

Abstract
Two decades following the discovery that α1-adrenoceptor antagonists suppress prostate tumor growth at the molecular and cellular level, the impact of α-blockade as re-purposed treatment strategy in the medical management of prostate cancer is gradually being recognized. Prostate cancer is the second most common cause of cancer deaths among males in the United States, yet the disease maintains inconsistent recommendations for prevention and screening. The functional relationship between α-adrenergic signaling and smooth muscle cells in the stroma of the prostate gland and the bladder neck empowered the use of α-adrenoceptor antagonists for the relief of urethral obstruction and clinical symptoms associated with benign prostatic hyperplasia (BPH). Adrenoceptors are G-protein-coupled receptors (GCPRs) that are functionally bound by catecholamines: epinephrine (ER) and norepinephrine (NE). The α1A adrenoceptor subtype is primarily responsible for smooth muscle contraction in the bladder neck and prostate gland. α1-adrenoceptor antagonists are clinically indicated as first-line therapies for the relief of BPH, hypertension, and post-traumatic stress disorder (PTSD). Compelling evidence from cellular and pre-clinical models have identified additional effects of α1-adrenoceptor antagonists regarding their ability to induce apoptosis-mediated suppression of prostate tumor growth and metastasis. Additionally, early epidemiologic data suggest that they may serve as a safe treatment to reduce the risk of prostate cancer. Optimization of quinazoline based compounds (doxazosin) to exploit pharmacologic targeting of tumor growth and vascularization revealed high efficacy of the lead novel compound DZ-50 against prostate tumors. This review discusses the experimental and pre-clinical evidence on the impact of α-blockade on prostate cancer.
Keywords: Alpha blockers, prostate cancer risk, sympathetic blockade
Introduction
The clinical burden of prostate cancer and benign prostatic hyperplasia (BPH)
Prostate cancer is the most frequently diagnosed cancer in males and the second leading cause of cancer deaths in males with an estimated 29,430 deaths in the United States for 2018, following only respiratory malignancies for mortality [1]. There is an estimated incidence of 164,690 new cases of prostate cancer in the United States for 2018 [1]. These cases account for approximately 19.2% of all estimated new cases of cancer in males in the United States. The five-year survival for patients with non-metastatic prostate cancer is 98.9% (measured between 2005 and 2011) but patients with metastatic prostate cancer on initial diagnosis (4% of prostate cancer patients) had only a 28.2% five-year survival [1]. The morbidity and mortality associated with advanced prostate cancer calls for preventive tools that reduce the likelihood of developing prostate cancer and impairing growth, metastases, and progression to therapeutic resistance. Despite the high morbidity of prostate cancer in the United States there are currently no drugs indicated for the prevention of prostate cancer in at-risk individuals. Recurrent prostate cancer progresses to resistance to androgen deprivation therapy (ADT) and/or taxane-based chemotherapeutic drugs [2]. The stromal microenvironment plays a major role in the progression of prostate cancer to ADT resistance by conferring a process called epithelial-to-mesenchymal transition (EMT) [2].
BPH is a benign proliferative pathology of the prostate glandular epithelium, connective tissue, and smooth muscle that affects males older than 30 years of age [3]. Histopathological evaluations demonstrate that 68% of men over the age of 50 show cellular changes associated with BPH [4]. Evidence derived from a population-based study has shown that 75% of men over the age of 70 describe at least one lower urinary tract symptom associated with BPH [5]. A contributor to the etiology of BPH is the reversion of the normal cellular environment to an embryologic growth phase where new epithelial gland formation and stromal cell inductive potential is aberrantly restarted from stem cell progenitors [6,7], and deregulated cell survival signaling [8]. The relationship between adrenergic signaling and smooth muscle cells of the stroma introduced the use of adrenoceptor antagonists for the relief of urethral obstruction in BPH [9]. The clinical utility of α1-adenoceptor antagonists has long been established as first-line therapy to relieve lower urinary tract symptoms (LUTS) secondary to BPH (BPH-LUTS) in aging men [10].
The economic impact of treatment of BPH, including direct costs (drugs, procedures, imaging, office visits), indirect costs (lost earnings), and intangible costs (pain and suffering) is approximately $4 billion in the United States alone [11]. Evaluation of claims data demonstrate BPH associated costs begin to rise as early as men in the 4th decade of life, with 4.7% of men between the ages of 45-54 seeking treatment, and 14.3% of men between 55-64 seeking care for this condition [12]. The relatively rapid onset of the BPH effects in aging males is indicated by the prostate growth doubling time of only 4.5 years between the ages of 31-50 and 10 years between ages 50-70 [3]. The estimated prevalence of BPH for males over the age of 30 in the United States for 2015 was 38,000,000 [4], with 12 million seeking active treatment. Of those actively managed, 54.8% choose drug management, primarily with α1-blockade [13]. The medical management of patients with BPH demonstrates an estimated 23% of all visits to urologic practices in the US, second only to urinary tract infections (UTIs) [13]. The impact of this prostatic condition at both the clinical and economical level is expected to grow as the population of the United States ages. By 2030, 20% of the male population in the United States is expected to be 65 years of age or older, with the 85 and older subset being the fastest growing portion of the population [14].
In this review we discuss the pre-clinical and epidemiological evidence (spanning 20 years) on the impact of α-blockade (in clinical use for the treatment of BPH) on the cellular landscape and clinical outcomes in prostate cancer progression to advanced disease.
The origins of the prostate gland
The prostate gland is a walnut-sized glandular organ found inferior to the urinary bladder and surrounds the proximal urethra that is found in human males [15]. Histologically the structure of the prostate gland is organized by two different schemas: “zones” or “lobes”. For this review we will be using the zone classification schema to better illustrate the localization of cell populations and their impact on BPH and prostate cancer. The four major zones of the prostate include the peripheral zone which makes up most of the glandular organ, the central zone that surrounds the ejaculatory ducts, the transition zone that surrounds the proximal urethra, and the anterior fibromuscular zone [16]. The function of the healthy prostate is the emission of prostatic fluid into the seminal fluid for the survival and function of spermatozoa in male ejaculate [17]. Prostatic fluid contains many proteins, including various enzymes that prolong the survival of spermatozoa in the ejaculate, prostate specific antigen (PSA), and highly concentrated zinc [17]. Despite the small size of the prostate, the gland is the site of major contributors to morbidity and mortality from diseases such as BPH and prostate cancer [17].
Embryologically the prostate gland arises from the primitive endoderm or ‘gut tube’. The hindgut region of the primitive endoderm swells into a larger hollow structure called the cloaca. The cloaca is separated into the ventral and dorsal outlets by the urorectal septum which give rise to the urogenital and anorectal structures, respectively. The ventral outlet is termed the primitive urogenital sinus. The urogenital sinus gives rise to the urinary bladder at the cranial end and the urethra caudally [17]. During the 10th week of human gestation, the prostate forms caudal to the bladder neck by an interplay of early epithelial and mesenchymal cells which both arise from the urogenital sinus. In an androgen independent process (occurs in both male and female embryos), the urogenital sinus gives rise to loose connective tissue composed of mesenchymal cells into distinct regions that will determine the lobes of the prostate [18]. The release of mesenchymal cells into these distinct regions is called “mesenchymal condensation” [18]. The mesenchymal cells ultimately differentiate into the prostate stromal cells, the supporting cells of the fibromuscular tissue within the prostate microenvironment [18]. In male embryos, the urogenital sinus-derived epithelial buds are stimulated in an androgen dependent process by mesenchymal-secreted fibroblast growth factors (FGFs), transcription factors, and transforming growth factor-β (TGF-β) to invade the mesenchymal condensations and grow into the early prostate epithelium [18,19]. The invading urogenital sinus epithelium differentiates into distinct epithelial cell populations of the prostate including the basal, intermediate, and luminal layers (Figure 1) [20]. The basal epithelial layer consists of non-secretory cells adjacent to the stroma, the luminal layer makes up the secretory columnar epithelium, and the intermediate layer is a population of epithelial cells with shared basal and luminal characteristics [20,21]. These epithelial cells make up the functioning luminal structure (prostate glands). The mesenchyme gives rise to distinct cell populations that occupy the prostate gland microenvironment: smooth muscle cells, fibroblasts, and endothelial cells (Figure 1).
Figure 1.
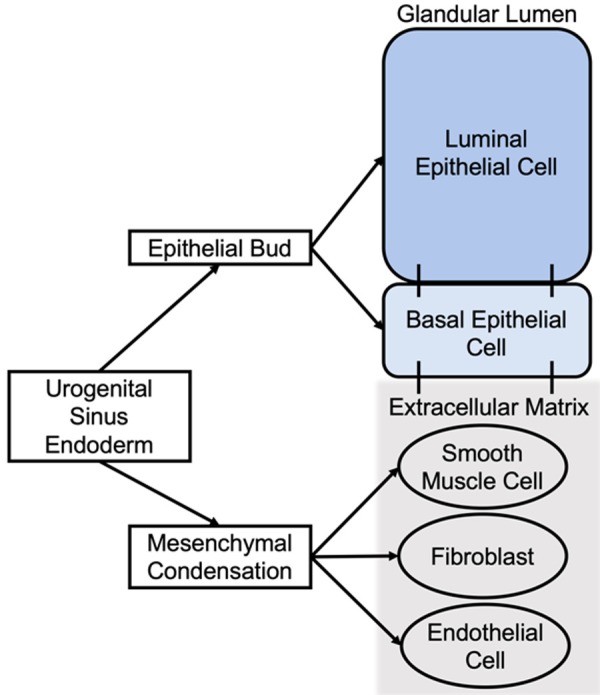
Embryologic order of development for prostate cells. Urogenital sinus gives rise to the epithelial and mesenchymal cells of the prostate. The epithelial bud differentiates into luminal, basal, and intermediate (not pictured) epithelium. The mesenchymal condensation gives rise to smooth muscle cells, fibroblasts, and endothelial cells that inhabit the extracellular matrix.
Neurologically, the prostate is innervated by both parasympathetic muscarinic and sympathetic adrenergic nerve plexuses from the pelvic nerve and hypogastric nerve, respectively. The glands of the prostate in the peripheral and central zones are innervated by muscarinic autonomic nerve plexuses, with high association between cholinergic signaling receptors and the epithelial lining, which drives secretions by the prostate [22]. While the stromal areas of the prostate gland also contain muscarinic autonomic neurons, the smooth muscle cells are predominantly autonomically innervated by adrenergic signaling [23]. Smooth muscle cells are primarily in the anterior fibromuscular portion of the prostate, and throughout the peripheral and central glandular zones of the organ [18]. Adrenergic innervation mediates the contraction of prostatic smooth muscle via norepinephrine [24].
Clinical recommendations for prostate cancer
Diagnosis
After the widespread implementation of PSA screening began in the 1990s, the rate of diagnosis of prostate cancer remains nearly twice the rate prior to the pre-PSA era, while the rate of mortality has slowly decreased since 1991, which suggests many of the cases diagnosed would otherwise be indolent and have no clinical significance [25]. While advanced stage prostate cancer carries significant mortality, there is growing evidence over the past decade that many low-grade diseases are best managed by observation [26].
Current recommendations by the American Urologic Association (AUA) advocate screening for prostate cancer by means of annual PSA and digital rectal exam in select groups of patients. This recommendation is not shared by all professional groups, as until recently the United States Preventive Services Task Force (USPSTF) provided a D rating (recommends against) for PSA-based screening for prostate cancer. This was recently updated in May 2018 to more closely mirror the AUA’s current recommendation, and the USPTF now recommends shared discussion with men between the ages of 55-69 to discuss the risks and benefits of PSA screening for prostate cancer. The overall benefit of prostate cancer screening is also a debated topic. Two large population-based randomized trials have been conducted with conflicting results. The European Randomized Study of Screening for Prostate Cancer (ERPSC) and the United States Prostate, Lung, Colorectal, and Ovarian (PLCO) are the two most cited studies. The ERPSC demonstrated a 21 percent prostate cancer mortality improvement in men obtaining routine PSA screening vs the unscreened control, however, the PLCO trial showed no survival benefit [27,28]. The controversy surrounding these trials is beyond the scope of this article. Prostate cancer diagnosis is confirmed with ultrasound-guided needle biopsy after an abnormal digital rectal exam and/or elevated PSA is noted [26]. Staging of prostate cancer is based on the American Joint Committee on Cancer Tumor/Node/Metastases (TNM) system that combines tumor size, lymph node involvement, metastases, PSA at time of diagnosis, T1-T4 stage, and Gleason score [29].
Prevention
Androgens are the major contributor to the development of malignancy in the prostate gland [30]. Several clinical trials have determined the relationship between 5α-reductase inhibitors and prostate cancer development, the most prominent being the PCPT and the REDUCE trials [31,32]. Reduced overall incidence of low grade prostate tumors (Gleason 5 or 6) was found in both trials; however there was no significant change in the mortality, and the rates of high grade prostate cancer (≥ Gleason grade 7) were unaffected and possibly increased, resulting in a black-box warning for 5α-reductase inhibitors by the FDA [33]. A recent case-control study supported this association, citing that use of finasteride, a common 5α-reductase inhibitor used to treat BPH, was associated with increasing the risk of developing high-grade (Gleason scores ≥8) prostate cancer and lower risk of developing low-grade (Gleason scores <8) prostate cancer, fueling the controversy on the use of 5α-reductase inhibitors as therapeutic strategy in high risk patients for prostate cancer [34].
A number of other agents have been evaluated for prevention of prostate cancer with limited evidence including metformin, statins, Selenium, Vitamin C, D, and E, Retinoids, and dietary phytoestrogen. For men already diagnosed with low grade disease (Gleason scores 5-6), the REDEEM (reduction by dutasteride of clinical progression events in expectant management) trial demonstrated dutasteride reduced the progression of low risk prostate cancer (HR 0.62, P=0.009) [35]. To date, no professional medical organization currently recommends chemoprevention for prostate cancer and all therapies studied to date are ineffective or experimental. While not associated with overall increased risk of prostate cancer, obesity is associated with higher-grade prostate tumors at diagnosis [36,37]. These effects are potentially mediated by recruitment of adipose stromal cells from areas of white fatty tissue to the tumor site where they induce the EMT phenotype [36]. This is a burgeoning area of prostate cancer research in the growing body of evidence that targeting EMT may prevent progression to therapeutic resistance.
Treatment
Treatment recommendations for localized prostate cancer are based on a risk stratification scheme that integrates serum PSA, Gleason score (grade), and clinical stage [26]. The recommendation for very low risk disease is active surveillance including routine PSA surveillance and ultrasound or MRI-guided imaging [26]. Options for low-risk disease include active surveillance or interventions including radical prostatectomy or radiotherapy [26]. With progression from low-risk localized to intermediate- or high-risk localized disease radical prostatectomy or radiation with ADT are recommended therapy options [26]. ADT is considered first line therapy for metastatic and some stages of biochemically (PSA) recurrent prostate cancer. ADT includes bilateral orchiectomy or drugs to eliminate testosterone production by manipulation of the hypothalamic, pituitary, and gonadal axis in combination with androgen receptor (AR) antagonists [26]. Prostate cancer patients may ultimately progress to castration resistant prostate cancer (CRPC) disease, defined as rising PSA in the setting of castrate levels of testosterone [2]. With emergence of CRPC, the treatment recommendation is continued ADT until progression to radiographic or symptomatic metastasis [38]; there has been no treatment, however, to increase cancer specific survival in CRPC. The PROSPER trial demonstrated patients treated with enzalutamide had a 71% lower risk of progression to metastatic CRPC (mCRPC) or death, while the SPARTAN trial demonstrated patients treated with apalutamide had an increased metastatic free survival and time to symptomatic progression when compared to placebo. With mCRPC the treatment recommendation is second generation antiandrogen (abiraterone) with prednisone or taxane chemotherapy with immunotherapy (docetaxel with sipuleucel-T) [38,39].
Adrenoceptor signaling and prostatic disease
Mechanism of α-adrenoceptor antagonism
The prostate gland is innervated by the autonomic nervous system, with the glandular epithelium largely muscarinic-innervated, and the stroma associated with adrenergic-innervation (responsive to adrenoceptor stimulation) [22,23]. Adrenoceptors are a class of G-protein coupled receptors (GCPRs) distributed throughout the body and constructed of seven transmembrane domains that are physiologically responsible for mediating responses to endogenous catecholamines, namely epinephrine and norepinephrine [40-42]. The adrenoceptors are found throughout the body in neuronal and non-neuronal tissues, serving as regulators of many autonomic nervous functions [40]. Adrenoceptors are divided into α (alpha) and β (beta) groups, where α-adrenoceptors mediate predominantly excitatory functions like smooth muscle contraction and vasoconstriction and β-adrenoceptors mediate predominantly muscular inhibitory functions like vasodilation, smooth muscle relaxation, and bronchodilation, as well as excitatory cardiac function [40]. The α-adrenoceptors are divided into α1 and α2, where the α1 group was originally characterized as a separate group by binding affinity to the quinazoline-based α-antagonist prazosin, which had a minimal effect when administered to the α2 group [40]. The α1-adrenoceptors can be further sub-classified into α1A, α1B, and α1D based on binding and functional studies [40]. Of interest, the α1A subtype is localized to the prostate, vas deferens, and urethra in humans providing a localized drug target for patients suffering urinary symptoms [23,40,43,44]. Compared to the normal adult prostate, α1-adrenoceptor mRNA and α1-adrenoceptors increase throughout in the aging gland and with BPH manifestation [45-48]. With aging, the topological distribution of the α1-adrenoceptors in the prostate surrounding the prostatic urethra, contributes to BPH-LUTS development [49]. Early pathophysiologic studies demonstrated prostatic tissue contraction when exposed to nonselective α-blocker inhibition [50], due to the abundance of α1A adrenoreceptors localized to the prostatic stroma [51]. Significantly, functional studies have demonstrated α1-adrenoceptor antagonists also relieve LUTS via mechanisms independent of prostatic muscle contraction [52].
α1-adrenoreceptor antagonists have long been used clinically as first-line therapy to relieve BPH-LUTS [10]. BPH is characterized by non-malignant proliferation of prostatic glandular epithelium and stroma (connective tissues, smooth muscle) resulting in obstruction of the prostatic urethra and bladder outlet. This obstruction causes LUTS that may be worsened by an increase in prostate smooth muscle tone [10,53]. α1-adrenoreceptor antagonists act to relieve BPH-LUTS by antagonizing the smooth muscle contractility induced by the catecholamine binding to α1A-adrenoreceptors in the prostate and bladder neck, providing relief of urinary obstruction by competitive inhibition [53]. Second-generation α-blockers (doxazosin, prazosin, and terazosin) are considered ‘non-uroselective’ where third-generation α-blockers (alfuzosin, tamsulosin, and silodosin) are considered ‘uroselective’ [54]. While both generations of α-adrenoceptor antagonists are indicated for first line treatment of BPH, second-generation α-blockers are highly associated with orthostatic hypotension, dizziness, tiredness, and ejaculatory problems due to less regionally selective α1-adrenoceptors antagonism [54,55]. The function of α1-adrenoceptors as regulators of basal vascular tone, smooth muscle contraction, and arterial blood pressure provides the basis for the α1-antagonists use as hypertension treatment. Current guidelines from the American College of Cardiology and American Heart Association recommend the second generation α1-adrenoceptor antagonists as second-line agents for the treatment of hypertension, especially in the setting of male patients with concurrent LUTS [56,57]. A review of the ALLHAT trial notes that patients with low risk for heart failure or young patients with hypertension with concomitant LUTS may benefit from monotherapy for both disease processes with a single agent [57].
Impact of α-adrenergic blockade on prostate cancer
The link between sympathetic nervous signaling and cancer neovascularization, metastasis, and survival has been well established in β-adrenergic signaling knockout models as well as epidemiologic cohort studies of β-blockade in patients [58]. The use of propranolol in vitro decreased viability and increased caspase activation in both hemangioblastoma and HeLa cell lines [59]. Treatment with propranolol decreased the hypoxia inducible factor (HIF) downstream transcription products, involved in angiogenesis, and extracellular matrix (ECM) degradation in HeLa cells, pointing to a mechanism underlying the anti-angiogenic effects of β-adrenergic blockade [59]. The in vivo silencing of β2 and β3 adrenoceptors in the prostate resulted in inhibition of angiogenic switch, mediated by pro-angiogenic factors, like vascular endothelial growth factor (VEGF) [60,61].
Novel anti-tumor action by quinazoline-based α1-antagonists
Quinazoline-based α1-adrenoceptor antagonists, doxazosin, prazosin, terazosin, and alfuzosin, are structural competitive antagonists to epinephrine and norepinephrine, the predominant ligands of α-adrenoceptors (Figure 2). The structures of α1-adrenoceptor antagonists confer the ability to selectively antagonize adrenoceptors via post-synaptic blockade, inhibiting smooth muscle contraction, an effect that spares central action on blood pressure and neuronal adrenergic function, resulting in an effective drug class with few adverse or severe side-effects [41,62,63]. Subsequent work in the 1990s identified additional non-target quinazoline derivative mechanisms of action by impacting tumor vascularity and growth dynamics. Our group pioneered evidence on the apoptotic action of doxazosin mediated by TGF-β signaling disruption against benign prostate epithelial and stromal cells in pre-clinical models as well as in clinical specimens [64,65]. Stimulation of α1-adrenoceptors with catecholamine ligands in prostate cancer epithelium promotes proliferation [66]. This response is mediated by induction of store-dependent Ca2+ entry resulting in activation of nuclear factor of activated T-cells (NFAT) [66]. Furthermore, there is a correlation between α1-adrenoceptor activation and expression of VEGF and HIF-1α expression (inducers of angiogenesis and tumor invasion) [67]. Binding of the α1-adrenoceptors induces a second-messenger pathway via cAMP resulting in downstream PKA/PI3K/Akt/p70S6K pathway activation, driving HIF-1/VEGF-mediated angiogenesis in prostate cancer [67]. However, some pro-apoptotic mechanisms of action of quinazoline derivatives like doxazosin and terazosin are independent of the α1-adrenoceptor antagonism action [68]. Prostate cancer cells lacking α1-adrenoceptor undergo apoptosis in response to quinazolines, evidence supporting the α1-adrenoceptor-independent action of apoptosis induction [69]. Moreover, the sulfonamide-based third generation α1-adrenoceptor antagonist tamsulosin (Figure 3), had no effect on prostate cancer cell apoptosis [70]. Besides prostate cancer cells, breast and urothelial cancer cells, bladder smooth muscle cells, cardiac myocytes, pituitary adenoma cells, vascular endothelial cells, and HeLa cells undergo apoptosis in response to doxazosin [71-78]. The results of the ALLHAT trial that quinazoline-derived doxazosin doubled the risk of congestive heart failure resulted in investigation of the adrenoceptor blockade-independent mechanism of action for the pro-apoptotic activity in cardiac myocytes by these drugs [57,73,79]. Quinazoline-derived α1-adrenoceptor antagonist doxazosin induced apoptotic gene expression profiles in murine cardiac myocytes [73]. Specifically, doxazosin increased transcriptional activation of gadd153, C/epbβ, and DOC-1 genes, a profile associated with the ER stress apoptotic response. Downstream effects include the phosphorylation of p38 MAPK, GADD153 nuclear translocation, and phosphorylation of focal adhesion kinase (FAK) [73].
Figure 2.
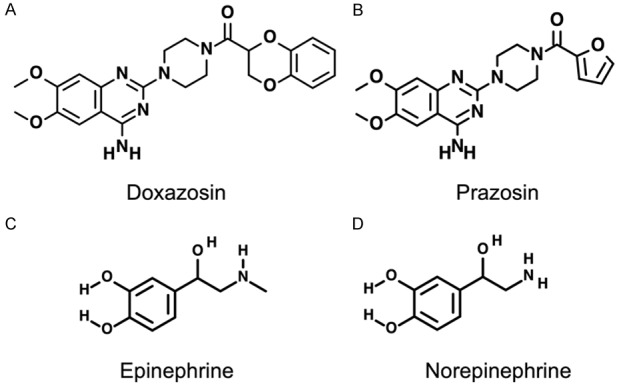
Structures of quinazoline-based α1-adrenoceptor antagonists doxazosin (A) and prazosin (B), and endogenous adrenergic agonists epinephrine (C) and norepinephrine (D).
Figure 3.
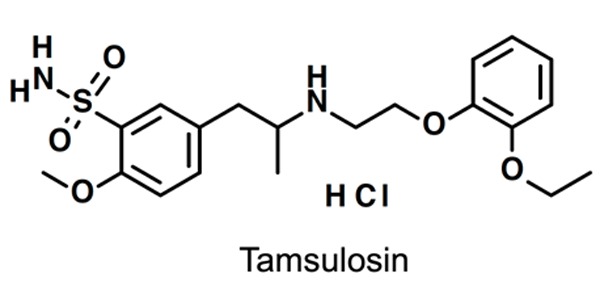
Structure of sulfonamide-based α1-adrenoceptor antagonist tamsulosin.
The process of EMT has been implicated as a contributor to the emergence of therapeutic resistance in advanced prostate cancer; however the current understanding of the impact of exposure to α1 blockade on EMT phenotypic landscape is limited. Anoikis is an apoptotic phenomenon that occurs when cells lose sufficient cell-cell or cell-matrix interactions [80,81]. Circumventing anoikis is a common loss of apoptotic control in therapeutically resistant prostate cancer that confers an aggressive metastatic phenotype, particularly among epithelial cancers that consequentially undergo EMT [2]. Quinazoline-based α1-adrenoceptor antagonists, doxazosin and terazosin, enhance prostate cancer cell and endothelial susceptibility to anoikis, resulting in decreased cell mobility, thus impairing neovascularization and metastasis [82,83]. Doxazosin induces apoptosis of prostate tumor cells via activation of the canonical TGF-β signaling, and caspase-mediated cell cleavage in vitro [84,85]. Prazosin was also found to exhibit a significant pro-apoptotic activity by induction of cell-cycle arrest [86]. Prazosin induces DNA strand breaks that result in cyclin-dependent kinase (CDK) phosphorylation, ultimately causing CDK1 inactivation leading to G2 checkpoint arrest [86]. Additionally, prazosin triggered caspase-mediated apoptosis in prostate cancer cell lines in vitro [86]. Oral administration of prazosin significantly reduced tumor mass in xenograft mice models [86]. Tamsulosin lacks the pro-apoptotic effect and demonstrates no induction of caspase-mediated cell death, unlike the quinazoline-based α1-adrenoceptor antagonists, suggesting the importance of structure in apoptotic induction [70,84]. All α1-adrenoceptor antagonists have been shown on a large-scale meta-analysis to have similar efficiency in the reduction of urinary symptoms and improvement in flow rates, with differences relating to the specific side effect profile [87-89].
Optimization of quinazoline compounds into directed anti-tumor therapies
DZ-50 is a quinazoline-derived α1-adrenoceptor antagonist (Figure 4A) synthesized by replacing the 2,3-dihydro-benzo[1,4]dioxane-carbonyl moiety of doxazosin with a biphenyl aryl sulfonyl substituent, and the methoxy side chains replaced with isopropyl propxy functions [91]. These structural changes in the quinazoline provide a cellular basis for targeting vascularity of pre-malignant or malignant prostate microenvironment with novel quinazoline derivatives [91]. DZ-50 significantly suppressed angiogenesis without increasing the apoptotic index in vivo [91], and exerted a metastasis impairing effect in prostate cancer pre-clinical models [91].
Figure 4.
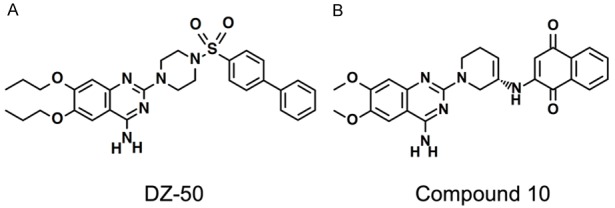
Structure of quinazoline-based modified prazosin derivative DZ-50 (A) and structure of quinazoline-based modified prazosin derivative Compound 10 (B).
More recent studies from our lab have demonstrated the ability of DZ-50 to mediate the mesenchymal-to-epithelial transition (MET) phenotypic change that reverts de-differentiated EMT phenotype of prostate cancer cells into the differentiated epithelial phenotype [92]. The changes in cell-cell junction by DZ-50 are mediated by disruption of TGF-β1 and insulin-like growth factor (IGF) signaling axis, specifically IGF binding protein 3 (IGFBP3) [92]. The TGF-β1 and IGF signaling axes have been linked to cancer progression to metastatic, treatment resistant disease by altering the microenvironment via differentiation of fibroblasts into cancer-associated fibroblasts (CAFs), facilitating tumor survival, growth, and neovascularization, consequential to a dedifferentiated phenotype [93-96]. The IGF-I axis is responsible for upregulation of zinc finger E-box-binding protein 1 (ZEB1), a protein that transcriptionally represses E-cadherin [96]. Treatment of prostate cancer cells in vitro with DZ-50 results in drug sequestration of IGFBP3, the serum carrier of IGF ligand and promoter of IGF action; this leads to EMT reversal (MET) and ultimately anoikis [92,96,97]. The in vitro evidence so far provides a basis for advancing DZ-50 to a clinical trial for patients with metastatic CRPC [2].
Quinazoline structural optimization led to potent pro-apoptotic effects in prostate cancer cells, via piperazine ring substitution to the prazosin quinazoline nucleus [98,99]. The most promising of the novel compounds from a recent study of quinazoline modification are compound 9 and compound 10 (Figure 4B), both demonstrated anti-proliferative effects in prostate cancer cells at a magnitude higher than doxazosin [99]. Compound 10, the S enantiomer of a prazosin quinazoline-quinone, was synthesized by substituting the piperazine side chain of prazosin with a 1,4-napthoquinone moiety [99]. The R enantiomer, compound 9, exhibits less binding affinity for the α1-adrenoceptor subtypes and lower apoptotic action, implicating the stereochemistry in the anti-cancer actions of the novel compounds [99].
Translational significance of cellular events
Based on the observed pro-apoptotic and anti-neovascularization actions of quinazoline-based α1-adrenoceptor antagonists summarized in Figure 5, investigators have completed retrospective risk analyses to elucidate the early translational evidence of these drugs as preventive tools. A 2018 meta-analysis by Cao et al. found non-significant association between the use of anti-adrenergic drugs and the risk of prostate cancer across 3 case-control studies (RR 1.22; 95% CI 0.76-1.96), and a significant decrease in risk across 2 cohort studies (RR 0.71; 95% CI 0.57-0.90) [100]. A case-control study by another team used the Finnish Cancer Registry and the national prescription database to observe the odds of developing prostate cancer for patients using α1-adrenoceptor antagonists for the treatment of LUTS (OR 1.79; 95% CI 1.67-1.91) [101]. However the exposed group of cases and controls were almost entirely users of sulfonamide-based α1-adrenoceptor antagonist tamsulosin (N=6,352) and only a small proportion represented quinazoline-based α1-antagonist alfuzosin (N=596) [101]. This is in accord with the pre-clinical evidence that only quinazoline-based antagonists confer the pro-apoptotic effect against prostate tumors, while the sulfonamide-derived α-adrenoceptor antagonist, tamsulosin, failed to exert such an effect [70].
Figure 5.
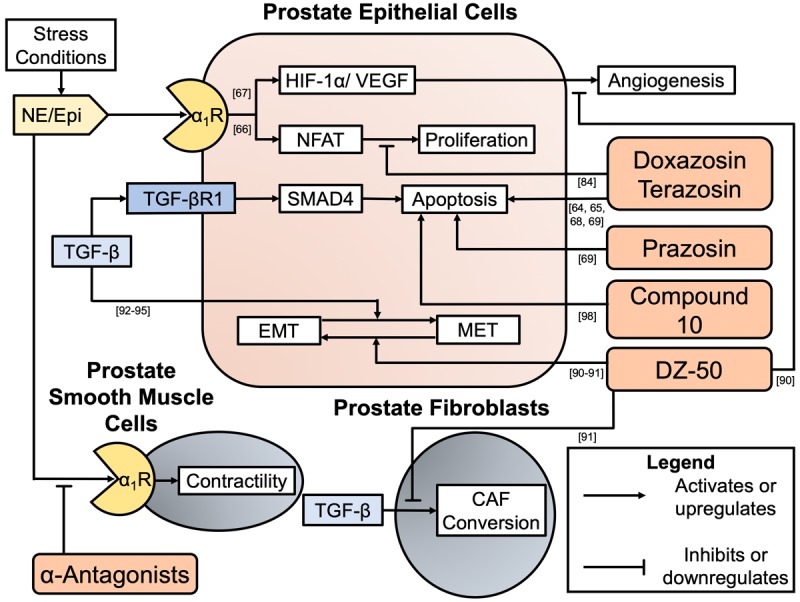
Summary of in vitro evidence demonstrating quinazoline-derived α1-adrenoceptor antagonist or modified quinazoline anti-cancer actions. Prostate epithelial-derived cancers and the microenvironment are influenced by α1-adrenoceptor antagonists. Blockade by α1-adrenoceptor antagonists at the smooth muscle cells in the prostate stroma mediate the anti-contractility benefits of the drugs when used for BPH-LUTS. Quinazoline derived α1-adrenoceptor antagonist doxazosin disrupts the proliferation axis induced by α1-adrenoceptor activation in a non-α1-adrenoceptor antagonism action ([84]). Quinazoline derived α1-adrenoceptor antagonists doxazosin, terazosin, and prazosin have demonstrated the ability to induce apoptosis in prostate cancer cells ([64,65,68,69] and [69], respectively). Novel quinazoline-derived compound titled ‘Compound 10’ demonstrates anti-cancer activity with pro-oxidant DNA fragmentation in prostate cancer cell lines ([99]). Novel quinazoline-derived compound titled ‘DZ-50’ demonstrates multiple anti-cancer actions including induction of MET (reversal of EMT), inhibition of TGF-β-induced microenvironment fibroblast changes, and the inhibition of angiogenesis ([91-92], [92], and [91], respectively).
More than a decade ago a retrospective study conducted by Harris et al. provided the first observational evidence that patients treated for hypertension or BPH with quinazoline-derived α1-adrenoceptor antagonists doxazosin, prazosin, and terazosin [102], exhibited a reduced risk ratio of 0.683 for developing prostate cancer [102]. This study revealed no effect on the overall survival in men with prostate cancer who were exposed to quinazoline-based α1-adrenoceptor antagonists [102]. Potentially limited by the population age with group median exposure age of 68-yrs (Table 1). A more recent case-control study by Van Rompay et al. examined the overall risk of prostate cancer among patients taking the α1-antagonists alfuzosin, doxazosin, prazosin, terazosin, or tamsulosin, and found a protective risk ratio of 0.89 (Table 1) [103]. The average age at prescription of α1-antagonists in this study was 66.7, with a mean follow-up time of 6.3 years (exposure to α1-antagonists) [103].
Table 1.
Clinical evidence on impact of quinazoline α1-antagonists on prostate cancer
| Study | Risk Ratio | Odds Ratio | Drug (s) | Drug Class | Source |
|---|---|---|---|---|---|
| Harris et al. | 0.683 (95% CI 0.532-0.8776) | N/A | Doxazosin, prazosin, terazosin | Quinazolines | [102] |
| Van Rompay et al. | 0.89 (95% CI 0.81-0.97) | N/A | Alfuzosin, doxazosin, prazosin, terazosin, or tamsulosin | Quinazolines (4), Sulfonamide (1) | [103] |
| Friedman et al. | N/A | 1.17, 1.22 (95% CI 1.11-1.24, 1.16-1.29) | Prazosin, terazosin | Quinazolines | [104] |
Epidemiological studies observing the relative odds or risk of developing prostate cancer amongst individuals treated with different α1-adrenoceptor antagonists.
A direct assessment of the link between norepinephrine signaling and cancer risk, by Friedman and colleagues, revealed both quinazoline-derived α1-adrenoceptor antagonists prazosin and terazosin increased the odds of developing prostate cancer (OR 1.17, 1.22; 95% CI 1.11-1.24, 1.16-1.29) [104]. These findings are challenged by evidence that terazosin showed statistically significantly reduction in the odds of developing colon and lung cancer with odds ratios of 0.86 and 0.89, respectively [104] (shown on Table 1). Users of naphthalene-based α1-adrenoceptor antagonist naftopidil had a risk ratio of 0.46 for developing prostate cancer when compared to users of sulfonamide-derived tamsulosin [105]. These findings drive the pursuit for further structural optimization of α1-adrenoceptor antagonists to mechanistically exploit the pathways causing prostate apoptosis towards the development of agents for prostate cancer prevention [2,91,92].
Conclusion
The epidemiologic data surrounding the use of quinazoline based α1-adrenoceptor antagonists as chemopreventive agents in prostate cancer is mixed and very limited. In order to further assess the role of these drugs as prostate cancer preventive agents requires a large-scale cohort of individuals exposed to quinazoline-derived antagonists for a more prolonged period than the existing studies. Given that the majority of studies investigating this relationship have a mean exposure age of more than 65 years old, the cohort should concern individuals treated at a younger age with α-antagonists for familial hypertension or BPH. As α-adrenoceptor antagonists are low risk drugs and widely available, a randomized controlled trial with a long-term cohort of young participants could provide the strongest evidence. The available in vitro and in vivo data provide an initial basis for quinazoline derivatives as cancer preventive agents. There is an immediate need for low risk and inexpensive chemo-preventative agents for prostate cancer, a major contributor of morbidity and mortality. The existing data support the design of prospective studies using quinazoline derivatives, like doxazosin (in clinical use) and novel drugs like DZ-50, as prostate cancer chemoprevention drugs.
Acknowledgements
The James F. Hardymon Endowment, the Randal Rowland Urology Research Fund, and the University of Kentucky Center for Clinical and Translational Science PSMR Fellowship. The authors would like to acknowledge Lorie Howard for her help in the submission process.
Disclosure of conflict of interest
None.
Abbreviations
- BPH
Benign Prostatic Hyperplasia
- LUTS
Lower Urinary Tract Symptoms
- EMT
Epithelial to mesenchymal transition
- TGF-β
Transforming Growth Factor-β
- ADT
Androgen Deprivation Therapy
- ECM
extracellular matrix
- AR
androgen receptor
- ZEB1
E-box-binding protein 1
- GCPRs
G-protein-coupled receptors
- PCPT
Prostate Cancer Prevention Trial
- REDUCE
Reduction of Prostate Cancer Events
- PROSPER
Safety and Efficacy Study of Enzalutamide in Patients With Non-metastatic Castration-Resistant Prostate Cancer
- SPARTAN
Selective Prostate Androgen Receptor Targeting with ARN-50
- CRPC
Castration Resistant Prostate Cancer
- NE
norepinephrine
- PSA
Prostate Specific Antigen
- MET
mesenchymal to epithelial transition
- AUA
American Urological Association
- MRI
Magnetic Resonance Imaging
- FDA
Food and Drug Administration
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Wade CA, Kyprianou N. Profiling prostate cancer therapeutic resistance. Int J Mol Sci. 2018;19 doi: 10.3390/ijms19030904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berry SJ, Coffey DS, Walsh PC, Ewing LL. The development of human benign prostatic hyperplasia with age. J Urol. 1984;132:474–479. doi: 10.1016/s0022-5347(17)49698-4. [DOI] [PubMed] [Google Scholar]
- 4.Carter HB, Coffey DS. The prostate: an increasing medical problem. Prostate. 1990;16:39–48. doi: 10.1002/pros.2990160105. [DOI] [PubMed] [Google Scholar]
- 5.Platz EA, Smit E, Curhan GC, Nyberg LM, Giovannucci E. Prevalence of and racial/ethnic variation in lower urinary tract symptoms and noncancer prostate surgery in U. S. men. Urology. 2002;59:877–883. doi: 10.1016/s0090-4295(01)01673-9. [DOI] [PubMed] [Google Scholar]
- 6.Isaacs JT. Prostate stem cells and benign prostatic hyperplasia. Prostate. 2008;68:1025–1034. doi: 10.1002/pros.20763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naslund MJ, Coffey DS. The differential effects of neonatal androgen, estrogen and progesterone on adult rat prostate growth. J Urol. 1986;136:1136–1140. doi: 10.1016/s0022-5347(17)45239-6. [DOI] [PubMed] [Google Scholar]
- 8.Kyprianou N, Tu H, Jacobs SC. Apoptotic versus proliferative activities in human benign prostatic hyperplasia. Hum Pathol. 1996;27:668–675. doi: 10.1016/s0046-8177(96)90396-2. [DOI] [PubMed] [Google Scholar]
- 9.Lepor H. Landmark studies impacting the medical management of benign prostatic hyperplasia. Rev Urol. 2003;5(Suppl 4):S34–41. [PMC free article] [PubMed] [Google Scholar]
- 10.Van Asseldonk B, Barkin J, Elterman DS. Medical therapy for benign prostatic hyperplasia: a review. Can J Urol. 2015;22(Suppl 1):7–17. [PubMed] [Google Scholar]
- 11.Taub DA, Wei JT. The economics of benign prostatic hyperplasia and lower urinary tract symptoms in the United States. Curr Urol Rep. 2006;7:272–281. doi: 10.1007/s11934-996-0006-0. [DOI] [PubMed] [Google Scholar]
- 12.Saigal CS, Joyce G. Economic costs of benign prostatic hyperplasia in the private sector. J Urol. 2005;173:1309–1313. doi: 10.1097/01.ju.0000152318.79184.6f. [DOI] [PubMed] [Google Scholar]
- 13.Amerson D, editor. Urolift for BPH: changing the game in BPH care; AACU state advocacy conference; Chicago, IL. 2015. [Google Scholar]
- 14.From the centers for disease control and prevention. Public health and aging: trends in aging--United States and worldwide. JAMA. 2003;289:1371–1373. [PubMed] [Google Scholar]
- 15.Price D, William Ashman D. The accessory reproductive glands of mammals. In: Young W, editor. Sex and internal secretions. Baltimore, MD: Williams and Wilkins; 1961. pp. 366–448. [Google Scholar]
- 16.Myers RP. Structure of the adult prostate from a clinician’s standpoint. Clin Anat. 2000;13:214–215. doi: 10.1002/(SICI)1098-2353(2000)13:3<214::AID-CA10>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 17.Wein AJ, Kavoussi LR, Peters CA. Campbell-Walsh Urology. Philadelphia, PA: Elsevier; 2015. [Google Scholar]
- 18.Thomson AA. Mesenchymal mechanisms in prostate organogenesis. Differentiation. 2008;76:587–598. doi: 10.1111/j.1432-0436.2008.00296.x. [DOI] [PubMed] [Google Scholar]
- 19.Donjacour AA, Thomson AA, Cunha GR. FGF-10 plays an essential role in the growth of the fetal prostate. Dev Biol. 2003;261:39–54. doi: 10.1016/s0012-1606(03)00250-1. [DOI] [PubMed] [Google Scholar]
- 20.De Marzo AM, Meeker AK, Epstein JI, Coffey DS. Prostate stem cell compartments: expression of the cell cycle inhibitor p27Kip1 in normal, hyperplastic, and neoplastic cells. Am J Pathol. 1998;153:911–919. doi: 10.1016/S0002-9440(10)65632-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Hayward S, Cao M, Thayer K, Cunha G. Cell differentiation lineage in the prostate. Differentiation. 2001;68:270–279. doi: 10.1046/j.1432-0436.2001.680414.x. [DOI] [PubMed] [Google Scholar]
- 22.Lepor H, Kuhar MJ. Characterization and localization of the muscarinic cholinergic receptor in human prostatic tissue. J Urol. 1984;132:397–402. doi: 10.1016/s0022-5347(17)49636-4. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi S, Tang R, Shapiro E, Lepor H. Characterization and localization of prostatic alpha 1 adrenoceptors using radioligand receptor binding on slide-mounted tissue section. J Urol. 1993;150:2002–2006. doi: 10.1016/s0022-5347(17)35954-2. [DOI] [PubMed] [Google Scholar]
- 24.Vaalasti A, Hervonen A. Autonomic innervation of the human prostate. Invest Urol. 1980;17:293–297. [PubMed] [Google Scholar]
- 25.Negoita S, Feuer EJ, Mariotto A, Cronin KA, Petkov VI, Hussey SK, Benard V, Henley SJ, Anderson RN, Fedewa S, Sherman RL, Kohler BA, Dearmon BJ, Lake AJ, Ma J, Richardson LC, Jemal A, Penberthy L. Annual report to the nation on the status of cancer, part II: recent changes in prostate cancer trends and disease characteristics. Cancer. 2018;124:2801–2814. doi: 10.1002/cncr.31549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanda MG, Cadeddu JA, Kirkby E, Chen RC, Crispino T, Fontanarosa J, Freedland SJ, Greene K, Klotz LH, Makarov DV, Nelson JB, Rodrigues G, Sandler HM, Taplin ME, Treadwell JR. Clinically localized prostate cancer: AUA/ASTRO/SUO guideline. part I: risk stratification, shared decision making, and care options. J Urol. 2018;199:683–690. doi: 10.1016/j.juro.2017.11.095. [DOI] [PubMed] [Google Scholar]
- 27.Schroder FH, Hugosson J, Carlsson S, Tammela T, Maattanen L, Auvinen A, Kwiatkowski M, Recker F, Roobol MJ. Screening for prostate cancer decreases the risk of developing metastatic disease: findings from the european randomized study of screening for prostate cancer (ERSPC) Eur Urol. 2012;62:745–752. doi: 10.1016/j.eururo.2012.05.068. [DOI] [PubMed] [Google Scholar]
- 28.Andriole GL, Crawford ED, Grubb RL 3rd, Buys SS, Chia D, Church TR, Fouad MN, Gelmann EP, Kvale PA, Reding DJ, Weissfeld JL, Yokochi LA, O’Brien B, Clapp JD, Rathmell JM, Riley TL, Hayes RB, Kramer BS, Izmirlian G, Miller AB, Pinsky PF, Prorok PC, Gohagan JK, Berg CD, Team PP. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med. 2009;360:1310–1319. doi: 10.1056/NEJMoa0810696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buyyounouski MK, Choyke PL, McKenney JK, Sartor O, Sandler HM, Amin MB, Kattan MW, Lin DW. Prostate cancer - major changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J Clin. 2017;67:245–253. doi: 10.3322/caac.21391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ricke EA, Williams K, Lee YF, Couto S, Wang Y, Hayward SW, Cunha GR, Ricke WA. Androgen hormone action in prostatic carcinogenesis: stromal androgen receptors mediate prostate cancer progression, malignant transformation and metastasis. Carcinogenesis. 2012;33:1391–1398. doi: 10.1093/carcin/bgs153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, Lieber MM, Cespedes RD, Atkins JN, Lippman SM, Carlin SM, Ryan A, Szczepanek CM, Crowley JJ, Coltman CA Jr. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349:215–224. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 32.Andriole GL, Bostwick DG, Brawley OW, Gomella LG, Marberger M, Montorsi F, Pettaway CA, Tammela TL, Teloken C, Tindall DJ, Somerville MC, Wilson TH, Fowler IL, Rittmaster RS REDUCE Study Group. Effect of dutasteride on the risk of prostate cancer. N Engl J Med. 2010;362:1192–1202. doi: 10.1056/NEJMoa0908127. [DOI] [PubMed] [Google Scholar]
- 33.Silberstein JL, Sartor O. Long-term survival of participants in the prostate cancer prevention trial. Asian J Androl. 2014;16:413–414. doi: 10.4103/1008-682X.122868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scailteux LM, Rioux-Leclercq N, Vincendeau S, Balusson F, Nowak E, Oger E. Use of 5alpha-reductase inhibitors for benign prostate hypertrophy and risk of high-grade prostate cancer: a French population-based study. BJU Int. 2019;123:293–299. doi: 10.1111/bju.14495. [DOI] [PubMed] [Google Scholar]
- 35.Fleshner NE, Lucia MS, Egerdie B, Aaron L, Eure G, Nandy I, Black L, Rittmaster RS. Dutasteride in localised prostate cancer management: the REDEEM randomised, double-blind, placebo-controlled trial. Lancet. 2012;379:1103–1111. doi: 10.1016/S0140-6736(11)61619-X. [DOI] [PubMed] [Google Scholar]
- 36.Su F, Ahn S, Saha A, DiGiovanni J, Kolonin MG. Adipose stromal cell targeting suppresses prostate cancer epithelial-mesenchymal transition and chemoresistance. Oncogene. 2018 doi: 10.1038/s41388-018-0558-8. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.American Cancer Society: Prostate Cancer Risk Factors. https://www.cancer.org/cancer/prostate-cancer/causes-risks-prevention/risk-factors.html.
- 38.Cookson MS, Roth BJ, Dahm P, Engstrom C, Freedland SJ, Hussain M, Lin DW, Lowrance WT, Murad MH, Oh WK, Penson DF, Kibel AS. Castration-resistant prostate cancer: AUA guideline. J Urol. 2013;190:429–438. doi: 10.1016/j.juro.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 39.Lowrance WT, Murad MH, Oh WK, Jarrard DF, Resnick MJ, Cookson MS. Castration-resistant prostate cancer: AUA guideline amendment 2018. J Urol. 2018;200:1264–1272. doi: 10.1016/j.juro.2018.07.090. [DOI] [PubMed] [Google Scholar]
- 40.Civantos Calzada B, Aleixandre de Artinano A. Alpha-adrenoceptor subtypes. Pharmacol Res. 2001;44:195–208. doi: 10.1006/phrs.2001.0857. [DOI] [PubMed] [Google Scholar]
- 41.Desiniotis A, Kyprianou N. Advances in the design and synthesis of prazosin derivatives over the last ten years. Expert Opin Ther Targets. 2011;15:1405–1418. doi: 10.1517/14728222.2011.641534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cotecchia S. The alpha1-adrenergic receptors: diversity of signaling networks and regulation. J Recept Signal Transduct Res. 2010;30:410–419. doi: 10.3109/10799893.2010.518152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marshall I, Burt RP, Chapple CR. Noradrenaline contractions of human prostate mediated by alpha 1A-(alpha 1c-) adrenoceptor subtype. Br J Pharmacol. 1995;115:781–786. doi: 10.1111/j.1476-5381.1995.tb15001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taniguchi N, Ukai Y, Tanaka T, Yano J, Kimura K, Moriyama N, Kawabe K. Identification of alpha 1-adrenoceptor subtypes in the human prostatic urethra. Naunyn Schmiedebergs Arch Pharmacol. 1997;355:412–416. doi: 10.1007/pl00004962. [DOI] [PubMed] [Google Scholar]
- 45.Yamada S, Ashizawa N, Ushijima H, Nakayama K, Hayashi E, Honda K. Alpha-1 adrenoceptors in human prostate: characterization and alteration in benign prostatic hypertrophy. J Pharmacol Exp Ther. 1987;242:326–330. [PubMed] [Google Scholar]
- 46.Hedlund H, Andersson KE, Larsson B. Alpha-adrenoceptors and muscarinic receptors in the isolated human prostate. J Urol. 1985;134:1291–1298. doi: 10.1016/s0022-5347(17)47714-7. [DOI] [PubMed] [Google Scholar]
- 47.White CW, Xie JH, Ventura S. Age-related changes in the innervation of the prostate gland: implications for prostate cancer initiation and progression. Organogenesis. 2013;9:206–215. doi: 10.4161/org.24843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moriyama N, Yamaguchi T, Takeuchi T, Sakamoto E, Ueki T, Tsujimoto G, Kawabe K. Semiquantitative evaluation of alpha1A-adrenoceptor subtype mRNA in human hypertrophied and non-hypertrophied prostates: regional comparison. Life Sci. 1999;64:201–210. doi: 10.1016/s0024-3205(98)00552-9. [DOI] [PubMed] [Google Scholar]
- 49.Nasu K, Moriyama N, Kawabe K, Tsujimoto G, Murai M, Tanaka T, Yano J. Quantification and distribution of alpha 1-adrenoceptor subtype mRNAs in human prostate: comparison of benign hypertrophied tissue and non-hypertrophied tissue. Br J Pharmacol. 1996;119:797–803. doi: 10.1111/j.1476-5381.1996.tb15742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caine M, Raz S, Zeigler M. Adrenergic and cholinergic receptors in the human prostate, prostatic capsule and bladder neck. Br J Urol. 1975;47:193–202. doi: 10.1111/j.1464-410x.1975.tb03947.x. [DOI] [PubMed] [Google Scholar]
- 51.Lepor H. Alpha blockers for the treatment of benign prostatic hyperplasia. Rev Urol. 2007;9:181–190. [PMC free article] [PubMed] [Google Scholar]
- 52.Lepor H. Pathophysiology of lower urinary tract symptoms in the aging male population. Rev Urol. 2005;7(Suppl 7):S3–S11. [PMC free article] [PubMed] [Google Scholar]
- 53.Kaplan SA. Current role of alpha-blockers in the treatment of benign prostatic hyperplasia. BJU Int. 2008;102(Suppl 2):3–7. doi: 10.1111/j.1464-410X.2008.08086.x. [DOI] [PubMed] [Google Scholar]
- 54.Rosenberg MT, Witt ES, Miner M, Barkin J. A practical primary care approach to lower urinary tract symptoms caused by benign prostatic hyperplasia (BPH-LUTS) Can J Urol. 2014;21(Suppl 2):12–24. [PubMed] [Google Scholar]
- 55.McVary KT, Roehrborn CG, Avins AL, Barry MJ, Bruskewitz RC, Donnell RF, Foster HE Jr, Gonzalez CM, Kaplan SA, Penson DF, Ulchaker JC, Wei JT. Update on AUA guideline on the management of benign prostatic hyperplasia. J Urol. 2011;185:1793–1803. doi: 10.1016/j.juro.2011.01.074. [DOI] [PubMed] [Google Scholar]
- 56.Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, DePalma SM, Gidding S, Jamerson KA, Jones DW, MacLaughlin EJ, Muntner P, Ovbiagele B, Smith SC Jr, Spencer CC, Stafford RS, Taler SJ, Thomas RJ, Williams KA Sr, Williamson JD, Wright JT Jr. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. J Am Coll Cardiol. 2018;71:e127–e248. doi: 10.1016/j.jacc.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 57.Davis BR, Cutler JA, Furberg CD, Wright JT, Farber MA, Felicetta JV, Stokes JD ALLHAT Collaborative Research Group. Relationship of antihypertensive treatment regimens and change in blood pressure to risk for heart failure in hypertensive patients randomly assigned to doxazosin or chlorthalidone: further analyses from the antihypertensive and lipid-lowering treatment to prevent heart attack trial. Ann Intern Med. 2002;137:313–320. doi: 10.7326/0003-4819-137-5_part_1-200209030-00006. [DOI] [PubMed] [Google Scholar]
- 58.Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, Frenette PS. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341:1236361. doi: 10.1126/science.1236361. [DOI] [PubMed] [Google Scholar]
- 59.Albinana V, Villar Gomez de Las Heras K, Serrano-Heras G, Segura T, Perona-Moratalla AB, Mota-Perez M, de Campos JM, Botella LM. Propranolol reduces viability and induces apoptosis in hemangioblastoma cells from von Hippel-Lindau patients. Orphanet J Rare Dis. 2015;10:118. doi: 10.1186/s13023-015-0343-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zahalka AH, Arnal-Estape A, Maryanovich M, Nakahara F, Cruz CD, Finley LWS, Frenette PS. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science. 2017;358:321–326. doi: 10.1126/science.aah5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 62.Taylor SH. Clinical pharmacotherapeutics of doxazosin. Am J Med. 1989;87:2S–11S. doi: 10.1016/0002-9343(89)90107-1. [DOI] [PubMed] [Google Scholar]
- 63.Cohen BM. Prazosin hydrochloride (CP-12,299-1), an oral anti-hypertensive agent: preliminary clinical observations in ambulatory patients. J Clin Pharmacol J New Drugs. 1970;10:408–417. [PubMed] [Google Scholar]
- 64.Yang G, Timme TL, Park SH, Wu X, Wyllie MG, Thompson TC. Transforming growth factor beta 1 transduced mouse prostate reconstitutions: II. Induction of apoptosis by doxazosin. Prostate. 1997;33:157–163. doi: 10.1002/(sici)1097-0045(19971101)33:3<157::aid-pros2>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 65.Kyprianou N, Litvak JP, Borkowski A, Alexander R, Jacobs SC. Induction of prostate apoptosis by doxazosin in benign prostatic hyperplasia. J Urol. 1998;159:1810–1815. doi: 10.1016/S0022-5347(01)63162-8. [DOI] [PubMed] [Google Scholar]
- 66.Thebault S, Flourakis M, Vanoverberghe K, Vandermoere F, Roudbaraki M, Lehen’kyi V, Slomianny C, Beck B, Mariot P, Bonnal JL, Mauroy B, Shuba Y, Capiod T, Skryma R, Prevarskaya N. Differential role of transient receptor potential channels in Ca2+ entry and proliferation of prostate cancer epithelial cells. Cancer Res. 2006;66:2038–2047. doi: 10.1158/0008-5472.CAN-05-0376. [DOI] [PubMed] [Google Scholar]
- 67.Park SY, Kang JH, Jeong KJ, Lee J, Han JW, Choi WS, Kim YK, Kang J, Park CG, Lee HY. Norepinephrine induces VEGF expression and angiogenesis by a hypoxia-inducible factor-1alpha protein-dependent mechanism. Int J Cancer. 2011;128:2306–2316. doi: 10.1002/ijc.25589. [DOI] [PubMed] [Google Scholar]
- 68.Kyprianou N, Benning CM. Suppression of human prostate cancer cell growth by alpha1-adrenoceptor antagonists doxazosin and terazosin via induction of apoptosis. Cancer Res. 2000;60:4550–4555. [PubMed] [Google Scholar]
- 69.Forbes A, Anoopkumar-Dukie S, Chess-Williams R, McDermott C. Relative cytotoxic potencies and cell death mechanisms of alpha1 -adrenoceptor antagonists in prostate cancer cell lines. Prostate. 2016;76:757–766. doi: 10.1002/pros.23167. [DOI] [PubMed] [Google Scholar]
- 70.Benning CM, Kyprianou N. Quinazoline-derived alpha1-adrenoceptor antagonists induce prostate cancer cell apoptosis via an alpha1-adrenoceptor-independent action. Cancer Res. 2002;62:597–602. [PubMed] [Google Scholar]
- 71.Hui H, Fernando MA, Heaney AP. The alpha1-adrenergic receptor antagonist doxazosin inhibits EGFR and NF-kappaB signalling to induce breast cancer cell apoptosis. Eur J Cancer. 2008;44:160–166. doi: 10.1016/j.ejca.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 72.Gan L, Zhu DX, Yang LP, Liu RS, Yan F, Zhang J. Involvement of transcription factor activator protein-2alpha in doxazosin-induced HeLa cell apoptosis. Acta Pharmacol Sin. 2008;29:465–472. doi: 10.1111/j.1745-7254.2008.00780.x. [DOI] [PubMed] [Google Scholar]
- 73.Eiras S, Fernandez P, Pineiro R, Iglesias MJ, Gonzalez-Juanatey JR, Lago F. Doxazosin induces activation of GADD153 and cleavage of focal adhesion kinase in cardiomyocytes en route to apoptosis. Cardiovasc Res. 2006;71:118–128. doi: 10.1016/j.cardiores.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 74.Keledjian K, Garrison JB, Kyprianou N. Doxazosin inhibits human vascular endothelial cell adhesion, migration, and invasion. J Cell Biochem. 2005;94:374–388. doi: 10.1002/jcb.20240. [DOI] [PubMed] [Google Scholar]
- 75.Siddiqui EJ, Shabbir M, Thompson CS, Mumtaz FH, Mikhailidis DP. Growth inhibitory effect of doxazosin on prostate and bladder cancer cells. Is the serotonin receptor pathway involved? Anticancer Res. 2005;25:4281–4286. [PubMed] [Google Scholar]
- 76.Fernando MA, Heaney AP. Alpha1-adrenergic receptor antagonists: novel therapy for pituitary adenomas. Mol Endocrinol. 2005;19:3085–3096. doi: 10.1210/me.2004-0471. [DOI] [PubMed] [Google Scholar]
- 77.Austin PF, Cook BL, Niederhoff RA, Manson SR, Coplen DE, Weintraub SJ. Inhibition of mitogenic signaling and induction of apoptosis in human bladder smooth muscle cells treated with doxazosin. J Urol. 2004;172:1662–1665. doi: 10.1097/01.ju.0000138524.18870.af. discussion 1666. [DOI] [PubMed] [Google Scholar]
- 78.Rodriguez-Feo JA, Fortes J, Aceituno E, Farre J, Ayala R, Castilla C, Rico L, Gonzalez-Fernandez F, Garcia-Duran M, Casado S, Lopez-Farre A. Doxazosin modifies Bcl-2 and Bax protein expression in the left ventricle of spontaneously hypertensive rats. J Hypertens. 2000;18:307–315. doi: 10.1097/00004872-200018030-00011. [DOI] [PubMed] [Google Scholar]
- 79.Gonzalez-Juanatey JR, Iglesias MJ, Alcaide C, Pineiro R, Lago F. Doxazosin induces apoptosis in cardiomyocytes cultured in vitro by a mechanism that is independent of alpha1-adrenergic blockade. Circulation. 2003;107:127–131. doi: 10.1161/01.cir.0000043803.20822.d1. [DOI] [PubMed] [Google Scholar]
- 80.Rennebeck G, Martelli M, Kyprianou N. Anoikis and survival connections in the tumor microenvironment: is there a role in prostate cancer metastasis? Cancer Res. 2005;65:11230–11235. doi: 10.1158/0008-5472.CAN-05-2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13:555–562. doi: 10.1016/s0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- 82.Tahmatzopoulos A, Kyprianou N. Apoptotic impact of alpha1-blockers on prostate cancer growth: a myth or an inviting reality? Prostate. 2004;59:91–100. doi: 10.1002/pros.10357. [DOI] [PubMed] [Google Scholar]
- 83.Tahmatzopoulos A, Rowland RG, Kyprianou N. The role of alpha-blockers in the management of prostate cancer. Expert Opin Pharmacother. 2004;5:1279–1285. doi: 10.1517/14656566.5.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Partin JV, Anglin IE, Kyprianou N. Quinazoline-based alpha 1-adrenoceptor antagonists induce prostate cancer cell apoptosis via TGF-beta signalling and I kappa B alpha induction. Br J Cancer. 2003;88:1615–1621. doi: 10.1038/sj.bjc.6600961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin SC, Chueh SC, Hsiao CJ, Li TK, Chen TH, Liao CH, Lyu PC, Guh JH. Prazosin displays anticancer activity against human prostate cancers: targeting DNA and cell cycle. Neoplasia. 2007;9:830–839. doi: 10.1593/neo.07475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chapple CR. Selective alpha 1-adrenoceptor antagonists in benign prostatic hyperplasia: rationale and clinical experience. Eur Urol. 1996;29:129–144. [PubMed] [Google Scholar]
- 88.Djavan B, Marberger M. A meta-analysis on the efficacy and tolerability of alpha1-adrenoceptor antagonists in patients with lower urinary tract symptoms suggestive of benign prostatic obstruction. Eur Urol. 1999;36:1–13. doi: 10.1159/000019919. [DOI] [PubMed] [Google Scholar]
- 89.Debruyne FM. Alpha blockers: are all created equal? Urology. 2000;56:20–22. doi: 10.1016/s0090-4295(00)00744-5. [DOI] [PubMed] [Google Scholar]
- 90.Srivastava K, Arora A, Kataria A, Cappelleri JC, Sadosky A, Peterson AM. Impact of reducing dosing frequency on adherence to oral therapies: a literature review and meta-analysis. Patient Prefer Adherence. 2013;7:419–434. doi: 10.2147/PPA.S44646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Garrison JB, Shaw YJ, Chen CS, Kyprianou N. Novel quinazoline-based compounds impair prostate tumorigenesis by targeting tumor vascularity. Cancer Res. 2007;67:11344–11352. doi: 10.1158/0008-5472.CAN-07-1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cao Z, Koochekpour S, Strup SE, Kyprianou N. Reversion of epithelial-mesenchymal transition by a novel agent DZ-50 via IGF binding protein-3 in prostate cancer cells. Oncotarget. 2017;8:78507–78519. doi: 10.18632/oncotarget.19659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barron DA, Rowley DR. The reactive stroma microenvironment and prostate cancer progression. Endocr Relat Cancer. 2012;19:R187–204. doi: 10.1530/ERC-12-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sampson N, Zenzmaier C, Heitz M, Hermann M, Plas E, Schafer G, Klocker H, Berger P. Stromal insulin-like growth factor binding protein 3 (IGFBP3) is elevated in the diseased human prostate and promotes ex vivo fibroblast-to-myofibroblast differentiation. Endocrinology. 2013;154:2586–2599. doi: 10.1210/en.2012-2259. [DOI] [PubMed] [Google Scholar]
- 95.Biernacka KM, Perks CM, Holly JM. Role of the IGF axis in prostate cancer. Minerva Endocrinol. 2012;37:173–185. [PubMed] [Google Scholar]
- 96.Graham TR, Zhau HE, Odero-Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, Liu T, Simons JW, O’Regan RM. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008;68:2479–2488. doi: 10.1158/0008-5472.CAN-07-2559. [DOI] [PubMed] [Google Scholar]
- 97.De Mellow JS, Baxter RC. Growth hormone-dependent insulin-like growth factor (IGF) binding protein both inhibits and potentiates IGF-I-stimulated DNA synthesis in human skin fibroblasts. Biochem Biophys Res Commun. 1988;156:199–204. doi: 10.1016/s0006-291x(88)80824-6. [DOI] [PubMed] [Google Scholar]
- 98.Giardina D, Martarelli D, Sagratini G, Angeli P, Ballinari D, Gulini U, Melchiorre C, Poggesi E, Pompei P. Doxazosin-related alpha1-adrenoceptor antagonists with prostate antitumor activity. J Med Chem. 2009;52:4951–4954. doi: 10.1021/jm8016046. [DOI] [PubMed] [Google Scholar]
- 99.Maestri V, Tarozzi A, Simoni E, Cilia A, Poggesi E, Naldi M, Nicolini B, Pruccoli L, Rosini M, Minarini A. Quinazoline based alpha1-adrenoreceptor antagonists with potent antiproliferative activity in human prostate cancer cell lines. Eur J Med Chem. 2017;136:259–269. doi: 10.1016/j.ejmech.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 100.Cao L, Zhang S, Jia CM, He W, Wu LT, Li YQ, Wang W, Li Z, Ma J. Antihypertensive drugs use and the risk of prostate cancer: a meta-analysis of 21 observational studies. BMC Urol. 2018;18:17. doi: 10.1186/s12894-018-0318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Murtola TJ, Tammela TL, Maattanen L, Hakama M, Auvinen A. Prostate cancer risk among users of finasteride and alpha-blockers - a population based case-control study. Eur J Cancer. 2007;43:775–781. doi: 10.1016/j.ejca.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 102.Harris AM, Warner BW, Wilson JM, Becker A, Rowland RG, Conner W, Lane M, Kimbler K, Durbin EB, Baron AT, Kyprianou N. Effect of alpha1-adrenoceptor antagonist exposure on prostate cancer incidence: an observational cohort study. J Urol. 2007;178:2176–2180. doi: 10.1016/j.juro.2007.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Van Rompay MI, Curtis Nickel J, Ranganathan G, Kantoff PW, Solomon KR, Lund JL, McKinlay JB. Impact of 5alpha-reductase inhibitor and alpha-blocker therapy for benign prostatic hyperplasia on prostate cancer incidence and mortality. BJU Int. 2018 doi: 10.1111/bju.14534. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Friedman GD, Udaltsova N, Habel LA. Norepinephrine antagonists and cancer risk. Int J Cancer. 2011;128:737–738. doi: 10.1002/ijc.25351. author reply 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yamada D, Nishimatsu H, Kumano S, Hirano Y, Suzuki M, Fujimura T, Fukuhara H, Enomoto Y, Kume H, Homma Y. Reduction of prostate cancer incidence by naftopidil, an alpha1 -adrenoceptor antagonist and transforming growth factor-beta signaling inhibitor. Int J Urol. 2013;20:1220–1227. doi: 10.1111/iju.12156. [DOI] [PubMed] [Google Scholar]