skimmer gas. Standards for elemental analyses were obtained from Inorganic Ventures (Christiansburg, VA) and digestion vessels, trace mineral grade nitric acid and hydrochloric acid were obtained from Fisher Scientific (Pittsburgh, PA). Briefly, samples were digested in 70% nitric acid at 60˚C for ≥12 hours. Pre-weighed samples were transferred to 15 mL tubes and 0.25 mL of 70% nitric acid was added. All samples were digested overnight at 60˚C. After digestion, all samples were diluted to 5 mL using 1% nitric acid with 0.5% hydrochloric acid and then analyzed by ICP-MS. For quality control, Bi, Sc, In, Li, Y, and Tb were used as internal standards for the ICP-MS.
NanoSight
Ultracentrifuged exosome samples were used for nanoparticle tracking analysis (NTA), as previously described (102). Briefly, isolated exosomes were resuspended in 500–1000 μL of PBS, from which approximately 300 μL was loaded into the sample chamber of an LM10 unit (NanoSight, Amesbury, UK) using a disposable syringe. Sample durations of 30–60 sec per sample were analyzed with NTA 2.3 software (NanoSight). Samples containing higher numbers of exosomes were diluted before the analysis and their relative concentrations were then calculated according to the dilution factor. The size distribution of exosomes (fig. S10) was also plotted using this method.
Mitochondrial dynamics analysis
A Seahorse XFe24 Analyzer was used to measure mitochondrial oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) using the Mito-Stress test following a previously published protocol (44). Primary mouse microglia were plated 90,000 cells/well of a 24-well, Seahorse plate. All treatments were done in serum-free medium. For the Mito-Stress test, 0.75 μM oligomycin, 1 μM FCCP, and 0.5 μM rotenone/antimycin were used. The Seahorse’s Mito-Stress report generator and cell phenotype report generator were used for analysis.
Statistical analysis
GraphPad 5.0 was used for statistical analysis. One-way ANOVA was used for comparing multiple groups with Tukey post analysis, unless otherwise mentioned. For comparing 2 groups, Student’s t-test was used.
Supplementary Material
Acknowledgments:
The authors acknowledge Dr. Chong Wang (Department of Statistics, Iowa State University) for assistance with the statistical analysis. The authors also acknowledge Gary Zenitsky for his assistance in editing this manuscript and Jyothsna Ramesh Kumar for her technical assistance.
Funding: This work was supported by National Institutes of Health grants (#ES026892 to AGK, and #NS088206 to AK).
Footnotes
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
Competing interests: A.G.K. and V.A. have an equity interest in PK Biosciences Corporation located in Ames, IA. The terms of this arrangement have been reviewed and approved by Iowa State University in accordance with its conflict of interest policies. The other authors declare that they have no competing interests.
Publisher's Disclaimer: This manuscript has been accepted for publication in Science Signaling. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencesignaling.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
References and Notes
- 1.Block ML, Zecca L, Hong JS, Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8, 57–69 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Kuhl AA, Erben U, Kredel LI, Siegmund B, Diversity of Intestinal Macrophages in Inflammatory Bowel Diseases. Front Immunol 6, 613 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wehrens EJ, Mijnheer G, Duurland CL, Klein M, Meerding J, van Loosdregt J, de Jager W, Sawitzki B, Coffer PJ, Vastert B, Prakken BJ, van Wijk F, Functional human regulatory T cells fail to control autoimmune inflammation due to PKB/c-akt hyperactivation in effector cells. Blood 118, 3538–3548 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Nagatsu T, Mogi M, Ichinose H, Togari A, Cytokines in Parkinson’s disease. J Neural Transm Suppl, 143–151 (2000). [PubMed] [Google Scholar]
- 5.Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S, Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22, 1763–1771 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh A, Langley MR, Harischandra DS, Neal ML, Jin H, Anantharam V, Joseph J, Brenza T, Narasimhan B, Kanthasamy A, Kalyanaraman B, Kanthasamy AG, Mitoapocynin Treatment Protects Against Neuroinflammation and Dopaminergic Neurodegeneration in a Preclinical Animal Model of Parkinson’s Disease. J Neuroimmune Pharmacol 11, 259–278 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghosh A, Tyson T, George S, Hildebrandt EN, Steiner JA, Madaj Z, Schulz E, Machiela E, McDonald WG, Escobar Galvis ML, Kordower JH, Van Raamsdonk JM, Colca JR, Brundin P, Mitochondrial pyruvate carrier regulates autophagy, inflammation, and neurodegeneration in experimental models of Parkinson’s disease. Sci Transl Med 8, 368ra174 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Ghosh A, Kanthasamy A, Joseph J, Anantharam V, Srivastava P, Dranka BP, Kalyanaraman B, Kanthasamy AG, Anti-inflammatory and neuroprotective effects of an orally active apocynin derivative in pre-clinical models of Parkinson’s disease. J Neuroinflammation 9, 241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gordon R, Neal ML, Luo J, Langley MR, Harischandra DS, Panicker N, Charli A, Jin H, Anantharam V, Woodruff TM, Zhou QY, Kanthasamy AG, Kanthasamy A, Prokineticin-2 upregulation during neuronal injury mediates a compensatory protective response against dopaminergic neuronal degeneration. Nat Commun 7, 12932 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milatovic D, Gupta RC, Yu Y, Zaja-Milatovic S, Aschner M, Protective effects of antioxidants and anti-inflammatory agents against manganese-induced oxidative damage and neuronal injury. Toxicol Appl Pharmacol 256, 219–226 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rees K, Stowe R, Patel S, Ives N, Breen K, Clarke CE, Ben-Shlomo Y, Non-steroidal anti-inflammatory drugs as disease-modifying agents for Parkinson’s disease: evidence from observational studies. Cochrane Database Syst Rev, CD008454 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Herrero MT, Estrada C, Maatouk L, Vyas S, Inflammation in Parkinson’s disease: role of glucocorticoids. Front Neuroanat 9, 32 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo H, Callaway JB, Ting JP, Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21, 677–687 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freeman LC, Ting JP, The pathogenic role of the inflammasome in neurodegenerative diseases. J Neurochem, (2015). [DOI] [PubMed] [Google Scholar]
- 15.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT, The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9, 857–865 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, de Bernard M, Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS One 8, e55375 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou R, Yazdi AS, Menu P, Tschopp J, A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Alfonso-Loeches S, Urena-Peralta JR, Morillo-Bargues MJ, Oliver-De La Cruz J, Guerri C, Role of mitochondria ROS generation in ethanol-induced NLRP3 inflammasome activation and cell death in astroglial cells. Front Cell Neurosci 8, 216 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, Brenker C, Nordhoff M, Mirandola SR, Al-Amoudi A, Mangan MS, Zimmer S, Monks BG, Fricke M, Schmidt RE, Espevik T, Jones B, Jarnicki AG, Hansbro PM, Busto P, Marshak-Rothstein A, Hornemann S, Aguzzi A, Kastenmuller W, Latz E, The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol 15, 727–737 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodier J, Manganese poisoning in Moroccan miners. Br J Ind Med 12, 21–35 (1955). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Archibald FS, Tyree C, Manganese poisoning and the attack of trivalent manganese upon catecholamines. Arch Biochem Biophys 256, 638–650 (1987). [DOI] [PubMed] [Google Scholar]
- 22.Sadek AH, Rauch R, Schulz PE, Parkinsonism due to manganism in a welder. Int J Toxicol 22, 393–401 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Fored CM, Fryzek JP, Brandt L, Nise G, Sjogren B, McLaughlin JK, Blot WJ, Ekbom A, Parkinson’s disease and other basal ganglia or movement disorders in a large nationwide cohort of Swedish welders. Occup Environ Med 63, 135–140 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kenangil G, Ertan S, Sayilir I, Ozekmekci S, Progressive motor syndrome in a welder with pallidal T1 hyperintensity on MRI: A two-year follow-up. Mov Disord 21, 2197–2200 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Bowler RM, Koller W, Schulz PE, Parkinsonism due to manganism in a welder: neurological and neuropsychological sequelae. Neurotoxicology 27, 327–332 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Milatovic D, Yin Z, Gupta RC, Sidoryk M, Albrecht J, Aschner JL, Aschner M, Manganese induces oxidative impairment in cultured rat astrocytes. Toxicol Sci 98, 198–205 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Sidoryk-Wegrzynowicz M, Lee E, Albrecht J, Aschner M, Manganese disrupts astrocyte glutamine transporter expression and function. J Neurochem 110, 822–830 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hazell AS, Desjardins P, Butterworth RF, Chronic exposure of rat primary astrocyte cultures to manganese results in increased binding sites for the ‘peripheral-type’ benzodiazepine receptor ligand 3H-PK 11195. Neurosci Lett 271, 5–8 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Dodd CA, Filipov NM, Manganese potentiates LPS-induced heme-oxygenase 1 in microglia but not dopaminergic cells: role in controlling microglial hydrogen peroxide and inflammatory cytokine output. Neurotoxicology 32, 683–692 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filipov NM, Seegal RF, Lawrence DA, Manganese potentiates in vitro production of proinflammatory cytokines and nitric oxide by microglia through a nuclear factor kappa B-dependent mechanism. Toxicol Sci 84, 139–148 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Sarkar S, Malovic E, Harischandra DS, Ngwa HA, Ghosh A, Hogan C, Rokad D, Zenitsky G, Jin H, Anantharam V, Kanthasamy AG, Kanthasamy A, Manganese exposure induces neuroinflammation by impairing mitochondrial dynamics in astrocytes. Neurotoxicology, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moreno JA, Sullivan KA, Carbone DL, Hanneman WH, Tjalkens RB, Manganese potentiates nuclear factor-kappaB-dependent expression of nitric oxide synthase 2 in astrocytes by activating soluble guanylate cyclase and extracellular responsive kinase signaling pathways. J Neurosci Res 86, 2028–2038 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moreno JA, Streifel KM, Sullivan KA, Legare ME, Tjalkens RB, Developmental exposure to manganese increases adult susceptibility to inflammatory activation of glia and neuronal protein nitration. Toxicol Sci 112, 405–415 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yin Z, Aschner JL, dos Santos AP, Aschner M, Mitochondrial-dependent manganese neurotoxicity in rat primary astrocyte cultures. Brain Res 1203, 1–11 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao F, Cai T, Liu M, Zheng G, Luo W, Chen J, Manganese induces dopaminergic neurodegeneration via microglial activation in a rat model of manganism. Toxicol Sci 107, 156–164 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Chang JY, Liu LZ, Manganese potentiates nitric oxide production by microglia. Brain Res Mol Brain Res 68, 22–28 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Filipov NM, Seegal RF, Lawrence DA, Manganese potentiates in vitro production of proinflammatory cytokines and nitric oxide by microglia through a nuclear factor kappa B-dependent mechanism. Toxicol. Sci. Oxford University Press 84, (2005). [DOI] [PubMed] [Google Scholar]
- 38.Kirkley KS, Popichak KA, Afzali MF, Legare ME, Tjalkens RB, Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. Journal of Neuroinflammation 14, 99 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang D, Zhang J, Jiang W, Cao Z, Zhao F, Cai T, Aschner M, Luo W, The role of NLRP3-CASP1 in inflammasome-mediated neuroinflammation and autophagy dysfunction in manganese-induced, hippocampal-dependent impairment of learning and memory ability. Autophagy 13, 914–927 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dick FD, De Palma G, Ahmadi A, Osborne A, Scott NW, Prescott GJ, Bennett J, Semple S, Dick S, Mozzoni P, Haites N, Wettinger SB, Mutti A, Otelea M, Seaton A, Soderkvist P, Felice A, Geoparkinson Study G, Gene-environment interactions in parkinsonism and Parkinson’s disease: the Geoparkinson study. Occup Environ Med 64, 673–680 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aboud AA, Tidball AM, Kumar KK, Neely MD, Ess KC, Erikson KM, Bowman AB, Genetic risk for Parkinson’s disease correlates with alterations in neuronal manganese sensitivity between two human subjects. Neurotoxicology 33, 1443–1449 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chuang YH, Lill CM, Lee PC, Hansen J, Lassen CF, Bertram L, Greene N, Sinsheimer JS, Ritz B, Gene-Environment Interaction in Parkinson’s Disease: Coffee, ADORA2A, and CYP1A2. Neuroepidemiology 47, 192–200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peres TV, Schettinger MR, Chen P, Carvalho F, Avila DS, Bowman AB, Aschner M, “Manganese-induced neurotoxicity: a review of its behavioral consequences and neuroprotective strategies”. BMC Pharmacol Toxicol 17, 57 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarkar S, Malovic E, Harishchandra DS, Ghaisas S, Panicker N, Charli A, Palanisamy BN, Rokad D, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG, Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinsons Dis 3, 30 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bordt EA, Polster BM, NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: a bipartisan affair? Free Radic Biol Med 76, 34–46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD, Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol 191, 5230–5238 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lopez-Armada MJ, Riveiro-Naveira RR, Vaamonde-Garcia C, Valcarcel-Ares MN, Mitochondrial dysfunction and the inflammatory response. Mitochondrion 13, 106–118 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Ayotte P, Plaa GL, Hepatic subcellular distribution of manganese in manganese and manganese-bilirubin induced cholestasis. Biochem Pharmacol 34, 3857–3865 (1985). [DOI] [PubMed] [Google Scholar]
- 49.Lai JC, Minski MJ, Chan AW, Leung TK, Lim L, Manganese mineral interactions in brain. Neurotoxicology 20, 433–444 (1999). [PubMed] [Google Scholar]
- 50.Liccione JJ, Maines MD, Selective vulnerability of glutathione metabolism and cellular defense mechanisms in rat striatum to manganese. J Pharmacol Exp Ther 247, 156–161 (1988). [PubMed] [Google Scholar]
- 51.Roth JA, Horbinski C, Higgins D, Lein P, Garrick MD, Mechanisms of manganese-induced rat pheochromocytoma (PC12) cell death and cell differentiation. Neurotoxicology 23, 147–157 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Wang W, Zhang F, Li L, Tang F, Siedlak SL, Fujioka H, Liu Y, Su B, Pi Y, Wang X, MFN2 couples glutamate excitotoxicity and mitochondrial dysfunction in motor neurons. J Biol Chem 290, 168–182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martorell-Riera A, Segarra-Mondejar M, Munoz JP, Ginet V, Olloquequi J, Perez-Clausell J, Palacin M, Reina M, Puyal J, Zorzano A, Soriano FX, Mfn2 downregulation in excitotoxicity causes mitochondrial dysfunction and delayed neuronal death. EMBO J 33, 2388–2407 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao N, Zhang Y, Liu Q, Xiang W, Mfn2 Affects Embryo Development via Mitochondrial Dysfunction and Apoptosis. PLoS One 10, e0125680 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang FL, Liu W, Hu JX, Erion JR, Ye J, Mei L, Xiong WC, VPS35 Deficiency or Mutation Causes Dopaminergic Neuronal Loss by Impairing Mitochondrial Fusion and Function. Cell Rep 12, 1631–1643 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Racette BA, Searles Nielsen S, Criswell SR, Sheppard L, Seixas N, Warden MN, Checkoway H, Dose-dependent progression of parkinsonism in manganese-exposed welders. Neurology 88, 344–351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee EY, Flynn MR, Du G, Lewis MM, Fry R, Herring AH, Van Buren E, Van Buren S, Smeester L, Kong L, Yang Q, Mailman RB, Huang X, T1 Relaxation Rate (R1) Indicates Nonlinear Mn Accumulation in Brain Tissue of Welders With Low-Level Exposure. Toxicol Sci 146, 281–289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hazuda DJ, Lee JC, Young PR, The kinetics of interleukin 1 secretion from activated monocytes. Differences between interleukin 1 alpha and interleukin 1 beta. J Biol Chem 263, 8473–8479 (1988). [PubMed] [Google Scholar]
- 59.Jo EK, Kim JK, Shin DM, Sasakawa C, Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol 13, 148–159 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J, Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J, Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lawana V, Singh N, Sarkar S, Charli A, Jin H, Anantharam V, Kanthasamy AG, Kanthasamy A, Involvement of c-Abl Kinase in Microglial Activation of NLRP3 Inflammasome and Impairment in Autolysosomal System. J Neuroimmune Pharmacol, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guilarte TR, Manganese and Parkinson’s disease: a critical review and new findings. Environ Health Perspect. 118, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guilarte TR, Manganese neurotoxicity: new perspectives from behavioral, neuroimaging, and neuropathological studies in humans and non-human primates. Front Aging Neurosci 5, 23 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guilarte TR, Gonzales KK, Manganese-Induced Parkinsonism Is Not Idiopathic Parkinson’s Disease: Environmental and Genetic Evidence. Toxicol Sci 146, 204–212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aschner M, Erikson KM, Herrero Hernandez E, Tjalkens R, Manganese and its role in Parkinson’s disease: from transport to neuropathology. Neuromolecular Med 11, 252–266 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Horning KJ, Caito SW, Tipps KG, Bowman AB, Aschner M, Manganese Is Essential for Neuronal Health. Annu Rev Nutr 35, 71–108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Verina T, Kiihl SF, Schneider JS, Guilarte TR, Manganese exposure induces microglia activation and dystrophy in the substantia nigra of non-human primates. NeuroToxicology. 32, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen CJ, Ou YC, Lin SY, Liao SL, Chen SY, Chen JH, Manganese modulates pro-inflammatory gene expression in activated glia. Neurochem Int 49, 62–71 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Crittenden PL, Filipov NM, Manganese-induced potentiation of in vitro proinflammatory cytokine production by activated microglial cells is associated with persistent activation of p38 MAPK. Toxicology in Vitro. 22, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Filipov NM, Dodd CA, Role of glial cells in manganese neurotoxicity. J. Appl. Toxicol. 32, (2011). [DOI] [PubMed] [Google Scholar]
- 72.Mao Z, Liu C, Ji S, Yang Q, Ye H, Han H, Xue Z, The NLRP3 Inflammasome is Involved in the Pathogenesis of Parkinson’s Disease in Rats. Neurochem Res 42, 1104–1115 (2017). [DOI] [PubMed] [Google Scholar]
- 73.Aflaki E, Moaven N, Borger DK, Lopez G, Westbroek W, Chae JJ, Marugan J, Patnaik S, Maniwang E, Gonzalez AN, Sidransky E, Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 15, 77–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rao KV, Norenberg MD, Manganese induces the mitochondrial permeability transition in cultured astrocytes. J Biol Chem 279, 32333–32338 (2004). [DOI] [PubMed] [Google Scholar]
- 75.Rakovic A, Grunewald A, Kottwitz J, Bruggemann N, Pramstaller PP, Lohmann K, Klein C, Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PLoS One 6, e16746 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rocha NM, Bulger DA, Frontini A, Titheradge H, Gribsholt SB, Knox R, Page M, Harris J, Payne F, Adams C, Sleigh A, Crawford J, Gjesing AP, Bork-Jensen J, Pedersen O, Barroso I, Hansen T, Cox H, Reilly M, Rossor A, Brown RJ, Taylor SI, McHale D, Armstrong M, Oral EA, Saudek V, O’Rahilly SI, Maher ER, Richelsen B, Savage DB, Semple RK, Human biallelic MFN2 mutations induce mitochondrial dysfunction, upper body adipose hyperplasia, and suppression of leptin expression. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang P, Yu T, Yoon Y, Mitochondrial clustering induced by overexpression of the mitochondrial fusion protein Mfn2 causes mitochondrial dysfunction and cell death. Eur J Cell Biol 86, 289–302 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Liu X, Yang J, Lu C, Jiang S, Nie X, Han J, Yin L, Jiang J, Downregulation of Mfn2 participates in manganese-induced neuronal apoptosis in rat striatum and PC12 cells. Neurochem Int, (2017). [DOI] [PubMed] [Google Scholar]
- 79.Wang W, Wang X, Fujioka H, Hoppel C, Whone AL, Caldwell MA, Cullen PJ, Liu J, Zhu X, Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat Med 22, 54–63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mecozzi VJ, Berman DE, Simoes S, Vetanovetz C, Awal MR, Patel VM, Schneider RT, Petsko GA, Ringe D, Small SA, Pharmacological chaperones stabilize retromer to limit APP processing. Nat Chem Biol 10, 443–449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Swarbrick JD, Shaw DJ, Chhabra S, Ghai R, Valkov E, Norwood SJ, Seaman MN, Collins BM, VPS29 is not an active metallo-phosphatase but is a rigid scaffold required for retromer interaction with accessory proteins. PLoS One 6, e20420 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Follett J, Norwood SJ, Hamilton NA, Mohan M, Kovtun O, Tay S, Zhe Y, Wood SA, Mellick GD, Silburn PA, Collins BM, Bugarcic A, Teasdale RD, The Vps35 D620N mutation linked to Parkinson’s disease disrupts the cargo sorting function of retromer. Traffic 15, 230–244 (2014). [DOI] [PubMed] [Google Scholar]
- 83.Vella LJ, Hill AF, Cheng L, Focus on Extracellular Vesicles: Exosomes and Their Role in Protein Trafficking and Biomarker Potential in Alzheimer’s and Parkinson’s Disease. Int J Mol Sci 17, 173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sullivan CP, Jay AG, Stack EC, Pakaluk M, Wadlinger E, Fine RE, Wells JM, Morin PJ, Retromer disruption promotes amyloidogenic APP processing. Neurobiol Dis 43, 338–345 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Russo I, Bubacco L, Greggio E, Exosomes-associated neurodegeneration and progression of Parkinson’s disease. Am J Neurodegener Dis 1, 217–225 (2012). [PMC free article] [PubMed] [Google Scholar]
- 86.Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM, Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gupta A, Pulliam L, Exosomes as mediators of neuroinflammation. J Neuroinflammation 11, 68 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Racette BA, Aschner M, Guilarte TR, Dydak U, Criswell SR, Zheng W, Pathophysiology of manganese-associated neurotoxicity. Neurotoxicology 33, 881–886 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dierschke K, Isaxon C, Andersson UB, Assarsson E, Axmon A, Stockfelt L, Gudmundsson A, Jonsson BA, Karedal M, Londahl J, Pagels J, Wierzbicka A, Bohgard M, Nielsen J, Acute respiratory effects and biomarkers of inflammation due to welding-derived nanoparticle aggregates. Int Arch Occup Environ Health, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lopez-Castejon G, Brough D, Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev 22, 189–195 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gordon R, Hogan CE, Neal ML, Anantharam V, Kanthasamy AG, Kanthasamy A, A simple magnetic separation method for high-yield isolation of pure primary microglia. J Neurosci Methods 194, 287–296 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sarkar S, Malovic E, Plante B, Zenitsky G, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG, Rapid and Refined CD11b Magnetic Isolation of Primary Microglia with Enhanced Purity and Versatility. J Vis Exp, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sarkar S, Malovic E, Sarda D, Lawana V, Rokad D, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG, Characterization and comparative analysis of a new mouse microglial cell model for studying neuroinflammatory mechanisms during neurotoxic insults. Neurotoxicology 67, 129–140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Panicker N, Saminathan H, Jin H, Neal M, Harischandra DS, Gordon R, Kanthasamy K, Lawana V, Sarkar S, Luo J, Anantharam V, Kanthasamy AG, Kanthasamy A, Fyn Kinase Regulates Microglial Neuroinflammatory Responses in Cell Culture and Animal Models of Parkinson’s Disease. J Neurosci 35, 10058–10077 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Crossgrove J, Zheng W, Manganese toxicity upon overexposure. NMR Biomed 17, 544–553 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li GJ, Choi BS, Wang X, Liu J, Waalkes MP, Zheng W, Molecular mechanism of distorted iron regulation in the blood-CSF barrier and regional blood-brain barrier following in vivo subchronic manganese exposure. Neurotoxicology 27, 737–744 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zheng W, Kim H, Zhao Q, Comparative toxicokinetics of manganese chloride and methylcyclopentadienyl manganese tricarbonyl (MMT) in Sprague-Dawley rats. Toxicological sciences : an official journal of the Society of Toxicology 54, 295–301 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gordon R, Singh N, Lawana V, Ghosh A, Harischandra DS, Jin H, Hogan C, Sarkar S, Rokad D, Panicker N, Anantharam V, Kanthasamy AG, Kanthasamy A, Protein kinase Cdelta upregulation in microglia drives neuroinflammatory responses and dopaminergic neurodegeneration in experimental models of Parkinson’s disease. Neurobiol Dis 93, 96–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Seo J, Ottesen EW, Singh RN, Antisense methods to modulate pre-mRNA splicing. Methods Mol Biol 1126, 271–283 (2014). [DOI] [PubMed] [Google Scholar]
- 100.Sharma S, Carlson S, Puttachary S, Sarkar S, Showman L, Putra M, Kanthasamy AG, Thippeswamy T, Role of the Fyn-PKCdelta signaling in SE-induced neuroinflammation and epileptogenesis in experimental models of temporal lobe epilepsy. Neurobiol Dis 110, 102–121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Langley M, Ghosh A, Charli A, Sarkar S, Ay M, Luo J, Zielonka J, Brenza T, Bennett B, Jin H, Ghaisas S, Schlichtmann B, Kim D, Anantharam V, Kanthasamy A, Narasimhan B, Kalyanaraman B, Kanthasamy AG, Mito-Apocynin Prevents Mitochondrial Dysfunction, Microglial Activation, Oxidative Damage, and Progressive Neurodegeneration in MitoPark Transgenic Mice. Antioxid Redox Signal, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Harischandra DS, Ghaisas S, Rokad D, Zamanian M, Jin H, Anantharam V, Kimber M, Kanthasamy A, Kanthasamy AG, Environmental neurotoxicant manganese regulates exosome-mediated extracellular miRNAs in cell culture model of Parkinson’s disease: Relevance to alpha-synuclein misfolding in metal neurotoxicity. Neurotoxicology, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.