

Abstract
Pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is associated with a high frequency of copy number alterations (CNAs) in IKZF1, EBF1, PAX5, CDKN2A/B, RB1, BTG1, ETV6, and/or the PAR1 region (henceforth: B-cell development genes). We aimed to gain insight in the association between CNAs in these genes, clinical outcome parameters, and cellular drug resistance. 71% of newly diagnosed pediatric BCP-ALL cases harbored one or more CNAs in these B-cell development genes. The distribution and clinical relevance of these CNAs was highly subtype-dependent. In the DCOG-ALL10 cohort, only loss of IKZF1 associated as single marker with unfavorable outcome parameters and cellular drug resistance. Prednisolone resistance was observed in IKZF1-deleted primary high hyperdiploid cells (~1500-fold), while thiopurine resistance was detected in IKZF1-deleted primary BCR-ABL1-like and non-BCR-ABL1-like B-other cells (~2.7-fold). The previously described risk stratification classifiers, i.e. IKZF1plus and integrated cytogenetic and CNA classification, both predicted unfavorable outcome in the DCOG-ALL10 cohort, and associated with ex vivo drug cellular resistance to thiopurines, or L-asparaginase and thiopurines, respectively. This resistance could be attributed to overrepresentation of BCR-ABL1-like cases in these risk groups. Taken together, our data indicate that the prognostic value of CNAs in B-cell development genes is linked to subtype-related drug responses.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common cancer diagnosed in children. The introduction of risk-adjusted treatment protocols has significantly improved survival rates, which nowadays is approaching 90% survival1–3. Outcome of B-cell precursor ALL (BCP-ALL) differs by genetic subtype, i.e. ETV6-RUNX1, high hyperdiploid, and TCF3-PBX1 cases have favorable prognosis, whereas BCR-ABL1, and KMT2A-rearranged BCP-ALL is associated with an unfavorable treatment outcome3. Approximately 25% of the patients has a genetically unclassified disease, which is defined as ‘B-other’. This heterogeneous group can be subdivided in BCR-ABL1-like patients and non-BCR-ABL1-like B-other patients4,5. Within the BCR-ABL1-like subtype intrachromosomal amplification of chromosome 21, dicentric chromosome (9;20), and kinase activating lesions are reported4–9. In non-BCR-ABL1-like B-other cases chromosomal translocations involving DUX4, ZNF384, and MEF2D were identified10–12. In addition to the major classifying abnormalities, secondary aberrations have been observed, including copy number alterations (CNAs) in genes involved in B-cell development (e.g. IKZF1, EBF1, PAX5, ETV6), cell cycle and proliferation (e.g. CDKN2A, CDKN2B, RB1, BTG1), and cytokine receptors (e.g. CRLF2)4,5,8,9,13–16. Interestingly, some of these genetic lesions (e.g. IKZF1) were shown to predict clinical outcome5,14,17. The Dutch Childhood Oncology Group (DCOG) implemented IKZF1 status as risk factor in the ongoing DCOG-ALL11 protocol. In addition, risk stratification strategies were designed by integration of CNA profiles and genetic subtypes18–20.
Cellular drug resistance is an important cause of relapse. Ex vivo drug resistance at diagnosis is associated with high risk of early treatment failures21–23. In addition, BCP-ALL cells at relapse are more resistant towards glucocorticoids, L-asparaginase, anthracyclines, and thiopurines24. IKZF1 deletions are reported to mediate resistance towards glucocorticoids, but the relationship between remaining CNAs and cellular drug resistance is yet unknown25–27. Therefore, we performed an explorative study, which aimed to gain insight in associations between CNAs, cellular drug resistance, and clinical outcome.
Results
A pediatric BCP-ALL cohort of 515 newly diagnosed cases, representing all major ALL subtypes, was screened for CNAs in eight genes involved in transcription of lymphoid genes and the differentiation and proliferation of precursor B-cells (henceforth: B-cell development genes; Supplementary Fig. 1). In total, 71% of the pediatric BCP-ALL cases harbored one or more CNAs in these B-cell development genes (Fig. 1). The CNA frequency differed between genetic BCP-ALL subtypes. The percentage of patients with one of more CNAs was the highest in BCR-ABL1-like cases and the lowest in TCF3-PBX1 cases (Supplementary Fig. 2A).
Figure 1.

CNA landscape of B-cell development genes in the different subtypes of pediatric BCP-ALL. CNA profile of 515 newly diagnosed pediatric BCP-ALL patients, representing all major BCP-ALL subtypes, was determined using MLPA. Association between CNAs and subtypes was studied using the Fisher Exact test. The proportion of patients per subtype with a specific CNAs is shown. CNAs tested included IKZF1 (A), EBF1 (B), PAX5 (C), ETV6 (D), CDKN2A/B (E), RB1 (F), BTG1 (G), PAR1 (H). **p ≤ 0.01, *p ≤ 0.05. del = deletion.
CNAs in B-cell transcription factors
IKZF1
Deletions of the transcription factor IKZF1 were detected in 20% of the BCP-ALL cases. This frequency differed between subtypes: IKZF1 deletions were enriched in BCR-ABL1 (65%) and BCR-ABL1-like (44%) cases, whereas deletions were low or absent in ETV6-RUNX1 (3%) and TCF3-PBX1 (0%), respectively (Fig. 1A; Supplementary Fig. 2B). In addition, 76% (78/102) of the cases with an IKZF1 deletion harbored CNAs in additional genes, which mainly involved PAX5 and CDKN2A/B (Fig. 2). This co-occurrence was subtype dependent: a strong association (OR >2, p < 0.001) was observed in BCR-ABL1, BCR-ABL1-like and B-other cases, whereas in high hyperdiploid cases IKZF1 deletions mainly occurred independent of CNAs in PAX5 and/or CDKN2A/B. Within the group of genetically unclassified patients, loss of IKZF1 associated with dicentric chromosome (9;20) and tyrosine kinase fusion genes (Supplementary Table 1).
Figure 2.

Co-occurence of CNAs in B-cell development genes in the different BCP-ALL subtypes. Heatmap of CNA profile of 515 newly diagnosed pediatric BCP-ALL patients, representing the major BCP-ALL subtypes. CNAs are shown per subtype. Colors indicate presence of a CNA and absence of CNAs is shown in white. The heatmap is sorted on IKZF1 deletions followed by CNAs in PAX5. Each column represents an individual patient. The co-occurrence between the different CNAs in all BCP-ALL cases was calculated using the Fisher Exact test. Odds ratios and p-values of significant associations are shown.
IKZF1-deleted cases more often showed high MRD levels (≥10−3) after induction therapy (TP1; p = 0.013), and intermediate MRD levels (10−4 ≤ MRD < 10−3) after the first consolidation course (TP2; p = 0.028), compared to IKZF1-wildtype cases (Fig. 3A). This association could be predominantly attributed to high MRD levels in BCR-ABL1-like and B-other cases, but was not observed in high hyperdiploid or ETV6-RUNX1 cases (Supplementary Figs 3–6). In addition, IKZF1-deleted cases more often suffered from a non-response or relapse compared to IKZF1-wildtype cases (5-year CIR: 30.4% versus 9.0%; p < 0.001; Fig. 4A,B), confirming previous findings5,14,17. An IKZF1 deletion remained predictive for an unfavorable outcome in DCOG-ALL10 cases treated in the medium risk arm (Fig. 4B), indicating that the prognostic value of IKZF1 is independent of the early treatment response monitored by MRD.
Figure 3.

The association between CNAs and MRD levels after induction therapy and the first consolidation course in newly diagnosed BCP-ALL. MRD levels of DCOG-ALL treated BCP-ALL cases (all risk groups) after induction (TP1; n = 183) and first consolidation course (TP2; n = 183). The percentage of cases with high (≥10−3), medium (10−4 ≤ MRD < 10−3), and undetectable MRD levels (<10−4) is depicted per CNA. The Fisher’s Exact test was applied to study associations between CNAs and MRD levels. **p ≤ 0.01, *p ≤ 0.05. del = deletion.
Figure 4.
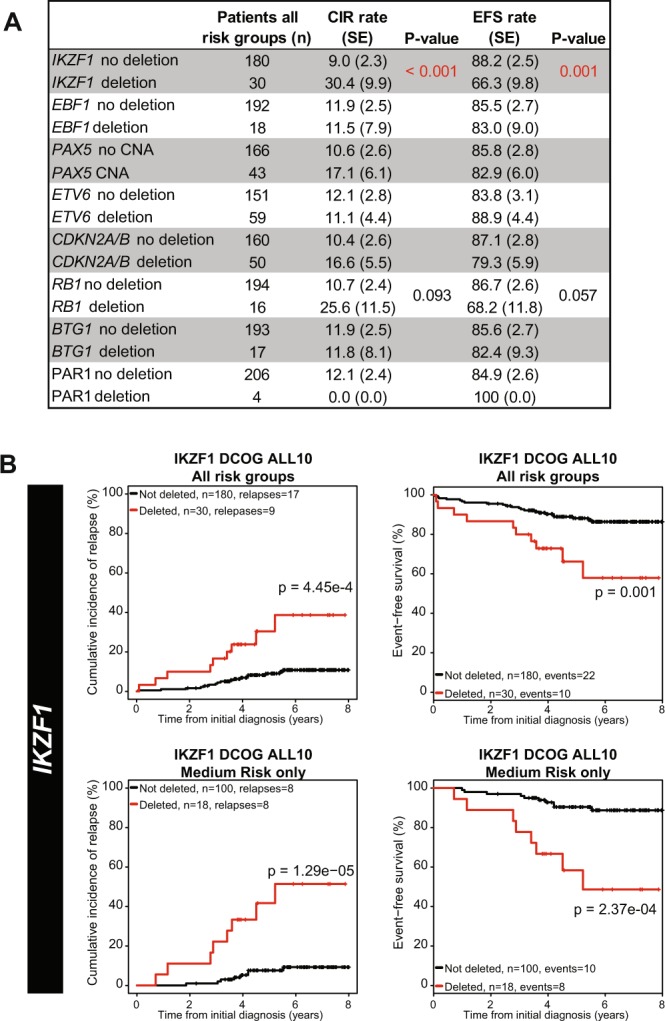
Prognostic value of CNAs in DCOG-ALL10 treated cases. (A) The association between CNAs in all risk groups and cumulative incidence of relapse (CIR) and event-free-survival (EFS) was examined. BCP-ALL patients (n = 210) were treated according to DCOG-ALL10 protocol. CIR was estimated using a competing risk model. Relapse and non-response were considered as event, and death as competing event. To test equality of the CIRs, the Gray’s test was applied. Non-response, relapse, and death were considered as events for EFS. EFS rates were determined using Cox regression, and compared using the Wald test. For reliable test results, groups should contain at least 5 cases. (B) CIR and EFS curves of cases without or with an IKZF1 deletion. Curves contain either all risk groups, or the medium risk arm only.
As cellular drug resistance might underlie this poor outcome, we examined the ex vivo efficacy of chemotherapeutic agents that are commonly used during induction and consolidation therapy. Primary BCP-ALL cells harboring IKZF1 deletions were more resistant to prednisolone and thiopurines compared to IKZF1 wildtype cells (p < 0.05; Fig. 5A,B). Resistance against these agents was subtype dependent, as visualized in Fig. 5B: prednisolone resistance was predominantly observed in high hyperdiploid cells (~1500-fold, p = 0.009), whereas thiopurine resistance (6-thioguanine (1.6 fold, p = 0.011) and 6-mercaptopurine (1.7 fold, p < 0.001)) was mainly identified in IKZF1-deleted BCR-ABL1-like and B-other cells (Fig. 5B). Moreover, high hyperdiploid cells with a deletion of IKZF1 were more resistant to L-asparaginase(Supplementary Fig. 8A).
Figure 5.

The association between CNAs and the ex vivo cellular drug response. (A) Leukemic cells were incubated for four days with a concentration range of prednisolone (µg/ml), vincristine (µg/ml), L-asparaginase (IU/ml), daunorubicin (µg/ml), 6-mercaptopurine (µg/ml), and 6-thioguanine (µg/ml), after which cell viability was measured using an MTT assay. The Mann-Whitney U test was applied to compare LC50-values. No association is depicted in grey, resistance in blue (p < 0.05, fold induction (FI) > 1), sensitive in green (p < 0.05, FI < 1), and not determined in white. The number of cases that were tested for prednisolone is depicted, and represent the maximum number of cases. For reliable test results, groups should contain at least 5 cases (groups ≤ 5 are depiced as ND). Results of single CNAs are depicted for all risk groups and for BCR-ABL1-like/B-other cells, high hyperdiploid cells, and ETV6-RUNX1 cells. In addition, associations between the risk classifiers IKZF1plus and integrated cytogenetic and CNA classification (poor risk) and cellular drug resistance are shown18,20. (B) LC50 values for prednisolone (µg/ml), 6-thiogunanine (µg/ml), and 6-mercaptopurine (µg/ml) of cases without or with IKZF1 deletion. Columns include all BCP-ALL subtypes (grey), BCR-ABL1-like/B-other cells (blue), and high hyperdiploid cells (green). The red line represent the median LC50 value in the each group. (C) LC50 values for prednisolone (µg/ml) of cases without or with PAX5 CNAs. Columns include all BCP-ALL subtypes (grey), BCR-ABL1-like/B-other cells (blue), high hyperdiploid cells (green), and ETV6-RUNX1 cells (yellow). The red line represent the median LC50 value in the each group. (D) LC50 values for prednisolone (µg/ml), vincristine (µg/ml), and daunorubicin (µg/ml) of primary leukemic cells without or with ETV6 deletion. Columns include all BCP-ALL subtypes (grey), BCR-ABL1-like/B-other cells (blue), high hyperdiploid cells (green), and ETV6-RUNX1 cells (yellow). The red line represent the median LC50 value in the each group. *p < 0.05, **p < 0.01; Mann-Whitney U test.
EBF1
The transcription factor EBF1 was deleted in a minority (6%) of the BCP-ALL cases (Fig. 1B). Deletions were enriched in BCR-ABL1-like cases (15%), but absent in KMT2A-rearranged and TCF3-PBX1 cases (Fig. 1B). Cases that harbored an EBF1 deletion showed a trend towards higher MRD levels after induction therapy compared to non-EBF1 deleted cases (OR = 5.16, p = 0.057; Fig. 3B). No association between loss of EBF1 and cellular drug resistance was observed, though drug resistance data were only available for six EBF1 deleted cases.
PAX5
CNAs of the transcription factor PAX5 were observed in 28% of the BCP-ALL cases (Fig. 1C). CNAs were detected throughout all BCP-ALL subtypes, although the frequency was relatively high in BCR-ABL1 (50%) and BCR-ABL1-like (46%) cases (Fig. 1C). The strong co-occurrence of PAX5 and CDKN2A/B CNAs (Fig. 2, OR = 6.36, p < 0.001) is likely caused by the high frequency of chromosome 9p deletions observed in these cases28; the chromosome arm on which PAX5 and CDKN2A/B are located. In correspondence, chromosome 9p deletions were observed in 51.8% (44/85) of these cases. Despite of the strong association between PAX5 CNAs and IKZF1 deletions (Fig. 2, OR = 2.94, p < 0.001), CNAs in PAX5 were not predictive for high MRD levels (Fig. 3C) nor a poor prognosis in pediatric BCP-ALL cases (Fig. 4A). Strikingly, leukemic cells harboring CNAs in PAX5 showed an increased sensitivity (~5.1 fold, p = 0.008) to prednisolone compared to PAX5-wildtype cells (Fig. 5C). This difference in sensitivity was only significant (p = 0.031) in ETV6-RUNX1 cases, but a similar pattern was also observed in the remaining BCP-ALL subtypes (Fig. 5C). Interestingly, this association depended on the IKZF1 status: cells with both an IKZF1 deletion and a CNA in PAX5 were equally sensitive to prednisolone as IKZF1 and PAX5 wildtype cells, whereas cells with only an IKZF1 deletion were more resistant to prednisolone (Supplementary Fig. 8B). These results suggest that CNAs in PAX5 might compensate for prednisolone resistance induced by loss of IKZF1.
ETV6
Deletions of the transcription factor ETV6 were detected in all BCP-ALL subtypes, but were especially enriched in ETV6-RUNX1 cases (71%; Fig. 1D). After induction therapy (TP1), ETV6-deleted cases more often showed low (<10−4) MRD levels compared to ETV6-wildtype cases (Fig. 3D; OR = 2.6, p = 0.02). However, this association was subtype dependent: in BCR-ABL1-like and B-other cases an adverse association between ETV6 deletions and MRD levels was observed (Supplementary Fig. 3). Prognosis of cases with loss of ETV6 was not different compared to ETV6-wildtype cases (Fig. 4). ETV6-deleted cells appeared to be more sensitive to prednisolone (~3.2 fold, p = 0.046), but more resistant to vincristine (~1.8 fold, p < 0.01) and daunorubicin (~1.9 fold, p = 0.028). Remarkably, loss of the wildtype ETV6 allele in ETV6-RUNX1-positve cells associated with a high sensitivity to vincristine instead of resistance (p = 0.013, Fig. 5D), suggesting that associations of vincristine resistance differ between genetic subtypes of ALL. Moreover, deletion of ETV6 was associated with L-asparaginase resistance in high hyperdiploid cells and high 6-thioguanine sensitivity in ETV6-RUNX1 cells (Supplementary Fig. 8C).
CNAs in cell cycle and proliferation genes
CDKN2A/B
Deletions of the cell cycle regulators CDKN2A and/or CDKN2B were often observed (33%) in the pediatric BCP-ALL cohort (Fig. 1E). Similar to PAX5, the deletions in CDKN2A/B were found in all BCP-ALL subtypes, but were especially enriched in BCR-ABL1 (65%, OR = 3.95, p = 0.003), BCR-ABL1-like (54%, OR = 2.88, p < 0.001), and B-other cases (OR = 1.67, p = 0.026). No association with clinical outcome parameters or cellular drug resistance was observed (Figs 3–5).
RB1
The cell cycle regulator RB1 was deleted in a minority (~7%) of the BCP-ALL cases (Fig. 1F) and deletions were detected in all BCP-ALL subtypes. Within the DCOG-ALL10 cohort, RB1-deleted cases showed a trend towards a poor event free survival (5- year EFS: 68.2% versus 86.7%, p = 0.057), which was caused by an unfavorable response in the medium risk (MR) treatment group (5-year EFS: 46.9% versus 88.3%, p = 0.003; Supplementary Fig. 7A). No association with MRD levels or cellular resistance to the tested drug panel was observed (Fig. 5).
BTG1
The anti-proliferative gene BTG1 was deleted in a minority (~8%) of the BCP-ALL cases. No deletions were detected in KMT2A-rearranged or TCF3-PBX1 cases, whereas the highest frequency was observed in BCR-ABL1 (30%) and ETV6-RUNX1 (16%) cases (Fig. 1G). Four out of five BTG1-deleted BCR-ABL1-like and B-other cases also harbored an IKZF1 deletion. These four cases all experienced an event and only the patient with wildtype IKZF1 remained in remission (Supplementary Fig. 7B). This finding underlines an earlier report, in which a cooperative effect of BTG1 and IKZF1 lesions in leukemogenesis was observed27.
CNAs in cytokine receptors
PAR1
Deletions in the pseudoautosomal region 1 (PAR1) were the least prevalent (~4%) in this pediatric BCP-ALL cohort. CNAs in this region indicate presence of interstitial deletions or a translocation, which both induce overexpression of CRLF229. Deletions of the PAR1 region were detected in BCR-ABL1-like (11%), B-other (10%), and high hyperdiploid cases (2%), but not in remaining BCP-ALL subtypes (Fig. 1H). Unfortunately, power was lacking to reliable study the association between deletions in the PAR1 region, MRD levels, clinical prognosis, and cellular drug resistance.
Taken together, with the exception of loss of the IKZF1 gene, none of the CNAs in the remaining B-cell development genes strongly associates with clinical outcome and cellular drug resistance as single marker. Our results show that the clinical value of CNAs in B-cell development genes is highly context dependent and differs between the diverse oncogenic drivers of pediatric BCP-ALL.
Risk stratification classifiers
In recent studies, IKZF1plus and integrated cytogenetic and CNA classification were shown to be prognostic classifiers18,20. In the DCOG-ALL10 cohort 12 of the 210 cases were classified as IKZF1plus20. The prognosis of IKZF1plus cases was unfavorable compared to cases with wildtype IKZF1 (Supplementary Fig. 9). Strikingly, no prednisolone resistance was observed in IKZF1plus cells, which could be explained by underrepresentation of high hyperdiploid cases in this group (n = 1, Fig. 5A). However, IKZF1plus cases did show ex vivo resistance to 6-thioguanine and 6-mercaptopurine, mainly caused by the high proportion of BCR-ABL1-like and B-other cases in this group.
Integration of cytogenetic and CNA data as reported by Moorman et al.18 identified cases with a genetic good and poor risk. Cases that were classified as poor risk showed an unfavorable 5-years EFS and CIR compared good risk cases, as shown in Supplementary Fig. 10A. These genetically poor risk cases showed high MRD levels after induction therapy and the first block of consolidation therapy, indicating a poor response to drugs that are used during these treatment phases (Supplementary Fig. 10B). Indeed, ex vivo cellular drug response data showed resistance of poor risk cells to L-asparaginase, 6-thioguanine, and 6-mercaptopurine (Fig. 5A, Supplementary Fig. 10C). Enrichment of BCR-ABL1-like cases could attribute to the thiopurine and L-asparaginase resistance observed in the poor risk group4.
Discussion
BCP-ALL cases harbor various genetic aberrations in genes involved in lymphoid maturation, cell cycle regulators, tumor suppressors, and tyrosine kinases. We performed an explorative study to gain insight in the association between CNAs in B-cell development genes, MRD levels, long-term prognosis, and cellular drug resistance. Interestingly, the distribution and clinical relevance of these CNAs was subtype-dependent. A high frequency of CNAs in these B-cell development genes was found in the poor prognostic subtypes BCR-ABL1, BCR-ABL1-like, and B-other. Cooperative lesions may favor the aggressive phenotype of a leukemia, such as exemplified by the synergistic effect between loss of IKZF1 and the BCR-ABL1 fusion gene in leukemogenesis30, and the antagonizing effect of IKZF1 deletions in the response to imatinib31. In contrast, the prognosis of ETV6-RUNX1, DUX4-rearranged, and ERG-deleted BCP-ALL is probably not affected by IKZF1 deletions, but numbers with IKZF1 deletions in these subtypes are low11,12,15,32,33. These observations indicate that the genetic context influences the functional effect of CNAs in B-cell development genes. The importance of the genetic context is exemplified by the fact that isolated deletions of BTG1 do not affect cellular drug resistance or the prognosis of BCP-ALL cases, whereas all four patients with concomitant loss of BTG1 and IKZF1 experienced an event. Moreover, combined loss BTG1 and IKZF1 was shown to enhance glucocorticoid resistance27. In contrast to BTG1-IKZF1 synergy, we observed that CNAs in PAX5 may counteract the effect of an IKZF1 deletion on prednisolone resistance. Various combinations of cooperative lesions may therefore have different effects on the pathobiology of B-cell precursor ALL cells.
In the present study we observed an association between deletion of IKZF1 and prednisolone resistance, especially in high hyperdiploid cells. In correspondence, a direct association has been demonstrated between IKZF1 deletion and glucocorticoid-induced cell death25,34. IKZF1 functions as a metabolic gatekeeper and consequently loss of IKZF1 results in increased intracellular ATP and glucose levels34. Interestingly, we previously observed a direct relation between an increased glycolytic rate and prednisolone resistance in primary BCP-ALL cells35,36. In these leukemic cells, inhibition of glycolysis restored the efficacy of prednisolone36. Hence, inhibition of glycolysis might also be a potential treatment strategy to re-sensitize IKZF1-deleted cells to prednisolone and should be explored in more detail in future studies, also in the context of BTG1 and PAX5.
In contrast to high hyperdiploid cells, deletion of IKZF1 was not linked to prednisolone resistance in primary BCR-ABL1-like and B-other ALL cells, suggesting that additional factors (e.g. differentiation stage, other oncogenic drivers) are important for the functional effect of a deletion of the IKZF1 gene in these type of cells. Instead of prednisolone resistance, we observed thiopurines resistance in these BCR-ABL1-like and B-other ALL cells. Thiopurine resistance might be caused by deficiencies in the DNA mismatch repair system and indeed DNA repair genes were reported to be downregulated in IKZF1-deleted cells37,38. Interestingly, this characteristic might offer opportunities to target these leukemic cells via the DNA mismatch repair apparatus, e.g. by PARP inhibitors, and warrants further studies.
Besides IKZF1, deletion of RB1 was predictive for a poor outcome in the MR-risk group of the DCOG-ALL10 cohort. RB1 deletions are known to be enriched in poor prognostic iAMP21 and hypodiploid cases, which might explain the unfavorable outcome of RB1-deleted cases13,39,40. However, the unfavorable outcome could not be explained by or cellular resistance against induction therapy drugs.
Recently, two independent studies showed that integration of genetic aberrations improved the risk stratification of BCP-ALL in children18,20. Both IKZF1plus and integrated cytogenetic and CNA classification predicted poor outcome in the DCOG-ALL10 cohort, and associated with drug resistance to thiopurines, or L-asparaginase and thiopurines, respectively. The cellular drug resistance could be attributed to overrepresentation of BCR-ABL1-like cases in these risk groups4. Taken together, our results suggest that the prognostic value of CNAs in B-cell development genes is linked to subtype-related drug resistance.
In the current study, we restricted our analyses to CNAs in eight genes that are recurrently deleted in pediatric BCP-ALL. However, additional genetic aberrations may be of importance for prognosis and cellular drug resistance and should be explored in future research. Moreover, as we performed an explorative study, it is of importance to confirm the associations that are proposed in the present paper in independent studies.
In conclusion, results obtained in the present study revealed that, with the exception of an IKZF1 deletion, none of the remaining CNAs as single marker associated both with an unfavorable clinical prognosis and cellular drug resistance. Our results indicate that the biological and clinical importance of CNAs in B-cell development genes (and presumably also other genetic aberrations) is highly context dependent and differs between the diverse oncogenic drivers of pediatric BCP-ALL. Functional studies that focus on potential causes of cellular drug resistance should therefore take the oncogenic driver and additional genetic aberrations into account.
Methods
Processing of primary patient material
Bone marrow and/or peripheral blood samples were obtained from children (1–18 years) with newly diagnosed ALL. Written informed consent was obtained from parents or guardians to use excess of diagnostic material for research purposes, as approved by the Medical Ethics Committee of the Erasmus Medical Center, The Netherlands. These studies were conducted in accordance with the Declaration of Helsinki. Mononuclear cells were isolated using Lymphoprep gradient separation and the leukemic blast percentage was determined microscopically by May-Grünwald Giemsa stained cytospin preparations, as described previously21. Samples were enriched to over 90% purity of leukemic cells by depletion of non-leukemic cells using immunomagnetic beads. Primary leukemic cells were maintained in RPMI-1640 Dutch modification supplemented with 20% fetal calf serum (Integro), with 0.1% insulin-transferrin-sodium selenite (Sigma), 0.4 mM glutamine (Invitrogen), 0.25 μg/ml gentamycine (Gibco), 100 IU/ml penicillin (Gibco), 100 μg/ml streptomycin (Gibco), 0.125 μg/ml fungizone (Gibco).
The major cytogenetic subtypes, i.e. high hyperdiploid (>50 chromosomes), ETV6-RUNX1, TCF3-PBX1, KMT2A-rearranged, BCR-ABL1, BCR-ABL1-like, and B-other (negative for all before mentioned genomic lesions), were determined using fluorescent in situ hybridization and (RT-)PCR. The 110-probeset gene expression classifier was used to identify BCR-ABL1-like cases4. Patients were treated according to the Dutch Childhood Oncology Group (DCOG)-ALL8, -ALL9, -ALL10, the EsPhALL protocol, or the COALL-06-97 and COALL-07-03 study protocols3,14,41,42. Patient characteristics were provided by the central study centers of DCOG, The Hague, the Netherlands and COALL, Hamburg, Germany. PCR-detected MRD was evaluated according to the EuroMRD guidelines3,43,44.
Multiplex Ligation-Dependent Probe Amplification
To identify genomic lesions in IKZF1, CDKN2A, CDKN2B, ETV6, PAX5, RB1, BTG1, EBF1, and the PAR1 region (CSF2RA/IL3RA/CRLF2), the SALSA P335 ALL-IKZF1 (a3) and the SALSA P202 Multiplex Ligation-dependent Probe Amplification (MLPA) assays (MRC-Holland, Amsterdam, Netherlands) were used as described previously14. In short, DNA fragments with incorporated FAM nucleotides were generated using 125 ng of genomic DNA, according to the manufacturer’s protocol. To quantify the amplified fragments, an ABI-3130 genetic analyzer (Applied Biosystems, Carlsbad, CA) was used. The manufacturer’s control probes were used to normalize peak intensities, as well as a synthetic control reference generated from five normal DNA samples in the same MLPA run (normal copy number = 0.75 ≤ peak ratio ≤ 2.0; deletions = peak ratio < 0.75; gain = peak ratio > 2.0). A deletion was defined by a peak ratio below 0.75 for at least one MLPA-probe per gene. CDKN2A/B deletions included loss of either CDKN2A or CDKN2B. The effect of intragenic amplifications and/or deletions in PAX5 were analyzed within one group, as they were predicted to be functionally equivalent18,45. Loss of the PAR1 region was defined by deletion of both IL3RA and CSF2RA probes while expression of the CRLF2 and SHOX-AREA probes was maintained. MLPA analyses were performed in 515 BCP-ALL cases, representing the major genetic subtypes in childhood ALL, i.e. 3.9% BCR-ABL1, 17.3% BCR-ABL1-like, 20.2% non-BCR-ABL1-like B-other, 28.7% ETV6-RUNX1, 24.5% high hyperdiploid, 1.6% KMT2A-rearranged, 3.9% TCF3-PBX1.
Clinical characteristics and statistics
To identify whether CNAs were underrepresented or enriched in a subtype, the Fisher’s exact test was applied using R software (version 3.2.1). Obtained odds ratios (ORs), 95% confidence interval, and p-values are reported. The Fisher’s exact test was also applied to compare minimal residual disease (MRD) levels after induction and first consolidation therapy between patients groups with CNAs and wildtype patients. Cumulative incidence of relapse (CIR) was estimated using a competing risk model and significance was determined using the Gray’s test. Relapse and non-response (counted as event at day 79) were considered as event, with death as competing event. Event-free survival (EFS) probabilities were estimated using cox regression and compared using the Wald test. Relapse, non-response, secondary malignancies and death were counted as events. Outcome analyses were performed in R (version 3.2.1), using the packages cmprsk version 2.2–746, mstate version 0.2.747 and survival version 2.38–448. Five-year EFS and CIR are reported. The DCOG-ALL10 trial is the most recently completed nationwide trial in which patients were risk-stratified by minimal residual disease (MRD) levels and for whom sufficient long-term follow-up data were available. Therefore, we restricted the analysis of associations between CNAs and clinical response parameters (MRD, clinical outcome) to this cohort. In addition, the genetic subtypes are represented with a distribution that is comparable to the general pediatric BCP-ALL population (excluding BCR-ABL1-positive cases since these patients are eligible for the EsPhALL protocol), i.e.12.2% BCR-ABL1-like, 13.9% non-BCR-ABL1-like B-other, 33.5% ETV6-RUNX1, 32.7% high hyperdiploid, 2.0% KMT2A-rearranged, and 5.7% TCF3-PBX1 positive cases. The clinical characteristics of this cohort are displayed in Supplementary Table 2.
Ex vivo drug resistance assays
Ex vivo cytotoxicity of prednisolone, vincristine, L-asparaginase, daunorubicin, 6-mercaptopurine, and 6-thioguanine was evaluated using 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT), as previously described21. In brief, cells were exposed to a concentration range of chemotherapeutics (prednisolone: 0.008 to 250 μg/mL; vincristine: 0.05 to 50 μg/mL; L-asparaginase: 0.003 to 10 IU/mL; daunorubicin: 0.002 to 2 μg/ml; 6-mercaptopurine: 15.6 to 500 μg/ml; and 6-thioguanine: 1.56 to 500 μg/ml) in a 96 wells plates for four days at 37 °C and 5% CO2. After four days of culture, samples were included if control wells harbored more than 70% leukemic cells and an optical density higher than 0.050 arbitrary units (adjusted for blank values). The concentration of drug lethal to 50% of the cells (LC50) was calculated. LC50-values were compared by the Mann-Whitney U test and adjusted for tied ranks if applicable.
Supplementary information
Acknowledgements
The authors thank all members of the Pediatric Oncology research laboratory of the Erasmus MC for their help in processing leukemic cell samples and performing ex vivo cytotoxicity assays. In addition, we would like to thank Arian van der Veer for performing the MLPA experiments. This work was supported by the VICI program grant 016.126.612 from Netherlands Organisation for Scientific Research (NWO), the Dutch Cancer Society grants AMC 2008–4265 and EMCR 2014–6998, the Kika Foundation (grant 132, 161, and 264) and the Pediatric Oncology Foundation Rotterdam.
Author Contributions
E.M.P.S., A.H.Q. and J.M.B. analyzed and interpreted data. A.B. performed experiments. R.P., H.A.d.G.K., V.d.H., M.A.H. and G.E. provided clinical characteristics, clinical outcome data, and interpreted data. R.P. and MLdB conceptualized the study, and interpreted data. E.M.P.S. and M.L.d.B. drafted the manuscript. The manuscript was revised and approved by all authors.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-41078-4.
References
- 1.Moorman AV. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood reviews. 2012;26:123–135. doi: 10.1016/j.blre.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- 3.Pieters R, et al. Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2016;34:2591–2601. doi: 10.1200/JCO.2015.64.6364. [DOI] [PubMed] [Google Scholar]
- 4.Den Boer ML, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10:125–134. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mullighan CG, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. The New England journal of medicine. 2009;360:470–480. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boer JM, et al. Tyrosine kinase fusion genes in pediatric BCR-ABL1-like acute lymphoblastic leukemia. Oncotarget. 2017;8:4618–4628. doi: 10.18632/oncotarget.13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imamura T, et al. Characterization of pediatric Philadelphia-negative B-cell precursor acute lymphoblastic leukemia with kinase fusions in Japan. Blood cancer journal. 2016;6:e419. doi: 10.1038/bcj.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts KG, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. The New England journal of medicine. 2014;371:1005–1015. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts KG, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153–166. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lilljebjorn H, et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nature communications. 2016;7:11790. doi: 10.1038/ncomms11790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yasuda T, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nature genetics. 2016;48:569–574. doi: 10.1038/ng.3535. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nature genetics. 2016;48:1481–1489. doi: 10.1038/ng.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moorman AV. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica. 2016;101:407–416. doi: 10.3324/haematol.2015.141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Veer A, et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood. 2013;122:2622–2629. doi: 10.1182/blood-2012-10-462358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harvey RC, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116:4874–4884. doi: 10.1182/blood-2009-08-239681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boer JM, et al. Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia. 2016;30:32–38. doi: 10.1038/leu.2015.199. [DOI] [PubMed] [Google Scholar]
- 17.Kuiper RP, et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia. 2010;24:1258–1264. doi: 10.1038/leu.2010.87. [DOI] [PubMed] [Google Scholar]
- 18.Moorman AV, et al. A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood. 2014;124:1434–1444. doi: 10.1182/blood-2014-03-562918. [DOI] [PubMed] [Google Scholar]
- 19.Gupta SK, Bakhshi S, Kumar L, Kamal VK, Kumar R. Gene copy number alteration profile and its clinical correlation in B-cell acute lymphoblastic leukemia. Leukemia & lymphoma. 2017;58:333–342. doi: 10.1080/10428194.2016.1193855. [DOI] [PubMed] [Google Scholar]
- 20.Stanulla M, et al. IKZF1(plus) Defines a New Minimal Residual Disease-Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2018;36:1240–1249. doi: 10.1200/JCO.2017.74.3617. [DOI] [PubMed] [Google Scholar]
- 21.Den Boer ML, et al. Patient stratification based on prednisolone-vincristine-asparaginase resistance profiles in children with acute lymphoblastic leukemia. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2003;21:3262–3268. doi: 10.1200/JCO.2003.11.031. [DOI] [PubMed] [Google Scholar]
- 22.Kaspers GJ, et al. Prednisolone resistance in childhood acute lymphoblastic leukemia: vitro-vivo correlations and cross-resistance to other drugs. Blood. 1998;92:259–266. [PubMed] [Google Scholar]
- 23.Pieters R, et al. Relation of cellular drug resistance to long-term clinical outcome in childhood acute lymphoblastic leukaemia. Lancet. 1991;338:399–403. doi: 10.1016/0140-6736(91)91029-T. [DOI] [PubMed] [Google Scholar]
- 24.Klumper E, et al. In vitro cellular drug resistance in children with relapsed/refractory acute lymphoblastic leukemia. Blood. 1995;86:3861–3868. [PubMed] [Google Scholar]
- 25.Marke R, et al. Tumor suppressor IKZF1 mediates glucocorticoid resistance in B-cell precursor acute lymphoblastic leukemia. Leukemia. 2016;30:1599–1603. doi: 10.1038/leu.2015.359. [DOI] [PubMed] [Google Scholar]
- 26.Imamura T, et al. IKZF1 deletion is enriched in pediatric B-cell precursor acute lymphoblastic leukemia patients showing prednisolone resistance. Leukemia. 2016;30:1801–1803. doi: 10.1038/leu.2016.128. [DOI] [PubMed] [Google Scholar]
- 27.Scheijen B, et al. Tumor suppressors BTG1 and IKZF1 cooperate during mouse leukemia development and increase relapse risk in B-cell precursor acute lymphoblastic leukemia patients. Haematologica. 2017;102:541–551. doi: 10.3324/haematol.2016.153023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwab CJ, et al. Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica. 2013;98:1081–1088. doi: 10.3324/haematol.2013.085175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russell LJ, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114:2688–2698. doi: 10.1182/blood-2009-03-208397. [DOI] [PubMed] [Google Scholar]
- 30.Virely C, et al. Haploinsufficiency of the IKZF1 (IKAROS) tumor suppressor gene cooperates with BCR-ABL in a transgenic model of acute lymphoblastic leukemia. Leukemia. 2010;24:1200–1204. doi: 10.1038/leu.2010.63. [DOI] [PubMed] [Google Scholar]
- 31.van der Veer A, et al. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood. 2014;123:1691–1698. doi: 10.1182/blood-2013-06-509794. [DOI] [PubMed] [Google Scholar]
- 32.Clappier E, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia. 2014;28:70–77. doi: 10.1038/leu.2013.277. [DOI] [PubMed] [Google Scholar]
- 33.Enshaei A, et al. Long-term follow-up of ETV6-RUNX1 ALL reveals that NCI risk, rather than secondary genetic abnormalities, is the key risk factor. Leukemia. 2013;27:2256–2259. doi: 10.1038/leu.2013.136. [DOI] [PubMed] [Google Scholar]
- 34.Chan LN, et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature. 2017;542:479–483. doi: 10.1038/nature21076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holleman A, et al. Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. The New England journal of medicine. 2004;351:533–542. doi: 10.1056/NEJMoa033513. [DOI] [PubMed] [Google Scholar]
- 36.Hulleman E, et al. Inhibition of glycolysis modulates prednisolone resistance in acute lymphoblastic leukemia cells. Blood. 2009;113:2014–2021. doi: 10.1182/blood-2008-05-157842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chouchana L, et al. Molecular insight into thiopurine resistance: transcriptomic signature in lymphoblastoid cell lines. Genome medicine. 2015;7:37. doi: 10.1186/s13073-015-0150-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iacobucci I, et al. IKAROS deletions dictate a unique gene expression signature in patients with adult B-cell acute lymphoblastic leukemia. PloS one. 2012;7:e40934. doi: 10.1371/journal.pone.0040934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrison CJ, et al. An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia. 2014;28:1015–1021. doi: 10.1038/leu.2013.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holmfeldt L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nature genetics. 2013;45:242–252. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Veerman AJ, et al. Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997-2004) Lancet Oncol. 2009;10:957–966. doi: 10.1016/S1470-2045(09)70228-1. [DOI] [PubMed] [Google Scholar]
- 42.Escherich G, et al. The long-term impact of in vitro drug sensitivity on risk stratification and treatment outcome in acute lymphoblastic leukemia of childhood (CoALL 06-97) Haematologica. 2011;96:854–862. doi: 10.3324/haematol.2010.039735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Velden VH, et al. Optimization of PCR-based minimal residual disease diagnostics for childhood acute lymphoblastic leukemia in a multi-center setting. Leukemia. 2007;21:706–713. doi: 10.1038/sj.leu.2404535. [DOI] [PubMed] [Google Scholar]
- 44.van der Velden VH, van Dongen JJ. MRD detection in acute lymphoblastic leukemia patients using Ig/TCR gene rearrangements as targets for real-time quantitative PCR. Methods in molecular biology. 2009;538:115–150. doi: 10.1007/978-1-59745-418-6_7. [DOI] [PubMed] [Google Scholar]
- 45.Familiades J, et al. PAX5 mutations occur frequently in adult B-cell progenitor acute lymphoblastic leukemia and PAX5 haploinsufficiency is associated with BCR-ABL1 and TCF3-PBX1 fusion genes: a GRAALL study. Leukemia. 2009;23:1989–1998. doi: 10.1038/leu.2009.135. [DOI] [PubMed] [Google Scholar]
- 46.Gray, R. J. cmprsk: Subdistribution Analysis of Competing Risks. R package version 2.2-6. http://CRAN.R-project.org/package=cmprsk (2013).
- 47.De Wreede LC, Fiocco M, Putter H. mstate: An R Package for the Analysis of Competing Risks and Multi-State Models. J Stat Softw. 2011;38:1–30. doi: 10.18637/jss.v038.i07. [DOI] [Google Scholar]
- 48.Therneau, T. A Package for Survival Analysis in S. R package version 2.36-12 (2012).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.