

Abstract
Both endocannabinoids and insulin regulate peripheral and cerebral glucose homeostasis via convergent signaling pathways that are impacted by diabetes. Here we asked how glucose metabolism and important facets of insulin signaling are affected in the forebrain of cannabinoid CB1 receptor knockout mice (CB1R-KO) and their wild-type (WT) littermates, seven weeks after the induction of insulinopenia/hyperglycemia (diabetes) with intraperitoneal streptozotocin injection. Sham-injected animals served as control. Diabetes caused milder weight loss in the WT mice compared to the phenotypically −11% leaner CB1R-KO, while hyperglycemia was similar. Resting [3H]deoxyglucose uptake was significantly reduced by −20% in acute ex vivo frontocortical and hippocampal slices obtained from both the sham-injected CB1R-KO and the diabetic WT mice. Surprisingly, the third cohort, the diabetic CB1R-KO showed no further impairment in glucose uptake, as compared to the sham-injected CB1R-KO. Depolarization-induced [3H]deoxyglucose uptake was proportional to the respective resting values only in the cortex in all four cohorts. The dissipative metabolism of [14C]-U-glucose remained largely unaffected in all cohorts of animals. However, diabetes reduced cortical CB1R density by −20%, as assessed by Western blotting. Albeit the changes in insulin signaling did not reflect the glucose uptake profile in each cohort, there were significant interactions between diabetes and genotype. In conclusion, a chronic decrease or lack of CB1R expression reduces glucose uptake in the mouse brain. Additionally, diabetes failed to cause further impairment in cerebral glucose uptake in the CB1R-KO. These suggest that diabetic encephalopathy may be in part associated with lower CB1R expression.
Keywords: cannabinoid CB1 receptor knockout, diabetes, frontal cortex, glucose uptake, hippocampus, insulin, streptozotocin
1. Introduction
Almost a century ago, it was discovered that diabetes was not only detrimental to peripheral nerves but could also cause cerebral neuropathy (Miles and Root 1922), i.e. diabetic encephalopathy (Reske-Nielsen et al. 1965). Diabetic encephalopathy is accompanied by attention and cognition deficits, resulting from complex neurophysiological and microanatomical alterations (Miles and Root 1922; Brands, 2004; Duarte 2015). Impaired brain glucoregulation could be one of them (de Ceballos and Köfalvi 2017).
Glucose is the primary energy source for the brain (Clarke and Sokoloff 1999; McCall 2004), but chronic hyperglycemia overactivates the sorbitol pathway, generates toxic glycation, oxidative stress and microvasculature damage with consequent neuroinflammation (Brands et al. 2004; McCall 2004; Duarte 2015). However, the proinsulin C-peptide also possesses important neuroprotecive roles (Sima et al. 2008).
Moreover, hyperglycemia augments phospholipase C (PLC) activity, resulting in increased cellular diacylglycerol content (Ramana et al. 2005). Importantly, PLCβ is a key enzyme in endocannabinoid signaling while diacylglycerol is the direct precursor of 2-arachidonoyl-glycerol (2-AG), a major endocannabinoid molecule (Harkany et al. 2008; Solymosi and Köfalvi 2017). Therefore, uncontrolled hyperglycemia may impair endocannabinoid signaling in nervous system tissues.
The latter may have further pathological consequences, since endocannabinoids regulate communication, proliferation, growth, migration and metabolism of brain cells, among other roles (Di Marzo et al. 2011; Solymosi and Köfalvi 2017). Since endocannabinoid production is associated with neural metabolic activity, it may also be linked with the modulation of glucose metabolism in these cells. Indeed, the activation of the cannabinoid CB2 receptor increases astrocytic and neuronal glucose uptake with relevance to Alzheimer’s disease (Köfalvi et al. 2016) while the activation of cannabinoid CB1 receptors (CB1Rs) reduces mitochondrial oxidative glucose metabolism (Bénard et al. 2012; Duarte et al. 2012).
CB1R can form functional heterodimers with the insulin and the insulin-like growth factor (IGF-1) receptors in neuronal cell culture (Dalton et al. 2012) and pancreatic β-cells (Kim et al. 2012). In addition, CB1R overactivation in adipocytes and liver causes peripheral insulin resistance (Jourdan et al. 2017; Sidibeh et al. 2017), and similarly in the brain, CB1Rs regulate insulin receptor-induced [3H]deoxyglucose uptake in the nucleus accumbens of the rat (Pinheiro et al. 2016). As a possible feed-back mechanism, insulin has been shown to induce 2-AG release and indirectly, activate CB1R in the ventral tegmental area (Labouèbe et al. 2013). These findings therefore suggest that chronic changes in cannabinoid or insulin receptor activity may perturb cerebral glucose homeostasis, especially, if the two conditions co-occur. Hence, they may contribute to diabetic encephalopathy and the detrimental hypoglycemia unawareness in type-1 diabetic patients (Cranston et al. 2001; Duarte 2015).
We now asked how chronic insulinopenic diabetes affects forebrain glucoregulation in CB1R knockout (KO) mice, as compared to their wild-type (WT) littermates. Glycemia and body weight changes were also compared. With the help of previously validated techniques we argue for an association between CB1R expression and diabetes-induced metabolic distress.
Materials and methods
Animals
All studies were conducted in accordance with the principles and procedures outlined as “3Rs” in the guidelines of EU (86/609/EEC), FELASA, and the National Centre for the 3Rs (the ARRIVE; Kilkenny et al. 2010), and were approved by the Animal Care Committee of the Center for Neuroscience and Cell Biology of the University of Coimbra, Portugal. We also applied the ARRIVE guideline for the design and execution of in vitro experiments (see below), as well as for data management and interpretation (McGrath et al. 2010).
Sixteen CB1 receptor null-mutant (knockout, CB1R-KO) male mice with CD-I background (Ledent et al. 1999) and 16 wild-type littermates (10-week old) were randomly stratified in 2 × 2 cohorts of 8 mice. Mice were raised in the laboratory of Dr. Catherine Ledent (IRIBHM, Université Libre de Bruxelles, Brussels B-1070), where they were genotyped before shipping. The mice arrived with 6 weeks of age and were kept at the conventional animal facility until use. The mice were housed with 12 h light on/off cycles under controlled temperature (23 ± 2 °C), and ad libitum access to food and water. All efforts were made to minimize the number of animals used and to minimize their stress and discomfort.
Materials
The antibodies against phospho-(p)-(Ser21) GSK3α, total and p(Ser9)-GSK3β, total and p(Ser473)-Akt, were purchased from Cell Signaling Technologies (Danvers, MA, USA). The antibodies against IRβ, IGF-1R, GLUT1 and GLUT4 were purchased from Santa Cruz Biotechnology (Santa Cruz, California, USA). The antibody against total GSK3α was obtained from UpState (Lake Placid, NY) and the antibody against β-actin was bought from BioLegend (San Diego, CA). The guinea-pig anti-CB1R antibody was obtained from Frontier Institute co., ltd. (Hokkaido, Japan). The chemifluorescence (ECF) reagent was acquired from GE Healthcare (Chalfont St. Giles, UK).
All other unspecified (in)organic reagents were from Merck (formerly, Calbiochem, Sigma-Aldrich, Merck-Millipore Corporation; Darmstadt, Germany).
Diabetes induction
Chronic insulinopenia (aka. unmedicated type-1 diabetes)/hyperglycemia was induced as described previously (Moura et al. 2014), following the protocol of Rossini et al. (1977). In brief, one cohort of 8 CB1R-KO mice and one cohort of 8 wild-type littermates (WT) were intraperitoneally injected with streptozotocin (50 mg/kg body weight) dissolved in citrate buffer (pH 4.5), during 5 consecutive days. The remaining cohorts of 8 WTs and 8 CB1R-KOs received only citrate buffer injection during 5 days. Four days after the last injection, blood glucose levels were measured by the Accu-Chek Aviva glucometer (Roche Diagnostics, Germany). Mice with blood glucose levels higher than 300 mg/dL were considered diabetic. All streptozotocin-injected mice became diabetic and none perished during the 7-week post-injection period. Blood glucose and body weight levels were regularly recorded in the diabetic cohorts (see Fig. 1). Seven weeks after the first injection, mice were deeply anesthetized (no reaction to tail pinch and handling while still breathing) with 2-bromo-2-chloro-1,1,1-trifluoroethane (halothane; 5%, 1 L/min flow rate) before decapitation with a scissor.
Fig. 1.

The effect of diabetes on body weight and plasma glucose levels in both genotypes. (A) In spite of being ~11% lighter than their wild-type (WT) littermates from the beginning, cannabinoid CB1 receptor global knockout (CB1R KO) mice lost significantly more body weight in both absolute and relative terms in the first month of hyperglycemia/insulinopenia, aka. STZ-induced type-1 diabetes. (B) Albeit the gradual development of hyperglycemia is virtually different in the two genotypes, neither the plateau nor the half maximum time to reach the plateau revealed significant difference, as assessed by the Student’s t-test. Symbols represent mean ± S.E.M. of 8 mice; *P < 0.05 vs. the preinjection value, as determined with one way ANOVA followed with Bonferroni’s multiple comparison post-hoc test; n.s., not significant.
In vitro tandem [3H]deoxyglucose/[14C]6-glucose uptake in brain slices
This assay was carried out as before (Lemos et al. 2012; Pinheiro et al. 2016), with slight modifications. Before decapitation with a scissor, the mice were deeply anesthetized with halothane vapor in air, using 1 mL liquid halothane in a 9000 cm3-volume box. The mice were killed around 2:00 PM each experimental day to reduce potential circadian hormonal effects, and their brain was immediately placed in ice-cold Krebs-HEPES assay medium of the following composition: (in mM: NaCl 133, KCl 3, KH2PO4 1.2, MgSO4 1.2, CaCl2 2.5, NHCO3 25, glucose 5.5, HEPES 10 [pH 7.4]). The pairs of hippocampi and the frontal cortices were rapidly dissected and 400 μm-thick transverse slices were cut parallel to the coronal plane with the help of a McIlwain tissue chopper. The slices were gently separated in ice-cold assay solution (carboxygenated with 95% O2 and 5% CO2), then transferred to a slice incubator using 4 chambers, each containing four 10-mm diameter baskets with 80-μm-pore nylon mesh bottom and maintained at 37°C in 50 mL of carboxygenated assay solution until the end of the experiment. Each basket contained either 6 hippocampal or 3 frontocortical slices of the same animal, and each chamber had slices from all the four cohorts to allow statistical comparison using repeated measures ANOVA. A sham experiment was also run on ice to measure the nonspecific external labeling.
After 1 h incubation under gentle gassing at 37°C, 263 μL of 3 M NaCl solution or KCl solution was added to one of the two chambers, to create the osmotic control and the KCl-depolarized conditions. Subsequently, 2-[3H](N)-deoxy-D-glucose ([3H]DG; 1 nM; specific activity; 60 Ci/mM; American Radiolabeled Chemicals, USA) and [14C]6-(uniformly labeled)-D-glucose (50 nM; 360 mCi/mM; Perkin Elmer, USA) were pipetted into the chambers. The idea behind this is that [3H]DG is a glucose analogue generally thought to represent only the net uptake process, taken that once it enters the cell it stays trapped phosphorylated. On the contrary, [14C]6-glucose is subject to metabolism, thus a difference between the final [3H] and [14C] contents in the same slice gives a rough readout of the dissipative glucose metabolism (Lemos et al. 2012).
Thirty min later, the baskets with the slices were transferred to large volumes of ice-cold assay medium for extensive washing, and then the slices were removed to 1 mL of NaOH (0.5 M). Once the slices were dissolved, 800 μL of these samples were counted for [3H] and [14C] by a dual-label counting protocol with the help of a 2900TR Tricarb β-counter (PerkinElmer). The remaining 200 μL from the samples were used for protein quantification with the bicinchoninic acid assay (BCA, Smith-assay) method (Smith et al. 1985). Original bath samples were also counted to measure the precise starting concentrations of the two tracers.
Western blotting
Remaining frontocortical slices from each mouse, not used for the glucose uptake experiments were homogenized with RIP A buffer (50 mM Tris/HCl [pH 8.0], 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 2 mM EDTA, proteases inhibitor cocktail, phosphatase inhibitor cocktail and 1 mM dithiothreitol) and cortex protein lysates were denatured at 95°C, for 5 min, in sample buffer (0.125 mM Tris [pH 6.8], 2% w/v SDS, 100 mM dithiothreitol, 10% glycerol and bromophenol blue). Thirty μg of total protein was resolved on 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% (w/v) fat-free dry milk in Tris-buffered saline containing 0.1% (v/v) Tween 20, for 1 h at room temperature.
After blocking and washing, membranes were incubated overnight at 4°C with the primary antibodies against the different proteins studied: IRβ (1:1000), IGF-1R (1:1000), GLUT1 (1:2000), GLUT4 (1:1000), p-GSK3α (1:1000), GSK3α (1:1000), p-GSK3β (1:1000), GSK3β (1:1000), p-Akt (1:500), Akt (1:1000), CB1R (1:1000), and β-actin (1:1000). After incubation, membranes were washed and incubated for 1 h at room temperature, with alkaline phosphatase-conjugated anti-rabbit, anti-guinea pig or anti-mouse antibody (Santa Cruz Biotechnology; all at 1:5000). The membranes were exposed to ECF reagent followed by scanning for blue excited fluorescence on the VersaDoc (Bio-Rad Laboratories, Amadora, Portugal). The generated signals were analyzed using the Image-Quant TL software.
Data handling
For data presentation and manipulation, we followed the rules of Curtis et al. (2015). All data represent the mean and S.E.M. of n ≥ 5 individual observations (animals). Means were tested for normality with the Kolmogorov-Smirnov test. Statistical significance was determined by one-way ANOVA of repeated measures followed by Bonferroni’s post-hoc test or two-tailed Student’s t-test, and P < 0.05 was accepted for significant difference, according to Curtis et al. (2015). Tests were performed and figures were prepared using the GraphPad Prism 5.0 software package
Results
Body weight and blood glucose changes
Long-term cerebral effects on glucose metabolism due to an insulinopenic/hyperglycemic (diabetic) state have not been compared between wild-type (WT) and CB1R knockout (CB1R-KO) mice. Therefore, we now assessed these effects seven weeks after the last streptozotocin injection. During this period, we monitored both body weight and blood glucose levels in all cohorts studied. The starting body weight of CB1R-KO mice was 10.7 ± 1.3% lower (n = 16; P < 0.001) than their (WT) littermates (Fig. 1A). Surprisingly, weight loss was more severe in the diabetic CB1R-KO mice, compared to the diabetic WT. To note, between the first and the fourth weeks after diabetes induction, the average weight loss was −6.5 ± 0.4% in the WT (−3.0 ± 0.2 g) vs. −8.5 ± 0.3% (−3.4 ± 0.1 g) in the CB1R-KO (P < 0.001 for both the absolute and the relative differences). Nevertheless, the CB1R-KO also recovered faster from weight loss since their body weight was not statistically different from their starting body weight, unlike the WT (Fig. 1A).
Normoglycemia of the WT mice (7.55 ± 0.63 mM) was not statistically different from that of the CB1R-KO (7.34 ± 0.51 mM) (n = 16; P > 0.05). After the last streptozotocin injection, the two genotypes had slightly different kinetics in the rise of their blood glucose levels (Fig. 1B). Fitting a hyperbolic function onto the data point of each rat, the WT cohort exhibited a plateau of 31.7 ± 2.3 mM vs. the CB1R-KO (27.0 ± 1.1) (n = 8; P = 0.09), while the time to reach half-maximum hyperglycemia was 2.3 ± 0.5 days for the WT and 1.5 ± 0.4 days for the CB1R-KO (P > 0.1) (Fig. 1B)
Glucose uptake in frontocortical and hippocampal slices
Resting glucose uptake, as calculated from the [3H]deoxyglucose uptake amounted to 66.3 ± 3.8 nmol/mg protein in the frontocortical slices and 65.5 ±3.1 nmol/mg protein in the hippocampal slices (WT sham; NaCl controls; Figs. 2A and 3A). On the other hand, diabetes significantly reduced the resting glucose uptake by −18.1 ± 3.8% in the frontal cortex (n = 8; P < 0.05; Fig. 2A WT STZ) and −12.1 ± 4.2% in the hippocampus (n = 8; P < 0.05; Fig. 3A WT STZ). Interestingly, there were similarly lower glucose uptake values observed in the frontal cortex (−21.8 ± 6.4%; P < 0.05) and the hippocampus (−17.0 ± 4.4%; P < 0.05) of the CB1R-KO sham animals (Figs. 2A and 3A; KO sham). Surprisingly, diabetes failed to further reduce resting glucose uptake in the CB1R-KO (Figs. 2A, 3A; KO STZ), as the frontocortical uptake remained 19.1 ± 4.0% (P < 0.05) and the hippocampal uptake 19.0 ± 3.95% smaller (P < 0.05) than in the WT sham. No statistical difference was revealed when comparing the two cohorts of CB1R-KOs.
Fig. 2.

Glucose uptake and metabolism in the frontal cortex. (A) Seven weeks of STZ-induced diabetes and the genetic deletion of the CB1R cause mutually occlusive impairment on both the resting and the 15 mM K+-depolarization induced uptake of glucose in the frontal cortex of male mice of the CD-I strain. For the resting condition, KCl was substituted with 15 mM extra NaCl. (B) Glucose metabolism was similar among the four cohorts both under resting (+15 mM NaCl) and depolarized conditions (+15 mM KCl). Bars represent mean + S.E.M. of 8 mice; *P < 0.05 when basal glucose uptake was compared to the WT sham, $P < 0.05 when the depolarized slices were compared to their respective resting control, and #P < 0.05 when the depolarized glucose uptake was compared to the depolarized WT sham slices, as determined with one way ANOVA followed with Bonferroni’s multiple comparison post-hoc test.
Fig. 3.
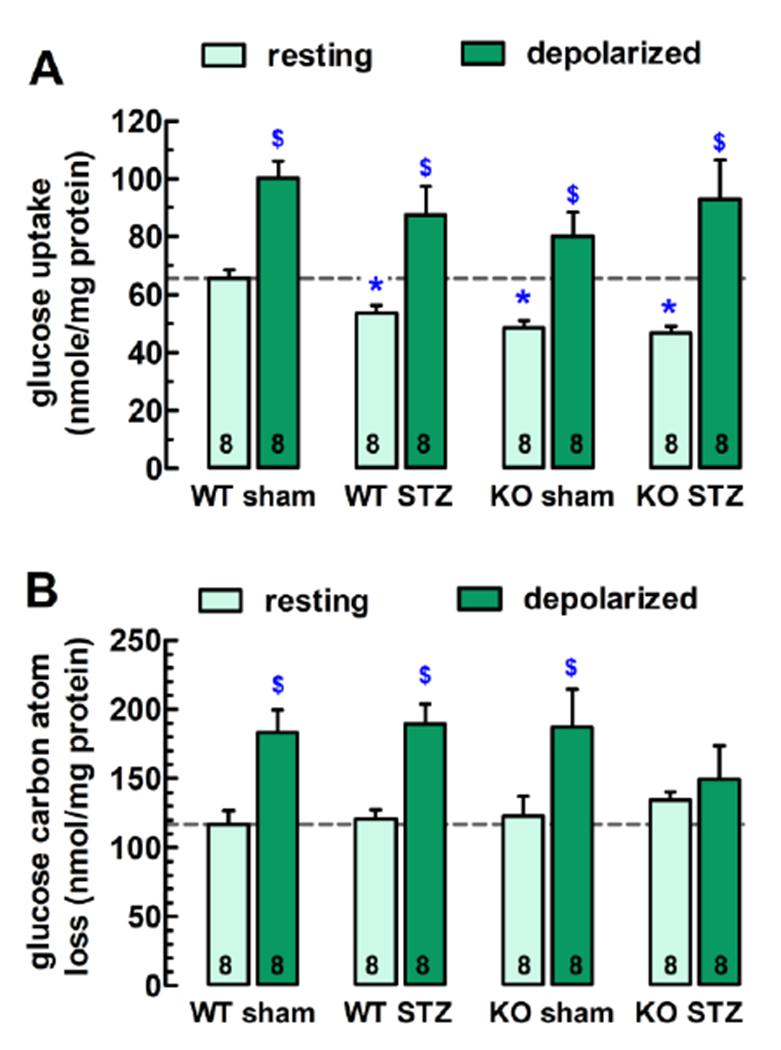
Glucose uptake and metabolism in the hippocampus. (A) Seven weeks of STZ-induced diabetes and the genetic deletion of the CB1R cause mutually occlusive impairment of the resting uptake of glucose in acute hippocampal slices of male mice. (B) While resting glucose metabolism was similar among the four cohorts, and 15 mM KCl-depolarization also triggered carbon atom dissipation of similar magnitudes in the WT sham, WT STZ and KO sham mice, it failed to stimulate glucose metabolism in the hippocampus of the KO STZ mice, pointing to a failure to cope with increased energy needs. For the resting condition, KCl was substituted with 15 mM extra NaCl. Bars represent mean + S.E.M. of 8 mice; *P < 0.05 when basal glucose uptake was compared to the WT sham, and $P < 0.05 when the depolarized slices were compared to their respective resting control, as determined with one way ANOVA followed with Bonferroni’s multiple comparison post-hoc test.
Depolarization with 15 mM K+ stimulated glucose uptake by 60.7 ± 12.9% in the frontal cortex (WT sham; Fig. 2A). While depolarization also stimulated glucose uptake in the remaining three cohorts, some difference between the cortex and the hippocampus were observed. In the cortex, the stimulated glucose uptake was strongly proportional in all four cohorts to their respective resting glucose uptake value, i.e. +66.4 ± 12.2% (P < 0.05; WT STZ), +68.3 ± 16.0% (P < 0.05; KO sham) and +65.1 ± 14.3% (P < 0.05; KO STZ) above resting uptake, and consequently, these stimulated nmol/mg (i.e. absolute) uptake values were also significantly different (P < 0.05) from those of the WT sham (Fig. 2A).
In regard to the hippocampal slices, glucose uptake was stimulated by 53.4 ± 8.9% in the WT sham (P < 0.05 vs. resting value; Fig. 3A). Depolarization stimulated glucose uptake was slightly greater in the diabetic WT cohort (+63.2 ± 18.4%; P < 0.05; WT STZ) and in the CB1R-KO sham (+65.0 ± 17.3%; P < 0.05; KO sham), but proportionally much larger in the diabetic CB1R-KO mouse slices (+99.0 ± 28.9%; P < 0.05; KO STZ), as compared to their respective resting values. Therefore, the absolute values of stimulated glucose uptake did not differ statistically among the four cohorts (P > 0.05; Fig. 3A)
Glucose metabolism in frontocortical and hippocampal slices
In the above uptake experiments, we simultaneously measured the accumulation of [3H] label associated with [3H]deoxyglucose and [14C] label derived from [14C]-U(uniformly labelled)-glucose. While the former is not metabolized, the latter is, and the discrepancy between the final [3H] and [14C] contents indicates a gross metabolism of glucose that dissipates carbon atoms e.g. in the form of [14C]O2 and [14C]lactate into the bath (for details see Lemos et al. 2012, and Methods).
The resting glucose-derived carbon loss amounted to 115.2 ± 9.5 nmol/mg in the frontocortical slices, and 117.1 ± 9.6 nmol/mg in the hippocampus of the WT sham (Figs. 3A, 3B). High-K+-depolarization stimulated carbon dissipation by 80.1 ± 17.8% (frontal cortex; P < 0.05) and by 56.4 ± 13.9% (hippocampus; P < 0.05) above the respective resting values (Figs. 3A, 3B). Neither the genetic deletion of the CB1R nor diabetes affected significantly the dissipation of glucose carbon atoms under either resting or depolarization in the frontocortical and hippocampal slices (Figs. 3A, 3B). The dissipative metabolism was also unaltered in the cortex of the diabetic CB1R-KO mice (Fig. 3A), however, their hippocampal slices were unable to correctly respond to high-K+ depolarization, as the [3H]/[14C] ratio remained similar to the resting value (Fig. 3B).
Diabetes decreases cortical CB1R density, and affects the insulin pathway
The above neurochemical data indicate that the lack of CB1Rs triggers a diabetic phenotype in cerebral glucoregulation. We hypothesized that diabetes may affect CB1R density to trigger impaired glucoregulation. Indeed, CB1R density in cortical membranes was 21.6 ± 9.1% smaller in diabetic mice (n = 8; P < 0.05; Fig. 4A,B). Unfortunately, there was not enough hippocampal tissue for both glucose uptake and Western blotting.
Fig. 4.

CB1R density and insulin signaling in WT and CB1R-KO animals with and without diabetes: (A) Representative Western blot images and (B) the resulting bar graph documenting that seven weeks of STZ-induced diabetes significantly reduced CB1R density in protein extracts from frontocortical slices of CD-I mice (note that 2 WT sham and 2 WT STZ mice are shown in the uppermost left panel). The CB1R-KO sham displayed significant reduction in insulin receptor β-chain density, while the diabetic CB1R-KO mice show marked increase in IGF-1R β-chain density accompanied with a reduction in Akt phosphorylation at Ser473. Neither the phosphorylation of glycogen synthase kinases nor the density of insulin independent (GLUT1) and insulin-dependent (GLUT4) glucose transporters were different throughout the cohorts. Normalization controls were as indicated below each protein of interest. Bars represent mean + S.E.M. of 5-8 mice; *P < 0.05 as determined with one way ANOVA followed with Bonferroni’s multiple comparison post-hoc test.
While diabetes did not significantly affect insulin receptor density in either WT or KO mice as compared to sham (n = 8; P > 0.05; Fig. 4A,B), the CB1R-KO mice had significantly less insulin receptor β-chain immunoreactivity in their frontal cortex (P < 0.05; InRβ in Fig. 4A,B). The insulin-like growth factor 1 receptor β density remained unaffected in both the diabetic and the CB1R-KO (n = 8; P > 0.05; IGF-1Rβ in Fig. 4A,B), however, the two conditions combined caused a 43.0 ± 11.0% increase in IGF-1Rβ density (n = 8; P < 0.05; Fig. 4A,B).
Akt phosphorylation at Ser473 followed a similar trend in that it became significantly different only in the diabetic CB1R-KO, with a marked 30.0 ± 4.1% drop in phosphorylation, as compared to WT sham (n = 5; P < 0.05; Fig. 4A,B). Nevertheless, the phosphorylation of the downstream kinases GSK3α at Ser21 and 3β at Ser9 remained unaffected in diabetic WT, CB1R-KO sham and diabetic CB1R-KO (n = 8; P > 0.05 for both; Fig. 4A,B). Similarly, there was no statistical difference throughout the cohorts in the density of glucose transporters 1 and 4.
Discussion
This study shed light on two major findings. First, chronic type-1 diabetes lowers glucose uptake in forebrain areas responsible for cognition, memory and mental health, even in the absence of circulating ketone bodies. Second, the lack of CB1Rs either in specific subcellular compartments or in the entirety of tissue seems to be the downstream link between hyperglycemia/insulinopenia and impaired cerebral glucoregulation.
We compared two different metabolic phenotypes, chronically type-1 diabetic mice and CB1R global KO mice with their sham-injected wild-type (WT) littermates. In the fourth cohort, we rendered the CB1R-KO mice also diabetic. There is robust literature on the roles of the CB1R in the control of central and peripheral energy metabolism (Matias et al. 2008; Piazza et al. 2017), including the regulation of glucose homeostasis (Bermúdez-Silva et al. 2016), brain energetics (Bénard et al. 2012; Duarte et al. 2012; Harkany and Horvath 2017) and insulin/IGF-1 signaling (Bouaboula et al. 1997; Dalton et al. 2012; Kim et al. 2012; Labouèbe et al. 2013). Furthermore, the blockade or the genetic deletion of CB1Rs reduces food intake and the mass of white adipose tissue (WAT), with a consequent increase in energy expanditure (Cota et al. 2003; Ravinet Trillou et al. 2004).
Even though CB1R-KO mice have a lean phenotype and smaller body weight due to reduced WAT mass (Cota et al. 2003; Ravinet Trillou et al. 2004), they lost significantly more weight both in relative and absolute terms in the first month of diabetes, as compared to their WT littermates. Streptozotocin-induced diabetes may atrophy skeletal muscle and WAT and decrease liver weight, both in rats (Pillion et al. 1988; Price et al. 1996) and in mice (Chen et al. 2009). Likely, diabetic CB1R-KO mice wasted more skeletal muscle protein than their WT littermates taken that their WAT reserve is already smaller, however this was not measured. We speculate that this could be due to loss of anabolic function of the CB1R in the skeletal muscle of the KO mice. Namely, CB1Rs can transactivate and phosphorylate the insulin receptor in the absence of its ligand (Dalton and Howlett 2012; Kim et al. 2012), which theoretically could attenuate wasting in diabetic WT mice.
The major aim of this study was to assess glucose uptake and metabolism in the frontal cortex and the hippocampus under chronic hyperglycemia/insulinopenia in the absence and the presence of the CB1R. Impaired cerebral glucoregulation contributes to or even triggers brain diseases (de Ceballos and Köfalvi 2017; Zilberter and Zilberter 2017). Encephalopathy is a possible consequence of both insulinopenia and impaired peripheral glucoregulation (Miles and Root 1922; Brands 2004; McCall 2004). Type-1 diabetes in humans is normally medicated with intermittent insulin administration that can make blood glucose levels oscillate with much greater amplitudes than in healthy subject. Hence, the chronic hyperglycemia/insulinopenia animal models have limited predictability for the human disease. Albeit more studies are necessary in diabetic patients, it appears that hypoglycemia-unaware patients also have compromised cerebral glucose uptake at euglycemia and hypoglycemia (Cranston et al. 2001) while they appear to have preserved glucose transport through the blood-brain barrier (Duarte 2015).
In streptozotocin-induced diabetic models (STZ-DM) in rats and mice, impairments in hippocampal neurogenesis, synaptic plasticity and learning, neuroinflammation, tau hyperphosphorylation and oxidative stress have been described (Stranahan et al. 2008; Duarte 2015: Elahi et al. 2018). No significant increase in cell death has been reported in the hippocampi of STZ-DM rats after one month with diabetes (Guven et al. 2009), even though such animals (in that case, rats) exhibited altered memory and learning deficits accompanied with strongly impaired expression of neural cell adhesion molecules in the hippocampus and cortex (Baydas et al. 2003). After two months of diabetes induction, we also found no significant change in vesicular GABA and glutamate transporter densities in the hippocampi of STZ-DM rats (Baptista et al. 2011), which together with the above data suggest that lower glucose uptake is unlikely a consequence of neuronal loss.
In accordance with our present ex vivo glucose uptake data, a previous in vivo study found a 24% decrease in frontocortical and a 48% decrease in hippocampal glucose uptake of systemically injected [3H]deoxyglucose, as assessed by post-mortem autoradiography in STZ-DM rats after 4 months with diabetes (Jakobsen et al. 1990). Another in vivo study obtained a similar 32% reduction in global cerebral [14C]-3-O-methylglucose uptake in STZ-DM rats 8 days after diabetes induction (Mooradian and Morin 1991). None of the two studies observed significant change in blood flow in the brain regions of interest. The question then is how does the long-term insulinopenia cause neuroglycopenia. Our ex vivo method has the advantage of eliminating the confounding factors in diabetes, including the possible differences in the permeability of the blood-brain barrier, in blood glucose levels and in blood flow. Consequently, the impairments detected in our assays are exclusive to the brain parenchyma.
At the first glance, it is logical to assume that the brain takes up less glucose when circulating insulin levels are low. Indeed, brain cell express insulin-sensitive glucose transporters including the GLUT3 and GLUT4, and insulin can stimulate glucose uptake in cultured astrocytes and neurons (Clarke et al. 1984; Uemura and Greenlee 2006; Bakirtzi et al. 2009; Pearson-Leary et al. 2018). However, we found that in experimental conditions similar to the present study, insulin (1 nM-1 μM) had negligible effect on glucose uptake in hippocampal and frontocortical slices (not shown), prepared from fed mice and fed, fasted and STZ-DM rats, which is in accordance with a previous study (Abdul-Ghani et al. 2007). Moreover, the STZ-DM model not only failed to reduce cerebral insulin content but also increased it 2-3 times within a month spent with diabetes (Havrankova et al. 1979). It is known now that there is not only a concentrating insulin transport into the brain but also brain cells themselves are capable of synthesizing insulin (Gashemi et al. 2013; Molnár et al. 2014). Therefore, it is less likely that cerebral insulinopenia causes reduced glucose uptake in the slices.
An alternative explanation for lower glucose uptake in the STZ-DM brain slices could be a lower density of the insulin receptor or hypophosphorylation of its downstream targets. Nevertheless, neither our results nor a previous milestone study (Havrankova et al. 1979) found significantly altered insulin receptor density in the cerebral cortex of STZ-DM mice and rats. Furthermore, we did not detect significant impairment in Akt or GSK3α/β phosphorylation in the STZ-DM mice. Taken that diabetes failed to reduce glucose uptake and metabolism in the CB1R-KO mice which already had an STZ-DM-like metabolic phenotype, its is safe to hypothesize that STZ-DM causes an impairment in the putative glucoregulator role of the CB1R as an intermediate step leading to cerebral hypometabolism. In line with this, Zhang et al. (2007) found that chronic hyperglycemia itself is sufficient to downregulate neuronal CB1R expression in a model of diabetic neuropathy.
The CB1R-insulin receptor interaction is further supported by our protein density data: while diabetes per se did not affect insulin receptor density, the genetic deletion of CB1R had already significantly reduced it. Furthermore, under insulinopenia in the CB1R-KO cortex, the phosphorylation of Akt at Ser473 was strongly diminished. Both insulin and IGF-1 enhance the expression of the monocarboxylate transporter 2 in neurons via the PI3K-Akt pathway, which is crucial for the astrocyte-neuron lactate shuttle (Chenal et al. 2008). Thus the markedly elevated IGF-1R density in the diabetic CB1R-KO mice suggests some kind of incomplete compensatory mechanism.
Glucose is the most abundant but not the only energy source for the brain. We found recently that ketone bodies in excess are preferred substrates over glucose for neuronal acetyl-coenzyme A generation in hippocampal neurons (Valente-Silva et al. 2015). Importantly, in STZ-DM rats, circulating ketone levels can go up to 160% after 4 weeks in diabetes (Hawkins et al. 1986). This leads to a greater cerebral uptake of ketone bodies (Hawkins et al. 1986), resulting in up to 60% increase in citrate levels that can inhibit phosphofructokinase, glycolysis, and consequently, the uptake and oxidation of glucose in the brain (Ruderman et al. 1974). Taken that the activation of CB1Rs in the outer leaflet of the mitochondrion (Bénard et al. 2012; Harkany and Horvath 2017) reduces intermediary metabolism in both neurons and astrocytes (Duarte et al. 2012), the lack of CB1Rs in the KO animal is expected to increase the velocity of the Krebs-Szentgyörgyi (aka. citrate) cycle. In the presence of enough ATP derived from ketone bodies, citrate would inhibit the most important enzyme in the glycolysis pathway, i.e. phosphofructokinase, leading to decreased glucose uptake. It is important to note that acute CB1R activation slowed the citrate cycle (Duarte et al. 2012) rather than reducing glucose uptake (Lemos et al. 2011; Köfalvi et al. 2016) in hippocampal slices.
Against this plausible explanation, in the absence of ketoacidosis, the lack of CB1Rs is expected to increase cerebral glucose oxidation, albeit acute CB1R blockade with the CB1R antagonist, AM251 did not affect the citrate cycle (Duarte et al. 2012). Also, CB1R activation triggers ketone body synthesis in astrocytes to support energy metabolism (Blázquez et al. 1999), hence the lack of CB1R should theoretically increase glucose utilization rather than reduce it.
Another possibility is that the lack of CB1Rs may hamper the trafficking or the glucoregulator function or both of the CB2R. Namely, the CB1R and the CB2R form a functional heterodimer in the brain (Callén et al. 2012) and there is evidence for concomitant reduction in both CB1R and CB2R density in disease conditions (Solymosi and Köfalvi 2017). Now taken that CB2R activation stimulates neuronal and astrocytic glucose uptake in the frontal cortex and the hippocampus by ~30% (Köfalvi et al. 2016), this process may become impaired in the CB1R-KO mice, and may lead to cerebral hypometabolism.
Finally, the last testable hypothesis is the change in the function of the cation pumps in diabetic encephalopathy. It has been shown before that the activity of Na+/K+-ATPases is decreased in both the peripheral nerves (Greene and Lattimer 1983; Catanzaro et al. 2013) and in the blood-brain barrier (Jakobsen et al. 1987). Importantly, Na+/K+-ATPases are the biggest consumers of ATP and consequently, glucose in the brain, even under resting conditions (Astrup et al. 1981; Attwell and Laughlin 2001). Remarkably, CB1R activation via Gi/o proteins stimulates the activity of Na+/K+>-ATPases in synaptoneurosomes (Araya et al. 2007). Furthermore, a solid body of evidence claims that insulin directly enhances Na+/K+-ATPase activity in skeletal muscle, liver, kidney, adipocytes, corneal endothelial cells, lymphocytes, among others (Hatou et al. 2010). Thus, it is quite possible that the action of both insulin and CB1R is necessary to exert a converging stimulation on neuronal Na+/K+-ATPases, and as a result, increased basal and K+-evoked energy expenditure.
In summary, reduced/absent CB1R expression is sufficient to mimic and to occlude insulinopenia/hyperglycemia-induced impairment in forebrain glucose metabolism. These findings are mirroring our skin ageing data in these four cohorts, namely that 7-week STZ-DM reduced CB1R density in the skin of WT mice. Also, collagen levels and antioxidant capacity were decreased, and a plethora of inflammatory markers were increased in the skin of the WT STZ, CB1R-KO, and KO STZ mice with similar amplitude, that is, reduced/absent CB1R expression was sufficient to mimic and to occlude insulinopenia/hyperglycemia-induced skin ageing (Leal et al. 2018). These results prompt the idea that the hypoactivity of the CB1R could be a druggable pharmacologic target to treat peripheral and central pathologies in type-1 diabetes. As an example, insulin exerts an important antidepressant role via IGF-1R activation in the brain (Mueller et al. 2018). Accordingly, de Morais and colleagues (2016) showed that a systemic injection of the endocannabinoid anandamide counteracts depressive-like behaviour in STZ-DM rats 4 weeks after diabetes induction, in a CB1R-dependent fashion. Hence, further studies are invited to compare the pathophysiological importance of the CB1R in the STZ-DM model, but especially when combined with insulin administration.
Highlights.
Hippocampal and frontocortical resting glucose use is 20% smaller in the CB1R-KO mice
KCl-stimulated glucose uptake was also lesser in frontocortical slices of the CB1R-KO
Seven weeks of type-1 diabetes mimicked the impaired glucose use found in the CB1R-KO
Diabetes also reduced frontocortical CB1R density, which could reduce glucose use
Indeed, diabetes failed to affect glucose uptake in the forebrain of the CB1R-KO mice
Acknowledgement
This study was supported by FEDER (QREN), through Programa Operacional Factores de Competitividade – COMPETE 2020 (POCI-01-0145-FEDER-007440), National funds via FCT – Fundação para a Ciência e a Tecnologia (AK: PTDC/SAU-OSM/105663/2008, PTDC/DTP-FTO/3346/2014; EC: EXCL/DTP-PIC/0069/2012) and P30 AG028718 and NIGMS_NIH P20GM109096 (EC), EFSD ERP Microvascular Novartis Pharma (EC), UID/NEU/04539/2013 (Strategic project) and HealthyAging2020 CENTRO-01-0145-FEDER-000012-N2323.
Abbreviations
- [3H]DG
[3H]-2-deoxy-D-glucose
- 2-AG
2-arachidonoyl-glycerol
- 3Rs
Replacement, Refinement and Reduction of Animals in Research
- ARRIVE
Animals in Research: Reporting In Vivo Experiments
- CB1R(s)
cannabinoid CB1 receptor(s)
- FELASA
Federation for Laboratory Animal Science Associations
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IGF-1R(s)
insulin-like growth factor-1 receptor(s)
- RIPA
radioimmunoprecipitation assay
- SDS-PAGE
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Compliance with ethical standards
Conflict of interest The authors declare no conflict of interests.
References
- Abdul-Ghani R, Qazzaz MM, Abdul-Ghani A (2007) Effect of insulin on 3H-deoxy-glucose uptake into brain slices and synaptosomal preparations from different brain regions. Eur J Sci Res 18:532–540 [Google Scholar]
- Araya KA, David Pessoa Mahana C, González LG (2007) Role of cannabinoid CB1 receptors and Gi/o protein activation in the modulation of synaptosomal Na+,K+- ATPase activity by WIN55,212-2 and delta9-THC. Eur J Pharmacol 572:32–39. 10.1016/j.ejphar.2007.06 [DOI] [PubMed] [Google Scholar]
- Astrup J, Sørensen PM, Sørensen HR (1981) Oxygen and glucose consumption related to Na+-K+ transport in canine brain. Stroke 12:726–730. 10.1161/01.STR.12.6.726 [DOI] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21:1133–1145. 10.1097/00004647-200110000-00001 [DOI] [PubMed] [Google Scholar]
- Bakirtzi K, Belfort G, Lopez-Coviella I, Kuruppu D, Cao L, Abel ED, Brownell AL, Kandror KV (2009) Cerebellar neurons possess a vesicular compartment structurally and functionally similar to Glut4-storage vesicles from peripheral insulin-sensitive tissues. J Neurosci 29:5193–5201. 10.1523/JNEUROSCI.0858-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista FI, Gaspar JM, Cristóvão A, Santos PF, Köfalvi A, Ambrósio AF (2011) Diabetes induces early transient changes in the content of vesicular transporters and no major effects in neurotransmitter release in hippocampus and retina. Brain Res 1383:257–269. 10.1016/j.brainres.2011.01.071 [DOI] [PubMed] [Google Scholar]
- Baydas G, Nedzvetskii VS, Nerush PA, Kirichenko SV, Yoldas T (2003) Altered expression of NCAM in hippocampus and cortex may underlie memory and learning deficits in rats with streptozotocin-induced diabetes mellitus. Life Sci 73:1907–1916 10.1016/S0024-3205(03)00561-7 [DOI] [PubMed] [Google Scholar]
- Bénard G, Massa F, Puente N, Lourenço J, Bellocchio L, Soria-Gómez E, Matias I, Delamarre A, Metna-Laurent M, Cannich A, Hebert-Chatelain E, Mulle C, Ortega-Gutiérrez S, Martín-Fontecha M, Klugmann M, Guggenhuber S, Lutz B, Gertsch J, Chaouloff F, López-Rodríguez ML, Grandes P, Rossignol R, Marsicano (2012) Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat Neurosci 15:558–564. 10.1038/nn.3053 [DOI] [PubMed] [Google Scholar]
- Bermudez-Silva FJ, Romero-Zerbo SY, Haissaguerre M, Ruz-Maldonado I, Lhamyani S, El Bekay R, Tabarin A, Marsicano G, Cota D (2016) The cannabinoid CB1 receptor and mTORCl signalling pathways interact to modulate glucose homeostasis in mice. Dis Model Mech 9:51–61. 10.1242/dmm.020750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blázquez C, Sánchez C, Daza A, Galve-Roperh I, Guzmán M (1999) The stimulation of ketogenesis by cannabinoids in cultured astrocytes defines carnitine palmitoyltransferase I as a new ceramide-activated enzyme. J Neurochem 72:1759–1768. 10.1046/j.1471-4159.1999.721759.x [DOI] [PubMed] [Google Scholar]
- Bouaboula M, Perrachon S, Milligan L, Canat X, Rinaldi-Carmona M, Portier M, Barth F, Calandra B, Pecceu F, Lupker J, Maffrand JP, Le Fur G, Casellas P (1997) A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1. Evidence for a new model of receptor/ligand interactions. J Biol Chem 272:22330–22339. 10.1074/jbc.272.35.22330 [DOI] [PubMed] [Google Scholar]
- Brands AMA, Kessels RPC, de Haan EHF, Kappelle LJ, Biessels GJ (2004) Cerebral dysfunction in type 1 diabetes: effects of insulin, vascular risk factors and blood-glucose levels. Eur J Pharmacol 490:159–168. 10.1016/j.ejphar.2004.02.053 [DOI] [PubMed] [Google Scholar]
- Callén L, Moreno E, Barroso-Chinea P, Moreno-Delgado D, Cortés A, Mallol J, Casadó V, Lanciego JL, Franco R, Lluis C, Canela El, McCormick PJ (2012) Cannabinoid receptors CBI and CB2 form functional heteromers in brain. J Biol Chem 287:20851–20865. 10.1074/jbc.Mlll.335273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Cao L, Ye J, Zhu D (2009) Upregulation of myostatin gene expression in streptozotocin-induced type 1 diabetes mice is attenuated by insulin. Biochem Biophys Res Commun 388:112–116. 10.1016/j.bbrc.2009.07.129 [DOI] [PubMed] [Google Scholar]
- Catanzaro O, Capponi JA, Michieli J, Michieli J, Labal E, Di Martino I, Sirois P (2013) Bradykinin B1 antagonism inhibits oxidative stress and restores Na+K+ ATPase activity in diabetic rat peripheral nervous system. Peptides 44:100–104. 10.1016/j.peptides.2013.01.019 [DOI] [PubMed] [Google Scholar]
- Chenal J, Pierre K, Pellerin L (2008) Insulin and IGF-1 enhance the expression of the neuronal monocarboxylate transporter MCT2 by translational activation via stimulation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin pathway. Eur J Neurosci 27:53–65. 10.1111/j.1460-9568.2007.05981.x [DOI] [PubMed] [Google Scholar]
- Clarke DD, Sokoloff L (1999) Circulation and Energy Metabolism of the BrainIn Siegel GJ, Agranoff BW, Albers RW, et al. (ed) Basic Neurochem Mol Cell Med Asp, 6th ed Lippincott-Raven, Philadelphia, pp 637–669 [Google Scholar]
- Clarke DW, Boyd FT, Kappy MS, Raizada MK (1984) Insulin binds to specific receptors and stimulates 2-deoxy-D-glucose uptake in cultured glial cells from rat brain. J Biol Chem 259:11672–11675. [PubMed] [Google Scholar]
- Cota D, Marsicano G, Tschöp M, Grübler Y, Flachskamm C, Schubert M, Auer D, Yassouridis A, Thöne-Reineke C, Ortmann S, Tomassoni F, Cervino C, Nisoli E, Linthorst AC, Pasquali R, Lutz B, Stalla GK, Pagotto U (2003) The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest 112:423–431. 10.1172/JCI17725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cranston I, Reed LJ, Marsden PK, Amiel SA (2001) Changes in regional brain (18)F-fluorodeoxyglucose uptake at hypoglycemia in type 1 diabetic men associated with hypoglycemia unawareness and counter-regulatory failure. Diabetes 50:2329–2336. 10.2337/diabetes.50.10.2329 [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA, Gilchrist A, Hoyer D, Insel PA, Izzo AA, Lawrence AJ, MacEwan DJ, Moon LD, Wonnacott S, Weston AH, McGrath JC (2015) Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172:3461–3471. 10.1111/bph.12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton GD, Howlett AC (2012) Cannabinoid CB1 receptors transactivate multiple receptor tyrosine kinases and regulate serine/threonine kinases to activate ERK in neuronal cells. Br J Pharmacol 165:2497–2511. 10.1111/j.1476-5381.2011.01455.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ceballos ML, Köfalvi A (2017) Boosting brain glucose metabolism to fight neurodegeneration? Oncotarget 8:14273–14274. 10.18632/oncotarget.15131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Morais H, de Souza CP, da Silva LM, Ferreira DM, Baggio CH, Vanvossen AC, Cristina de Carvalho M, da Silva-Santos JE, Bertoglio LJ, Cunha JM, Zanoveli JM (2016) Anandamide reverses depressive-like behavior, neurochemical abnormalities and oxidative-stress parameters in streptozotocin-diabetic rats: Role of CB1 receptors. Eur Neuropsychopharmacol 26:1590–1600. 10.1016/j.euroneuro.2016.08.007 [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Piscitelli F, Mechoulam R (2011) Cannabinoids and endocannabinoids in metabolic disorders with focus on diabetes. Handb Exp Pharmacol 203:75–104. 10.1007/978-3-642-17214-4_4 [DOI] [PubMed] [Google Scholar]
- Duarte JMN (2015) Metabolic Alterations Associated to Brain Dysfunction in Diabetes. Aging Dis 6:304–321. 10.14336/AD.2014.1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte JMN, Ferreira SG, Carvalho RA, Cunha RA, Köfalvi A (2012) CB1 receptor activation inhibits neuronal and astrocytic intermediary metabolism in the rat hippocampus. Neurochem Int 60:1–8. 10.1016/j.neuint.2011.10.019 [DOI] [PubMed] [Google Scholar]
- Elahi M, Hasan Z, Motoi Y, Matsumoto SE, Ishiguro K, Hattori N (2016) Region-specific vulnerability to oxidative stress, neuroinflammation, and tau hyperphosphorylation in experimental diabetes mellitus mice. J Alzheimers Dis 51:1209–1224. 10.3233/JAD-150820 [DOI] [PubMed] [Google Scholar]
- Ghasemi R, Haeri A, Dargahi L, Mohamed Z, Ahmadiani A (2013) Insulin in the brain: sources, localization and functions. Mol Neurobiol 47:145–171. 10.1007/sl2035-012-8339-9 [DOI] [PubMed] [Google Scholar]
- Greene DA, Lattimer SA (1983) Impaired rat sciatic nerve sodium-potassium adenosine triphosphatase in acute streptozocin diabetes and its correction by dietary myo-inositol supplementation. J Clin Invest 72:1058–1063. 10.1172/JCI111030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guven A, Yavuz O, Cam M, Comunoglu C, Sevi’nc O (2009) Central nervous system complications of diabetes in streptozotocin-induced diabetic rats:a histopathological and immunohistochemical examination. Int J Neurosci 119:1155–1169 [DOI] [PubMed] [Google Scholar]
- Hatou S, Yamada M, Akune Y, Mochizuki H, Shiraishi A, Joko T, Nishida T, Tsubota K (2010) Role of insulin in regulation of Na+-/K+-dependent ATPase activity and pump function in corneal endothelial cells. Invest Ophthalmol Vis Sci 51:3935–3942. 10.1167/iovs.09-4027 [DOI] [PubMed] [Google Scholar]
- Harkany T, Horvath TL (2017) (S) Pot on Mitochondria: Cannabinoids Disrupt Cellular Respiration to Limit Neuronal Activity. Cell Metab 25:8–10. 10.1016/j.cmet.2016.12.020 [DOI] [PubMed] [Google Scholar]
- Havrankova J, Roth J, Brownstein MJ (1979) Concentrations of insulin and insulin receptors in the brain are independent of peripheral insulin levels. Studies of obese and streptozotocin-treated rodents. J Clin Invest 64:636–642. 10.1172/JCI109504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RA, Mans AM, Davis DW (1986) Regional ketone body utilization by rat brain in starvation and diabetes. Am J Physiol 250:E169–178. https://doi:10.1152/ajpendo.1986.250.2.E169 [DOI] [PubMed] [Google Scholar]
- Jakobsen J, Knudsen GM, Juhler M (1987) Cation permeability of the blood-brain barrier in streptozotocin-diabetic rats. Diabetologia 30:409–413. 10.1007/BF00292543 [DOI] [PubMed] [Google Scholar]
- Jakobsen J, Nedergaard M, Aarslew-Jensen M, Diemer NH (1990) Regional brain glucose metabolism and blood flow in streptozocin-induced diabetic rats. Diabetes 39:437–440. 10.2337/diab.39.4.437 [DOI] [PubMed] [Google Scholar]
- Jourdan T, Nicoloro SM, Zhou Z, Shen Y, Liu J, Coffey NJ, Cinar R, Godlewski G, Gao B, Aouadi M, Czech MP, Kunos G (2017) Decreasing CB1 receptor signaling in Kupffer cells improves insulin sensitivity in obese mice. Mol Metab 6:1517–1528. 10.1016/j.molmet.2017.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG; NC3Rs Reporting Guidelines Working Group (2010) Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160:1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Lao Q, Shin YK, Carlson OD, Lee EK, Gorospe M, Kulkarni RN, Egan JM (2012) Cannabinoids induce pancreatic β-cell death by directly inhibiting insulin receptor activation. Sci Signal 5:ra23 10.1126/scisignal.2002519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köfalvi A, Lemos C, Martín-Moreno AM, Pinheiro BS, García-García L, Pozo MA, Valério-Femandes Â, Beleza RO, Agostinho P, Rodrigues RJ, Pasquaré SJ7, Cunha RA6, de Ceballos ML, (2016) Stimulation of brain glucose uptake by cannabinoid CB2 receptors and its therapeutic potential in Alzheimer’s disease. Neuropharmacology 110:519–529. 10.1016/j.neuropharm.2016.03.015 [DOI] [PubMed] [Google Scholar]
- Labouèbe G, Liu S, Dias C, Zou H, Wong JC, Karunakaran S, Clee SM, Phillips AG, Boutrel B, Borgland SL (2013) Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat Neurosci 16:300–308. 10.1038/nn.3321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F, Böhme GA, Imperato A, Pedrazzini T, Roques BP, Vassart G, Fratta W, Parmentier M (1999) Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science 283:401–404 10.1126/science.283.5400.401 [DOI] [PubMed] [Google Scholar]
- Leal EC, Moura LIF, Pirzgalska RM, Marques-da-Silva D, Ledent C, Köfalvi A, Carvalho C (2018) Diabetes Decreases Cannabinoid Receptor 1 Expression Promoting Early Skin Ageing submitted [Google Scholar]
- Lemos C, Valério-Fernandes Â, Ghisleni GC, Ferreira SG, Ledent C, de Ceballos ML, Köfalvi A (2012) Impaired hippocampal glucoregulation in the cannabinoid CB1 receptor knockout mice as revealed by an optimized in vitro experimental approach. J Neurosci Methods 204:366–373. 10.1016/j.jneumeth.2011.11.028 [DOI] [PubMed] [Google Scholar]
- Matias I, Di Marzo V, Köfalvi A (2008) Endocannabinoids in Energy Homeostasis and Metabolic Disorders In: Köfalvi A (ed) Cannabinoids and the Brain Springer; US, pp 277–316 10.1007/978-0-387-74349-3_14 [DOI] [Google Scholar]
- McCall AL (2004) Cerebral glucose metabolism in diabetes mellitus. Eur J Pharmacol 490:147–158. 10.1016/j.ejphar.2004.02.052 [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright CL (2010) Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160:1573–1576. 10.1111/j.1476-5381.2010.00873.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles WR, Root HF (1922) Psychologic tests applied to diabetic patients. Arch Intern Med 30:767–777. 10.1001/archinte.1922.00110120086003 [DOI] [Google Scholar]
- Molnár G, Faragó N, Kocsis ÁK, Rózsa M, Lovas S, Boldog E, Báldi R, Csajbók É, Gardi J, Puskás LG, Tamás G (2014) GABAergic neurogliaform cells represent local sources of insulin in the cerebral cortex. J Neurosci 34:1133–1137. 10.1523/JNEUROSCI.4082-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooradian AD, Morin AM (1991) Brain uptake of glucose in diabetes mellitus: the role of glucose transporters. Am J Med Sci 301:173–177. 10.1097/00000441-199103000-00004 [DOI] [PubMed] [Google Scholar]
- Moura LIF, Dias AMA, Suesca E, Casadiegos S, Leal EC, Fontanilla MR, Carvalho L, de Sousa HC, Carvalho E (2014) Neurotensin-loaded collagen dressings reduce inflammation and improve wound healing in diabetic mice. Biochim Biophys Acta 1842:32–43. 10.1016/j.bbadis.2013.10.009 [DOI] [PubMed] [Google Scholar]
- Mueller PL, Pritchett CE, Wiechman TN, Zharikov A, Hajnal A (2018) Antidepressant-like effects of insulin and IGF-1 are mediated by IGF-1 receptors in the brain. Brain Res Bull 143:27–35. 10.1016/j.brainresbull.2018.09.017 [DOI] [PubMed] [Google Scholar]
- Pearson-Leary J, Jahagirdar V, Sage J, McNay EC (2018) Insulin modulates hippocampally-mediated spatial working memory via glucose transporter-4. Behav Brain Res 338:32–39. 10.1016/j.bbr.2017.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza PV, Cota D, Marsicano G (2017) The CB1 Receptor as the Cornerstone of Exostasis. Neuron 93:1252–1274. 10.1016/j.neuron.2017.02.002 [DOI] [PubMed] [Google Scholar]
- Pillion DJ, Jenkins RL, Atchison JA, Stockard CR, Clements RS Jr, Grizzle WE (1988) Paradoxical organ-specific adaptations to streptozotocin diabetes mellitus in adult rats. Am J Physiol 254:E749–755. 10.1152/ajpendo.1988.254.6.E749 [DOI] [PubMed] [Google Scholar]
- Pinheiro BS, Lemos C, Neutzling Kaufmann F, Marques JM, da Silva-Santos CS, Carvalho E, Mackie K, Rodrigues RJ, Cunha RA, Köfalvi A (2016) Hierarchical glucocorticoid-endocannabinoid interplay regulates the activation of the nucleus accumbens by insulin. Brain Res Bull 124:222–230. 10.1016/j.brainresbull.2016.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price SR, Bailey JL, Wang X, Jurkovitz C, England BK, Ding X, Phillips LS, Mitch WE (1996) Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription. J Clin Invest 98:1703–1708. 10.1172/JCI118968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramana KV, Friedrich B, Tammali R, West MB, Bhatnagar A, Srivastava SK(2005) Requirement of aldose reductase for the hyperglycemic activation of protein kinase C and formation of diacylglycerol in vascular smooth muscle cells. Diabetes 54:818–829. 10.2337/diabetes.54.3.818 [DOI] [PubMed] [Google Scholar]
- Ravinet Trillou C, Delgorge C, Menet C, Arnone M, Soubrié P (2004) CBI cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int J Obes Relat Metab Disord 28:640–648. 10.1038/sj.ijo.0802583 [DOI] [PubMed] [Google Scholar]
- Reske-Nielsen E, Lundbæk K, Rafaelsen OJ (1966) Pathological changes in the central and peripheral nervous system of young long-term diabetics. Diabetologia 1:233–241. 10.1007/BF01257917 [DOI] [PubMed] [Google Scholar]
- Rossini AA, Like AA, Chick WL, Appel MC, Cahill GF Jr (1977) Studies of streptozotocin-induced insulitis and diabetes. Proc Natl Acad Sci USA 74:2485–2489. 10.1073/pnas.74.6.2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderman NB, Ross PS, Berger M, Goodman MN (1974) Regulation of glucose and ketone-body metabolism in brain of anaesthetized rats. Biochem J 138:1–10. 10.1042/bjl380001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidibeh CO, Pereira MJ, Lau Börjesson J, Kamble PG, Skrtic S, Katsogiannos P, Sundbom M, Svensson MK, Eriksson JW (2017) Role of cannabinoid receptor 1 in human adipose tissue for lipolysis regulation and insulin resistance. Endocrine 55:839–852. 10.1007/s12020-016-1172-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima AAF, Zhang W, Li Z, Kamiya H (2008) The effects of C-peptide on type 1 diabetic polyneuropathies and encephalopathy in the BB/Wor-rat. Exp Diabetes Res 2008:230458 10.1155/2008/230458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85. 10.1016/0003-2697(85)90442-7 [DOI] [PubMed] [Google Scholar]
- Solymosi K, Köfalvi A (2017) Cannabis: A Treasure Trove or Pandora’s Box? Mini Rev Med Chem 17:1223–1291 10.2174/1389557516666161004162133 [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP (2008) Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci 11:309–317. 10.1038/nn2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura E, Greenlee HW (2006) Insulin regulates neuronal glucose uptake by promoting translocation of glucose transporter GLUT3. Exp Neurol 198:48–53. 10.1016/j.expneurol.2005.10.035 [DOI] [PubMed] [Google Scholar]
- Valente-Silva P, Lemos C, Köfalvi A, Cunha RA, Jones JG (2015) Ketone bodies effectively compete with glucose for neuronal acetyl-CoA generation in rat hippocampal slices. NMR Biomed 28:1111–1116. 10.1002/nbm.3355 [DOI] [PubMed] [Google Scholar]
- Zhang F, Hong S, Stone V, Smith PJW (2007) Expression of cannabinoid CB1 receptors in models of diabetic neuropathy. J Pharmacol Exp Ther 323:508–515. 10.1124/jpet.107.128272 [DOI] [PubMed] [Google Scholar]
- Zilberter Y, Zilberter M (2017) The vicious circle of hypometabolism in neurodegenerative diseases: Ways and mechanisms of metabolic correction. J Neurosci Res 95:2217–2235. 10.1002/jnr.24064 [DOI] [PubMed] [Google Scholar]