

Abstract
Parkinson disease (PD) is a neurodegenerative disease with motor as well as non-motor symptoms, including gastrointestinal dysfunction. In humans, these precede the motor symptoms by decades. Previously developed and characterized transgenic mice expressing the mutant human α-synuclein gene (SNCA) (either A53T or A30P), but not the endogenous mouse Snca, serve as models for familial PD. These animals demonstrate both robust abnormalities in enteric nervous system (ENS) function as well as synuclein-immunoreactive aggregates in ENS ganglia by 3 months of age, recapitulating early gastrointestinal abnormalities seen before the gait impairment characteristics of human and murine PD. Posiphen is a translational inhibitor of α-synuclein that targets the 5’ untranslated region (UTR) of SNCA mRNA and could be a potential drug for the treatment of PD. However, its efficacy in ameliorating symptoms of PD has not yet been evaluated. Here, we used these transgenic mouse models to investigate the efficacy of Posiphen in reversing the gastrointestinal dysfunction. We show that Posiphen normalizes the colonic motility of both transgenic mouse models, although it did not affect the Whole Gut Transit Time (WGTT). Pharmacokinetics studies revealed that Posiphen is more abundant in the brain than in blood, in agreement with its lipophilicity, and the main metabolite is N8-NorPosiphen, a molecule with similar properties as Posiphen. The brain Posiphen levels necessary to effect optimal function were calculated and compared with efficacious brain levels from previous studies, showing that a 2-3 mM concentration of Posiphen and metabolites is sufficient for functional efficacy. Finally, 10 mg/kg Posiphen reduced α-synuclein levels in the gut of hSNCAA53T mice treated for twenty-one weeks, while 50 and 65 mg/kg Posiphen reduced α-synuclein levels in the brain of hSNCAA53T mice treated for twenty-one days. In conclusion, this is the first study showing the preclinical efficacy of Posiphen in normalizing the colonic motility in mouse models of gastrointestinal dysfunction in early PD. This result is in agreement with the ability of Posiphen to reach the nervous system, and its mechanism of action, the translational inhibition of α-synuclein expression. These significant findings support further development of Posiphen as a drug for the treatment of PD.
Keywords: Posiphen, α-synuclein, Parkinson’s disease, gastrointestinal dysfunction, colonic motility
Introduction
PD is the second most common neurodegenerative disease after Alzheimer’s disease (AD), with a prevalence in the USA of 0.3%, rising to 1.5% in the over 55 age group [1]. PD affects the central, peripheral and enteric nervous systems [2-6]. Gastrointestinal dysfunction is a particularly common non-motor abnormality in PD, documented in over 80% of patients [7-9]. Symptoms include dysphagia, gastroparesis, prolonged gastrointestinal transit time, constipation and difficulty with defecation [10]. Gastrointestinal dysfunction can precede the onset of motor symptoms in PD patients by decades [11,12].
The discovery of the α-synuclein gene (SNCA), and its association with autosomal dominant PD [13], has provided researchers with an important insight into the pathogenesis of PD. The product of the SNCA gene is a 15 kDa protein expressed primarily in the nervous system and hematopoietic lineages. Although its physiological roles are not yet fully understood, evidence linking it with PD has been continuously mounting since its original discovery. Five missense mutations in the SNCA gene cause autosomal dominant PD [14-16]. In addition, duplication or triplication of the normal gene also causes a heritable form of the disease [17].
Transgenic mice, expressing mutant forms of α-synuclein have been utilized as mouse models of PD. It has been previously shown that mice containing the entire human SNCA gene, expressing either A53T or A30P mutant hSNCA, associated with familial PD in humans, but lacking endogenous mSnca, displayed robust abnormalities in ENS function by 3 months of age. The abnormalities reached maximum severity by 6 months of age, which persisted to 18 months of age [18]. However, only the A53T line developed abnormal motor behavior without detectable non-enteric autonomic abnormalities, olfactory dysfunction or dopaminergic deficits, Lewy body inclusions or neurodegeneration. The early ENS dysfunction in the absence of major central nervous system pathology mimics what is seen early in human PD patients, where ENS dysfunction has been reported to precede the more classical motor symptoms by years to decades [7-9,19]. Therefore, these animals can serve as an in vivo model of early PD.
One approach to lessen α-synuclein-induced pathogenesis is to reduce the synthesis of the protein. Posiphen is an orally available small molecule drug that inhibits the translation of α-synuclein through a novel mechanism of action. Posiphen increases the affinity of Iron Regulatory Protein-1 (IRP1) to the Iron-Response Element (IRE) of the 5’UTR of SNCA mRNA, thus preventing the association of the mRNA with the ribosome and repressing translation [20-22]. This is supported by the fact that Posiphen also suppresses the translation of another molecular target, amyloid precursor protein (APP), the mRNA of which contains an IRE homologous to the one in SNCA mRNA [23-26]. APP is involved in Alzheimer’s disease (AD) pathogenesis, through its own action as well as the action of its metabolic fragments.
The effects of Posiphen treatment have been studied in the context of AD. Posiphen treatment reduced APP and/or Aβ42 levels in various systems, including in vitro - in human neuroblastoma cell cultures and rodent primary neurons, and in vivo - in the brain of transgenic mice over-expressing the human APP gene with the Swedish mutation K670N/M671L (APPSWE), a model of early-onset AD [20,27,28]. Neurotrophic and neuroprotective functions have also been described, presumably secondary to the reduction of APP and Aβ42 levels [28,29]. Furthermore, the translational inhibition of APP by Posiphen normalized impairments in spatial working memory, contextual fear learning, and synaptic function in human transgenic APPSWE/presenilin-1 (APP/PS1) mice [30]. Most importantly, in a phase I clinical trial, Posiphen treatment was well tolerated and reduced the level of soluble APP (sAPP) fragments, Aβ42 and tau in the cerebrospinal fluid (CSF) of mildly cognitively impaired (MCI) patients [31].
Posiphen treatment also reduced levels of α-synuclein in human neuroblastoma cell lines and rodent primary neurons [20,21,32]. This makes Posiphen a promising drug candidate for the treatment of PD. However, the effects of Posiphen treatment in animal models of PD have not yet been studied.
The goal of this study is to establish preclinical efficacy of Posiphen in a PD mouse model overexpressing the full-length human SNCA gene under the endogenous promoter. To this end, we used the previously characterized transgenic mice, hSNCAA53T and hSNCAA30P [18], as models for gastrointestinal dysfunction that is commonly seen in the early stages of PD. We tested if various doses of Posiphen treatment, delivered by intraperitoneal (IP) injection once daily, ameliorated colonic motility dysfunction, and the duration of its effect when Posiphen treatment was removed. We also tested whether WGTT and the motor functions of hSNCAA53T mice were altered after Posiphen treatment.
A secondary goal was to examine the pharmacokinetics of Posiphen in the hSNCAA53T mice, to allow us to make comparisons with the pharmacokinetics in humans [31] and the APP/PS1 mouse [30]. Specifically, we tested the distribution of Posiphen and its metabolites in the mouse brain and blood. By comparing these concentrations, which were achieved by the effective Posiphen dose of 10 mg/kg IP once a day, to the ones reported in [31] and [30], we were able to extrapolate an effective Posiphen dose for humans.
Finally, we examined the effect of Posiphen treatment on the α-synuclein protein levels in the gut and brain of hSNCAA53T mice. For this purpose we used conventional Western blotting and ELISA. We should mention that conventional Western blotting for α-synuclein has been shown to be problematic, due to the fact that α-synuclein monomers get easily detached from the blotting membrane during the washing steps [33]. Due to this effect and/or other technical difficulties, we had trouble reproducing our initial Western blot results. However, we also performed ELISA, and the data are in good agreement with the initial Western blot.
Materials and methods
Animal care
Mice were given free access to food and drinking water. All animals were maintained at 24°C with 55% relative humidity on a 12-h light cycle (0700-1900-h). Care was in accordance with the guidelines of University of California San Francisco Animal Care and Use Committee.
Transgenic mice
Two human synuclein transgenic lines were used: 1) double-PAC-transgenic (SNCA A53T)+/+; Snca -/-, expressing A53T mutant hSNCA homozygous for both insertion sites of the PAC and deficient in endogenous mSnca, and 2) double-PAC-transgenic (SNCA A30P)+/+; Snca -/-, expressing A30P mutant hSNCA homozygous for both insertion sites of the PAC and deficient in endogenous mSnca [18] (where PAC is P1-derived artificial chromosomes). The nomenclature for these lines will be abbreviated throughout this paper to hSNCAA53T and hSNCAA30P respectively. Wild type Snca +/+ and α-synuclein knock out mice Snca -/- were used as controls for these experiments [18]. Both the transgenic and control mice share a similar genetic background (129S6/SvEvTac_FVB/N_C57/BL6), thus minimizing differences that could be attributed to the genetic background or to strain differences. Only male mice were used.
Drug substance
Posiphen, (3aR)-1, 3a, 8-trimethyl-1, 2, 3, 3a, 8, 8a-hexahydropyrrolo[2,3-b]indol-5-yl phenyl-carbamate tartrate (IND#72,654) was manufactured to GMP requirements by Rhodia (Boulogne-Billancourt, France).
Standards
Posiphen and its metabolites, N1- and N8-NorPosiphen, were synthesized by Chemtos (16713 Picadilly Ct, Round Rock, TX 78664) to greater than 99.9% purity.
Posiphen administration
Posiphen doses were made fresh daily. Posiphen was dissolved in saline (0.9% sodium chloride) to various concentrations so that each mouse received the drug in a volume of 0.1 ml. The 0 mg/kg dose was 0.1 ml of saline only. Male mice were injected once daily by IP injection starting at 6 weeks of age. Three treatment paradigms were followed, depending on the experiment: 0, 3 or 10 mg/kg daily for 7 months before stopping treatment; 0 or 10 mg/kg daily for 21 days or 21 weeks; 0, 5, 20, 35, 50, or 65 mg/kg daily for 21 days.
Harvesting of tissues
Mice were bled, by submandibular bleeding into EDTA di-potassium salt SAFE-T-FILL blood collection tubes (Ram Scientific Inc) and snap frozen. The brain was harvested and divided into 5 parts; cerebellum and each hemisphere cut in half transversely were collected and snap frozen on dry ice, to use in pharmacokinetics and pharmacodynamics analyses, respectively. The gut was washed with PBS to remove fecal matter and the descending colon was harvested and used for Western blots.
Colonic motility
Colonic motility was measured by the bead expulsion test as previously described [18]. Briefly, a glass bead (diameter, 3 mm) was inserted through the anus with a polished glass rod and pushed into the colon for a distance of 2 cm. The time required for expulsion of the glass bead was recorded.
Whole gut transit time (WGTT)
WGTT was measured by oral gavage of 0.3 ml of 6% (w/v) carmine dye in 0.5% methylcellulose (Sigma) to each mouse as previously described [18]. The time taken from the administration of carmine until the first appearance of one red fecal pellet was recorded.
Rotarod test
The Ugo Basile rotarod test was used to test motor ability of hSNCAA53T mice as described previously [18]. Tests were carried out at 7 months of age. Snca +/+ and Snca -/- mice were used as controls. Mice were tested using four trials per day on three consecutive days. The rod accelerated from 4 to 40 rpm over 5 minutes, and the time each mouse stayed on the Rotarod before falling off was recorded.
Posiphen and metabolite pharmacokinetic assays
Concentrations of Posiphen, N1-NorPosiphen and N8-NorPosiphen in mouse blood and brain (cerebellum) samples were determined by LC-MS/MS at Alliance Pharma (Malvern, PA).
Analysis was conducted on an HPLC system consisting of two PE Series 200 micropumps (Wellesley, MA) and a CTC Leap auto-sampler (Carrboro, NC) connected to an Applied Biosystems API4000 triple quadruple mass spectrometer (Foster City, CA), operated in the MRM mode with a turbo ion spray interface. Chromatographic separation was achieved on a Phenomenex Synergi Polar RP, 100 × 2.0 mm id, 2.5 µm column. The mobile phases were 0.1% formic acid in water (A) or 0.1% formic acid in methanol (B). Stable deuterated (d5) internal standards were used for each analyte, except in the case of N1-NorPosiphen where N8-NorPosiphen-d5 was used as the internal standard.
Blood samples were prepared for analysis by acetonitrile precipitation and centrifuged, the supernatant was dried under nitrogen and the dried samples were reconstituted with 10:90:0.1 methanol: water: formic acid, vortexed and analyzed. Brain samples were sonicated in acetonitrile, and, thereafter, treated as described for blood.
Calibration ranges for each analyte ranged from 1000 ng/ml to 1 ng/ml, in blood and brain matrices. The detection limit was 0.025 ng/ml.
Gut Western blotting
Protein from the gut was extracted as previously described [18]. 10 μg of descending colon protein was loaded per well on 10% SDS-PAGE gels (BioRad). After electrophoresis, transfer to a polyvinylidene difluoride membrane followed. The presence of α-synuclein was assayed with rabbit anti-α-synuclein (Assay Designs) antibody that recognizes both human and mouse α-synuclein. Bands were visualized with horseradish peroxidase (HRP)-coupled goat anti-rabbit IgG (GE Healthcare) and the chemiluminescent substrate ECL system used for detection (GE Healthcare Cell Signaling). Mouse anti-β-actin (Calbiochem) was used as a loading control.
Brain Western blotting
Mice were treated IP with 0, 5, 20, 35, 50, or 65 mg/kg of Posiphen for three weeks, sacrificed, and their brains were harvested. Post nuclear homogenates were prepared after pre-lysis of brain tissue in sterile phosphate buffer and centrifugation at 10,000 g for 10 min to pellet the nuclei. Cytoplasmic protein lysates of isolated brain cortex tissue were then homogenized in midRIPA buffer (25 mM Tris pH 7.4, 1% NP40, 0.5% sodium deoxycholate, 15 mM NaCl, protease inhibitors, RNase inhibitor and 10 μM DTT, and protease inhibitor PMSF and leupeptin as present in the buffer). Western blotting for α-synuclein was performed using mouse monoclonal anti-α-synuclein (BD Biosciences (Clone 42/α-synuclein)), and anti-β-actin as described [20,21]. The blots were developed using chemiluminescence (Pierce), visualized with a PhosphoImager (BioRad, Hercules, CA), and the bands were quantified using QuantityOne software (BioRad).
Enzyme-linked immunosorbent assay (ELISA)
A human α-synuclein ELISA kit (KHB0061, Invitrogen, Inc.) was used, and the manufacturer’s instructions were followed. Briefly, brain lysates were prepared in a lysis solution with protease inhibitor (10 μl/ml) added according to the manufacturer’s instructions. Samples were incubated on a plate shaker for 1 h, before removing 20 μl of lysate. The cell lysates were transferred to 500 μl Eppendorf tubes and centrifuged (Eppendorf Centrifuge 5417C) for 10 min at 13,000 rpm. 12 μl of the supernatant was added to 48 μl of standard diluent buffer (1:5), and these samples were then added to the ELISA plate wells. α-synuclein standard curve wells were prepared according to manufacturer’s instructions. Next, 50 μl of anti-α-synuclein detection antibody solution was added to each well, and the plate was tapped to mix. The plate was then covered with a tape seal and incubated for 3 h at room temperature (RT). Wells were washed four times with wash buffer, and 100 μl of anti-rabbit IgG conjugated to HRP antibody solution was added to each well. The plate was covered and incubated for 30 min at RT. Wells were washed four times with wash buffer and 100 μl of Stabilized Chromogen was added to each well. The plate was then sealed and incubated in the dark at RT for 30 min. Finally, 100 μl of Stop Solution was added to each well, the plate was tapped to mix, and absorbance was read at 450 nm using an Envision plate reader (Perkin Elmer).
Statistics
Unpaired nonparametric two-tailed Mann-Whitney U test was used to determine statistical significance when the data were not normally distributed or could only be ranked. Unpaired, two-tailed Student’s t-test was used when N was too small to test for normality, but there was no outlier. Repeated measures two-way ANOVA, with Tukey’s multiple comparisons test was used for the Rotarod data. Kruskal-Wallis test with Dunn’s multiple comparisons test was used to compare the weights. All data were analyzed by GraphPad Prism 6 statistical software (GraphPad Software).
Results
Gastrointestinal dysfunction studies
In this study, we set out to investigate the effect of translational inhibition of α-synuclein by Posiphen on gastrointestinal dysfunction, specifically colonic motility and WGTT, of hSNCAA53T and hSNCAA30P transgenic mice. These, as well as control wild type Snca +/+ and knockout Snca -/- mice were injected IP once daily, starting at 6 weeks of age, with either 0, 3 or 10 mg/kg of Posiphen in saline. We tested only male mice for all groups, since the hSNCAA53T and hSNCAA30P males previously exhibited an extended colonic motility time phenotype that was consistently robust compared to females [18]. We assessed the colonic motility by measuring the time required to expel a glass bead inserted into the colon at a distance of 2 cm above the anus at 4 and 7 months of age (shown in Figure 1A and 1B for hSNCAA53T and hSNCAA30P, respectively). Note the different time scales in Figure 1A and 1B; hSNCAA30P mice display longer bead expulsion times than hSNCAA53T, which is the opposite than what was reported in [18]. At 4 and 7 months of age the bead expulsion times were similar for control wild type Snca +/+ and knockout Snca -/- male mice. Figure 1A shows that, as predicted, vehicle-treated hSNCAA53T mice displayed statistically significantly increased bead expulsion time, in comparison to vehicle-treated control mice (not indicated on graph). The hSNCAA53T mice that received treatment with either 3 or 10 mg/kg of Posiphen displayed a statistically significant decrease in bead expulsion time at both 4 and 7 months of age, when compared to vehicle-treated hSNCAA53T. Both doses had a comparable effect. Similarly, Figure 1B shows that the expulsion time of vehicle-treated hSNCAA30P mice was increased, as compared with vehicle-treated control mice. Treatment of hSNCAA30P mice with 3 mg/kg Posiphen statistically significantly reduced the latency as compared to treatment with vehicle at 7 months, while the reduction did not reach significance at 4 months. However, treatment of hSNCAA30P mice with 10 mg/kg Posiphen statistically significantly reduced the latency as compared to treatment with vehicle at both time points. As seen, at 4 months, but not at 7 months, the effect of Posiphen on the colonic motility of hSNCAA30P is dose-dependent. These results show that Posiphen treatment of hSNCAA53T and hSNCAA30P transgenic mice can decrease the latency time of colonic motility.
Figure 1.
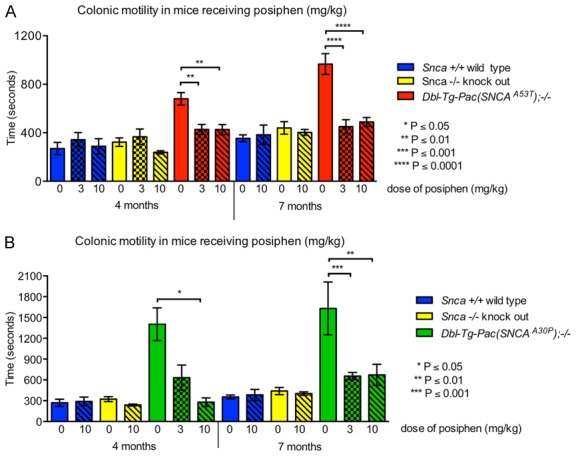
Posiphen decreases latency time of colonic motility in two mouse models of PD. (A) hSNCA A53T, Snca +/+, and Snca -/-, and (B) hSNCA A30P, Snca +/+, and Snca -/- are compared. Daily IP injection of 3 or 10 mg/kg Posiphen starting at six weeks of age until four or seven months of age decreases the prolonged bead expulsion times in hSNCA A53T and hSNCA A30P mice, in comparison to their saline-treated counterparts (0 mg/kg); statistically significant differences are indicated on the graphs. Mean and SEM are shown. N = on average 9 mice per group. Unpaired, two-tailed Mann-Whitney tests were performed. SEM; standard error of the mean.
Next, we asked what would happen to the colonic motility latency time after the Posiphen treatments were terminated. We tested all treatment groups for colonic motility at 9 and 16 weeks after the termination of vehicle or Posiphen treatments, and results are shown in Figure 2A and 2B. As expected, the bead expulsion times of control wild type Snca +/+ and knockout Snca -/- male mice at 9 weeks after treatment discontinuation were comparable between those groups that had previously received vehicle vs. Posiphen. Therefore, these control groups were not tested again at 16 weeks. At 9 and 16 weeks after treatment discontinuation, hSNCAA53T animals that had previously received 10 mg/kg of Posiphen showed comparable expulsion times to the mice that had previously received vehicle. The ones that had received 3 mg/kg displayed significantly shorter expulsion time at 9 weeks, but a statistically insignificant trend for prolonged expulsion time at 16 weeks after the end of treatment, in comparison to the ones that had received vehicle (Figure 2A). In case of the hSNCAA30P mice, after 9 weeks without treatment, the expulsion times in 10 mg/kg Posiphen treated mice were still statistically significantly decreased, while the 3 mg/kg Posiphen treated mice displayed a trend of reduced expulsion time, which did not reach statistical significance, in comparison to the vehicle control. After 16 weeks without treatment, the 3 mg/kg Posiphen group displayed statistically significantly reduced bead expulsion times, while the 10 mg/kg Posiphen group was comparable to the vehicle control (Figure 2B). Collectively, these data demonstrate that, in hSNCAA30P mice, the beneficial effects of Posiphen on colonic motility extend to at least 9 weeks after the end of treatment.
Figure 2.
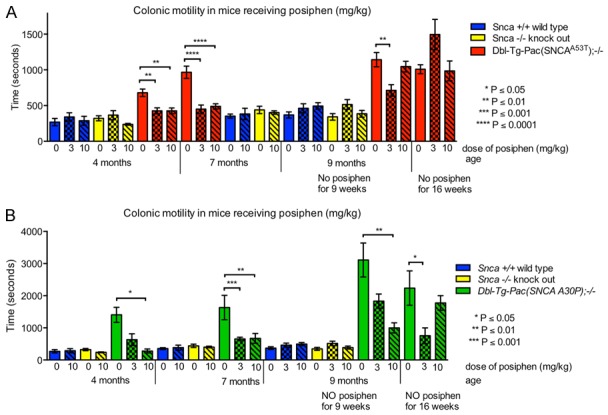
Colonic motility after Posiphen discontinuation. (A) hSNCA A53T, Snca +/+, and Snca -/-, and (B) hSNCA A30P, Snca +/+, and Snca -/- are compared. Mice were treated with daily IP injections of 0, 3 or 10 mg/kg Posiphen in saline from six weeks to seven months of age, at which point the treatment stopped. Nine and sixteen weeks after the discontinuation of treatment, the mice described in Figure 1 were tested again with the bead expulsion test to assess their colonic motility. The statistically significant differences found between saline-treated vs. Posiphen-treated hSNCA A53T and hSNCA A30P mice after treatment discontinuation are indicated on the graphs. Mean and SEM are shown. N = on average 10 mice per group. Unpaired, two-tailed Mann-Whitney tests were performed. SEM; standard error of the mean.
Another assay that can be used to examine gastrointestinal dysfunction is the WGTT test. hSNCAA53T mice have been previously shown to display prolonged WGTT at as early as 3 months of age [18]. We therefore examined the WGTT of 7-month-old hSNCAA53T mice that received 0, 3 or 10 mg/kg IP injections of Posiphen starting 6 weeks of age, but we did not see any differences in the transit times among these treatment groups (Figure 3). However, we should note that the WGTT of seven month old hSNCAA53T mice here (between 200-250 minutes) is much lower than the WGTT of hSNCAA53T mice reported in [18] (between 400-500 minutes). Although control Snca +/+ and Snca -/- mice were not used here, in [18], the WGTT of control Snca +/+ and Snca -/- mice was about 200-250 minutes. Therefore, it is possible that, in the present study, the vehicle-treated hSNCAA53T mice did not display an increased WGTT, and, thus lost the WGTT phenotype described in [18].
Figure 3.

WGTT in hSNCA A53T male mice treated with daily IP injections of 0, 3 or 10 mg/kg Posiphen in saline from six weeks to seven months of age. Posiphen did not affect the WGTT of hSNCA A53T mice, as determined by unpaired, two-tailed t-tests. Mean and SEM are shown. N is indicated on the graph. SEM; standard error of the mean.
Motor function studies
hSNCAA53T mice have been previously shown to display reduced motor function starting at 6 months of age [18]. Here, we tested the motor function of 7 month old hSNCAA53T mice that received 0, 3 or 10 mg/kg IP injections of Posiphen daily starting at 6 weeks of age, using the accelerating Rotarod test (Figure 4). Contrary to previous findings [18], the hSNCAA53T mice did not show motor dysfunction on the Rotarod test (Figure 4A). The performance of vehicle-treated hSNCAA53T mice was not significantly different than the one of the vehicle-treated controls. Figure 4B shows that, at the last Rotarod trial (day 3 - trial 4), there is also no difference in motor coordination between vehicle- and Posiphen-treated groups.
Figure 4.
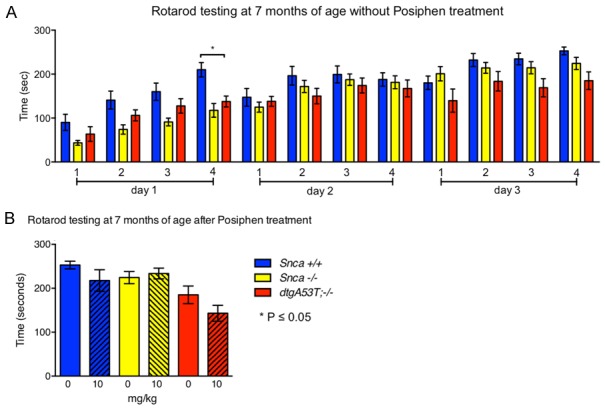
Motor function assessment by rotarod testing. Mice treated with daily IP injections of 0 or 10 mg/kg Posiphen starting at six weeks until seven months of age were assessed by rotarod testing at seven months of age. Repeated measures two-way ANOVA, with Tukey’s multiple comparisons test, was performed for all treatment groups. (A) Rotarod performance of the saline treated animals is shown. The hSNCA A53T genotype did not affect the time the mice stayed on the accelerating rotator in a statistically significant manner, as compared to the control mice, with the exception of a single difference between hSNCA A53T vs. Snca +/+ (day 1 - test 4). (B) Rotarod performance of the 0 and 10 mg/kg Posiphen-treated mice at the last rotarod trial (day 3 - test 4). Posiphen treatment has no effect on motor function of either Snca +/+, Snca -/- or hSNCA A53T mice. Mean and SEM are shown. N = on average 13 per group for (A), and 11 per group for (B). SEM; standard error of the mean.
Body weight
To monitor if Posiphen has any effects on body weight, vehicle- and Posiphen-treated mice were weighed after treatment at 7 months of age. Posiphen treatment with either the 3 or 10 mg/kg IP dose did not significantly alter the weight of mice of any genotype at 7 months of age (Figure 5). However, while Posiphen did not alter the weights, the hSNCAA53T mice weigh less than the control mice.
Figure 5.
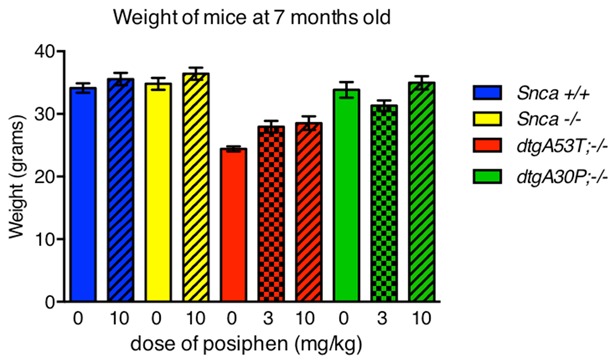
Weights of mice at seven months of age, after daily IP injections of 0, 3, or 10 mg/kg Posiphen starting at six weeks old. Posiphen treatment did not affect the weight of treated mice, as determined by Kruskal-Wallis test with Dunn’s multiple comparisons test. Mean and SEM are shown. N = on average 12 per group. SEM; standard error of the mean.
Pharmacokinetics
Posiphen has been previously shown to be more abundant in brain tissue than plasma [30,31]. Here, we examined the distribution of Posiphen and its two main metabolites, N1-NorPosiphen and N8-NorPosiphen, in the brain and blood of Snca +/+, Snca -/- and hSNCAA53T mice injected with 10 mg/kg Posiphen IP daily for 21 days, starting at 6 weeks of age. The levels of these molecules in the different tissues were determined by LC-MS/MS and are shown in Figure 6. As predicted, the concentration of Posiphen and its metabolites is roughly 6 times higher in brain than blood. In the brain and blood of control wild type Snca +/+, knockout Snca -/- mice, and hSNCAA53T mice, N8-NorPosiphen is the most abundant metabolite, followed by Posiphen and N1-NorPosiphen at comparable levels. Also, the metabolites are found in highest levels in hSNCAA53T mice, followed by the Snca -/- mice, and then by Snca +/+ mice, which display the lowest levels.
Figure 6.
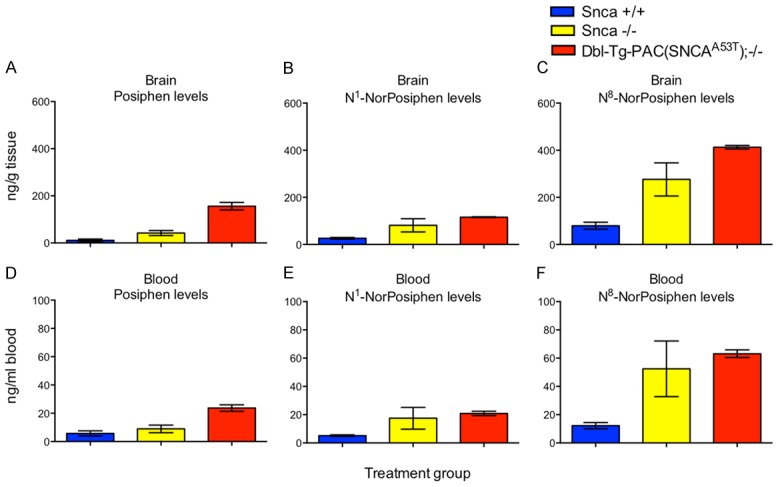
Distribution of Posiphen and its metabolites in brain and blood of Snca +/+, Snca -/-, and hSNCA A53T mice. Mice were treated with daily IP injections of 10 mg/kg Posiphen starting at six weeks old for 21 days. Concentrations of Posiphen (A, D) and its main metabolites N1-NorPosiphen (B, E) and N8-NorPosiphen (C, F) in brain cerebellum (A-C) and blood (D-F) were measured by LC-MS/MS. Mean and SEM are shown. N = 3 per group. SEM; standard error of the mean.
Next, we questioned whether the efficacious Posiphen levels that resulted in the reduced latency time of colonic motility in hSNCAA53T and hSNCAA30P mice in the present study are comparable to the efficacious Posiphen levels that rescued impairments in learning and memory in the transgenic AD mouse model, APP/PS1 [30]. Therefore, we compared the levels of Posiphen and its metabolites in the brain of hSNCAA53T mice treated daily with 10 mg/kg Posiphen IP for 21 days, starting at 6 weeks of age (Figure 7A) with those in brains of APP/PS1 mice treated daily per os (PO) with 25 mg/kg Posiphen for 14 days, starting at 3 months of age (Figure 7B, reproduced with permission [30]. Copyright 2018, Elsevier Inc.). As can be seen, the levels of all molecules are very similar between the two models. In both cases, N8-NorPosiphen is the most abundant form, while N1-NorPosiphen levels are similar to Posiphen. Importantly, the concentration of Posiphen in the brain of both models is roughly 155 ng/g of tissue (or 460 nM). N1-NorPosiphen and N8-NorPosiphen have also been shown to reduce the levels of α-synuclein and APP in primary neuron cultures [20,32] and should therefore be taken into consideration when calculating the effective drug brain levels. The total concentration of metabolites, which indicates the total efficacious drug level in the brains of the transgenic mouse models, is 684 ng/g in case of hSNCAA53T mice and 977 ng/g in case of APP/PS1 mice, corresponding to 2 and 3 mM.
Figure 7.
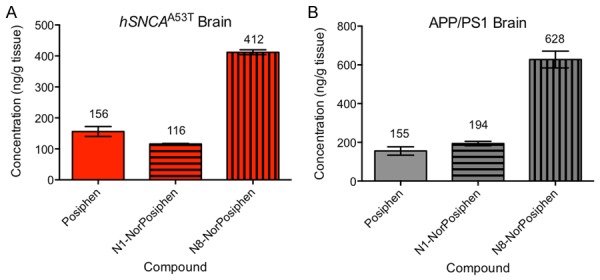
Concentration of Posiphen and metabolites in the brain of transgenic mouse models of PD (A) and AD (B), where Posiphen was effective in ameliorating impairments in colonic motility and memory, respectively. (A) hSNCA A53T mice were treated with 10 mg/kg Posiphen IP for 3 weeks, starting at 6 weeks of age. (B) APP/PS1 transgenic mice (carrying the human genes for APP and PS1 (Presenilin 1) with mutations associated with familial AD) were treated with 25 mg/kg PO for 2 weeks, starting at 3 months of age (adapted from [30]). Concentrations of Posiphen and metabolites were measured in the cerebellum by LC-MS/MS in both cases. Mean and SEM are shown. The numbers above the columns are the means, in ng/g. N = 3 per group. PO; per os. SEM; standard error of the mean.
Pharmacodynamics
Finally, to correlate the effect on colonic motility with the mechanism of action, we tested the effect of Posiphen treatment on the levels of α-synuclein. First we examined the gut. Figure 8 shows that treatment of hSNCAA53T mice with 10 mg/kg IP Posiphen for 21 weeks statistically significantly reduced the levels of the protein in the gut, as compared to levels in hSNCAA53T mice treated with vehicle. Treatment for 21 days was not enough to reduce α-synuclein levels, suggesting that Posiphen acts over a longer period of time in that tissue. β-actin was used as a loading control, resulting in multiple bands (not shown). Semi-quantization results were similar with and without normalization with the main bands; therefore, we present data without normalization. However, these Western blot results were variable and should be interpreted with caution.
Figure 8.
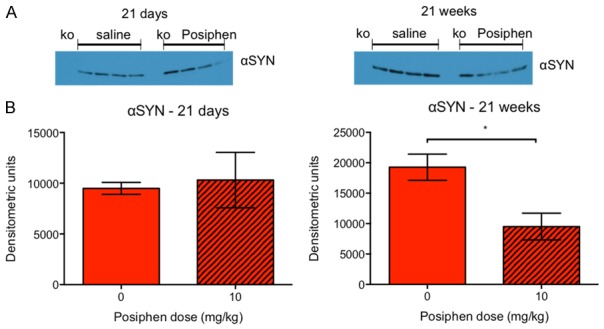
α-synuclein gut levels following treatment of hSNCA A53T mice with 10 mg/kg Posiphen for 21 weeks or 21 days, starting at six weeks of age. A. α-synuclein Western blot of gut samples of Snca -/- mice, and hSNCA A53T mice treated with saline or 10 mg/kg Posiphen (ko; knock out Snca -/- mice, as negative control). B. Semi-quantization of the α-synuclein Western blot for hSNCA A53T mice. 21 weeks but not 21 days of Posiphen treatment significantly reduced α-synuclein levels, by 50.6%. *P ≤ 0.05, by unpaired, two-tailed t-test. Mean and SEM are shown. N = 4 per group. αSYN; α-synuclein. SEM; standard error of the mean.
Last but not least, we examined the dose-response effect of Posiphen on α-synuclein brain levels in hSNCAA53T mice. Mice were treated with 0, 5, 20, 35, 50 or 65 mg/kg IP Posiphen for 21 days, and their brain extracts were used to measure α-synuclein by Western blot and ELISA (Figure 9). The Western blot and ELISA presented in Figure 9A-C, respectively, are the first ones performed, by a blinded investigator. Since the three blots comparing 0 vs. 65 mg/kg, 5 vs. 50 mg/kg and 20 vs. 35 mg/kg (Figure 9A) were run separately, without a common sample, the data could not be normalized between blots. Therefore, they were plotted separately (Figure 9B). A dose-response effect is indicated, since the percentage of reduction in α-synuclein levels is proportional to the dose difference. 50 mg/kg Posiphen treatment statistically significantly reduces α-synuclein levels as compared to the 5 mg/kg dose, while the 65 mg/kg vs. saline does not reach significance, due to high variability in the 65 mg/kg group. To substantiate the data we also ran an α-synuclein ELISA (Figure 9C) of the same treatment groups in duplicates, and we showed that Posiphen reduces α-synuclein hSNCAA53T brain levels in a dose-dependent manner. The reduction plateaus at about 28%. Mice treated with 65 and 50 mg/kg Posiphen display statistically significantly reduced α-synuclein levels, while 35 mg/kg display a trend for reduction, compared to saline-treated controls.
Figure 9.
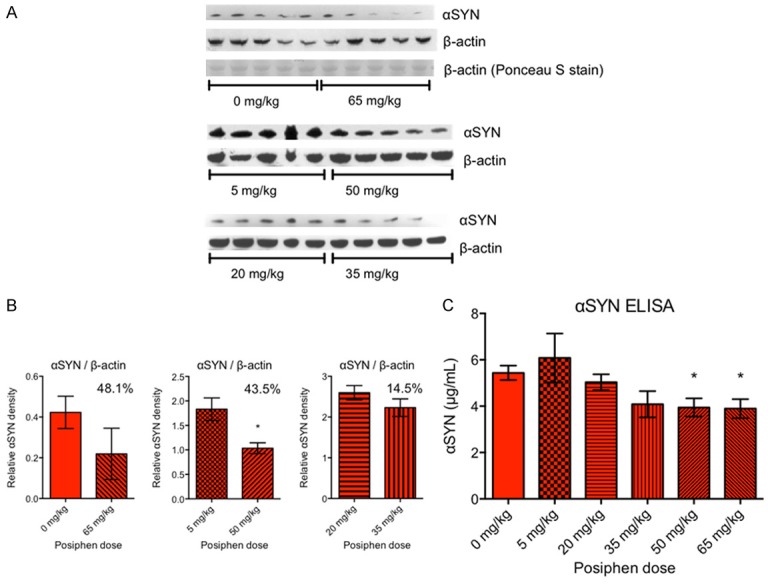
Daily IP Posiphen treatment of hSNCA A53T transgenic mice for 21 days, starting at six weeks of age, reduces α-synuclein brain levels in a dose-dependent manner. (A) α-synuclein Western blot of mouse brain extracts, following treatment with 0, 5, 20, 35, 50 or 65 mg/kg Posiphen in saline. β-actin was used as loading control. In the first blot, where β-actin signals were variable, Ponceau S stain indicates equal loading. (B) Semi-quantization of the Western blots in (A) after normalization with β-actin. Since the three blots comparing 0 vs. 65 mg/kg, 5 vs. 50 mg/kg and 20 vs. 35 mg/kg were run separately, without a common sample, they were plotted separately. Unpaired, two-tailed t-tests were performed for each pair, and significance is indicated on the graphs. *P ≤ 0.05. The numbers in the plots indicate the percent reduction of the α-synuclein level by the highest Posiphen dose, as compared to the lowest, in each pair. Data without normalization with β-actin look similar (not shown), with 50.6%, 33.7%, and 12.2% reduction, respectively, and unpaired two-tailed t-tests as follows: 0 vs. 65 mg/kg; P = 0.0858 (trend that does not reach significance), and 5 vs. 50 mg/kg; **P ≤ 0.01. C) α-synuclein ELISA of brain extracts from the same treatment groups, showing that the two highest doses reduced α-synuclein levels by roughly 28%. Unpaired, two-tailed t-tests were performed between 0 mg/kg and the various Posiphen doses. *P ≤ 0.05. In case of 35 vs. 0 mg/kg, there is a trend that does not reach significance: P = 0.0683. Mean and SEM are shown. N = 5 mice per group. αSYN; α-synuclein. SEM; standard error of the mean.
The overall agreement between the Western blot with the ELISA indicates that the results presented in Figure 9 are correct. We should mention that at 65 mg/kg we observed severe cholinomimetic side effects in the treated animals, in agreement with similar side effects seen in the APP/PS1 mice treated with 75 mg/kg [30]. So, this dose should not be used in future studies.
Discussion
Here, we studied for the first time the effects of translational inhibition of α-synuclein by Posiphen in the hSNCAA53T and hSNCAA30P mouse models of early gastrointestinal dysfunction in PD. We showed that chronic treatment with Posiphen prevents the slowing in colonic motility, thus demonstrating the therapeutic efficacy of Posiphen in both models. Interestingly, the beneficial effect of Posiphen on colonic motility extends to several months after the termination of treatment, especially in the case of hSNCAA30P mice. This may be indicative of the time it takes α-synuclein to accumulate and cause gastrointestinal dysfunction in hSNCAA30P mice after the discontinuation of translational inhibition by Posiphen.
Previously the early PD phenotype was more pronounced in hSNCAA53T mice than in hSNCAA30P mice [18], leading us to use hSNCAA53T mice here for the WGTT and Rotarod testing. In case of the WGTT, as mentioned, Posiphen does not affect the transit time in hSNCAA53T mice, which might be because in the present study, the vehicle-treated hSNCAA53T mice did not display an increased WGTT to begin with. Similarly, here, the hSNCAA53T mice did not display a reduction in motor function seen in [18], as compared to control mice. Finally, as mentioned, here the hSNCAA53T mice display shorter bead expulsion times than hSNCAA30P, which was the opposite in [18].
Taken together, the phenotype of hSNCAA53T mice here is overall milder than in [18]. What we have here is a very early model of PD, where alpha-synuclein accumulates in the enteric nervous system and causes defects in gut motility, but only shows minor accumulation in the brain and does not yet cause noticeable defects in motor symptoms.
Through our pharmacokinetics studies, we are able to compare the distribution of Posiphen and its metabolites in brain and blood of the hSNCAA53T mice, with that in APP/PS1 mice and humans [30,31]. The brain/blood ratio is similar as in those studies. A comparison of IP to PO administration has previously shown similar brain/blood ratio and similar Posiphen and metabolite ratios [30]. Here we compared the brain concentrations resulting from different doses and routes of administration (10 mg/kg IP vs. 25 mg/kg PO) in the PD and AD animal models, respectively, which have functional efficacy in common. We found that the concentration of Posiphen and its metabolites in the brain is 2-3 mM. In the phase I clinical study, the 4 × 60 mg/day treatment, which reduced the level of sAPP fragments, Aβ42, tau and phospho-tau in the CSF of MCI patients, resulted in a Cmax of 175 ng/ml or 0.52 mM of the sum of Posiphen, N1-NorPosiphen and N8-NorPosiphen in human plasma. Since brain concentrations were determined in that study to be about 8 times higher than plasma concentrations for Posiphen, we can extrapolate the unknown human brain concentrations to be roughly 4.15 mM [31], which is higher than 2-3 mM and therefore we can expect Posiphen to be efficacious at lower doses. Therefore, we suggest that lower doses can be tested in future clinical studies, since they are predicted to be effective, and less probable to cause side effects.
Furthermore, lower doses do not seem to affect the health of mice, as indicated by the comparison of body weights between vehicle- and Posiphen-treated groups (Figure 5). However, at the high dose of 65 mg/kg, the treated animals displayed severe cholinomimetic side effects. Accordingly, we used much lower doses to conduct the colonic motility studies.
Posiphen’s metabolites, N1-NorPosiphen and N8-NorPosiphen, have also been shown to reduce the levels of APP and α-synuclein in primary neuron cultures. However, whereas Posiphen and N8-NorPosiphen do not have acetyl-cholinesterase inhibitor activity, N1-NorPosiphen and N1, N8-BisNorPosiphen do have some acetyl-cholinesterase inhibitor activity [20,32]. Since N1, N8-BisNorPosiphen is a minor metabolite in animals and humans [31], the N1-NorPosiphen is the metabolite that determines its highest tolerable dose.
At the effective mouse brain concentration mentioned (155 ng/g), achieved previously in APP/PS1 mice by the 25 mg/kg PO dose, Posiphen only showed a statistically insignificant trend for reduction of APP brain levels by 21%. At higher doses, it reduced APP and Aβ levels by about 50% [30]. Similarly, here, the highest Posiphen doses (50 and 65 mg/kg) resulted in the most pronounced reduction of α-synuclein levels in the brains of hSNCAA53T mice (Figure 9). The middle Posiphen dose (35 mg/kg) resulted in a statistically insignificant trend for reduction, as compared to saline treatment, and as determined by ELISA. The lower Posiphen doses (5 and 20 mg/kg), which would be comparable to the doses that were effective in restoring colonic motility (3 and 10 mg/kg), barely reduced α-synuclein brain levels in hSNCAA53T mice. This suggests that even a small reduction in the levels of neurotoxic aggregating proteins in the brain is biologically significant. Finally, the expression of α-synuclein in the gut was significantly reduced after treatment with the low dose of 10 mg/kg Posiphen for 21 weeks (by approximately 50%), but not after 21 days of treatment.
The difficulty we had in reproducing our first Western blot results in both gut and brain may stem from the fact that α-synuclein monomers tend to detach from the blotting membrane easily [33]. This probably yields variable signals depending on the length of incubation, buffer used and washing steps, and on the distance of each lane and band from the edge of the membrane. However, recent improvements in the α-synuclein Western blot methodology result in stronger, more reproducible detection, presumably by increasing the hydrophobicity of the protein and its retention on the membrane [33-36]. Thus, in future studies these improved methods should be employed for α-synuclein semi-quantization by Western blot.
In conclusion, this is the first study showing the preclinical efficacy of Posiphen in normalizing the colonic motility in two mouse models of gastrointestinal dysfunction in early PD. This is the second disease after AD [30] where Posiphen’s preclinical efficacy is demonstrated. We also examined the pharmacokinetics and pharmacodynamics of Posiphen and found similar results as in [30]. Our data here and in [30] are in agreement with the ability of Posiphen to reach the nervous system, and its mechanism of action, the translational inhibition of α-synuclein and APP expression. Interestingly, both α-synuclein and APP are neurotoxic aggregating proteins that display several similar features, including the fact that iron levels regulate their expression [25,26,37,38], and the several ways they contribute to neurodegeneration: by impairing axonal transport [39-41] and synaptic transmission [42,43], causing inflammation [44-46], forming aggregates, and, finally, leading to nerve cell death [47,48]. Furthermore, both proteins are implicated in the pathogenesis of both PD and AD [49-52], and often patients present mixed PD and AD pathologies [53]. Since Posiphen can reduce the expression of both α-synuclein and APP, future studies are warranted to further investigate its beneficial effects in the preclinical and clinical settings and develop Posiphen as a drug for the treatment of both PD and AD.
Acknowledgements
This work was supported by a grant from the Michael J. Fox Foundation and QR Pharma, Inc. We thank Adam Mehring for performing the α-synuclein ELISA.
Disclosure of conflict of interest
None.
References
- 1.Nussbaum RL, Ellis CE. Alzheimer’s disease and Parkinson’s disease. N Engl J Med. 2003;348:1356–1364. doi: 10.1056/NEJM2003ra020003. [DOI] [PubMed] [Google Scholar]
- 2.Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rüb U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages) J Neurol. 2002;249(Suppl 3) doi: 10.1007/s00415-002-1301-4. III/1-5. [DOI] [PubMed] [Google Scholar]
- 3.Halliday GM, Del Tredici K, Braak H. Critical appraisal of brain pathology staging related to presymptomatic and symptomatic cases of sporadic Parkinson’s disease. J Neural Transm Suppl. 2006:99–103. doi: 10.1007/978-3-211-45295-0_16. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- 5.Wolters EC, Braak H. Parkinson’s disease: premotor clinico-pathological correlations. J Neural Transm Suppl. 2006:309–319. doi: 10.1007/978-3-211-45295-0_47. [DOI] [PubMed] [Google Scholar]
- 6.Djaldetti R, Lev N, Melamed E. Lesions outside the CNS in Parkinson’s disease. Mov Disord. 2009;24:793–800. doi: 10.1002/mds.22172. [DOI] [PubMed] [Google Scholar]
- 7.Fasano A, Visanji NP, Liu LWC, Lang AE, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2015;14:625–639. doi: 10.1016/S1474-4422(15)00007-1. [DOI] [PubMed] [Google Scholar]
- 8.Jost WH, Eckardt VF. Constipation in idiopathic Parkinson’s disease. Scand J Gastroenterol. 2003;38:681–686. doi: 10.1080/00365520310003200. [DOI] [PubMed] [Google Scholar]
- 9.Jost WH. Gastrointestinal dysfunction in Parkinson’s Disease. J Neurol Sci. 2010;289:69–73. doi: 10.1016/j.jns.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 10.Natale G, Pasquali L, Ruggieri S, Paparelli A, Fornai F. Parkinson’s disease and the gut: a well known clinical association in need of an effective cure and explanation. Neurogastroenterol Motil. 2008;20:741–749. doi: 10.1111/j.1365-2982.2008.01162.x. [DOI] [PubMed] [Google Scholar]
- 11.Abbott RD, Petrovitch H, White LR, Masaki KH, Tanner CM, Curb JD, Grandinetti A, Blanchette PL, Popper JS, Ross GW. Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology. 2001;57:456–462. doi: 10.1212/wnl.57.3.456. [DOI] [PubMed] [Google Scholar]
- 12.Ashraf W, Pfeiffer RF, Park F, Lof J, Quigley EM. Constipation in Parkinson’s disease: objective assessment and response to psyllium. Mov Disord. 1997;12:946–951. doi: 10.1002/mds.870120617. [DOI] [PubMed] [Google Scholar]
- 13.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 14.Corti O, Lesage S, Brice A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol Rev. 2011;91:1161–1218. doi: 10.1152/physrev.00022.2010. [DOI] [PubMed] [Google Scholar]
- 15.Lesage S, Anheim M, Letournel F, Bousset L, Honoré A, Rozas N, Pieri L, Madiona K, Dürr A, Melki R, Verny C, Brice A French Parkinson’s Disease Genetics Study Group. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol. 2013;73:459–471. doi: 10.1002/ana.23894. [DOI] [PubMed] [Google Scholar]
- 16.Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, Schapira AH. A novel α-synuclein missense mutation in Parkinson disease. Neurology. 2013;80:1062–1064. doi: 10.1212/WNL.0b013e31828727ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 18.Kuo YM, Li Z, Jiao Y, Gaborit N, Pani AK, Orrison BM, Bruneau BG, Giasson BI, Smeyne RJ, Gershon MD, Nussbaum RL. Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated alpha-synuclein gene mutations precede central nervous system changes. Hum Mol Genet. 2010;19:1633–1650. doi: 10.1093/hmg/ddq038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jost WH. Gastrointestinal motility problems in patients with Parkinson’s disease. Effects of antiparkinsonian treatment and guidelines for management. Drugs Aging. 1997;10:249–258. doi: 10.2165/00002512-199710040-00002. [DOI] [PubMed] [Google Scholar]
- 20.Mikkilineni S, Cantuti-Castelvetri I, Cahill CM, Balliedier A, Greig NH, Rogers JT. The anticholinesterase phenserine and its enantiomer posiphen as 5’untranslated-region-directed translation blockers of the Parkinson’s alpha synuclein expression. Parkinsons Dis. 2012;2012:142372. doi: 10.1155/2012/142372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rogers JT, Mikkilineni S, Cantuti-Castelvetri I, Smith DH, Huang X, Bandyopadhyay S, Cahill CM, Maccecchini ML, Lahiri DK, Greig NH. The alpha-synuclein 5’untranslated region targeted translation blockers: anti-alpha synuclein efficacy of cardiac glycosides and Posiphen. J Neural Transm (Vienna) 2011;118:493–507. doi: 10.1007/s00702-010-0513-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olivares D, Huang X, Branden L, Greig NH, Rogers JT. Physiological and pathological role of alpha-synuclein in Parkinson’s disease through iron mediated oxidative stress; the role of a putative iron-responsive element. Int J Mol Sci. 2009;10:1226–1260. doi: 10.3390/ijms10031226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaw KT, Utsuki T, Rogers J, Yu QS, Sambamurti K, Brossi A, Ge YW, Lahiri DK, Greig NH. Phenserine regulates translation of beta -amyloid precursor protein mRNA by a putative interleukin-1 responsive element, a target for drug development. Proc Natl Acad Sci U S A. 2001;98:7605–7610. doi: 10.1073/pnas.131152998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Venti A, Giordano T, Eder P, Bush AI, Lahiri DK, Greig NH, Rogers JT. The integrated role of desferrioxamine and phenserine targeted to an iron-responsive element in the APP-mRNA 5’-untranslated region. Ann N Y Acad Sci. 2004;1035:34–48. doi: 10.1196/annals.1332.003. [DOI] [PubMed] [Google Scholar]
- 25.Cahill CM, Lahiri DK, Huang X, Rogers JT. Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim Biophys Acta. 2009;1790:615–628. doi: 10.1016/j.bbagen.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho HH, Cahill CM, Vanderburg CR, Scherzer CR, Wang B, Huang X, Rogers JT. Selective translational control of the alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J Biol Chem. 2010;285:31217–31232. doi: 10.1074/jbc.M110.149161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lahiri DK, Chen D, Maloney B, Holloway HW, Yu Q, Utsuki T, Giordano T, Sambamurti K, Greig NH. The experimental Alzheimer’s disease drug posiphen [(+)-phenserine] lowers amyloid-beta peptide levels in cell culture and mice. J Pharmacol Exp Ther. 2007;320:386–396. doi: 10.1124/jpet.106.112102. [DOI] [PubMed] [Google Scholar]
- 28.Lilja AM, Röjdner J, Mustafiz T, Thomé CM, Storelli E, Gonzalez D, Unger-Lithner C, Greig NH, Nordberg A, Marutle A. Age-dependent neuroplasticity mechanisms in Alzheimer Tg2576 mice following modulation of brain amyloid-β levels. PLoS One. 2013;8:e58752. doi: 10.1371/journal.pone.0058752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lilja AM, Luo Y, Yu Q, Röjdner J, Li Y, Marini AM, Marutle A, Nordberg A, Greig NH. Neurotrophic and neuroprotective actions of (-)- and (+)-phenserine, candidate drugs for Alzheimer’s disease. PLoS One. 2013;8:e54887. doi: 10.1371/journal.pone.0054887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teich AF, Sharma E, Barnwell E, Zhang H, Staniszewski A, Utsuki T, Padmaraju V, Mazell C, Tzekou A, Sambamurti K, Arancio O, Maccecchini ML. Translational inhibition of APP by Posiphen: efficacy, pharmacodynamics, and pharmacokinetics in the APP/PS1 mouse. Alzheimers Dement (N Y) 2018;4:37–45. doi: 10.1016/j.trci.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maccecchini ML, Chang MY, Pan C, John V, Zetterberg H, Greig NH. Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid-β peptide and τ levels: target engagement, tolerability and pharmacokinetics in humans. J Neurol Neurosurg Psychiatry. 2012;83:894–902. doi: 10.1136/jnnp-2012-302589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu QS, Reale M, Kamal MA, Holloway HW, Luo W, Sambamurti K, Ray B, Lahiri DK, Rogers JT, Greig NH. Synthesis of the Alzheimer drug Posiphen into its primary metabolic products (+)-N1-norPosiphen, (+)-N8-norPosiphen and (+)-N1, N8-bisnorPosiphen, their inhibition of amyloid precursor protein, α-Synuclein synthesis, interleukin-1β release, and cholinergic action. Antiinflamm Antiallergy Agents Med Chem. 2013;12:117–128. doi: 10.2174/1871523011312020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee BR, Kamitani T. Improved immunodetection of endogenous α-synuclein. PLoS One. 2011;6:e23939. doi: 10.1371/journal.pone.0023939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newman AJ, Selkoe D, Dettmer U. A new method for quantitative immunoblotting of endogenous α-synuclein. PLoS One. 2013;8:e81314. doi: 10.1371/journal.pone.0081314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasaki A, Arawaka S, Sato H, Kato T. Sensitive western blotting for detection of endogenous Ser129-phosphorylated α-Synuclein in intracellular and extracellular spaces. Sci Rep. 2015;5:14211. doi: 10.1038/srep14211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Preterre C, Corbillé AG, Balloy G, Letournel F, Neunlist M, Derkinderen P. Optimizing Western Blots for the detection of endogenous α-Synuclein in the enteric nervous system. J Parkinsons Dis. 2015;5:765–772. doi: 10.3233/JPD-150670. [DOI] [PubMed] [Google Scholar]
- 37.Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, Greig NH, Lahiri DK, Tanzi RE, Bush AI, Giordano T, Gullans SR. An iron-responsive element Type II in the 5’-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J Biol Chem. 2002;277:45518–45528. doi: 10.1074/jbc.M207435200. [DOI] [PubMed] [Google Scholar]
- 38.Koukouraki P, Doxakis E. Constitutive translation of human α-synuclein is mediated by the 5’-untranslated region. Open Biol. 2016;6:160022. doi: 10.1098/rsob.160022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, Xia W, Villar A, Campbell WA, Kulnane LS, Nixon RA, Lamb BT, Epstein CJ, Stokin GB, Goldstein LS, Mobley WC. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 40.Volpicelli-Daley LA. Effects of α-synuclein on axonal transport. Neurobiol Dis. 2017;105:321–327. doi: 10.1016/j.nbd.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 41.Overk CR, Masliah E. Pathogenesis of synaptic degeneration in Alzheimer’s disease and Lewy body disease. Biochem Pharmacol. 2014;88:508–516. doi: 10.1016/j.bcp.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phan JA, Stokholm K, Zareba-Paslawska J, Jakobsen S, Vang K, Gjedde A, Landau AM, Romero-Ramos M. Early synaptic dysfunction induced by α-synuclein in a rat model of Parkinson’s disease. Sci Rep. 2017;7:6363. doi: 10.1038/s41598-017-06724-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stéphan A, Laroche S, Davis S. Generation of aggregated beta-amyloid in the rat hippocampus impairs synaptic transmission and plasticity and causes memory deficits. J Neurosci. 2001;21:5703–5714. doi: 10.1523/JNEUROSCI.21-15-05703.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T. Inflammation in Alzheimer’s disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol. 2009;87:181–194. doi: 10.1016/j.pneurobio.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 45.Sondag CM, Dhawan G, Combs CK. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflammation. 2009;6:1. doi: 10.1186/1742-2094-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoenen C, Gustin A, Birck C, Kirchmeyer M, Beaume N, Felten P, Grandbarbe L, Heuschling P, Heurtaux T. Alpha-Synuclein proteins promote pro-inflammatory cascades in microglia: stronger effects of the A53T mutant. PLoS One. 2016;11:e0162717. doi: 10.1371/journal.pone.0162717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carter J, Lippa CF. Beta-amyloid, neuronal death and Alzheimer’s disease. Curr Mol Med. 2001;1:733–737. doi: 10.2174/1566524013363177. [DOI] [PubMed] [Google Scholar]
- 48.Yasuda T, Nakata Y, Mochizuki H. α-Synuclein and neuronal cell death. Mol Neurobiol. 2013;47:466–483. doi: 10.1007/s12035-012-8327-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, Lee EB, Van Deerlin VM, Lopez OL, Kofler JK, Nelson PT, Jicha GA, Woltjer R, Quinn JF, Kaye J, Leverenz JB, Tsuang D, Longfellow K, Yearout D, Kukull W, Keene CD, Montine TJ, Zabetian CP, Trojanowski JQ. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol. 2017;16:55–65. doi: 10.1016/S1474-4422(16)30291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Toledo JB, Gopal P, Raible K, Irwin DJ, Brettschneider J, Sedor S, Waits K, Boluda S, Grossman M, Van Deerlin VM, Lee EB, Arnold SE, Duda JE, Hurtig H, Lee VM, Adler CH, Beach TG, Trojanowski JQ. Pathological α-synuclein distribution in subjects with coincident Alzheimer’s and Lewy body pathology. Acta Neuropathol. 2016;131:393–409. doi: 10.1007/s00401-015-1526-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gallardo G, Schlüter OM, Südhof TC. A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nat Neurosci. 2008;11:301–308. doi: 10.1038/nn2058. [DOI] [PubMed] [Google Scholar]
- 52.Eisele YS, Duyckaerts C. Propagation of Aß pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016;131:5–25. doi: 10.1007/s00401-015-1516-y. [DOI] [PubMed] [Google Scholar]
- 53.Rabinovici GD, Carrillo MC, Forman M, DeSanti S, Miller DS, Kozauer N, Petersen RC, Randolph C, Knopman DS, Smith EE, Isaac M, Mattsson N, Bain LJ, Hendrix JA, Sims JR. Multiple comorbid neuropathologies in the setting of Alzheimer’s disease neuropathology and implications for drug development. Alzheimers Dement (N Y) 2017;3:83–91. doi: 10.1016/j.trci.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]