

Abstract
All-trans-retinoic acid (atRA), the active metabolite of vitamin A, is a critical signaling molecule during embryonic and fetal development and is necessary for maternal health. Fetal exposure to endogenous atRA is tightly regulated during gestation in a tissue specific manner and maternal exposure to exogenous retinoids during pregnancy is teratogenic. The clearance of atRA is primarily mediated by the cytochrome P450 (CYP) 26 enzymes, which play an essential role in controlling retinoid gradients during organogenesis. We hypothesized that CYP26 enzymes in the human fetal liver also function as a protective barrier to prevent maternal atRA reaching fetal circulation. Using human fetal liver tissue, we found that the mRNA of CYP26A1 and CYP26B1 enzymes is expressed in the human fetal liver. However, based on inhibition studies, metabolite profiles and correlation of atRA metabolism with testosterone hydroxylation, clearance of atRA in the fetal livers was mediated by CYP3A7. Based on in vitro-to-in vivo scaling, atRA clearance in the fetal liver was quantitatively minimal, thus providing an insufficient maternal-fetal barrier for atRA exposure.
Introduction
All-trans-retinoic acid (atRA) is the key active metabolite of vitamin A (retinol). atRA is a critical morphogen and a signaling molecule during embryonic and fetal development. Carefully controlled concentration gradients of atRA in the developing embryo are needed to mediate organogenesis and other developmental processes1–3. atRA and its isomer 13-cisRA are well characterized teratogens in humans and in model species, and dosing of pregnant mothers with these retinoids results in severe fetal malformations4–6. Maternal deficiency of vitamin A is also associated with fetal malformations7,8. It is generally believed that atRA concentrations in specific cells in the developing embryo are controlled by gestational age, cell-type specific expression of atRA-synthesizing aldehyde dehydrogenase 1A (ALDH1A) enzymes and atRA metabolizing cytochrome P450 (CYP) family 26 (CYP26) enzymes, resulting in the necessary atRA gradients that regulate differentiation, proliferation and apoptosis of cells within the embryo3,9. The fetus appears to be dependent on a maternal supply of retinol, as evidenced by placental expression of Stra6, the uptake transporter for retinol10, and the severe birth defects observed in individuals carrying Stra6 mutations11. Similarly, studies in several model species have shown that disruption of atRA synthesis or metabolism via knocking out ALDH1A2, ALDH1A3, CYP26A1 or CYP26B1 in the embryo/fetus is detrimental, resulting in severe malformations and unviable offspring9,12. As such, it is expected that xenobiotics or environmental and genetic factors that alter the activity of these enzymes will result in teratogenicity due to too low or excessive exposure to atRA in the fetus. However, at present the processes that prevent fetal exposure to maternal circulating atRA to allow the fetus to independently regulate its retinoid gradients are not known.
In the adult human liver, atRA has been shown to be mainly metabolized by CYP enzymes CYP26A1, CYP26B1, CYP3A and CYP2C813,14. While CYP3A and CYP2C8 mainly hydroxylate atRA at the C4-position forming 4-OH-RA, CYP26 enzymes oxidize atRA at several sites resulting in the formation of 4-OH-RA as well as 16- and 18-OH-RA15,16. The 4-OH-RA is then sequentially oxidized to 4-oxo-RA by an unknown alcohol dehydrogenase17. In adult human liver, CYP26A1 has been shown to be the main atRA hydroxylase likely regulating atRA clearance13. CYP26A1 and CYP26B1 have also been shown to play an important role in regulating atRA homeostasis during mouse development, and these enzymes are expressed during mouse development in a tissue and gestational age dependent manner9,18,19. However, information regarding CYP26 activity or expression in human fetal organs is limited, and it is unknown whether the same CYP26 enzymes that appear predominant in metabolizing atRA in adult human liver also regulate atRA clearance in the fetal liver. In fact, clear dichotomy of expression of CYP enzymes between fetal and adult livers has been shown for the CYP3A family of enzymes20–22. While CYP3A4 and CYP3A5 are the main CYP3A enzymes in adult human liver, they are not well expressed in the fetal liver. Instead, CYP3A7 is the main human fetal liver CYP3A isoform20–22. CYP3A7 also metabolizes atRA13,23 and before the identification of CYP26 enzymes, CYP3A7 was suggested as the main fetal liver atRA hydroxylase limiting fetal exposure to atRA23. Although CYP26 mRNA has been previously detected in human fetal liver tissues24–26, the quantitative importance of the CYP26 enzymes and CYP3A7 in modulating maternal-fetal transfer of atRA and fetal atRA clearance is not known. Based on the existing data that show the importance of CYP26 enzymes in atRA metabolism in adult liver and the importance of tight regulation of retinoid gradients in the developing fetus, we hypothesized that CYP26A1 plays an important role in fetal atRA clearance, and that the fetal liver limits maternal-fetal transfer of atRA. The aims of this study were to determine whether CYP26 enzymes are important in atRA clearance in human fetal liver, whether CYP3A7 contributes to atRA clearance in human fetal liver and to determine the efficiency of the fetal liver in eliminating atRA that passes to the fetus from maternal circulation.
Results
Detection of CYP26 mRNA in human fetal livers
The mRNA expression of CYP26A1, CYP26B1 and CYP26C1 together with CYP3A7 was measured in fetal livers from 18 individual donors (Fig. 1) and in five control adult human livers (data not shown). CYP26A1 and CYP26B1 mRNAs were detected and quantified in 14 and 11 of the 18 fetal livers, respectively, while CYP26C1 mRNA was not detected in any of the 18 fetal livers. Considerable inter-individual variability, up to 125-fold, was observed in the expression of CYP26A1 in the fetal livers while the expression of CYP26B1 was less variable (Fig. 1). Two of the fetal livers had no detectable expression of any CYP26 mRNAs and in both of these livers robust CYP3A7 mRNA expression was detected. In fact, CYP3A7 expression was relatively high (Ct values 26–34) in 17 of the 18 fetal livers. One fetal liver had very low CYP3A7 expression (Ct value 38) and this liver showed the highest CYP26A1 expression among all 18 fetal livers. The overall Ct values of CYP26A1 were higher in the fetal livers (one with Ct value 32 and others 37–38) than in the adult livers analyzed (Ct 27–36) while CYP26B1 Ct values were generally lower in the fetal livers (Ct 36–39) than adult livers (CYP26B1 was only detectable in one adult liver). Due to differences in housekeeping gene expression between adult and fetal livers, no quantitative comparisons were made between fetal and adult liver mRNA expression. CYP26C1 mRNA was not detected in any of the adult human livers while CYP3A7 mRNA was detected in 3 of the 5 adult livers (Ct values 30–37).
Figure 1.
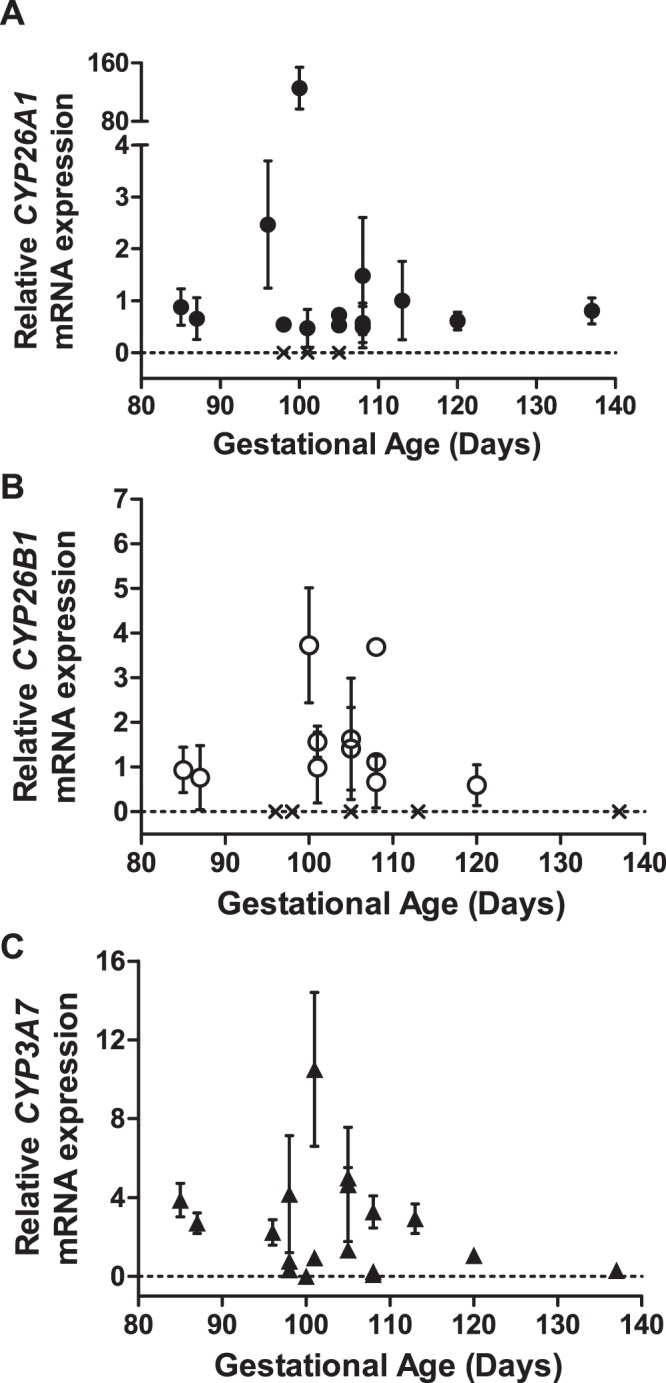
Expression of CYP26A1 (A, closed circles), CYP26B1 (B, open circles) and CYP3A7 (C, closed triangles) mRNA in human fetal livers. The mRNA expression in each fetal liver was measured as duplicates and repeated in three separate days. The data shown is the mean ± S.D. from the three different experiments. The symbol “X” indicates a sample with no detectable mRNA expression. The mRNA expression of CYP26A1 and CYP26B1 was undetectable in 4 and 7 of all 18 fetal livers, respectively, while CYP3A7 mRNA expression was detected in all fetal livers.
atRA metabolism in human fetal livers
To explore whether the metabolites formed from atRA in fetal liver resembled either the metabolite profile observed from atRA with recombinant CYP3A7 or CYP26 enzymes, the metabolites formed from atRA by CYP3A7, CYP26A1 and CYP26B1 and by fetal liver S9 fractions were characterized (Fig. 2). Consistent with previous data, recombinant CYP26A1 and CYP26B1 hydroxylated atRA at multiple sites, while CYP3A7 only formed 4-OH-atRA and 4-oxo-atRA. The metabolite profile in human fetal livers was similar to that observed with CYP3A7. Of the atRA metabolites, only the formation of 4-OH-RA and its metabolite 4-oxo-RA was observed (Fig. 2). No formation of the CYP26 specific metabolite 16-OH-RA was observed in fetal livers. The fetal liver metabolite profile corresponded to that observed with CYP3A7 and suggests lack of significant CYP26 contribution to atRA metabolism in fetal liver.
Figure 2.

Identification of the metabolites formed from atRA by recombinant CYP26A1, CYP26B1, CYP3A7 and by human fetal livers. Panels A–D show representative chromatograms of metabolite standards (A) and incubations with recombinant CYP26A1 (B) CYP26B1 (C) and CYP3A7 (D). Panels E and F show the metabolite formation in S9 fractions from two representative human fetal livers (hFL). The individual donors are indicated with the sample number (18 and 38 listed in Table 1). The incubations were conducted as described in materials and methods. The m/z transitions of 315 > 253 Da (4-OH-RA and 18-OH-RA; black line), 313 > 269 Da (4-oxo-RA; blue line) and 315 > 241 Da (16-OH-RA; red line) were monitored by LC-MS/MS and the observed peaks are labeled in each panel.
The metabolism of atRA was quantified in 27 individual fetal livers from gestational ages between 72 and 137 days (Table 1, Fig. 3). The metabolite formation velocity ranged from 24 ± 11 to 124 ± 7 pmol/min/mg protein for 4-OH-RA, from 1.6 ± 0.1 to 16 ± 1 pmol/min/mg protein for 4-oxo-RA and from 28 ± 11 to 134 ± 8 pmol/min/mg protein for the sum of 4-OH-RA and 4-oxo-RA. There was no correlation between gestational age and the formation of 4-OH-RA (r2 = 0.04, p = 0.33), 4-oxo-RA (r2 = 0.02, p = 0.49), or their sum (r2 = 0.04, p = 0.33), and the metabolite formation velocity did not differ between weeks of gestation (Fig. 3, Table 2). There were no significant differences in metabolite formation between fetal sexes or maternal races (Table 2).
Table 1.
Donor characteristics and fetal liver physiological parameters used.
| Fetal Liver code | Gestational Age (Day) | Sex Of fetus | Race | Yield of S9 protein as mg/g fetal liver | Fetal liver weight (g) | Umbilical vein flow (mL/min) |
|---|---|---|---|---|---|---|
| FL 30 | 72 | M | n/a | 27 | 0.4 | 0.45 |
| FL 20 | 74 | M | n/a | 70 | 0.5 | 0.45 |
| FL 21 | 74 | n/a | Asian | 79 | 0.5 | 0.45 |
| FL 36 | 80 | n/a | Caucasian | 43 | 0.9 | 0.78 |
| FL 39 | 80 | F | Asian/Hispanic | 53 | 0.9 | 0.78 |
| FL 24 | 85 | n/a | Asian | 75 | 1.2 | 1.6 |
| FL 37 | 85 | M | n/a | 43 | 1.2 | 1.6 |
| FL 14 | 87 | F | n/a | 47 | 1.4 | 1.6 |
| FL 12 | 91 | F | Asian | 52 | 1.9 | 2.6 |
| FL 31 | 96 | M | Caucasian | 46 | 2.6 | 2.6 |
| FL 13 | 98 | M | n/a | 32 | 3.0 | 4.8 |
| FL 27 | 98 | F | Caucasian | 36 | 3.0 | 4.8 |
| FL 18 | 98 | n/a | Asian | 33 | 3.0 | 4.8 |
| FL 34 | 100 | M | n/a | 48 | 3.4 | 4.8 |
| FL 40 | 101 | M | Caucasian | 68 | 3.7 | 4.8 |
| FL 33 | 101 | M | n/a | 93 | 3.7 | 4.8 |
| FL 15 | 101 | n/a | n/a | 37 | 3.7 | 4.8 |
| FL 23 | 105 | M | Alaskan | 46 | 4.7 | 7.8 |
| FL 38 | 105 | F | Caucasian | 63 | 4.7 | 7.8 |
| FL 19 | 108 | F | Hispanic | 60 | 5.6 | 7.8 |
| FL 28 | 108 | F | Asian | 67 | 5.6 | 7.8 |
| FL 29 | 108 | F | n/a | 42 | 5.6 | 7.8 |
| FL 26 | 113 | n/a | n/a | 44 | 7.4 | 11.1 |
| FL 25 | 113 | M | n/a | 57 | 7.4 | 11.1 |
| FL 17 | 115 | n/a | n/a | 47 | 8.3 | 11.1 |
| FL 16 | 120 | M | Caucasian | 42 | 10.7 | 15.6 |
| FL 22 | 137 | M | Alaskan | 62 | 23.2 | 26.8 |
Figure 3.

Quantitative analysis of atRA and testosterone (TST) metabolism in human fetal livers. Formation of 4-OH-RA (A), 4-oxo-RA (C) and the sum of 4-OH-RA and 4-oxo-RA (E) in individual human fetal livers from different gestational ages is shown. The insets list the correlation analysis between gestational age and product formation velocity. Panel D shows the correlation between atRA oxidation and 6βOH-TST formation in individual human fetal livers. Panel E shows the intrinsic clearance of atRA oxidation in the fetal livers (Clint,FL) based on whole organ scaling from the S9 fraction data as described in the methods section. The calculated extraction ratio of atRA at different gestational ages is shown in panel F for individual human fetal livers.
Table 2.
Descriptive analysis of atRA oxidation in the different groups of fetal livers included in the analysis.
| 4-OH-RA formation (pmol/min/mg protein) | 4-oxo-RA formation (pmol/min/mg protein) | 4-OH-RA+ 4-oxo-RA formation (pmol/min/mg protein) | 4-oxo-RA/4-OH-RA (ratio) | |
|---|---|---|---|---|
| gestational week 10–12 (n = 5) | 61 ± 23 | 5.2 ± 3.8 | 66 ± 25 | 0.09 ± 0.04 |
| gestational week 12–14 (n = 8) | 75 ± 34 | 7.1 ± 3.7 | 82 ± 37 | 0.10 ± 0.05 |
| gestational week 14–16 (n = 9) | 73 ± 30 | 6.9 ± 4.3 | 80 ± 33 | 0.09 ± 0.03 |
| gestational week 16–20 (n = 5) | 77 ± 24 | 6.6 ± 4.6 | 84 ± 28 | 0.08 ± 0.04 |
| Female Fetus (n = 8) | 80 ± 25 | 7.5 ± 3.6 | 87 ± 28 | 0.09 ± 0.03 |
| Male Fetus (n = 11) | 75 ± 27 | 7.4 ± 4.7 | 83 ± 31 | 0.10 ± 0.05 |
| Alaskan (n = 2) | 106 ± 9 | 14.8 ± 1.5 | 121 ± 10 | 0.14 ± 0.00 |
| Asian (n = 6) | 76 ± 35 | 6.4 ± 3.6 | 82 ± 38 | 0.09 ± 0.04 |
| Caucasian (n = 6) | 73 ± 21 | 5.7 ± 3.1 | 79 ± 24 | 0.08 ± 0.03 |
There were no significant differences in atRA oxidation rates between gestational age groups, sex, or race.
Identification of CYPs that metabolize atRA in human fetal livers
To quantify which CYP enzymes contribute to atRA clearance in fetal liver, selective inhibitors of CYP3A7 and CYP26 were used. Fluconazole has been previously shown not to inhibit CYP2616. The inhibition of CYP3A7 mediated 4-OH-atRA formation by fluconazole (300 μM) was confirmed using recombinant CYP3A7. Inhibition of CYP26A1 by talarozole (200 nM) was reproduced with recombinant CYP26A1 (Fig. 4). The specificity of talarozole (200 nM) towards CYP26 was confirmed using recombinant CYP3A7. Talarozole did not significantly inhibit CYP3A7 mediated 4-OH-RA formation (Fig. 4). Together these data show that fluconazole and talarozole can be used as specific inhibitors of CYP3A7 and CYP26 respectively. In human fetal liver S9 fractions, atRA metabolite formation was inhibited 30–60% by fluconazole and 30–40% by talarazole (Fig. 4), suggesting that both CYP26 enzymes and CYP3A7 contribute to atRA clearance in human fetal liver. Ketoconazole inhibits both CYP26 enzymes and CYP3A716. Consistent with this inhibition profile, ketoconazole caused a >95% decrease in atRA metabolite formation. However, these inhibition experiments do not unequivocally define which CYP enzyme is predominant in atRA clearance in human fetal liver. Therefore, to further define the importance of CYP3A7 in atRA clearance in fetal liver, correlation analysis between atRA metabolite formation and the formation of the CYP3A7 specific testosterone metabolite 6βOH-testosterone was conducted (Fig. 3). atRA oxidation (the sum of 4-OH-RA and 4-oxoRA formation) and 6βOH-testosterone formation from testosterone correlated significantly in the fetal livers tested (r2 = 0.58, p < 0.05) suggesting that CYP3A7 plays a major role in atRA metabolism in human fetal liver.
Figure 4.
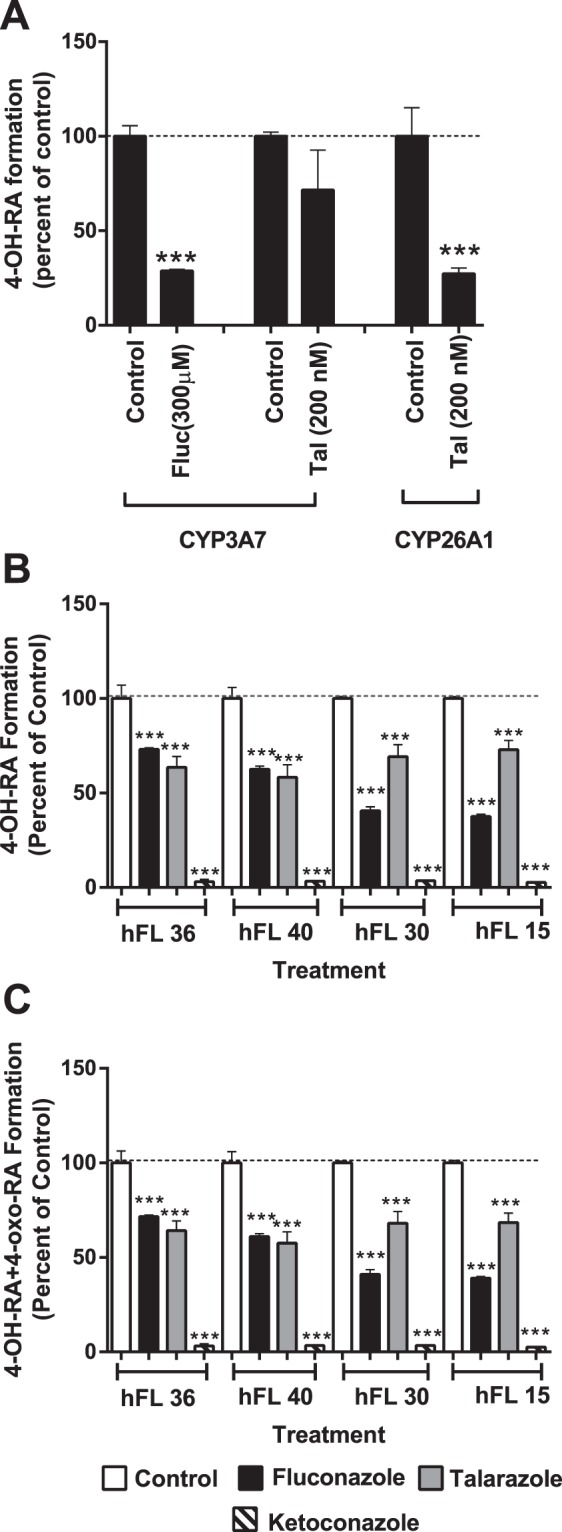
Inhibition of atRA metabolism by specific CYP inhibitors. Panel A shows the inhibition of 4-OH-RA formation by CYP3A7 inhibitor fluconazole (fluc, 300 μM) and the CYP26 inhibitor talarozole (tal, 200 nM). atRA concentrations were 10 μM and 500 nM in the CYP3A7 and CYP26 incubations. Panels B and C show the percentage inhibition of 4-OH-RA (B) and 4-OH-RA + 4-oxo-RA (C) formation in four representative human fetal livers (hFL, numbers correspond to sample identifiers) by fluconazole (black bars), talarozole (grey bars), and ketoconazole (striped bars) in comparison to vehicle controls (white bars). The stars (***) indicate a significant difference between the incubations with the indicated inhibitor and vehicle control. The numbers indicate the donor codes for the fetal liver donors.
Prediction of atRA clearance in the human fetal liver
To define the quantitative role of the fetal liver in atRA clearance and in potentially serving as a barrier to maternal-fetal retinoid transfer, the overall organ clearance and extraction ratio of atRA metabolism was calculated for the fetal liver for the gestational ages studied. The overall intrinsic clearance of atRA by the whole fetal liver (ClintFL) increased with gestational age due to the growth of the fetal liver. The intrinsic clearance per mg S9 protein was independent of gestational age (Fig. 3). The predicted fraction of atRA removed from fetal circulation by the fetal liver (extraction ratio) was very low, ranging from 0.01 to 0.05 (Fig. 3F). This suggests minimal extraction of maternal atRA by the fetal liver.
Discussion
atRA is a key developmental morphogen, and distinct concentration gradients of atRA within the developing embryo and fetus are crucial for regulation of cellular differentiation1,2,7. Embryonic development requires gestational age and tissue-type specific regulation of atRA concentrations2,3,9. Based on this, we hypothesized that a barrier, such as the fetal liver or the placenta, exists between the mother and the fetus to prevent maternal endogenous atRA passing to the fetus and enabling autonomous regulation of fetal atRA concentrations. Previous studies have shown CYP26 mRNA expression in fetal liver24–26. Hence, we hypothesized that the CYP26 enzymes, which are generally believed to be the main human retinoic acid hydroxylases14,27, would constitute a maternal-fetal barrier for maternal atRA. Based on the mRNA analysis of individual fetal livers, however, CYP26 enzymes appeared relatively insignificant in the human fetal liver both in terms of observed mRNA expression and apparent activity. At the same time, CYP3A7 mRNA was abundant in the fetal livers, in agreement with past studies20,21. The finding of low but detectable expression of CYP26A1 mRNA in the human fetal livers is similar to prior findings. One study showed low to undetectable CYP26A1 mRNA in the human fetal liver and relatively high CYP26A1 mRNA in human fetal cephalic tissue25. A second study in a single donor showed weak detection in a single donor26. The detection of CYP26B1 mRNA in a subset of the fetal livers is consistent with the prior detection of CYP26B1 mRNA in a single donor of human fetal liver26. The mRNA expression of CYP26A1 and CYP26B1 observed in the adult liver in this study agrees with previous reports showing that CYP26A1 is the predominant CYP26 enzyme in adult liver and CYP26B1 is either undetectable or has very low expression13,15,26,28. In contrast to the previous single donor analysis however26, CYP26C1 mRNA was not detected in adult or fetal livers. The detection of high CYP26A1 mRNA in one fetal liver that had very low CYP3A7 expression is of particular interest. If CYP3A7 is predominantly responsible for atRA clearance in the fetal liver, low expression or lack of this CYP would result in increased atRA concentrations in fetal liver. These increased atRA concentrations should in turn induce the expression of CYP26A1 in the fetal liver leading to the observed mRNA expression pattern. Although this observation is from a single donor, it is consistent with the prevailing notion that CYP26A1 expression is responsive to atRA concentrations and its expression is induced by increased atRA concentrations17,28.
Despite the detection of CYP26 isoform mRNA in human fetal livers, all the data presented here (the metabolite profiles, CYP inhibition data and correlation of 4-OH-RA formation with 6βOH-TST formation) support the role of CYP3A7 as the main human fetal liver atRA hydroxylase with only a minor contribution of CYP26 enzymes. This was surprising, as in adult human liver CYP26A1 was previously found to be the main atRA hydroxylase despite the activity of CYP3A4 and CYP3A5 as atRA hydroxylases13. Yet, even in the adult human liver, CYP3A4 and CYP3A5 expression did significantly correlate with atRA hydroxylation activity. In particular in livers with low CYP26A1 expression, CYP3A enzymes were likely to be significant atRA hydroxylases13. Although CYP3A7 constitutes 30–85% of the fetal liver CYPs29 we expected that the >1,000 fold higher intrinsic clearance of atRA by the CYP26 enzymes in comparison to CYP3A713,15,30 would translate to an important contribution of CYP26s to fetal liver atRA clearance, even if their expression was much lower than CYP3A7. However, the data collected does not support this hypothesis and we conclude that CYP3A7 is the main human fetal liver atRA hydroxylase. This finding is likely to translate to other RA isomers, 13-cisRA and 9-cisRA as well, as they have similar clearance profiles by CYP26s as atRA30 and are metabolized by CYP3A723. The lack of change in atRA and testosterone oxidation activity with gestational age observed in this study is consistent with previous reports that have shown relatively stable CYP3A7 expression across gestational ages20,21. The data is, however, discrepant with the study that showed higher atRA hydroxylation rates in three fetal livers from gestational days 96–109 when compared to livers from days 54–89 of gestation23. The previously observed change with gestational age is likely due to the small sample size that did not completely capture inter-individual variability in atRA clearance.
This is the first study to scale the observed microsomal or S9 protein activity to the entire fetal liver. This scaling shows that the metabolic capacity of the fetal liver towards atRA is unlikely to contribute significantly to fetal atRA clearance, and thus this metabolism is not quantitatively sufficient to protect the developing fetus. While the intrinsic clearance of atRA metabolism per mg of fetal liver protein did not change with gestational age, our analysis shows that when the growth of the liver and increase in umbilical blood flow is accounted for, the overall metabolic clearance of atRA increases significantly with gestational age. However, the extraction ratio is unchanged with gestational age, and the predicted extraction ratio (0.01–0.05) suggested that maximum 5% of maternal atRA could be extracted by fetal liver at any gestational age. The scaling methods used here, the analysis of the extraction ratio and the prediction of the overall fetal liver metabolic clearance are likely useful for future estimations of the role of the fetal liver in clearance of therapeutic drugs and toxins which are metabolized by fetal liver CYPs or glucuronidation enzymes.
Collectively, the data presented here are in agreement with the current consensus2,3,9 that fetal tissue exposure to atRA and tissue atRA concentrations and signaling are regulated at the individual organ level and not via a maternal-fetal barrier. It is also important to note that atRA signaling is most significant during early embryogenesis, a window of gestation that cannot be feasibly studied in human tissues. As such, concordance of the findings from this study with animal models will need to be established in studies in model organisms at gestational time periods after main sensitivity periods to atRA. During the early gestation, the placenta is likely to play a critical role in protecting the embryo, and atRA metabolism and transport in the human placenta requires further study. Interestingly in our preliminary studies, we did not observe any formation of atRA metabolites in human placental S9 preparations (data not shown) suggesting a lack of placental metabolic barrier. Further work is needed to define potential active transport mechanisms in the placenta that modulate species differences in fetal exposure to teratogenic retinoids4–6 and maternal-fetal transfer of retinoids during sensitive periods of development.
Methods
Chemicals and Reagents
atRA, ketoconazole, fluconazole, and talarazole were purchased from Sigma-Aldrich (St. Louis, MO). 4-oxo-RA-d3 and atRA-d5 were purchased from Toronto Research Chemicals (North York, Ontario). 4-OH-RA, 4-oxo-RA, and 18-OH-RA were synthesized as previously described15,31. Optima LC/MS-grade water, optima LC/MS-grade acetonitrile, ethanol and ethyl acetate were purchased from Fisher Scientific (Pittsburg, PA). Recombinant CYP3A7 supersomes coexpressed with P450 reductase were purchased from BD Gentest. Recombinant CYP26A1 and CYP26B1 were expressed in baculovirus infected insect cells and membrane fractions prepared via ultracentrifugation as previously described15,32.
Collection of Human livers
The study was approved by the Institutional Review Board (IRB) at the University of Washington and the studies were conducted in accordance with the guidance of Office of Human Research Protections. Human fetal liver tissues (n = 27) ranging from reported gestational days 67 to 137 were collected by the Birth Defects Laboratory at the University of Washington and flash frozen upon collection using liquid nitrogen and stored at −80 °C until ready for use. The gestational days were based on self-reports and exact dates of conception are not known. Donated tissues were from elective abortions and all donor moms signed informed consent for donating the tissues. Tissues from fetuses whose mothers had known drug use were excluded from this study. Adult liver tissues (n = 5) were from de-identified donors from University of Washington human liver bank.
Analysis of CYP26 mRNA expression
To explore the presence of CYP26 enzymes in human fetal liver, mRNA was extracted from 18 fetal livers and five adult human livers as previously described28. 50–70 mg of liver was homogenized in 2 ml Omni Hard Tissue Homogenizing tubes containing 1.4 mm ceramic beads and 1 ml of TRI reagent (Invitrogen; Grand Island, NY, USA). Homogenization was conducted using Omni Bead Ruptor 24 (Omni International; Kennesaw, GA, USA) and mRNA was extracted with TRI reagent according to the manufacturer’s recommendations. Total RNA was quantified using a Nanodrop 2000c Spectrophotometer (Thermo Fisher Scientific, Waltham, MA). cDNA was synthesized from 1 μg mRNA using TaqMan reverse transcription reagents (Applied Biosystems, Carlsbad, CA). The mRNAs of CYP26A1, CYP26B1 and CYP26C1 were quantified as target genes, CYP3A7 as a control gene, and β-actin, GAPDH and GUSB were evaluated as housekeeping genes. Based on the variability in the gene expression, β-actin was chosen as the housekeeping gene. mRNA expression was quantified using StepOnePlusTM q-RT-PCR (Applied Biosystems; Carlsbad, CA, USA). All samples were analyzed in duplicates and the q-RT-PCR repeated on three separate occasions. For samples that were undetected in one of the three experiments (5 samples for CYP26A1, 4 for CYP26B1 and 1 for CYP3A7) a Ct value of 40 was assigned to the undetected run and the mean of the three experiments was calculated. For samples that were undetected in two of the three experiments, samples were considered as target gene undetected (2 samples for CYP26A1 and 6 for CYP26B1). The relative abundance of CYP26A1, CYP26B1, CYP26C1 and CYP3A7 mRNA expression was analyzed by the ∆∆Ct method using β-actin as a housekeeping gene and the data are presented as a fold difference in comparison to the mean value for each gene. No comparisons for expression between genes and between adult and fetal livers were done. Human primer and probe pairs for CYP26A1 (Hs01075675_m1, FAM), CYP26B1 (Hs01011223_m1, FAM), CYP26C1 (Hs01595345_m1), CYP3A7 (Hs00426361_m1, FAM), ACTB (Hs01060665_g1, FAM), GUSB (Hs00939627_m1, FAM) and GAPDH (Hs02786624_g1, FAM) were obtained from Applied Biosystems (Carlsbad, CA, USA).
Preparation of liver S9 fractions
Human fetal liver S9 fractions containing cytosol, cell membranes including microsomes, and small mitochondria and other small cell organelles were prepared to evaluate atRA metabolism in human fetal liver. For this 300 µL of 50 mM Potassium phosphate (KPi) buffer (pH 7.4) containing 250 mM sucrose, 1 mM EDTA and 1 mM PMSF was added to 0.1–0.3 g of fetal liver sample and the tissue was homogenized in 2 mL Omni Hard Tissue Homogenizing tubes containing 1.4 mm ceramic beads using a 2*20 sec cycles with an Omni Bead Ruptor 24 containing dry ice in acetone (Omni International, Kennesaw, GA). The homogenates were then centrifuged at 9,000 g for 20 min to pellet cell nuclei, large organelles and unbroken cells and the resulting supernatant (S9 fraction) was stored at −80 °C. The overall protein concentration was determined using albumin as the calibration standard and a Pierce BCA Protein Assay (Thermo Fisher Scientific, Inc., Rockford IL).
Evaluation of atRA metabolism via in vitro Incubations
The metabolism of atRA in human fetal livers and the enzymes responsible for atRA metabolism were first qualitatively evaluated by standard incubation methods as previously described by us13,15,33. The specific metabolites formed from atRA by recombinant CYP26A1, CYP26B1 and CYP3A7 in comparison to human fetal liver S9 fractions from representative donors was assessed. 5 pmols of CYP26A1, CYP26B1 and CYP3A7 in 1 mL of 100 mM Potassium phosphate (KPi) buffer (pH 7.4) were incubated for 2 mins (CYP26A1) and 10 mins (CYP26B1 and CYP3A7) with 5 µM atRA at 37 °C, respectively. The incubations were quenched with ethyl acetate, metabolites extracted as previously described15 and the product formation was then measured by LC-MS/MS as described below. For fetal liver S9 incubations two representative livers were chosen based on the mRNA expression levels of CYP3A7 and CYP26A1. In brief, 0.3 mg S9 protein in 1 mL 100 mM KPi buffer were incubated at 37 °C with 5 µM atRA. After a pre-incubation of 5 min the reactions were initiated with the addition of NADPH (1 mM final concentration) and allowed to proceed for 10 min.
The formation of atRA metabolites was measured in individual human fetal liver S9 fractions from 27 donors as previously described33. All of the incubations were conducted under confirmed linear range of time and protein content. Of the atRA metabolites monitored, only 4-OH-RA and 4-oxo-RA were detected in fetal liver S9 fractions and therefore the formation of 4-OH-RA and 4-oxo-RA was quantified in the incubations. To correct the metabolic rates for the sequential formation of 4-oxo-RA from 4-OH-RA, the amount of 4-oxo-RA formed was added to the amount of 4-OH-RA formed for analysis of 4-OH-RA formation rate. For each fetal liver, atRA (500 nM) was incubated with 0.1 mg S9 protein/mL of 100 mM KPi buffer (pH 7.4) in 37 °C. After a pre-incubation of 10 min the reactions were initiated with the addition of NADPH (1 mM final concentration) and allowed to proceed for 10 min. The reactions were terminated with 3 mL ethyl acetate, internal standard (atRA-d5, 50 nM) was added and the metabolites extracted by liquid-liquid extraction. The ethyl acetate layer was collected and dried under a stream of nitrogen, and the dry residue reconstituted in 100 µL acetonitrile for MS/MS analysis.
To determine the relative contributions of CYP26 and CYP3A7 enzymes in atRA oxidation in the fetal livers, CYP selective inhibitors were used to inhibit the target enzymes in incubations with S9 fractions from four representative donors. Fluconazole was chosen as the CYP3A7 specific inhibitor as it has been shown to not inhibit CYP26A116. Talarozole was chosen as the CYP26 inhibitor based on previous characterization of its potency towards CYP26A1 and CYP26B134. Ketoconazole was included in the analysis as it is a well characterized pan-CYP inhibitor with high potency both towards CYP3A7 and CYP26s16,34. The inhibition of atRA metabolism by fluconazole (with CYP3A7) and by talarozole (with CYP26A1 and CYP3A7) was confirmed as previously described33. In brief, for fluconazole inhibition, atRA (10 µM) was incubated for 10 min with CYP3A7 (5 pmol/mL) in the presence and absence of fluconazole (300 µM) and the percent decrease in 4-OH-RA and 4-oxo-RA formation was quantified. For talarozole inhibition, atRA (500 nM) was incubated for 10 mins with CYP3A7 (5 pmol/mL) or 2 mins with CYP26A1 (2 pmol/mL with added 4 pmol/mL P450 reductase) in the presence and absence of talarozole (200 nM). All of the samples were extracted and analyzed by HPLC-MS/MS. Based on the data collected using recombinant enzymes, fluconazole was used at 200 µM to selectively inhibit CYP3A7 and talarozole was used at 200 nM to selectively inhibit CYP26 in human fetal liver S9 fractions from four representative donors. In addition, ketoconazole was tested at 10 µM concentration as a pan-CYP inhibitor but it is also likely more potent inhibitor of CYP3A7 than fluconazole. atRA concentration was 500 nM. Incubations were performed as described above for fetal livers and analyzed for metabolite formation by HPLC-MS/MS. The percent inhibition was calculated by comparing the metabolite formation velocity in the presence of the inhibitor to vehicle control.
LC-MS/MS methods of quantification of retinoid metabolites
The formation of atRA metabolites was measured using an Agilent 1290 Infinity UHPLC (Agilent Technologies, Santa Clara, CA) with an Agilent Zorbax C18 column (3.5 µm, 2.1 mm × 100 mm) and coupled to an AB Sciex API 5500 Q/LIT mass spectrometer (AB Sciex, Framingham, MA) as previously reported15 with minor modifications on the chromatography33. In brief, analytes were separated using a gradient elution as follows: starting from 10:90 acetonitrile: aqueous to 1:1 acetonitrile: aqueous over 0.5 min then increased to 85:15 acetonitrile: aqueous over 4 min and then after a 0.1 min hold increased to 95:5 acetonitrile: aqueous held for 2.5 min. The aqueous phase contained 0.1 % formic acid throughout. Analytes were detected using negative ion electrospray and monitoring MS/MS transitions of m/z 299 → 255 Da (atRA), m/z 315 → 253 Da (4-OH-RA), m/z 313 → 269 Da (4-oxo-RA), m/z 315 → 241 Da (16-OH-RA) and m/z 316 → 272 Da (4-oxo-RA-d3). Retinoid concentrations were quantified using Analyst software and peak height ratios between the analyte and the internal standard (4-oxo-RA-d3).
Characterization of CYP3A7 specific activity and Testosterone Metabolism in fetal livers
Testosterone hydroxylation (formation of 6βOH-testosterone) was used as a CYP3A7 specific probe reaction and analyzed as previously described33. All incubations were performed in triplicate. Testosterone (at 100 µM, final concentration) was incubated with 0.2 mg hFL S9 protein/mL of 100 mM KPi buffer (pH 7.4). All incubations were performed using 96-well plates with a total volume of 0.1 mL per well. The mixtures were pre-incubated for 10 minutes and reactions were initiated by the addition of NAPDH (1 mM final concentration). After 10 minutes 80 µL of the incubations were added to 80 µL ice-cold acetonitrile to quench the reaction. The samples were then centrifuged at 4 °C for 20 min at 612 g, and an aliquot of the supernatant was collected for LC-MS/MS analysis. 6βOH-testosterone was quantified based on a standard curve of 6βOH-TST (10 to 500 nM). 6βOH-TST was analyzed using a Shimadzu XR DGU-20A5 UFLC (Shimadzu Scientific Instruments, Columbia, MD) coupled to an AB Sciex 3200 Mass Spectrometer (AB Sciex, Framingham, MA). Chromatography was done using a Zorbax SB-C18 column (5 µm, 2.1 × 50 mm, Agilent Technologies, Palo Alto, CA) and gradient elution (0.3 mL/min) from initial 5:95 acetonitrile: aqueous 0.1% formic acid held for 2 min and the increased to 100% acetonitrile over 2 min and held for 1.5 min before returning to initial conditions for a re-equilibration period of 3.5 min. 6βOH-TST was detected using a mass transition of m/z 305 → 287 Da and positive ion electrospray at source parameters of 5500 V and 450 °C. Metabolite formation was quantified based on peak height and linear standard curve using Analyst software.
In vitro to in vivo Scaling of atRA metabolism and Statistical Analysis
Statistical analyses were performed using Prism v.5 (GraphPad Software, Inc., La Jolla, CA). Correlation between metabolite formation velocity from atRA and testosterone as substrates in human fetal livers was tested using linear regression. Differences between atRA metabolite formation in the presence and absence of inhibitors were tested by one-way analysis of variance. Differences in atRA metabolism between fetal livers collected at gestational weeks 10–12, 12–14, 14–16, and 16–20, between different genders of the fetus, and different races were tested by one-way analyses of variance coupled with Bonferroni’s Multiple Comparison Test. A p value < 0.05 was considered significant for all statistical analyses.
The overall clearance of atRA by fetal liver was predicted using standard in vitro-to-in vivo scaling methods35,36. First, the overall intrinsic clearance (Clint) of atRA metabolism in the fetal liver was calculated by summing 4-OH-RA and 4-oxo-RA formation from atRA in the fetal liver S9 fraction incubations for each individual donor and the Clint scaled to the whole liver using Eq. 1 as described previously for adult liver13:
| 1 |
in which FL refers to fetal liver and the total g of fetal liver (liver weight) is specified for the specific gestational ages in Table 1. The hepatic extraction ratio (ERFL) by fetal liver for atRA was calculated from Eq. 2 based on the well-stirred model of the liver37,38:
| 2 |
in which fetal liver blood flow is calculated as 0.5 times the blood flow of the umbilical vein (Qumbilical vein) based on the physiology that half of the umbilical vein flow goes to fetal liver and the rest goes to the fetal heart39. The Qumbilical vein for each gestational age is listed in Table 1. The plasma unbound fraction of atRA (fu) used was 0.01 based on previous report13.
Acknowledgements
These studies were supported in part by grants from the National Institutes of Health R01GM111772 (NI), T32 GM007750 (ART), P01 DA032507 (NI, GZ), and NIH award number 5R24 HD000836 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Author Contributions
All three authors wrote the manuscript, and designed the research, Dr Ariel Topletz and Dr Guo Zhong conducted the experiments and all three authors wrote the manuscript. Parts of the work in this manuscript were included in Dr Ariel Topletz’s PhD thesis from University of Washington as partial fulfillment of the requirements for her PhD degree.
Competing Interests
Nina Isoherranen holds a US Patent number: 9963439 on CYP26 Inhibitors. She has received honorariums from Pfizer and Genentech and is a consultant for Boehringer Ingleheim. Ariel Topletz is currently an employee of Seattle Genetics.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Duester G. Retinoic acid synthesis and signaling during early organogenesis. Cell. 2008;134:921–931. doi: 10.1016/j.cell.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCaffery PJ, Adams J, Maden M, Rosa-Molinar E. Too much of a good thing: Retinoic acid as an endogenous regulator of neural differentiation and exogenous teratogen. Eur. J. Neurosci. 2003;18:457–472. doi: 10.1046/j.1460-9568.2003.02765.x. [DOI] [PubMed] [Google Scholar]
- 3.Rhinn M, Dolle P. Retinoic acid signalling during development. Development. 2012;139:843–858. doi: 10.1242/dev.065938. [DOI] [PubMed] [Google Scholar]
- 4.Creech Kraft J, Löfberg B, Chahoud I, Bochert G, Nau H. Teratogenicity and placental transfer of all-trans-, 13-cis-, 4-oxo-all-trans-, and 4-oxo-13-cis-retinoic acid after administration of a low oral dose during organogenesis in mice. Toxicol. Appl. Pharmacol. 1989;100:162–76. doi: 10.1016/0041-008X(89)90099-9. [DOI] [PubMed] [Google Scholar]
- 5.Collins MD, Tzimas G, Hummler H, Burgin H, Nau H. Comparative Teratology and Transplacental Pharmacokinetics of All-trans-Retinoic Acid, 13-cis-Retinoic Acid, and Retinyl Palmitate Following Daily Administrations in Rats. Toxicol. Appl. Pharmacol. 1994;127:132–144. doi: 10.1006/taap.1994.1147. [DOI] [PubMed] [Google Scholar]
- 6.Nau H. Teratogenicity of isotretinoin revisited: Species variation and the role of all-trans-retinoic acid. J. Am. Acad. Dermatol. 2001;45:183–187. doi: 10.1067/mjd.2001.113720. [DOI] [PubMed] [Google Scholar]
- 7.Zile MH. Function of Vitamin A in Vertebrate Embryonic Development. J. Nutr. 2001;131:705–708. doi: 10.1093/jn/131.3.705. [DOI] [PubMed] [Google Scholar]
- 8.Clagett-Dame M, Knutson D. Vitamin a in reproduction and development. Nutrients. 2011;3:385–428. doi: 10.3390/nu3040385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pennimpede T, et al. The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res. A. Clin. Mol. Teratol. 2010;88:883–894. doi: 10.1002/bdra.20709. [DOI] [PubMed] [Google Scholar]
- 10.Kawaguchi R, et al. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science. 2007;315:820–825. doi: 10.1126/science.1136244. [DOI] [PubMed] [Google Scholar]
- 11.Pasutto F, et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am. J. Hum. Genet. 2007;80:550–60. doi: 10.1086/512203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niederreither K, et al. Genetic evidence that oxidative derivatives of retinoic acid are not involved in retinoid signaling during mouse development. Nat. Genet. 2002;31:84–88. doi: 10.1038/ng876. [DOI] [PubMed] [Google Scholar]
- 13.Thatcher JE, Zelter A, Isoherranen N. The relative importance of CYP26A1 in hepatic clearance of all-trans retinoic acid. Biochem. Pharmacol. 2010;80:903–912. doi: 10.1016/j.bcp.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ross AC, Zolfaghari R. Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annu. Rev. Nutr. 2011;31:65–87. doi: 10.1146/annurev-nutr-072610-145127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Topletz AR, et al. Comparison of the function and expression of CYP26A1 and CYP26B1, the two retinoic acid hydroxylases. Biochem. Pharmacol. 2012;83:149–163. doi: 10.1016/j.bcp.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thatcher JE, et al. Substrate specificity and ligand interactions of CYP26A1, the human liver retinoic acid hydroxylase. Mol. Pharmacol. 2011;80:228–239. doi: 10.1124/mol.111.072413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Topletz AR, et al. Induction of CYP26A1 by Metabolites of Retinoic Acid: Evidence That CYP26A1 Is an Important Enzyme in the Elimination of Active Retinoids. Mol. Pharmacol. 2015;87:430–441. doi: 10.1124/mol.114.096784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacLean G, et al. Cloning of a novel retinoic-acid metabolizing cytochrome P450, Cyp26B1, and comparative expression analysis with Cyp26A1 during early murine development. Mech. Dev. 2001;107:195–201. doi: 10.1016/S0925-4773(01)00463-4. [DOI] [PubMed] [Google Scholar]
- 19.Abu-Abed S, et al. Differential expression of the retinoic acid-metabolizing enzymes CYP26A1 and CYP26B1 during murine organogenesis. Mech. Dev. 2002;110:173–177. doi: 10.1016/S0925-4773(01)00572-X. [DOI] [PubMed] [Google Scholar]
- 20.Leeder JS, et al. Variability of CYP3A7 Expression in Human Fetal Liver. J. Pharmacol. Exp. Ther. 2005;314:626–635. doi: 10.1124/jpet.105.086504. [DOI] [PubMed] [Google Scholar]
- 21.Stevens, J. C. et al. Developmental Expression of the Major Human Hepatic CYP3A Enzymes. 307, 573–582 (2003). [DOI] [PubMed]
- 22.Betts, S., Bj, L., Rane, A. & Ekstr, L. Expression of CYP3A4 and CYP3A7 in Human Foetal Tissues and its Correlation with Nuclear Receptors. 261–266, 10.1111/bcpt.12392 (2015). [DOI] [PubMed]
- 23.Chen H, Fantel AG, Juchau MR. Catalysis of the 4-hydroxylation of retinoic acids by cyp3a7 in human fetal hepatic tissues. Drug Metab. Dispos. 2000;28:1051–7. [PubMed] [Google Scholar]
- 24.Trofimova-Griffin M, Juchau MR. Expression of Cytochrome P450RAI (CYP26) in Human Fetal Hepatic and Cephalic Tissues. Biochem. Biophys. Res. Commun. 1998;491:487–491. doi: 10.1006/bbrc.1998.9659. [DOI] [PubMed] [Google Scholar]
- 25.Trofimova-griffin ME, Brzezinski MR, Juchau MR. Patterns of CYP26 expression in human prenatal cephalic and hepatic tissues indicate an important role during early brain development. Dev. Brain Res. 2000;120:7–16. doi: 10.1016/S0165-3806(99)00185-6. [DOI] [PubMed] [Google Scholar]
- 26.Xi J, Yang Z. Expression of RALDHs (ALDH1As) and CYP26s in human tissues and during the neural differentiation of P19 embryonal carcinoma stem cell. Gene Expr. Patterns. 2008;8:438–442. doi: 10.1016/j.gep.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Thatcher JE, Isoherranen N. The role of CYP26 enzymes in retinoic acid clearance. Expert opin drug metab toxicol. 2009;5:875–886. doi: 10.1517/17425250903032681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tay S, Dickmann L, Dixit V, Isoherranen N. A comparison of the roles of peroxisome proliferator-activated receptor and retinoic acid receptor on CYP26 regulation. Mol Pharmacol. 2010;77:218–227. doi: 10.1124/mol.109.059071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Wildt SN, Kearns GL, Leeder JS, van den Anker JN. Cytochrome P450 3A. Clin. Pharmacokinet. 1999;37:485–505. doi: 10.2165/00003088-199937060-00004. [DOI] [PubMed] [Google Scholar]
- 30.Zhong G, Ortiz D, Zelter A, Nath A, Isoherranen N. CYP26C1 Is a Hydroxylase of Multiple Active Retinoids and Interacts with Cellular Retinoic Acid Binding Proteins. Mol. Pharmacol. 2018;93:489–503. doi: 10.1124/mol.117.111039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samokyszyn VM, et al. 4-Hydroxyretinoic acid, a novel substrate for human liver microsomal UDP-glucuronosyltransferase(s) and recombinant UGT2B7. J. Biol. Chem. 2000;275:6908–6914. doi: 10.1074/jbc.275.10.6908. [DOI] [PubMed] [Google Scholar]
- 32.Lutz JD, et al. Expression and functional characterization of cytochrome P450 26A1, a retinoic acid hydroxylase. Biochem. Pharmacol. 2009;77:258–268. doi: 10.1016/j.bcp.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Topletz, A. R. The Relative Importance of CYP26A1 and CYP26B1 in Mediating Retinoid Homeostasis: Studies on the Formation, Elimination and Biological Activity of All-trans-Retinoic Acid Metabolites. (University of Washington, 2013).
- 34.Diaz P, et al. Development and Characterization of Novel and Selective Inhibitors of Cytochrome P450 CYP26A1, the Human Liver Retinoic Acid Hydroxylase. J. Med. Chem. 2016;59:2579–2595. doi: 10.1021/acs.jmedchem.5b01780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwatsubo T, et al. Prediction of in vivo drug metabolism in the human liver from in vitro metabolism data. Pharmacol. Ther. 1997;73:147–171. doi: 10.1016/S0163-7258(96)00184-2. [DOI] [PubMed] [Google Scholar]
- 36.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27:1350–1359. [PubMed] [Google Scholar]
- 37.Wilkinson GR, Shand DG. Commentary: a physiological approach to hepatic drug clearance. Clin. Pharmacol. Ther. 1975;18:377–90. doi: 10.1002/cpt1975184377. [DOI] [PubMed] [Google Scholar]
- 38.Nies AS, Shand DG, Wilkinson GR. Altered Hepatic Blood Flow and Drug Disposition1. Clin. Pharmacokinet. 1976;1:135–155. doi: 10.2165/00003088-197601020-00005. [DOI] [PubMed] [Google Scholar]
- 39.Whitaker, K. B. Comprehensive perinatal & pediatric respiratory care. (Delmar Publishers, 1997).
- 40.Schiessl B, et al. 3-Dimensional sonographic volumetry of fetal brain, liver and myocardial mass–interdisciplinary clinical validation of the method and application in fetuses with and without structural heart disease. Z. Geburtshilfe Neonatol. 2011;215:60–8. doi: 10.1055/s-0030-1268431. [DOI] [PubMed] [Google Scholar]
- 41.Gielchinsky Y, Zvanca M, Minekawa R, Persico N, Nicolaides KH. Liver volume in trisomy 21 and euploid fetuses at 11 to 13 weeks. Prenat. Diagn. 2011;31:28–32. doi: 10.1002/pd.2633. [DOI] [PubMed] [Google Scholar]
- 42.Sutton MS, et al. Changes in placental blood flow in the normal human fetus with gestational age. Pediatr. Res. 1990;28:383–7. doi: 10.1203/00006450-199010000-00016. [DOI] [PubMed] [Google Scholar]