

Abstract
Background:
Anxiety disorders and major depressive disorder (MDD) have been associated with increased and blunted HPA axis reactivity to social stress. However, research focusing on associations between HPA axis responses to stress and symptoms of anxiety and depression among individuals without a diagnosis remains an understudied area of research.
Methods:
One hundred forty-three adults (52% female) completed the Trier Social Stress est (TSST). Symptoms of depression and anxiety were assessed prior to the TSST using the anxiety and depression subscales of the Hospital Anxiety and Depression Scale (HADS). HPA axis responses were assessed by measuring salivary cortisol at baseline and following the TSST. Reactivity to and recovery from stress were assessed using multilevel growth modeling controlling for age, BMI, and sex among the full sample and a subset of cortisol responders (n=72).
Results:
Anxiety symptoms were positively associated with flatter recovery slopes among the full sample (t(122.3)=2.082, p=.039). Among cortisol responders, depression symptoms were associated with steeper reactivity (t(63.32)=2.53, p=.026) and recovery (t(58.75)=−2.20, p=.03). Anxiety symptoms were associated with marginally flatter reactivity (t(64.00)=−1.97, p=.053) and significantly flatter recovery (t(59.22)=2.29, p=.025).
Conclusion:
Symptoms of anxiety and depression among individuals without a psychiatric diagnosis are associated with blunted and exaggerated cortisol responses to and recovery from stress. Such patterns could indicate increased risk for unhealthy HPA axis dysregulation, allostatic load, and disease.
Keywords: Depression, anxiety, cortisol, HPA axis, TSST, stress
2. Introduction
The ability to rapidly and adaptively respond to environmental threats and stressors is critical to survival. Interactive physiological responses involving the hypothalamic-pituitary-adrenal (HPA) axis and autonomic nervous systems influence metabolic, immune, and cardiovascular parameters in order to aid the body in responding appropriately to environmental demands (McEwen, 1998). This process of achieving stability through change, first introduced by Sterling and Eyer (Sterling & Eyer, 1988), is known as ‘allostasis’. While highly adaptive in the short-term, chronic exposure to stress mediators can lead to ‘allostatic load’—wear and tear of the body characterized by dysregulated physiological responses to stress—and disease (McEwen, 2008). One such mediator is the glucocorticoid (GC) cortisol, which is the end product of the HPA axis. Cortisol serves a variety of crucial roles in promoting allostasis, including mediating and suppressing healthy stress responses (Sapolsky, Romero, & Munck, 2000). However, chronic exposure to GCs can lead to structural changes in the brain regions responsible for modulating the stress response (e.g. the hippocampus) and may contribute to the pathophysiology of anxiety and mood disorders (McEwen, 2003, 2004; McEwen & Gianaros, 2010).
In order to investigate how mood and other psychiatric disorders are associated with HPA axis stress responses, many researchers have utilized lab-based social stress paradigms in conjunction with salivary cortisol sampling. Meta-analytic studies have indicated that major depressive disorder (MDD) is associated with blunted cortisol responses to laboratory stressors (Burke, Davis, Otte, & Mohr, 2005), and that this association may be sex-dependent, with women exhibiting blunted and men elevated responses (Zorn et al., 2017). Although many studies have evaluated cortisol responses in the context of clinical depression, relatively few have investigated associations between cortisol responses and sub-clinical depressive symptomology. Results have been mixed. One recent study of healthy older adults found that depressive symptoms predicted elevated cortisol responses to social stress among healthy older adults (Puig-Perez, Villada, Pulopulos, Hidalgo, & Salvador, 2016). Conversely, a larger study, also consisting of older adults, reported decreased reactivity with increasing depressive symptoms (de Rooij, Schene, Phillips, & Roseboom, 2010). Moreover, these associations may be sex-dependent: another study indicated that women exhibit blunted reactivity/recovery patterns in response to an interpersonal stressor while men exhibit steeper reactivity and recovery slopes (Powers, Laurent, Gunlicks-Stoessel, Balaban, & Bent, 2016). Overall, the association between depressive symptoms and cortisol responses among individuals without a diagnosed depressive mood disorder remains unclear.
Anxiety disorders have also been associated with differences in cortisol responses to laboratory social stressors; however, results have been inconsistent (Elnazer & Baldwin, 2014). For example, one study found that social phobia was associated with increased cortisol reactivity (Furlan, DeMartinis, Schweizer, Rickels, & Lucki, 2001). Meanwhile, other studies have failed to detect differences in a variety of physiological responses, including cortisol, between those with social phobia and healthy controls (Espín, Marquina, Hidalgo, Salvador, & Gómez-Amor, 2016; Klumbies, Braeuer, Hoyer, & Kirschbaum, 2014). As with MDD, there is evidence that the effect of anxiety disorders on cortisol responses are sex-dependent, with women exhibiting attenuated and men elevated cortisol output following acute stress (Zorn et al., 2017). he relationship between anxiety symptoms and cortisol responses to stress among individuals without a clinical diagnoses of an anxiety disorder remains understudied; however, there is evidence suggesting that recent anxiety symptoms are associated with blunted cortisol responses across a range of individuals including undergraduates of both sexes (Crisan, Vulturar, Miclea, & Miu, 2016), adult men (Brooks & Robles, 2009), and older adults of both sexes (de Rooij et al., 2010). Conversely, another study found that women with higher sub-clinical symptoms exhibited steeper reactivity and recovery slopes in response to an interpersonal stressor compared to men (Powers et al., 2016).
Although there is a relative dearth of research focusing on the role of comorbidity in the relationship between depression/anxiety and cortisol responses to stress, anxiety and mood disorders are well known to be highly comorbid (Brown, Campbell, Lehman, Grisham, & Mancill, 2001), and there is evidence that comorbidity may alter such associations. Specifically, comorbid depression and anxiety disorders have been associated with increased adrenocorticotropic hormone (ACTH) reactivity relative to pure MDD or anxiety disorders (Young, Abelson, & Cameron, 2004). Another study indicated that increased cortisol reactivity was present in pure anxiety disorders only, with participants with comorbid MDD exhibiting no increased reactivity relative to healthy controls (Yoon & Joormann, 2012). Still other studies found no significant effect of comorbidity (Burke et al., 2005; Steudte-Schmiedgen et al., 2017; Wichmann, Kirschbaum, Böhme, & Petrowski, 2017). More research is needed to elucidate the influence, if any, of comorbidity on the on the relationship between depression/anxiety and cortisol responses.
In summary, existing research suggests that both anxiety and depressive disorders may influence cortisol responses to stress. These associations may be dependent on sex and comorbidity status. While the majority of studies have focused on populations with clinical diagnoses of anxiety disorders and/or MDD, relatively few have focused on anxiety and depressive symptomology among individuals without a diagnosis of depression. Moreover, few studies have analyzed both reactivity and recovery trajectories in the context of depression, instead focusing exclusively on reactivity or overall response ‘output’ (e.g. area under the curve). This is common among studies utilizing laboratory-based stressors and cortisol sampling, and while there has been an increase in studies examining recovery, there remains a dearth of research explicitly analyzing reactivity and recovery (Ji, Negriff, Kim, & Susman, 2016). Therefore, the aim of the present study was to examine how anxiety and depressive symptoms are associated with cortisol reactivity and recovery trajectories among a healthy sample of adults without a depression or anxiety disorder diagnoses. Given the effects of sex and comorbidity status observed in the clinical literature, we also explored interactions between anxiety and depressive symptoms and those symptoms and sex in predicting cortisol responses. Finally, we conducted these analyses using both our full sample and a subset of cortisol responders only.
3. Material and Methods
3.1. Participants
The sample consisted of 149 adult participants from three studies carried out in the Laboratory for Biological Health Psychology between 2009 and 2017. Data from study 1 has been reported previously (e.g. Gianferante et al., 2014; Kuras et al., 2017). All participants were in good health and were recruited from Brandeis University and the greater Boston, area via flyers and local advertisements. All participants were screened for medical and psychological conditions via telephone prior to being invited to participate. Exclusion criteria included self-reported current or past history of chronic cardiovascular or psychiatric illness—including depression and anxiety disorders—at the time of screening, current tobacco use, use of hormonal contraception or any other drug that may alter hormonal responses, previous experience with the stress protocol, and a body mass index (BMI) outside the range of 18–35 kg/m2. Female participants were all tested during the luteal phase of their menstrual cycle to minimize the impact of menstrual cycle variation in stress hormones. No participants took part in more than one study. Six participants were excluded from analyses: one due to extremely high baseline cortisol concentration (>6 SD above the mean), two due to highly atypical spikes in cortisol indicative of measurement error, and three due to missing questionnaire data. The final sample consisted of 143 adults (52% female), with age ranges varying by study (Study 1: n=81, range=18–65, mean=37.2, SD=18.1; Study 2: n=16, range=18–29, mean=21.6, SD=3.4; Study 3: n=46, range=18–22, mean=18.8, SD=1.1).
3.2. Procedures
All three studies utilized similar procedures and took place in the same laboratory. The protocols for studies 1 and 3 incorporated a repeated stress testing component; however, as repeated stress testing was not of interest to this study, only data and procedural details related to the initial stress exposure are presented here. All participants were invited to the laboratory in the afternoon between 13:30 and 18:30 to minimize the impact of diurnal variations in hormone concentrations. Participants were instructed to refrain from eating or drinking anything aside from water in the hour prior to arrival. The specific procedures of the study were described in detail, and informed consent was obtained. Participants in studies 1 and 2 were compensated $100 and $50, respectively, and those in study 3 received university credit. The Brandeis Institutional Review Board approved all procedures in all studies.
After consent was obtained, participants in studies 1 and 2 had an intravenous catheter placed in the non-dominant arm for the collection of blood samples. No biological measures derived from blood samples are the subject of this study. Participants then provided an initial saliva sample and were seated comfortably in an quiet waiting room, where they completed a battery of questionnaires. Procedure schedules specific to each study are summarized in Figure 1 Generally, a resting period was followed by exposure to the Trier Social Stress Test (Kirschbaum, Pirke, & Hellhammer, 1993). Participants provided saliva samples immediately before and at similar intervals immediately after, through 60 minutes post-TSST. Saliva was collected using Salivette collection devices (Sarstedt, Newton, NC). Details on collection and storage of Salivettes are described below.
Figure 1.
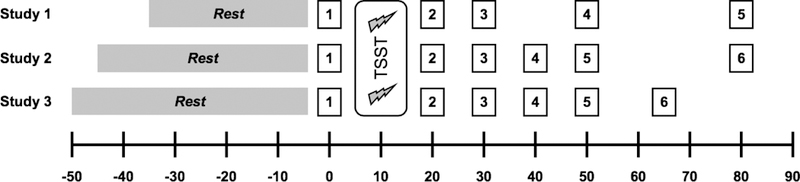
Study timelines. Time (in min) is in relation to TSST onset. Saliva sampling time points are represented as numbered boxes.
3.3. Measures
Anxiety and depressive symptoms were assessed prior to the TSST using the Hospital Anxiety and Depression Scale (HADS; Zigmond & Snaith, 1983). The scale consists of 14 items measuring symptom severity on a scale of 0–3 with subscales for anxiety (HADS_A) and depression (HADS_D), and a range of possible scores for each subscale of 0–21. The subscales were designed to distinguish between the two sets of symptoms while avoiding ‘noise’ from somatic or serious mental disorders (Zigmond & Snaith, 1983). The scale performs well in identifying symptom severity, and demonstrates good internal consistency across subscales on average (Chronbach’s alpha = .83 and .82 for HADS-A and HADS-D, respectively; Bjelland, Dahl, Haug, & Neckelmann, 2002) and acceptable in our sample (Cronbach’s alpha = .79 and .80 for HADS-A and HADS-D, respectively). Cut-off scores of 8+ for both subscales demonstrate the optimal balance between sensitivity and specificity for identifying cases of anxiety disorders and depression, with sensitivity and specificity of approximately .8 for both subscales (Bjelland et al., 2002).
3.4. HPA axis stress responses
HPA axis responses to stress were assessed by measuring salivary free cortisol. Saliva samples were collected at the time points outlined in Figure 1, and were immediately stored at −30C until processing. Prior to analysis, samples were thawed and centrifuged at 2000×g and 20C for 10 minutes. Cortisol was measured using a competitive chemiluminescence immunoassay (IBL-International, Toronto, ON, Canada). All samples were assayed in duplicate, with intra- and inter-assay CV’s below 10%.
3.5. Statistical analyses
All statistical analyses were conducted using version 3.5.0 (R Development Core Team, 2018). Multilevel modeling were fitted using the lme4 package (Bates, Mächler, Bolker, & Walker, 2015), with p-values supplied by the lmerTest package (Kuznetsova, Brockhoff, & Christensen, 2017), and plotting created using the ggplot2 package (Wickham, 2009).
Cortisol concentrations were log-transformed to adjust for skewness. Cortisol peaks were identified following a procedure adapted from Lopez-Duran and colleagues (2014). Peaks were initialized as the first post-stress sample that was followed by a decline or a “plateau” (defined as a subsequent sample <10% above the selected sample). If any subsequent samples were >10% above the previously defined peak, that sample was selected as the peak. Because detecting differences in cortisol responses to a stressor logically necessitates the success of that stressor eliciting a response in the first place, it is necessary to utilize a reasonable, biologically plausible threshold to differentiate cortisol “responders” from “non-responders” (Miller, Plessow, Kirschbaum, & Stalder, 2013). Cortisol responders were defined as those with raw baseline-to-peak increases greater than 1.5 nmol/l following thresholds described by Robert Miller and colleagues (2013). Baseline cortisol was defined as the sample taken immediately prior to TSST onset. For non-responders, “peak” times and samples were then defined as the mode peak time of the responders (30 minutes post-TSST onset).
Preliminary analyses included t-tests to identify differences by sex or responder status, and Pearson correlations to identify bi-variate associations among HADS subscales, cortisol baseline and BTP, and covariates. The data structure utilized in the multilevel models has been summarized previously by others (e.g. Lopez-Duran et al., 2014).
The efficacy of the TSST was assessed using an unconditional growth multilevel model, incorporating a reactivity slope (time in minutes from baseline to peak in relation to peak) and recovery slope (time in minutes from peak to final sample in relation to peak) simultaneously. Therefore, the reactivity slope variable is equal to 0 during the recovery phase, the recovery phase is equal to 0 during the reactivity phase, and the peak is represented as the intercept when both the reactivity and recovery slopes are equal to 0. If the TSST is efficacious in eliciting a cortisol response,the reactivity slope should be significant and positive, indicating that cortisol concentrations increased in response to the TSST. Likewise, a significant and negative recovery slope would be indicative of a typical cortisol response. Both reactivity and recovery slopes were included in addition to intercepts as random effects, with individual participants treated as the grouping variable. Because reactivity, recovery, and peaks are all correlated, the covariances of these random effects were estimated in all models as well.
A series of conditional growth models were then fit to address research aims. In order to evaluate the relationships between depression and anxiety symptoms and cortisol reactivity to and recovery from stress, we first fit a model that included HADS_A and HADS_D, sex (female), age, BMI, and dummy-coded study variables as fixed effects (Model 1).
Although our sample did not include anyone with a diagnosed psychiatric disorder, some participants reported scores greater than the recommended threshold for identifying possible caseness of anxiety or depression disorders. In order to test whether the relationships between HADS subscales and reactivity and/or recovery varied among those having HADS subscale scores above the threshold, we first constructed a dummy variable identifying observations that had any HADS subscale greater than or equal to the recommended threshold of 8 (i.e. “HADS8”). We then included that variable in three-way interactions with HADS subscales, reactivity, and recovery in Model 2. Those interactions were not significant, indicating that the relationships between HADS subscales and cortisol reactivity and recovery did not differ among those with scores above the threshold. As a result, the three-way interactions were dropped from subsequent models.
Given existing evidence that comorbidity may influence HPA axis responses to stress, we then tested whether the relationship between anxiety and cortisol reactivity and recovery was moderated by anxiety (and vice versa) by computing a model containing three-way interactions of HADS_A, HADS_D, and reactivity/recovery (Model 3). Finally, in order to investigate the role of sex in the relationships between depression, anxiety and cortisol reactivity and recovery we fit a model incorporating three-way interactions of HADS subscales, reactivity/recovery, and sex (Model 4). The statistical models for these analyses are as follows:
Unconditional Growth Model
Model 1
Model 2
Model 3
Model 4
All models were applied to both the full sample and the cortisol responder subset. HADS subscales, BMI, and age were standardized to have means of 0 and standard deviations of 1 (i.e. “z-score”) in order to facilitate model convergence. Results were considered significant at the p <= .05 level, with p-values for multilevel model fixed effects calculated using Satterthwaite’s approximation of degrees of freedom.
4. Results
4.1. Descriptive statistics
Seventy-two (50.3%) of participants had baseline-to-peak concentrations greater than 1.5 nmol/l and were classified as cortisol responders. A greater proportion of males responded to the TSST compared to females (58% vs. 43.2%); however, this difference was not statistically significant (χ2(1)=3.10, p=.078). Descriptive statistics by responder type and sex are summarized in Table 1. Mean HADS_A scores were slightly above the threshold of 8 recommended (Mean = 8.5) to identify caseness of anxiety among the full sample and responders, indicating that on average participants in our sample were showed symptoms of mild anxiety (Stern, 2014). Responders were younger on average than non-responders, though this difference did not reach statistical significance (t(141)=1.8, p=.07). Body mass index, log baseline cortisol and HADS subscales did not differ significantly on average between responders and non-responders (all p’s >.3). Males had significantly higher BMI on average overall (t(141)=2.85, p=.005), but not among responders (t(70)=1.73, p=.09). Females scored significantly higher on the HADS_A compared to males on average overall (t(141)=2.68, p=.008); however, the mean difference did not reach statistical significance among the responder subset (t(70)=1.86, p=.06). Age, log-transformed baseline cortisol, and HADS_D scores did not differ significantly by sex among the entire sample or the responder subset (all p’s>.1).
Table 1.
Descriptive statistics of study variables by responder status and sex.
| Total Sample | Responders | Non-Responders | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T | M | F | T | M | F | T | M | F | |
| N | 143 | 69 | 74 | 72 | 40 | 32 | 71 | 29 | 42 |
| 48.3% | 51.7% | 50.3% | 55.6% | 44.4% | 49.7% | 40.8% | 59.2% | ||
| Age | 29.5 | 29 | 30.1 | 27.1 | 29.7 | 23.9 | 32 | 28 | 34.7 |
| ±16.3 | ±15.9 | ±16.8 | ±15.2 | ±16.4 | ±13.1 | ±17.1 | ±15.4 | ±17.9 | |
| BMI | 24.1 | 25 | 23.2 | 24 | 24.7 | 23.1 | 24.1 | 25.4 | 23.3 |
| ±3.8 | ±3.9 | ±3.5 | ±3.8 | ±3.9 | ±3.6 | ±3.8 | ±3.9 | ±3.5 | |
| Cortisol | 11.5 | 11.6 | 11.4 | 11.1 | 11.3 | 11 | 11.9 | 12.1 | 11.8 |
| baseline | |||||||||
| ±7 | ±6.3 | ±7.6 | ±6.2 | ±5.8 | ±6.8 | ±7.7 | ±7.1 | ±8.2 | |
| Cortisol BTP | 4.2 | 5.6 | 2.9 | 11.4 | 12.1 | 10.4 | −3 | −3.3 | −2.8 |
| ±10.6 | ±10.9 | ±10.1 | ±10.1 | ±9.5 | ±11 | ±4 | ±4.7 | ±3.5 | |
| HAD_S | 8.5 | 7.5 | 9.3 | 8.8 | 8 | 9.7 | 8.1 | 6.9 | 9 |
| ±4.2 | ±4.3 | ±3.9 | ±4 | ±4.6 | ±2.9 | ±4.3 | ±3.8 | ±4.5 | |
| HADS_D | 6.3 | 5.7 | 6.8 | 6.3 | 5.6 | 7.3 | 6.2 | 5.9 | 6.5 |
| ±4.6 | ±4.4 | ±4.7 | ±4.4 | ±4.4 | ±4.2 | ±4.8 | ±4.5 | ±5.1 | |
Note: T=total; M=male; F=female; Cortisol baseline and BTP (baseline-to-peak) are in nmol/l. The first two rows show N’s and percentages, other numbers are means and standard deviations (±).
4.2. Correlations
The HADS_A subscale was highly positively correlated with HADS D for both the full sample and responder subset(Full sample: r=.64, p<.001; Responders: r=.69, p<.001). Baseline-to-peak cortisol calculated using log-transformed values was positively correlated with HADS_D among responders (r=.24, p=.04) but not the full sample (r=.11, p=.21) or HADS_A for either sample (Full sample: r=.08, p=.29, Responders: r=.02, p=.85). All other Pearson correlations of study variables for the full sample and responder subset are summarized in Table 2
Table 2.
Pearson correlations of study variables.
| Age | BMI | Cortisol baseline |
Cortisol BTP | HADS_A | |
|---|---|---|---|---|---|
| BMI | 0.29*** | ||||
| 0.35** | |||||
| Cortisol | |||||
| baseline | −0.43*** | −0.12 | |||
| −0.23† | −0.20† | ||||
| Cortisol BTP | −0.02 | 0.02 | −0.29*** | ||
| 0.03 | 0.09 | −0.423*** | |||
| HADS_A | −0.03 | 0.05 | −0.06 | 0.07 | |
| −0.14 | 0.04 | −0.01 | 0.02 | ||
| HADS_D | 0.08 | 0.16† | −0.02 | 0.11 | 0.64*** |
| 0.01 | 0.16 | −0.13 | 0.24* | 0.69*** |
Notes:
p < .1.
p < .05.
p < .01.
p < .001.
Shaded correlations are calculated from the full sample, unshaded regions are among responders only. Cortisol baseline is in units of log-transformed nmol/l. Cortisol BTP (baseline-to-peak) was calculated using log-transformed nmol/l concentrations.
4.3. Unconditional growth models
Cortisol responses by study and responder status are depicted in Figure 2. The unconditional growth model of the full sample indicates that the TSST elicited a robust cortisol response (Reactivity β =.007, t(141.3)=4.19, p<.001; Recovery β =−.010, t(135.0)=−14.66, p<.001). The unconditional growth model of the cortisol responder subset indicated steeper reactivity slopes and similar recovery slopes compared to the full sample (Reactivity β =.024, t(71.0)=12.31, p<.001; Recovery β =−.014, t(60.4)=−15.25, p<.001).
Figure 2.
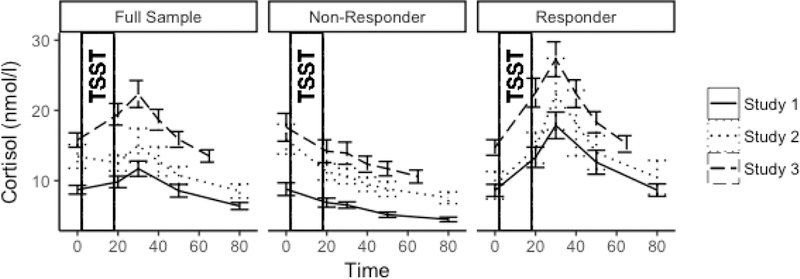
Average cortisol response by responder status and study. Error bars represent standard error of the mean.
4.4. Conditional growth models
All conditional growth models for both the full sample and responder subset are summarized in Table 3. Interactions with reactivity and recovery reflect how a predictor is associated with reactivity or recovery slopes. positive interaction with reactivity indicates that the predictor is associated with a steeper reactivity slope; conversely, a positive interaction with recovery indicates that the predictor is associated with less steep (i.e. flatter) recovery slope. Fixed effects that are not interactions represent how the peak (i.e. intercept) is associated with the predictor, holding all other variables constant.
Table 3.
Conditional model summaries among the full sample and responder subset.
| Full Sample | Responders | |||||||
|---|---|---|---|---|---|---|---|---|
| M1 | M2 | M3 | M4 | M1 | M2 | M3 | M4 | |
| (Intercept) | 2.375*** | 2.595*** | 2.346*** | 2.377*** | 2.804*** | 3.096*** | 2.737*** | 2.808*** |
| (0.097) | (0.022) | (0.105) | (0.097) | (0.117) | (0.295) | (0.139) | (0.119) | |
| Reactivity | 0.012*** | 0.016* | 0.01** | 0.011*** | 0.028*** | 0.033*** | 0.026*** | 0.028*** |
| (0.003) | (0.007) | (0.003) | (0.003) | (0.003) | (0.009) | (0.004) | (0.003) | |
| Recovery | −0.012*** | −0.011*** | −0.011*** | −0.012*** | −0.015*** | −0.015*** | −0.014*** | −0.015*** |
| (0.001) | (0.003) | (0.001) | (0.001) | (0.001) | (0.004) | (0.002) | (0.001) | |
| Female | −0.293* | −0.256* | −0.289* | −0.289* | −0.275† | −0.182 | −0.232 | −0.302* |
| (0.114) | (0.118) | (0.115) | (0.115) | (0.145) | (0.154) | (0.153) | (0.147) | |
| HADS_A | −0.099 | 0.310 | −0.102 | −0.035 | −0.091 | 0.612* | −0.093 | −0.166 |
| (0.073) | (.215) | (0.073) | (0.101) | (0.096) | (0.276) | (0.096) | (0.128) | |
| HADS_D | 0.177* | −0.100 | 0.157† | 0.118 | 0.152 | −0.465 | 0.105 | 0.206 |
| (0.077) | (0.241) | (0.081) | (0.112) | (0.1) | (0.313) | (0.113) | (0.15) | |
| HADS8 | −0.292 | −0.537† | ||||||
| (0.220) | (0.287) | |||||||
| HADS_A:HADS8 | −0.421† | −0.635* | ||||||
| (0.236) | (0.311) | |||||||
| HADS_D:HADS8 | 0.300 | 0.681* | ||||||
| (.255) | (0.332) | |||||||
| Reactivity:Female | −0.008* | −0.008* | −0.008* | −0.005 | −0.004 | −0.006 | ||
| (0.004) | (0.004) | (0.004) | (0.004) | (0.004) | (0.004) | |||
| Reactivity:HADS_A | 0.000 | 0.000 | 0.000 | −0.005† | −0.006† | −0.008* | ||
| (0.002) | (0.002) | (0.003) | (0.003) | (0.003) | (0.004) | |||
| Reactivity:HADS_D | 0.004 | 0.003 | 0.001 | 0.007* | 0.006† | 0.008† | ||
| (0.003) | (0.003) | (0.004) | (0.003) | (0.003) | (0.004) | |||
| Reactivity:HADS8 | −0.008 | −0.008 | ||||||
| (0.007) | (0.009) | |||||||
| Recovery:Female | 0.003† | 0.003† | 0.003† | 0.004* | 0.004† | 0.005** | ||
| (0.001) | (0.001) | (0.001) | (0.002) | (0.002) | (0.002) | |||
| Recovery:HADS_A | 0.002* | 0.002* | 0.002† | 0.003* | 0.003* | 0.004* | ||
| (0.001) | (0.001) | (0.001) | (0.001) | (0.001) | (0.002) | |||
| Recovery:HADS_D | −0.002† | −0.001 | −0.002 | −0.003* | −0.002 | −0.003 | ||
| (0.001) | (0.001) | (0.001) | (0.001) | (0.001) | (0.002) | |||
| Recovery:HADS8 | −0.000 | 0.002 | ||||||
| (0.003) | (0.004) | |||||||
| Reactivity:HADS_A:HADS_D | 0.002 | 0.001 | ||||||
| (0.002) | (0.002) | |||||||
| Recovery:HADS_A:HADS_D | −0.001 | −0.001 | ||||||
| (0.001) | (0.001) | |||||||
| Reactivity:HADS_A:Female | 0.000 | 0.011† | ||||||
| (0.005) | (0.006) | |||||||
| Reactivity:HADS_D:Female | 0.004 | −0.002 | ||||||
| (0.005) | (0.005) | |||||||
| Recovery:HADS_A:Female | −0.001 | −0.004 | ||||||
| (0.002) | (0.003) | |||||||
| Recovery:HADS_D:Female | 0.001 | 0 | ||||||
| (0.002) | (0.002) | |||||||
| Reactivity:HADS_A:HADS8 | −0.003 | −0.008 | ||||||
| (0.008) | (0.010) | |||||||
| Reactivity:HADS_D: HADS8 | 0.002 | 0.006 | ||||||
| (0.009) | (0.010) | |||||||
| Recovery:HADS_A: HADS8 | −0.002 | 0.005 | ||||||
| (0.003) | (0.004) | |||||||
| Recovery:HADS_D: HADS8 | 0.002 | −0.009† | ||||||
| (0.003) | (0.005) | |||||||
Notes:
p < .1.
p < .05.
p < .01.
p < .001.
Numbers in parentheses are standard errors. HADS_A and HADS_D scores are standardized with a mean of 0 and standard deviation of 1. All models include z-scores for Age and BMI as well as dummy-coded study variables predicting intercepts, reactivity and recovery (not summarized).
4.4.1. Full sample
Among the full sample, females had significantly lower peaks than males (Female β = −.29, t(135)=−2.566, p=.011). The interaction of female sex and reactivity was statistically significant, such that females had less steep reactivity slopes compared to males (Female×Reactivity β=−.008,t(135)=−2.203, p=.029). The interaction of female sex and recovery showed a similar pattern—such that females had less steep recovery slopes than men—but did not reach statistical significance (Female×Recovery β =.003, t(126.5)=1.839, p=.068).
Neither of the interactions of HADS subscales and reactivity were statistically significant (HADS_A×Reactivity β =−.0003, t(134.8)=.013, p=.99; HADS_D×Reactivity β =.004, t(134.1)=1.497, p=.14). The interaction of HADS_A and recovery was significant, such that greater HADS scores were associated with less steep recovery slopes (HADS_A×Recovery β =.0019, t(122.3)=2.082, p=.039). The relationship between HADS_D and recovery exhibited an opposite pattern, with higher scores predicting steeper recovery; however, the interaction did not reach statistical significance (HADS_D×Recovery β =−.0017, t(119.6)=−1.845, p=.068). HADS_A was not associated with peak cortisol concentrations (HADS_A β =−.099, t(135.1)=−Depression, Anxiety, and Cortisol Responses Fiksdal 14 1.354, p=.178), while higher HADS_D scores were associated with higher peaks (HADS_D β =.177, t(135)=2.298, p=.023).
Model 2 revealed that having HADS_A or HADS_D scores greater than or equal to 8 did not moderate HADS subscale interactions with reactivity (Reactivity×HADS_A×HADS8 β = − .003, t(131.2) = −.33, p= .74; Reactivity×HADS×HADS8β=.002,t(131.3)=.21,p=.83)or recovery (Recovery×HADS_A × HADS8 β = −.002, t(118.9)=−.80, p= .42; Recovery×HADS_D× HADS8 β = −.002, t(119.6)= .552, p= .58).
Model 3 did not reveal statistically significant three-way interactions of HADS A, HADS_D and reactivity (Reactivity×HADS_A×HADS D β = .002, t(133)=1.125, p=.26) or HADS_A, HADS_D and recovery (Recovery×HADS ×HADS_D β = −.0006, t(116.1)=−.883, p=.38). Likewise, in Model 4 sex did not moderate associations between HADS subscales and reactivity (Reactivity×HADS_A×Female β = .0004, t(132.4)=−.093, p=.93; Reactivity×HADS_D×Female β = .0041, t(131.7)=.881, p=.38) or recovery (Recovery×HADS_A ×Female β = −.0012, t(121.5)=−.648, p=.52; Recovery×HADS_D×Female β = .0007,t(119.3)=.422, p=.67).
4.4.2. Responder subset
Among responders, sex was not associated with peaks or reactivity slopes (Female β = − .275, t(63.96)=−1.904, p=.06; Female×Reactivity β =−.005, t(63.76)=−1.098, p=.28). The female sex by recovery interaction was significant, such that females had less steep recovery slopes compared to men (Female×Recovery β =.004, t(54.6)=2.363, p=.02).
The reactivity by HADS_A nteraction approached but did not reach statistical significance, such that higher HADS_A scores were marginally associated with less steep reactivity slopes (Reactivity×HADS_A β = −.006, t(64.00)=−1.97, p=.053). This pattern was similar for recovery and statistically significant, such that higher HADS_A scores predicted less steep recovery slopes (Recovery×HADS_A β = .003, t(59.22)=2.29, p=.025). HADS_A scores did not predict peak log cortisol concentrations (HADS_A β =−.09, t(64.08)=−.96, p=.34).
The reactivity by HADS_D interaction was statistically significant, such that higher HADS_D scores were associated with steeper reactivity slopes (Reactivity×HADS_D β = .007, t(63.32)=2.53, p=.026). Similarly, HADS_D significantly moderated recovery such that greater HADS_D scores predicted steeper recovery slopes (Recovery×HADS_D β = −.003, t(58.75)=− 2.20, p=.03). HADS_D did not predict log cortisol peaks (HADS D β =.15, t(64.08)=1.538, p=.13). Predicted log cortisol trajectories by HADS A, HADS D, and sex are depicted in Figure 3.
Figure 3.
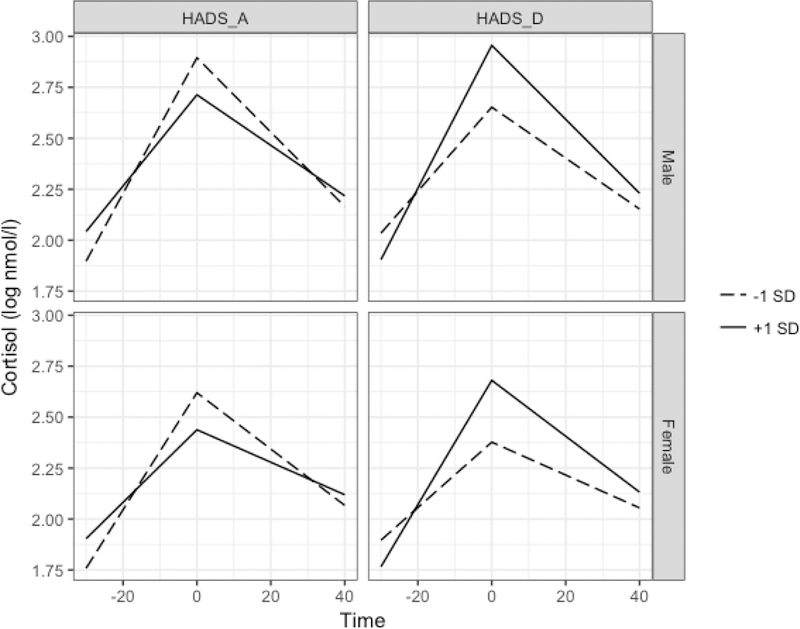
Predicted cortisol values based on Model 1 among cortisol responders by HADS subscale values and sex. All other covariates held constant at sample means. Greater HADS_A was associated with blunted responses and recovery, while greater HADS_D predicted steeper response and recovery slopes.
Model 2 revealed that having HADS_A or HADS_D scores greater than or equal to 8 did not moderate HADS subscale interactions with reactivity (Reactivity×HADS A×HADS8 β = −.008, t(60.3) = −.79, p= .43; Reactivity×HADS D×HADS8 β = .006, t(60.3)=.57, p=.57) or recovery (Recovery×HADS_A×HADS8 β = .005, t(52.4)=1.21, p= .23; Recovery×HADS_D×HADS8 β = −.009, t(56.9)= −1.90, p= .06).
Model 3 did not reveal statistically significant three-way interactions of HADS_A,HADS_D and reactivity (Reactivity×HADS_A×HADS_D β = .001, t(63.36)=.72, p=.48) or HADS_A, HADS_D and recovery (Recovery×HADS_A×HADS_D β = −.001, t(56.71)=−1.21, p=.23). Likewise, in Model 4 sex did not moderate associations between HADS subscales and reactivity (Reactivity×HADS_A×Female β = .011, t(60.85)=1.835, p=.07; Reactivity×HADS ×Female β = −.002, t(60.42)=−.36, p=.72) or recovery (Recovery×HADS_A×Female β = −.005, t(57.9)=−1.439, p=.16; Recovery×HADS_D×Female β = −.0004, t(57.33)=−.16, p=.87).
5. Discussion
The present study sought to examine the relationships between non-somatic symptoms of depression and anxiety and cortisol responses to and recovery from acute stress among healthy adults without a diagnosis of anxiety or mood disorders. Among a sample of cortisol responders and non-responders, we found that females exhibited blunted responses to stress, with lower peaks and more shallow reactivity slopes. Females exhibited flatter recovery slopes on average, though this association did not reach statistical significance. Among the full sample, anxiety was significantly associated with flatter recovery slopes. This association formed the tail end of an overall pattern consisting of increased anxiety symptoms predicting blunted reactivity and lower peaks, though those predictors did not reach statistical significance. Individuals with higher depressive symptoms exhibited an opposite pattern of responses on average, with higher peaks and steeper reactivity and recovery slopes; however, the coefficients predicting reactivity and recovery did not reach statistical significance. Given that the magnitude of the slopes of HADS_A and HADS_D predicting reactivity and recovery were similar, the smaller standard error of the HADS_A×Recovery slope accounts for the difference in statistical significance.
Among responders, we found similar patterns of anxiety and depressive symptoms predicting cortisol reactivity and recovery, but of greater magnitude compared to the full sample. Anxiety symptoms were marginally associated with blunted reactivity and significantly associated with flatter recovery, while depressive symptoms significantly predicted steeper reactivity and recovery slopes. Anxiety and depressive symptoms did not predict cortisol peaks among responders. Females had marginally lower peaks, and significantly flatter recovery slopes, but not reactivity slopes.
Having a HADS subscale value greater than or equal to the recommended cutoff of 8 for identifying possible caseness of anxiety or depression did not moderate any of the relationships between HADS subscales and reactivity or recovery in either the full sample or responder subset. Likewise, anxiety and depressive symptoms did not interact to predict peaks, reactivity or recovery slopes, nor did sex moderate any relationships between anxiety and depressive symptoms and the cortisol response among either the full sample or responder subset.
Our findings add to the limited existing research indicating that depressive symptoms can be associated with elevated cortisol responses to stress among individuals without psychiatric diagnoses (Powers et al., 2016; Puig-Perez et al., 2016). Our results also indicate that previously observed relationships between anxiety symptoms and blunted cortisol responses to stress are not limited to men or older adults (Brooks & Robles, 2009; de Rooij et al., 2010). The opposite relationship observed between anxiety and depressive symptoms and cortisol responses also sheds light on why previous findings have been inconsistent and why individuals with comorbid anxiety and depressive disorders fail to exhibit altered HPA axis responses while patterns emerge in isolation. Given that anxiety and depressive symptoms are highly correlated, it is possible that the individual effects of each symptom type cancel each other out when they occur at a similar magnitude. The differences in magnitude of the associations between depressive and anxiety symptoms and the cortisol response between the full sample and responder subset highlights the importance of taking into account responder status when evaluating predictors of reactivity and recovery. Even though the patterns of associations were similar, including non-responders in the analysis diluted the magnitude of associations. Thus, it appears that including the responses of non-responders in analyses may add noise to models assessing relationships with cortisol responses and obscure important findings. Our results also highlight another important emergent pattern: anxiety and depressive symptomology may influence the cortisol stress response in different or opposite directions than their respective psychiatric disorders. Our research supports existing literature suggesting that sub-clinical anxiety and depression are associated with blunted and heightened responses, respectively. Conversely, clinically MDD has thus far generally shown an opposite pattern, while results have been inconsistent in the context of diagnosed anxiety disorders. More observational, experimental, and longitudinal research will be needed to tease apart how depressive and anxiety symptom severity and clinical diagnoses impact HPA axis responses to stress.
The results of the present study fit into a larger pattern of associations depression symptoms and HPA axis responses being inconsistent or in different directions compared to diagnosed MDD. This pattern is in contrast to the literature concerning cardiovascular responses, which generally show consistent and congruent associations with depressive symptoms and MDD (Carroll, Ginty, Whittaker, Lovallo, & de Rooij, 2017). In the context of depression, alterations to the HPA axis response may depend on a variety of factors. For example, chronicity has been shown to influence the directionality of associations between depression symptoms and cortisol responses to stress in a curvilinear fashion, with recent-onset predicting elevated and chronic symptoms predicting blunted responses(Booij,Bouma,de Jonge, Ormel, & Oldehinkel, 2013; Zorn et al., 2017). There is evidence that alterations of HPA axis responses associated with depression may also dissipate in conjunction with remission (Zorn et al., 2017). Conversely, alterations to HPA axis responses may precede future re-occurrence of depression (Calhoun et al., 2012). aken together the findings of the present study and others portray a dynamic and interactive relationship between HPA axis responses and depression. Initial plasticity of the HPA axis may give way to alterations that are more consistent in directionality and more permanent in nature as symptom duration and severity increases. Unfortunately, HPA axis responses have not been studied as extensively in the context of anxiety symptoms and disorders, and the evidence that exists is mixed. Nevertheless, it is possible that such inconsistencies also stem from a similarly dynamic and interactive relationship between anxiety and HPA axis functioning. More longitudinal studies will be needed to tease apart how cortisol responses to stress and symptoms of anxiety and depression are associated with the development of HPA axis dysregulation and psychopathology.
Our results suggest that the altered cortisol responses associated with anxiety and depressive symptoms, independent of a clinical diagnosis, could potentially play a role in patterns of HPA axis dysregulation characteristic of allostatic overload. Indeed, depressive symptoms have been associated with indices of allostatic load (Kobrosly, van Wijngaarden, Seplaki, Cory-Slechta, & Moynihan, 2014). Moreover, allostatic load, MDD, and anxiety disorders are thought to alter brain structures responsible for the modulation of the HPA axis (e.g. hippocampus shrinkage), which in turn contributes to the dysregulation of other allostatic systems leading to increased risk of major disease (McEwen, 2003). Such alterations are thought to occur gradually, and the role of depression appears to depend on the duration of symptoms (Bremner et al., 2000; McEwen, 2003). While there is a relatively large amount of research focusing on depression and its relationship with allostatic load and its role in altering brain structures and regulating the HPA axis, less is known about anxiety symptoms and associated disorders in this context (Gale et al., 2015; Juster, McEwen, & Lupien, 2010). Regardless, our results suggest that both depressive and anxiety symptoms—independent of diagnosis—may be enough to begin potentially harmful alterations to HPA axis functioning in response to acute stress.
This study has some important limitations that should be considered. First, although the we sought to investigate anxiety and depressive symptoms among individuals without psychiatric illness, the average scores of the HADS anxiety subscale for our sample were slightly above the cut-off recommended for caseness of anxiety disorders, suggesting that our sample was mildly anxious (Stern, 2014). Second, the HADS does not evaluate symptom duration, which may play an important role in how anxiety and/or depression impacts HPA axis functioning (McEwen, 2003). Third, while two of the studies included in the dataset recruited adults of all ages, a majority of our sample were undergraduate students in an academically rigorous university setting which may limit the generalizability of the findings. Finally, many of our central findings were more pronounced among or limited to cortisol responders. While this reduction in effect size among the full sample could be the result of noise added by non-responders (as discussed above), it is also possible that the study simply did not have the statistical power to detect differences. However, this explanation fails to explain why the effects were more pronounced among the smaller subset. Regardless, we were unable to evaluate how symptoms of anxiety and/or depression were associated with HPA axis response to and recovery from acute stress among those who did respond to the TSST.
6. Conclusions
In summary, we found that anxiety symptoms were associated with blunted responses to and recovery from an acute psychosocial stressor, while depressive symptoms were associated with steeper response and recovery slopes. These patterns of associations were stronger among cortisol responders. These findings add to a growing body of research demonstrating heterogeneity of associations between HPA axis responses to stress and symptoms of anxiety and depression, and highlight the importance of taking responder status into account when analyzing cortisol responses to and recovery from stress. These results also indicate that anxiety or depressive symptoms—independent of a diagnosed disorder—may be enough to alter HPA axis functioning, which in turn may contribute to allostatic load and disease. Longitudinal approaches and research incorporating measures of symptom duration may help explain the emerging contradictory associations between cortisol responses and sub-clinical symptomology vs. clinical psychiatric disorders.
Highlights.
Greater symptoms of anxiety were significantly associated with flatter recovery slopes among the full sample and cortisol responders
Greater symptoms of anxiety were marginally associated with blunted cortisol responses among cortisol responders
Greater symptoms of depression significantly predicted steeper cortisol response and recovery slopes among cortisol responders
9. Acknowledgement
This research was supported by the American Federation of Aging Research (NR), and by training grants from the National Institute of Health (T32-084907 DG). We also want to thank research assistants in the Laboratory for Biological Health Psychology at Brandeis University for their help with data collection.
8 Role of the funding source
The study was partially funded by a research grant from the American Federation for Aging Research (AFAR), by a training grant from the National Institute of Health (T32-084907) supporting DG, and by the Swiss National Science Foundation (SNF) supporting MVT. None of the funding sources did have any role in study design, analyses, and interpretation of data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement:
The authors have no conflicts of interest to declare.
10 References
- Bates D, Mächler M, Bolker B, & Walker S (2015). Fitting Linear Mixed-Effects Models Using lme4. Journal of Statistical Software, 67(1), 1–48. [Google Scholar]
- Bjelland I, Dahl AA, Haug TT, & Neckelmann D (2002). The validity of the Hospital Anxiety and Depression Scale. An updated literaturereview. JPsychosomRes,52(2), 69–77. [DOI] [PubMed] [Google Scholar]
- Booij SH, Bouma EM, de Jonge P, Ormel J, & Oldehinkel AJ (2013). Chronicity of depressive problems and the cortisol response to psychosocial stress in adolescents: the TRAILS study. Psychoneuroendocrinology, 38(5), 659–666. doi: 10.1016/j.psyneuen.2012.08.004 [DOI] [PubMed] [Google Scholar]
- Bremner JD, Narayan M, Anderson E, Staib LH, Miller HL, & Charney DS (2000). Hippocampal volume reduction in major depression. Am J Psychiatry, 157(1), 115–118. doi: 10.1176/ajp.157.1.115 [DOI] [PubMed] [Google Scholar]
- Brooks KP, & Robles TF (2009). Recent depressive and anxious symptoms predict cortisol responsestostressinmen. Psychoneuroendocrinology, 34(7), 1041–1049. doi: 10.1016/j.psyneuen.2009.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TA, Campbell LA, Lehman CL, Grisham JR, & Mancill RB (2001). Current and lifetime comorbidity of the DSM-IV anxiety and mood disorders in a large clinical sample. J Abnorm Psychol, 110(4), 585–599. [DOI] [PubMed] [Google Scholar]
- Burke HM, Davis MC, Otte C, & Mohr DC (2005). Depression and cortisol responses to psychological stress: a meta-analysis. Psychoneuroendocrinology, 30(9), 846–856. doi: 10.1016/j.psyneuen.2005.02.010 [DOI] [PubMed] [Google Scholar]
- Calhoun D, Franklin JC, Adelman CB, Guerry JD, Hastings PD, Nock MK, & Prinstein MJ (2012). Biological and cognitive responses to an in vivo interpersonal stressor: Longitudinal associations with adolescent depression. International Journal of Cognitive Therapy, 5(3), 283–299. doi: 10.1521/ijct.2012.5.3.283 [DOI] [Google Scholar]
- Carroll D, Ginty AT, Whittaker AC, Lovallo WR, & de Rooij SR (2017). The behavioural, cognitive, and neural corollaries of blunted cardiovascular and cortisol reactions to acute psychological stress. Neurosci Biobehav Rev, 77, 74–86. doi: 10.1016/j.neubiorev.2017.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisan LG, Vulturar R, Miclea M, & Miu AC(2016).Reactivity to Social Stress in Subclinical Social Anxiety: Emotional Experience, Cognitive Appraisals, Behavior, and Physiology. Front Psychiatry, 7, 5. doi: 10.3389/fpsyt.2016.00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij SR, Schene AH, Phillips DI, & Roseboom TJ (2010). Depression and anxiety: Associations with biological and perceived stress reactivity to a psychological stress protocol in a middle-aged population. Psychoneuroendocrinology, 35(6), 866–877. doi: 10.1016/j.psyneuen.2009.11.011 [DOI] [PubMed] [Google Scholar]
- Elnazer HY, & Baldwin DS (2014). Investigation of cortisol levels in patients with anxiety disorders: a structured review. Curr Top Behav Neurosci, 18, 191–216. doi: 10.1007/7854_2014_299 [DOI] [PubMed] [Google Scholar]
- Espín L, Marquina M, Hidalgo V, Salvador A, & Gómez-Amor J (2016). No effects of psychosocial stress on memory retrieval in non-treated young students with Generalized Social Phobia. Psychoneuroendocrinology, 73, 51–62. doi: 10.1016/j.psyneuen.2016.07.211 [DOI] [PubMed] [Google Scholar]
- Furlan PM, DeMartinis N, Schweizer E, Rickels K, & Lucki I (2001). Abnormal salivary cortisol levels in social phobic patients in response to acute psychological but not physical stress. Biol Psychiatry, 50(4), 254–259. [DOI] [PubMed] [Google Scholar]
- Gale CR, Batty GD, Cooper SA, Deary IJ, Der G, McEwen BS, & Cavanagh J (2015). Reaction time in adolescence, cumulative allostatic load, and symptoms of anxiety and depression in adulthood: the West of Scotland Twenty-07 Study. Psychosom Med, 77(5), 493–505. doi: 10.1097/psy.0000000000000189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianferante D, Thoma MV, Hanlin L, Chen X, Breines JG, Zoccola PM, & Rohleder N (2014). Post-stress rumination predicts HPA axis responses to repeated acute stress. Psychoneuroendocrinology, 49, 244–252. doi: 10.1016/j.psyneuen.2014.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Negriff S, Kim H, & Susman EJ (2016). A study of cortisol reactivity and recovery among young adolescents: Heterogeneity and longitudinal stability and change. Dev Psychobiol, 58(3), 283–302. doi: 10.1002/dev.21369 [DOI] [PubMed] [Google Scholar]
- Juster RP, McEwen BS, & Lupien SJ (2010). Allostatic load biomarkers of chronic stress and impact on health and cognition. Neurosci Biobehav Rev, 35(1), 2–16. doi: 10.1016/j.neubiorev.2009.10.002 [DOI] [PubMed] [Google Scholar]
- Kirschbaum C, Pirke KM, & Hellhammer DH (1993). The ‘Trier ocial Stress Test’--a tool for investigating psychobiological stress responses in a laboratory setting. Neuropsychobiology, 28(1–2), 76–81. doi: 10.1159/000119004 [DOI] [PubMed] [Google Scholar]
- Klumbies E, Braeuer D, Hoyer J, & Kirschbaum C (2014). The reaction to social stress in social phobia: discordance between physiological and subjective parameters. PLoS One,9(8),e105670.doi: 10.1371/journal.pone.0105670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobrosly RW, van Wijngaarden E, Seplaki CL, Cory-Slechta DA, & Moynihan J (2014). Depressive symptoms are associated with allostatic load among community-dwelling older adults. Physiol Behav, 123, 223–230. doi: 10.1016/j.physbeh.2013.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuras YI, McInnis CM, Thoma MV, Chen X, Hanlin L, Gianferante D, & Rohleder N (2017). Increased alpha-amylase response to an acute psychosocial stress challenge in healthy adults with childhood adversity. Dev Psychobiol, 59(1), 91–98. doi: 10.1002/dev.21470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova A, Brockhoff PB, & Christensen RHB (2017). lmerTest Package: Tests in Linear Mixed Effects Models. Journal of Statistical Software, 82(13), 1–26. [Google Scholar]
- Lopez-Duran NL, Mayer SE, & Abelson JL (2014). Modeling neuroendocrine stress reactivity in salivary cortisol: adjusting for peak latency variability. Stress, 17(4), 285–295. doi: 10.3109/10253890.2014.915517 [DOI] [PubMed] [Google Scholar]
- McEwen BS (1998). Protective and damaging effects of stress mediators. N Engl J Med,338(3), 171–179. doi: 10.1056/NEJM199801153380307 [DOI] [PubMed] [Google Scholar]
- McEwen BS (2003). Mood disorders and allostatic load. Biol Psychiatry, 54(3), 200–207. [DOI] [PubMed] [Google Scholar]
- McEwen BS (2004). Protection and damage from acute and chronic stress: allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann N Y Acad Sci, 1032, 1–7. doi: 10.1196/annals.1314.001 [DOI] [PubMed] [Google Scholar]
- McEwen BS (2008). Central effects of stress hormones in health and disease: Understanding the protective and damaging effects of stress and stress mediators. Eur J Pharmacol, 583(2–3), 174–185. doi: 10.1016/j.ejphar.2007.11.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, & Gianaros PJ (2010). Central role of the brain in stress and adaptation: links o socioeconomic status, health, and disease. Ann Y Acad Sci, 1186, 190–222. doi: 10.1111/j.1749-6632.2009.05331.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R, Plessow F, Kirschbaum C, & Stalder T (2013). Classification criteria for distinguishing cortisol responders from nonresponders to psychosocial stress: evaluation of salivary cortisol pulse detection in panel designs. Psychosom Med, 75(9), 832–840. doi: 10.1097/PSY.0000000000000002 [DOI] [PubMed] [Google Scholar]
- Powers SI, Laurent HK, Gunlicks-Stoessel M, Balaban S, & Bent E (2016). Depression and anxiety predict sex-specific cortisol responses to interpersonal stress. Psychoneuroendocrinology, 69, 172–179. doi: 10.1016/j.psyneuen.2016.04.007 [DOI] [PubMed] [Google Scholar]
- Puig-Perez S, Villada C, Pulopulos MM, Hidalgo V, & Salvador A (2016). How are neuroticism and depression related to the psychophysiological stress response to acute stress in healthy older people? Physiol Behav, 156, 128–136. doi: 10.1016/j.physbeh.2016.01.015 [DOI] [PubMed] [Google Scholar]
- R Development Core Team. (2018). R: A Language and Environment for Statistical Computing Retrieved from https://www.R-project.org/
- Sapolsky RM, Romero LM, & Munck AU (2000). How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev, 21(1), 55–89. doi: 10.1210/edrv.21.1.0389 [DOI] [PubMed] [Google Scholar]
- Sterling P, & Eyer J (1988). Allostasis: A new paradigm to explain arousal pathology. In Handbook of life stress, cognition and health (pp. 629–649). Oxford, England: John Wiley & Sons. [Google Scholar]
- Stern AF (2014). The hospital anxiety and depression scale. Occup Med (Lond), 64(5), 393–394. doi: 10.1093/occmed/kqu024 [DOI] [PubMed] [Google Scholar]
- Steudte-Schmiedgen S, Wichmann S, Stalder T, Hilbert K, Muehlhan M, Lueken U, & Beesdo-Baum K (2017). Hair cortisol concentrations and cortisol stress reactivity in generalized anxiety disorder, major depression and their comorbidity. Journal of Psychiatric Research, 84, 184–190. doi: 10.1016/j.jpsychires.2016.09.024 [DOI] [PubMed] [Google Scholar]
- Wichmann S,Kirschbaum C,Böhme C, & Petrowski K (2017). Cortisol stress response in post-traumatic stress disorder, panic disorder, and major depressive disorder patients. Psychoneuroendocrinology, 83, 135–141. doi: 10.1016/j.psyneuen.2017.06.005 [DOI] [PubMed] [Google Scholar]
- Wickham H (2009). ggplot2: Elegant Graphics for Data Analysis: Springer-Verlag New York. [Google Scholar]
- Yoon KL, & Joormann J (2012). Stress reactivity in social anxiety disorder with and without comorbid depression. J Abnorm Psychol, 121(1), 250–255. doi: 10.1037/a0025079 [DOI] [PubMed] [Google Scholar]
- Young EA, Abelson JL, & Cameron OG (2004). Effect of comorbid anxiety disorders on the hypothalamic-pituitary-adrenal axis response to a social stressor in major depression. Biol Psychiatry, 56(2), 113–120. doi: 10.1016/j.biopsych.2004.03.017 [DOI] [PubMed] [Google Scholar]
- Zigmond AS, & Snaith RP (1983). The hospital anxiety and depression scale. Acta Psychiatr Scand, 67(6), 361–370. [DOI] [PubMed] [Google Scholar]
- Zorn JV, Schur RR, Boks MP, Kahn RS, Joels M, & Vinkers CH (2017). Cortisol stress reactivity across psychiatric disorders: A systematic review and meta-analysis. Psychoneuroendocrinology, 77, 25–36. doi: 10.1016/j.psyneuen.2016.11.036 [DOI] [PubMed] [Google Scholar]