

Abstract
Polymeric micelles are of increasing interest as drug delivery vehicles since they can accumulate in tumor tissue through EPR effect and deliver their hydrophobic cargo. The pharmacology can be visualized and quantified noninvasively by molecular imaging techniques. Here, a novel, fast and efficient technique for radiolabeling various HPMA-LMA based micellar aggregates with hydrophobic oxine-complexes of the trivalent radiometals 68Ga and 111In was investigated. The radiometal-oxine complexes resemble the hydrophobic drug 111In[In]-oxine considered for the diagnosis of infection and inflammation. Promising in vitro stability lead to in vivo evaluation in healthy mice in terms of quantitative ex vivo organ distribution. The results show that while the hydrophobic radiometal-oxine complexes were safely encapsulated in aqueous saline, they left the polymeric micelles slowly in contact with blood serum and more rapidly in vivo. Due to the similarity between the radiometal complexes and hydrophobic drugs transported in the polymeric micelles this has significant implications for further strategies on transport mechanisms of hydrophobically encapsulated drugs.
Keywords: HPMA, micelles, PET, SPECT, 68Ga, 111In, oxine complexes, PK-46
Introduction
Polymeric micelles are of increasing interest as drug delivery vehicles since they can accumulate in tumor tissue through EPR effect and deliver their hydrophobic cargo. The pharmacology can be visualized and quantified noninvasively by molecular imaging techniques. Here, a novel, fast and efficient technique for radiolabeling various HPMA-LMA based micellar aggregates with hydrophobic oxine-complexes of the trivalent radiometals 68Ga and 111In was investigated. The radiometal-oxine complexes resemble the hydrophobic drug 111In[In]-oxine considered for the diagnosis of infection and inflammation [1]. Due to the similarity between the radiometal complexes and hydrophobic drugs transported in the polymeric micelles those studies should have significant implications for further strategies on transport mechanisms of hydrophobically encapsulated drugs.
Micelles
Micelles and especially polymeric micelles find recently a lot of interest as nanoparticular drug carriers [2,3]. They offer the possibility to solubilize hydrophobic drugs in their hydrophobic core and increase thereby their bioavailability and their circulation time in blood. If the drug gets safely encapsulated in the micelle, it may thus get possible to transport it preferably into a tumor (or other tissue with an open vasculature) by the EPR-effect [4], which requires long circulation. As outlined in a recent review by Tallelli et al. [2] the permanent inclusion of drugs in polymeric micelles is, however, not trivial [5-7] as micelles are dynamic systems, which rearrange permanently and which allow a diffusion of the hydrophobic cargo within the core. So it could also be shown that a hydrophobic compound solubilized in the core can be transferred to cellular membranes just by close contact without the need of an uptake of the nanoparticle [8]. Nevertheless, there are very positive examples of nanoparticular formulations like Abraxane, which show improved pharmacokinetic properties although the formulation disintegrates quickly after intravenous injection and transfers the cargo to endogenous albumin [9-11].
Polymeric micelles, which are made by the self-assembly of amphiphilic block copolymers, offer a lot of variability, so there is much room to increase (or decrease) their stability, which determines their fate in the body. Their critical micelle concentration (CMC) is typically low and can be varied by the hydrophilic/hydrophobic balance (or simply the length) of both blocks. In addition the dynamic of the core can be modified by the core material, which may be crystallizable or crosslinkable [2,12]. In this regard block-copolymers from poly[N-(2-hydroxypropyl)methacrylamide] (polyHPMA) and polylaurylmethacrylate (polyLMA) have been reviewed quiet recently [13]. These block copolymers can be made by controlled radical polymerization (RAFT-polymerization), their core can be made crosslinkable [12] and their hydrophilic corona can be easily functionalized. As their hydrophilic corona is made of polyHPMA, a lot can be learned from studies with this polymer [14]. In addition, it was found that amphiphilic block copolymers of this type behave very differently from chemically comparable statistical copolymers [15,16]. While the block copolymers do not interact with serum proteins and are rarely taken up by cells in an unspecific manner [17-19], the statistical copolymer interacts with certain proteins and it can penetrate into the cellular membrane [8,16,20,21]. In this regard, such copolymers offer a basis to study structure property relations with regard to encapsulation and transport of hydrophobic drugs in the body.
Micelle-supported delivery of Me(III)-oxines
Oxine complexes of gallium radioisotopes (66Ga, 67Ga, 68Ga) and 111In have been used for decades for the labeling of white blood cells [22-27]. Recently, the tris(8-quinolinolato)gallium(iii) complex) (gallium-oxine), trade name KP-46, a drug considered for the treatment of malignancies, found a lot of interest [28-32].
In order to provide a prolonged availability and an enhanced therapeutic index of the Ga-oxine complexes, drug-delivery strategies may be applied. While there have been recent studies to improve the preparation of the 68Ga[Ga]-oxine complex for labeling of blood cells [24], there are no studies to adopt drug delivery systems for KP-46 itself.
Radiolabeling for imaging
To select promising candidates at an early stage of research and development, imaging approaches are essential [33]. By visualizing pharmacokinetics in vivo, the influence of specific characteristics such as charge or hydrophilic-lipophilic balance affecting the behaviour of carrier systems can be studied, which is necessary to optimize the preparation and formulation of carrier systems and to guide the way to a new generation of nanomedicines. Positron emission tomography (PET) and single photon emission computed tomography (SPECT) are both non-invasive, quantitative and whole body molecular imaging techniques that provide information of the in vivo behaviour of the labeled biomolecule [34]. In the present study, one PET (68Ga) and one SPECT radionuclide (111In) were used. The encapsulation of 68Ga or 111In into polymeric micelles should allow it thus to study both (i) the stability of these polymeric micelles, (ii) their body distribution and finally, (iii) the fate of an interesting new drug, KP-46 in particular.
The challenge consists in selectively incorporating them into the micelles and to design micelles, which allow for a controlled release. Accordingly, both the structure of the nanocarrier micellar system and the radiolabeling strategy need to be designed to guarantee a stable radiolabeled compound for in vivo applications.
The chemistry to incorporate such radionuclides can be divided into two general types depending on the characteristics of the nuclide: covalent and ionic. In covalent chemistry, examples of such nuclides are 18F [35,36] 76Br and 123/124I [37], the nuclide is incorporated through a radiohalogen-carbon bond. In most cases, those nuclides do not significantly alter the structure or the pharmacokinetics of the radiolabeled compound, but the chemical procedures are often lengthy, tedious and providing low yields. In contrast, radiometals are introduced in terms of coordination chemistry [38-40]. Bifunctional chelators are needed when metallic radioisotopes such as 68Ga, 111In etc. are involved for direct, covalent attachment to micelle’s components. Bifunctional chelators offer both a metal binding moiety, which allows complexation of the radionuclide in its cavity, and an extra-functional group for covalent attachment of the chelator to the biomolecule of interest [41]. For stable complexation, chelating agents based on polyamino carboxylic acids e.g. diethylene triamine pentaacetic acid (DTPA) or 1,4,7,10-tetraazacyclododecane-N,N”,N’’’,N’’’’-tetraacetic acid (DOTA) are commonly used in radiopharmaceutical chemistry, as they provide high thermodynamic stability of the complex. Radiolabeling typically is fast and proceeds in aqueous solution. Typically, the chelating agent itself is rather large, bulky and charged and as a result, it may influence the particle structure and consequently its biological behaviour. It requires considerable synthetic efforts to link these chelating agents to polymers.
In this context, we have developed a novel radiolabeling strategy that applies to micelles. It takes advantage of the micelles capacity to spontaneously incorporate hydrophobic compounds like drugs [21], but also hydrophobic radiometal-chelate complexes. Thus, a hydrophobic *Me(III)-ligand complex may be introduced into the micellar core with no requirement of previous modification of the polymers (incorporation of radionuclide or a chelator).
If this hydrophobic chelator stays stable in the micellar core in vitro, it shall be possible to study the body distribution of the loaded micelles. If the loading is less stable, e.g. due to various interactions with the environment, the body distribution of the complex will be similar to the body distribution of a drug of comparable hydrophobicity. This methodology is most known in the context of labeling white blood cells. 111In[In]-oxine is a well-established hydrophobic complex and routinely applied in the clinics [42,43]. In analogy, the positron emitter 68Ga appears to be a candidate for quantitative PET imaging and a similar complex, 68Ga[Ga]-oxine, was prepared. For the micelles, different structures of the well-known biocompatible HPMA polymer [14] modified with the hydrophobic moiety LMA have been studied [13,14,16,18,44] as well as the stability of the labeled micelles in presence of human serum and saline.
Generally speaking - as indium or gallium-oxines are finally just hydrophobic compounds - these kind of studies can shed light on the fate of other drugs of a comparable hydrophobicity in the body. We believe that the use of radiolabeled surrogates, which can be quantitatively investigated in vitro, in vivo and ex vivo, can contribute a substantial information database in terms of structure (micelles) versus pharmacology (load of drugs, controlled release of drugs).
Materials and methods
Materials
All chemicals were analytical or pure reagent grade and used as received unless otherwise specified.
Deionized Milli-Q water (18.2 MΩ•cm; Millipore) was used in all organic reactions. Dioxane was distilled over a sodium/potassium composition. Lauryl methacrylate was distilled to remove the stabilizer and stored at -18°C. 1H-NMR spectra were obtained by a Bruker AC 300 spectrometer at 300 MHz, 19F-NMR analysis was carried out with a Bruker DRX-400 at 400 MHz. All measurements were accomplished at room temperature.
The synthesized polymers were dried at 40°C under vacuum overnight, followed by size exclusion chromatography (SEC). SEC was performed in tetrahydrofuran (THF) or hexafluoroispropanol (HFIP) as solvent and polystyrene resp. polymethylmethacrylate as external standard and toluene as internal standard, using following equipment: pump PU 1580, auto sampler AS 1555, UV detector UV 1575 and RI detector RI 1530 from Jasco. Columns were used from MZ Analysentechnik, 300×8.0 mm: MZ-Gel SDplus 106 Å 5 µm, MZ-Gel SDplus 104 Å 5 µm and MZ-Gel SDplus 102 Å 5 µm. SEC data were evaluated by using the software PSS WinGPC Unity (Polymer Standard Service Mainz, Germany). The flow rate was set to 1 mL/min with a temperature of 25°C.
Commercial 68Ge/68Ga generators based on TiO2 were obtained from Cyclotron Co. Ltd (Obninsk, Russia). The cation exchange resins AG 50W-X8 (- 400 mesh), AG 50W-X4 (200 - 400 mesh) and AG 50W-X8 (200 - 400 mesh) were obtained from Bio-Rad (Munich, Germany). 111In was purchased from Mallinckrodt. HiTrap™ Desalting Columns were purchased from GE-Healthcare Europe GmbH (Freiburg, Germany). TraceSelect water (Sigma-Aldrich, Germany) was used for all aqueous radiolabeling solutions.
Thin layer chromatography (TLC) was performed on silica-gel (silica-gel 60 F254; MERCK, Darmstadt, Germany) coated aluminium TLC-sheets and instant thin layer chromatography (iTLC). They were analysed using an instant imager (Instant Imager, Canberra Packard, Schwadorf, Austria). RadioHPLC using HiTrapTM column (5 mL, G-25 sephadex) was used to quantify the purity of the compounds. HPLC was performed using Hitachi L-7100 pump system coupled with UV (Hitachi L-7400) and radiometric (Gamma Raytest) detectors. Isocratic elution (100% water) and flow rate of 0.2 mL/ min.
Synthesis of polymers
Synthesis of 4-cyano-4-((thiobenzoyl)sulfanyl)pentanoic acid
The 4-cyano-4-((thiobenzoyl)sulfanyl)pentanoic acid was used as the chain transfer agent (CTA) and synthesized according to the literature in a three step reaction [45].
Synthesis of pentafluorophenylmethacrylate (PFPMA)
PFPMA was prepared according to the literature [46].
Synthesis of the reactive ester random copolymer
RAFT polymerization of pentafluorophenylmethacrylate (PFPMA) and lauryl methacrylate (LMA) with 4-cyano-4-((thiobenzoyl)sulfanyl)pentanoic acid (CTA) was carried out in a schlenk tube. For this purpose, 4 g of PFPMA and 1.2 g of LMA were dissolved in 5 ml of absolute dioxane and 8.5 mg of CTA and 1.9 mg of 2,2’-Azobis-(4-methoxy)-2,4-dimethyl)valeronitrile (ABNDM) were added. After three-freeze-vacuum-thaw cycles, the mixture was immersed in an oil bath at 40°C and stirred overnight. Afterwards the polymer solution was precipitated three times in n-hexane, centrifuged und dried under vacuum at 40°C over night. A slightly pink powder was obtained. Yield: 65%. 1H NMR (300 MHz, CDCl3): δ [ppm] 4.00 (br, 2H), 2.48-0.83 (m, 5H). 19F NMR (400 MHz, CDCl3): δ [ppm] -151.5 - -153.1 (br, 2F), -157.9 - -158.2 (br, 1F), -162,9 - -163.4 (br, 2F).
Synthesis of the reactive ester homo polymer
The PFPMA-macro-CTA was synthesized by RAFT polymerization of PFPMA with CTA under schlenk conditions. The reaction vessel was loaded with 4 g of PFPMA, 40 mg of CTA, 4 mg of ABNDM and 5 ml of absolute dioxane. Following three freeze-vacuum-thaw cycles, the tube was immersed into an oil bath at 40°C and stirred overnight. After the polymerization the polymer solution was precipitated three times in n-hexane, isolated by centrifugation and dried overnight under vacuum at 40°C. In the end a slightly pink powder was obtained. Yield: 62%. 1H NMR (300 MHz, CDCl3): δ [ppm] 2.1 - 2.5 (br, 2H), 1.3 - 1.6 (m, 3H). 19F NMR (400 MHz, CDCl3): δ [ppm] -151.5 - -153.1 (br, 2F), -157.9 - -158.2 (br, 1F), -162,9 - -163.4 (br, 2F).
Synthesis of the reactive ester block copolymer
0.5 g of the macro-CTA obtained after homo polymerization of PFPMA was dissolved in 4 ml of absolute dioxane. Afterwards 0.3 g of LMA and 1.4 mg of ABNDM were added and mixed. After three freeze-vacuum-thaw cycles, the tube was immersed into an oil bath at 40°C and stirred for three days. Afterwards poly (PFPMA)-b-poly (LMA) was precipitated three times from dioxane into ethanol, isolated by centrifugation and dried overnight under vacuum at 40°C. In the end a slightly pink powder was obtained. Yield: 59%. 1H NMR (300 MHz, CDCl3): δ [ppm] 3.9 (br, 2H), 2.4 - 0.9 (m, 5H). 19F NMR (400 MHz, CDCl3): δ [ppm] -151.5 - -153.1 (br, 2F), -157.9 - -158.2 (br, 1F), -162,9 - -163.4 (br, 2F).
Removal of the dithioester end groups
The dithiobenzoate end groups were removed by adding a solution of 25 equivalents ABNDM in absolute dioxane to a block copolymer solution. After heating at 50°C for 12 h, the colourless solution was precipitated in ethanol and collected by centrifugation. The copolymer was dried under vacuum at 40°C overnight and a colourless powder was obtained. Yield: 95%. The absence of the dithiobenzoate end group was confirmed by UV/Vis spectroscopy by the absence of the peak at 302 nm.
Synthesis of poly (HPMA)-random-poly (LMA)
A total of 40 mg of the poly (PFPMA)-ran-poly (LMA) precursor polymer without dithioester end groups, was dissolved in 4 ml absolute dioxane and 1.5 ml absolute dimethylsulfoxide under argon atmosphere. 121 μL of 2-hydroxypropylamine and the equivalent molar amount of triethylamine were added. The mixture was stirred at 50°C for three days. Afterwards the solution was diluted with Milli-Q water and dialysed for 3 days using Spectra/Por membranes (MWCO 3500 g/mol) and changing the water every 12 hours. The resulting solution was freeze dried to obtain a colourless powder. Yield: 90%. 1H NMR (400 MHz, DMSO-d6): δ [ppm] 7.4 - 7.2 (br, -NH), 4.7 (br, 1H), 3.7 - 3.4 (br, 2H), 2.9 (br, 2H), 2.3 - 0.8 (br, 5H).
Synthesis of poly (HPMA)-block-poly (LMA)
A total of 100 mg of the poly (PFPMA)-b-poly (LMA) precursor polymer without dithioester end groups, was dissolved in 4 ml absolute dioxane and 1.5 ml absolute dimethylsulfoxide under argon atmosphere. 262 μL of 2-hydroxypropylamine and the equivalent molar amount of triethylamine were added. The mixture was stirred at 50°C for three days. Afterwards the solution was diluted with Milli-Q water and dialysed for 3 days using Spectra/Por membranes (MWCO 3500 g/mol) and changing the water every 12 hours. The resulting solution was freeze dried to obtain a colourless powder. Yield: 90%. 1H NMR (400 MHz, DMSO-d6): δ [ppm] 7.4 - 7.2 (br, -NH), 4.7 (br, 1H), 3.7 - 3.4 (br, 2H), 2.9 (br, 2H), 2.3 - 0.8 (br, 5H).
Synthesis of PEGylated poly (HPMA)-block-poly (LMA)
A total of 100 mg of the poly (PFPMA)-b-poly (LMA) precursor polymer without dithioester end groups, was dissolved in 4 ml absolute dioxane and 1.5 ml absolute dimethylsulfoxide under argon atmosphere. 53 mg of PEG2kDa with amine functionality and an equivalent amount of triethylamine was added. The colourless solution was stirred at 50°C for 24 hours. Afterwards 120 μL of 2-hydroxypropylamine and the equivalent molar amount of triethylamine were added and stirred at 50°C for another three days. The polymer solution was diluted with Milli-Q water and dialysed for 3 days using Spectra/Por membranes (MWCO 3500 g/mol) and changing the water every 12 hours. The resulting solution was freeze dried to obtain a colourless powder. Yield: 93%. 1H NMR (400 MHz, DMSO-d6): δ [ppm] 7.4 - 7.2 (br, -NH), 4.7 (br, 1H), 3.7 - 3.4 (br, 2H, 4H PEG), 3.3 (s, 2H PEG), 2.9 (br, 2H), 2.3 - 0.8 (br, 5H).
Synthesis of natGa[Ga]-Oxine
8-Hydroxiquinoline (3.5 eq) was dissolved in 10% acetic acid solution and Ga(NO3)3 (1 eq) was slowly added. The solution was stirred for 10 minutes and then heated up to 80°C under reflux. After few minutes, pH was adjusted to 7 with concentrated NH4OH. The yellow suspension was refluxed for 1 hour. The yellow precipitate was filtered off, washed with hot water and dried at 100°C.
Synthesis of 68Ga[Ga]-oxine and 111In[In]-oxine
The 68Ge/68Ga generator was eluted using 5 mL of 0.1 N HCl and the 68Ga was online trapped on a cation exchange resin. 68Ga was post-processed as previously described [47,48]. 400 µL of the acetone post-processed 68Ga eluate were added to 1 mL NaOAc 1 M solution together with 10-15 µL of oxine in ethanol (20 mg/mL). 111In was obtained as 111InCl3 in 0.02 N HCl solution from Mallinckrodt. Aliquots of these solutions were added to 1 mL of NaOAc (1 M) together with 10-15 µL of oxine in ethanol (20 mg/mL). The reaction mixture was stirred for 10 minutes at room temperature for both reactions. Final pH was of 6.6-6.8. 68Ga[Ga]-oxine and 111In[In]-oxine were analysed by iTLC: CHCl3/MeOH (95:5).
Micelle formation
Different amounts of polymer (3 mg - 7.5 mg) and of cold Ga-oxine (1.5 mg - 5.5 mg) were dissolved in 100 µL DMSO and added to the 68Ga-oxine NaOAc solution. The mixed solution was slowly dropped into 1 mL of NaCl 0.9%. The micelles formed spontaneously within 1 minute. After micelle formation, purification using Sephadex G-25 size exclusion chromatography with saline as eluent was performed. Micelle formation was analysed by TLC using citrate buffer 0.1 M pH=4.0 as a mobile phase.
Characterization of nanoparticles by dynamic light scattering (DLS)
For dynamic light scattering experiments micelle solutions were prepared with concentrations as described. After transfer to a dustfree flowbox, all samples were filtered (MillexHV 0.20 µm) into dust free cylindrical scattering cells (Suprasil, 20 mm diameter, Hellma, Mühlheim, Germany). Then, dynamic light scattering (DLS) measurements were performed using a Uniphase He/Ne Laser (632.8 nm, 22 mW), a ALV-SP125 Goniometer, a ALV/High QE APD-Avalanche photo-diode with fibre optical detection, an ALV5000/E/PCI-correlator and a Lauda RC-6 thermostat unit at 20°C. Angular dependent measurements of typically 15° steps were carried out in the range 30°-150°. For data evaluation experimental intensity correlation functions were transformed into amplitude correlation functions applying the Siegert relation extended to include negative values after baseline subtraction by calculation g1(t)=SIGN(G2(t))*SQRT(ABS((G2(t) - A)/A). All field correlation functions usually showed monomodal decay and were fitted by a sum of two exponentials g1(t)=a * exp(-t/b)+c * exp(-t/ d) to take polydispersity into account. Average apparent diffusion coefficients Dapp were calculated by applying q2 Dapp=(a * b-1 + c * d-1)/(a+c) resulting in an angular-dependent diffusion coefficient Dapp or reciprocal hydrodynamic radius <1/Rh, app >, according to formal application of Stokes-Einstein law. By extrapolation of <1/Rh, app > to q=0 z-average hydrodynamic radii Rh = <1/Rh >z-1 were obtained (uncorrected for c-dependency). The µ2 values, which result from the cumulate analysis at an angle of 90° allow an estimation of the polydispersity of nanoparticles [49]. Samples with µ2 values of 0.05 can be roughly considered as monodisperse, values greater than 0.2 can be estimated as polydisperse [50].
In vitro stability
Stability of purified micelles was tested in fresh human serum and isotonic NaCl solution, both at a 1:4 v/v dilution. The samples were incubated at 37°C and at various time points, aliquots were taken and analysed by TLC using citrate buffer 0.1 M pH 4.0 as a mobile phase.
In vivo PET/MR measurements
68G[Ga]-oxine was injected i.v. in anaesthetized mice (C57BL6/J, male, 6 weeks, Janvier) via the retro-orbital venous plexus (ID: 6.3±3.5 MBq). After an uptake period of 30 min mice were measured by FDG-PET/MRI (Mediso, Budapest, Hungary). Mice were anesthetized with 2% isoflurane and MRI measurements (Material Map for coregistration of the PET scan; 3D Gradient Echo External Averaging (GRE-EXT), Multi Field of View (FOV); slice thickness: 0.6 mm; TE: 2 ms; TR: 15 ms; flip angle: 25 deg) were performed followed by a 30 min static PET scan. PET data were reconstructed with Teratomo 3D (4 iterations, 6 subsets, voxel size 0.4 mm), coregistered to the MR and analyzed by Pmod software (version 3.6). Ex vivo biodistribution was measured as described below for 111In-oxine 1 hour pi.
Ex vivo evaluation of biodistribution
111In[In]-oxine micelles (1.5 mg in 1 mL of isotonic saline) and 111In[In]-oxine (0.15 mg/mL) to serve as a control were injected i.v. in anaesthetized healthy mice (C57BL6/J, male, 6 weeks, from Janvier) via the retro-orbital venous plexus, with a mean activity of 1.6±0.3 MBq for micelles and of 1.6±0.4 MBq for the 111In[In]-oxine. After 1, 4 and 24 hours, the animals were sacrificed and different organs and blood were removed. The tissue samples were weighed and the 111In activity in the organs was directly measured in a γ-counter (2470 WIZARD2 Automatic Gamma Counter, PerkinElmer). All animal experiments had previously been approved by the regional animal ethics committee and were conducted in accordance with the German Law for Animal Protection and the UKCCCR Guidelines [51].
Ex vivo metabolism evaluation
Blood samples (ca. 100 µl), collected during the biodistribution were mixed with heparin solution (150 µl) to avoid coagulation. Blood samples were weighed and activity was measured in a γ-counter. 500 µl of PBS were added and the blood was centrifuged at 4000 rpm for 5 min in order to separate blood cells and plasma. The plasma fractions were weighed and activity was measured in a γ-counter. Proteins and micelles were precipitated by adding 200 µl of acetonitrile to the plasma fraction. Thereby the micellar structures remain in solution. To separate proteins from plasma water and free polymer the samples were centrifuged at 4000 rpm for 5 min. The supernatants were weighed followed by determination of the activity in a γ-counter. The percentage of radioactivity in the blood cells, protein and plasma water plus micelles fractions was calculated by subtracting the activities of the supernatants from the activity of the whole blood sample.
Results
Synthesis of oxine complexes
The Ga-oxine cold complex was obtained as a yellow powder after purification by precipitation in diethyl ether. IR signal of -OH group of oxine was not detectable for the gallium complex which is an indicator of successful formation of the complex.
68Ga[Ga]-oxine complex was quantitatively (>96%) formed within 15 minutes at room temperature. Complex formation yields were analysed by iTLC using a mixture of CHCl3/MeOH (95:5) as eluent (Rf (68Ga)=0.0, Rf (68Ga-oxine)=0.9) [26]. The specific activity of 68Ga was of 155 MBq/µmol oxine. Same protocol was applied to 111In[In]-oxine.
Synthesis of polymers
Poly[N-(2-hydroxypropyl)methacrylamide] (pHPMA) is a hydrophilic polymer under investigation for its use in polymer-drug conjugates since a long time [14]. Being hydrophilic, it can serve as a micellar stealth corona, while it can also be modified with hydrophobic moieties such as laurylmethacrylate (LMA), to serve as a hydrophobic micellar core in which hydrophobic drugs can be solubilized and retained [2,4,12,13,15,21,44,48,49]. Here we combined HPMA and LMA moieties both as statistical and block copolymers to vary the micellar structure and to modify thereby the inclusion efficiency for hydrophobic drugs. Additionally, the block copolymer was modified with longer PEG chains to increase shielding efficacy and stabilization further [52] (Figure 1).
Figure 1.

Synthesis of HPMA based polymer in grey PEG moiety, in blue hydrophilic HPMA unit, in red hydrophobic LMA unit.
Random copolymers contain many independent hydrophobic groups spread throughout the hydrophilic polymer chain. As result the structure of the micelles in aqueous solution are not well defined [13,16] as hydrophobic and hydrophilic groups are always mixed to some extend (e.g. some hydrophobic groups will always be present in the corona increasing the interaction with hydrophobic regions in proteins or cellular membranes, Figure 2A). However, the statistical copolymers can solubilize hydrophobic compounds well.
Figure 2.
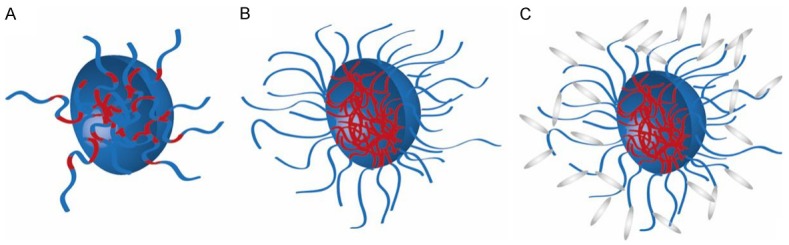
Schematic structures of polymeric micelles formed by (A) Random polymer (B) Block copolymer (C) PEGylated block copolymer.
In block copolymers both blocks can be well separated into hydrophilic and hydrophobic regions. Thereby the hydrophilic exterior provides steric stabilization/shielding against plasma proteins and cellular membranes. The hydrophobic core enables the facile entrapment of lipophilic drugs [13,19,49] (Figure 2B). Increasing the size and the hydrophilicity of the HPMA corona by incorporation of highly hydrophilic PEG chains is a concept to increase stabilization of the micellar structure. In addition, PEG is known for its shielding efficacy towards opsonic proteins [52] (Figure 2C).
The amphiphilic p(HPMA-co-LMA) copolymers were synthesized by controlled radical polymerization with lauryl methacrylate (LMA) monomers having 20-23 mol % of hydrophobic lauryl side chains. Their molecular weight and polydispersity are shown in Table 1.
Table 1.
Chemical analytics of the three HPMA polymers used
| Polymer | Mn PFPMA-LMA Precursora | Đa | % LMAa | Mn HPMA-LMA Polymerb |
|---|---|---|---|---|
| Random | 68.400 g/mol | 1.2 | 20 | 44.400 g/mol |
| Block | 26.800 g/mol | 1.3 | 23 | 17.800 g/mol |
| Block-PEG | 26.800 g/mol | 1.3 | 23 | 63.600 g/mol |
Mn (number average of the molecular weight), Đ (dispersity, ratio of Mw and Mn) and the molar ratio between PFPMA and LMA were determined by SEC in THF using PS standards (size exclusion chromatography);
Mn of the final polymers was measured by SEC in HFIP using PMMA standards.
Micelle formation
For all polymer systems, micelles were formed by slowly dropping the polymer solution in DMSO into the aqueous media under constant stirring. This leads for the block-PEG polymeric micelles to a radius of 18 nm as determined by dynamic light scattering (see Figure 3). This value corresponds to results described previously [12] for micelles from similar polymers. The structure of such micelles can be imaged by cryo-TEM as depicted in Figure 4.
Figure 3.

Dynamic light scattering of Block PEG micelles. (A) Micelles with Ga-oxine as cargo (B) Micelles Ga-oxine as cargo.
Figure 4.

Cryo TEM picture of micelles prepared from p(HPMA)-b-p(LMA) polymers.
A loading of these polymeric micelles with hydrophobic compounds is generally possible. Thereby the size of the micellar aggregates increases [13,17,21]. To load them with a hydrophobic Ga- or In-oxine we decided to dilute the “hot” compound, which is used only in trace amounts with “cold” oxine to obtain a more stable hydrophobic micellar core. For this purpose, the hydrophobic “cold” oxines were dissolved together with the polymer in DMSO and dropped into the “hot” aqueous 68Ga[Ga]- or 111In[In]-oxine solution to prepare micelles with hydrophobic oxines as core. Thereby it turned out that a weight ratio of 4 to 3, polymer to Ga- or In-oxine is optimal for incorporation of the oxines into the micelles. For 68Ga[Ga]-oxine, e.g. incorporation rates of about 79±4% for the random copolymer, 92±2% for the block copolymer and 97±1% for the PEGylated block copolymer could be obtained.
The stability of the micelles thus formed could be verified by radioactive thin layer chromatography (TLC) and by HPLC (see Figure 5). TLC shows that no oxines leek out of the micelles (Figure 5C) and HPLC shows (Figure 5A, 5B) that the radioactivity is completely localized in the micelles, with no larger aggregates and no low molar mass compounds available.
Figure 5.
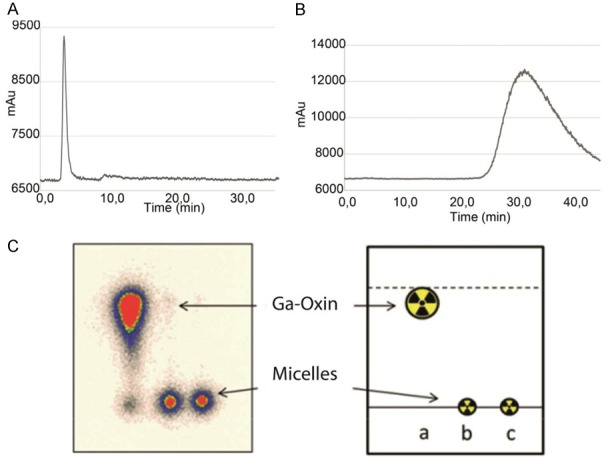
Radio HPLC chromatograms (A) of labeled purified micelles (B) 68Ga-oxine (C) radio TLC.
Purification through size exclusion columns was performed for the three type of polymeric micelles affording >97% of purified labeled micelles in all cases. Thereby 0.9% NaCl solution was used as eluent. Results were analysed by TLC using citrate buffer 0.1 M as mobile phase (micelles Rf=0.0 68Ga/111In-oxine Rf=0.9). Radio HPLC was performed with a 5 mL size exclusion column using Milli-Q water as a mobile phase at a flow rate of 0.2 ml/min. Retention time of labeled micelles was 3.8 min with 99% purity. A broad tailored signal with peak maximum at 33 min was observed for 68Ga[Ga]-oxine complex. Both chromatograms are shown in Figure 5A-C.
By loading the micelles their hydrodynamic radius (Rh) increased as measured by dynamic light scattering (Figure 3). E.g. for the Block-PEG polymer (this polymer was also used for in vivo measurements) it increased from 18 nm for the unloaded system to 24 nm for the system loaded with In-oxine.
In vitro stability
After administration, the polymer micelles are exposed first to blood, which may influence and alter their physicochemical properties due to interactions with blood proteins or other serum components [13,18]. While the loaded micelles are stable in isotonic NaCl (see TLC and HPLC), the addition of blood serum adds hydrophobic compartments, into which the hydrophobic oxines can diffuse. Therefore, the release of radioactivity was measured in vitro as an indicator for the stability of the 68Ga[Ga]-oxine or 111In[In]-oxine-labeled micelles (Figure 6). Both oxines gave rather comparable results, with the 111In-oxine allowing a characterization over a wider time range. Generally the random polymeric micelles showed to be the least stable, while the PEGylated-block micelles had the highest stability.
Figure 6.

Stability of (A) 111In[In]-oxine (n=3) and (B) 68Ga[Ga]-oxine (n=1) radiolabeled micelles in fresh human serum (n=3).
In detail, in human serum media, only 56±3% of random micelles were still intact after 30 minutes of incubation, while block and PEGylated-block micelles showed 63±2% and 91±3% respectively at the same time point (Figure 6). After 1 hour incubation, block micelles had released 48±1% of the radioactive 68Ga complex, while for the PEGylated-block micelles only 14±4% of the activity was released from the micelle core (Figure 6). Additional stability analysis after 1 day incubation were performed in the PEGylated micelles labeled with 111In[In]-oxine with >80% micelles still intact.
Biodistribution
The favorable loading capacity and stability presented by the PEGylated block-copolymer micelles claimed for in vivo evaluation to gain knowledge of their biodistribution and whether the stability presented in vitro would persist. Due to the long half-life of 111In, 111In-labeled oxine as well as 111In[In]-oxine labeled micelles were employed to prepare a pharmacological profile over a long period up to 24 h.
The ex vivo biodistribution of 111In[In]-oxine labeled micelles did not show specific uptake in any organ and values remained quite constant for each organ during the three time points analyzed (Figure 7). High uptake was observed in the lungs (31.2±2.9% ID/g), spleen (17.8±2.3% ID/g) and kidneys (21.7±1.6% ID/g) after 1 h p.i., and it barely changed after 4 and 24 h p.i.. High retention was also observed in blood after 1 h (25.9±2.8% ID/g) and 4 h (26.4±0.7% ID/g) p.i., which decreased to 18.3% ID/g±1.3 after 24 h p.i.. Uptake in liver and heart did not show remarkable variations in the three time points, with a mean value of 9.3±1.2% ID/g and 8.9±0.7% ID/g, respectively. Slight bone accumulation is also observed.
Figure 7.
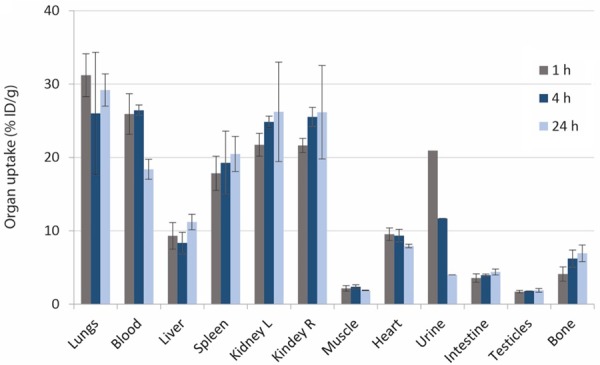
Ex vivo organ biodistribution of 111In[In]-oxine radiolabeled micelles from the PEGylated block copolymers in healthy mice (n=3) after 1 h, 4 h and 24 h p.i.
Control experiments were performed with the 111In[In]-oxine complex (Figure 8). High retention in the lungs was detected at all time points (>40% ID/g). High uptake of ca. 28% was observed in the blood after 1 and 4 h p.i., which decreased to ca. 18% after 24 h p.i.. Uptake of ca. 20% ID/g in the kidneys remained constant during the three time points analyzed. Slight increase of activity over time was observed in spleen, liver and heart, with maximum values of 14.8±1.9% ID/g, 6.1±0.4% ID/g and 12.0±0.4% ID/g respectively, after 24 h p.i..
Figure 8.
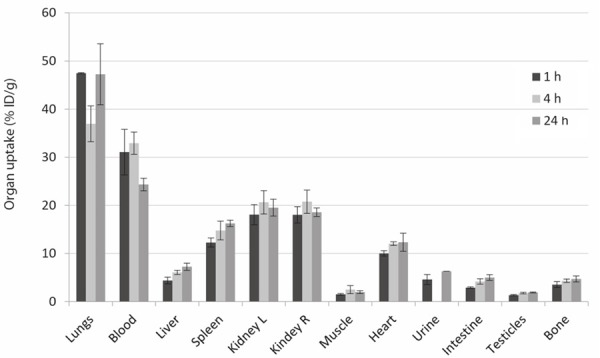
Ex vivo organ biodistribution of the 111In[In]-oxine complex (no micelles) in healthy mice (n=3) after 1 h, 4 h and 24 h p.i.
A similar pharmacological pattern of 111In[In]-oxine-labeled micelles and 111In[In]-oxine itself is observed in most organs: a small uptake increase over time in spleen and kidneys, a high retention in lungs and a decrease in blood uptake after 24 h p.i..
Since we could not find major differences in the biodistribution of micelles and oxine, only the latter one, i.e. 68Ga[Ga]-oxine, was additionally analyzed by in vivo PET in order to visualize its biodistribution. The PET/MR data revealed comparable uptake of 68Ga-oxine in liver, heart, lung and kidneys (Figure 9A). The ex vivo biodistribution data 1 hour p.i. confirmed the PET data with highest values for liver, heart, lung and kidneys (Table 2). They where comparable to the biodistribution data of 111In-labeled oxine at the same time point. Only the liver showed a distinct higher value in the 68Ga[Ga]-oxine group compared to 111In-labeled oxine.
Figure 9.
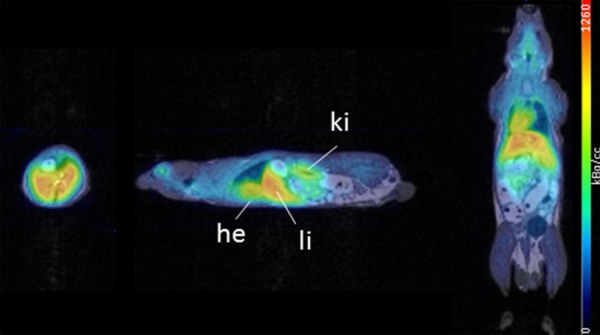
In vivo organ biodistribution of the 68Ga[Ga]-oxine complex (no micelles) in healthy mice 1 h p.i. PET/MR axial (left), sagittal (middle) and coronal (right) slices.
Table 2.
Ex vivo organ distribution (n=2) of the 68Ga[Ga]-oxine complex in healthy mice 1
| Organ | 68Ga[Ga]-oxine | SD |
|---|---|---|
| Lung | 19.20 | 1.73 |
| Blood | 9.55 | 0.63 |
| Liver | 17.48 | 2.07 |
| Spleen | 5.54 | 1.02 |
| Kidney l. | 21.18 | 0.40 |
| Kidney r. | 20.23 | 1.47 |
| Muscle | 1.64 | 0.05 |
| Heart | 19.44 | 5.16 |
| Urine | 4.79 | 0.18 |
| Intestine | 6.09 | 0.13 |
| Testicle | 0.93 | 0.02 |
| Bone | 3.38 | 0.71 |
Ex vivo metabolism
In order to investigate the in vivo stability of the labeled micelles and 111In[In]-oxine, the blood samples obtained during organ biodistribution were separated into different fractions: a) blood cells, b) proteins and c) plasma water plus polymer. Figure 10A and 10B show the amount of radioactivity in the different fractions with reference to the total amount of radioactivity in the blood samples of 111In[In]-oxine and 111In[In]-oxine-labeled micelles. 111In[In]-oxine remains constantly concentrated in the blood cell fraction at all time points with values >90%. 67.4±2.4% of the radiolabeled micelles are found after 1 h p.i. in the blood cells fraction and only 30.1±1.9% in the fraction containing proteins and micelles, which decreased to 21.1±1.4% and 15.1±1.5% after 4 and 24 h p.i. respectively, while it increased in the blood cells fraction up to 77.4±1.4% and 82.4±1.5% after 4 h and 24 h. The concentration in plasma water remained constant at low values below 2.5% during the complete observation period of 24 h.
Figure 10.
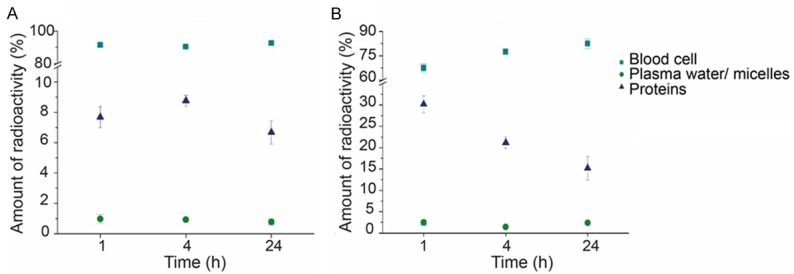
A. Distribution of 111In[In]-oxine in the blood for up to 24 h (n=3). B. Distribution of 111In[In]-oxine micelles in the blood for up to 24 h (n=3). Data is expressed as percentage of the amount of radioactivity that was present in the whole blood sample (mean ± SEM).
Discussion
The different structures of the polymers chosen were of interest as previous reports have demonstrated the capacity of those HPMA micellar structures as drug delivery systems [13,21]. One of the key findings of the present study is, that HPMA-based micelles can efficiently be labeled with hydrophobic 111In[In]-oxine and 68Ga[Ga]-oxine. Interestingly, the encapsulation of 68Ga-oxine into the micellar core showed different values depending on the structure of the polymer used. The lowest capability to incorporate the hydrophobic species into the micellar core was observed for the random copolymer structure. Its nature leads to conformations where hydrophobic LMA can be close to the surface of the core-shell of the micelles leading to a micellar aggregation formation, which promote dynamic changes of the micellar structure, and consequently the release of the incorporated drug [16]. This phenomenon is also observed in the lower stability of the radioactive micelles in human serum. It can be assumed that the hydrophobic moieties closer to the surface facilitate hydrophobic interactions with serum proteins and therefore 68Ga[Ga]- or 111In[In]-oxine is released from the micellar core more easily. These observations correspond to the fact that the statistical copolymers possess -in general- an efficient uptake, but also release for hydrophobic drugs [13,21,44].
HPMA-LMA block copolymer micelles, having a better-defined structure, facilitate a more stable incorporation of the hydrophobic complex into the core. Moreover, as the presence of hydrophobic LMA moieties in the micellar surface is minor when compared to random structures, the interactions between LMA and proteins in human serum decrease. This fact is translated in a higher in vitro stability compared to the micellar structures from statistical copolymers.
In order to increase stability further, the thickness of the hydrophilic corona had to be increased to keep the hydrophobic cargo further away from hydrophobic structures in plasma proteins. For this purpose, PEG moieties were incorporated into the hydrophilic corona of the block copolymer. This increased corona, including the PEG chains forms a protective layer that will reduce the interactions between micelles and serum components. This is due to the fact that PEG is only an acceptor for H-bonding whereas poly(HPMA) is both a donor and an acceptor [2]. Such micelles revealed higher loading capacity of 68Ga[Ga]-oxine, with quantitative incorporation and improved stability over time. In addition, 111In[In]-oxine could be stored for 24 hours in plasma with minimal loss.
After successful results obtained concerning the loading capacity and stability of the PEGylated micelles, biodistribution studies were performed in healthy mice to evaluate their in vivo behaviour. Due to the short half-life of 68Ga it was only used for PET-measurements and ex vivo biodistribution of oxine after 1 hour p.i.. 111In was employed instead, to allow evaluation over a longer time frame, up to 24 h by ex vivo biodistribution. No major differences in biodistribution were obvious comparing oxine-labeled micelles with the pure oxine assuming that the micelles are not stable in vivo. Furthermore, we could also find no big differences between the biodistribution of 68Ga[Ga]-oxine and 111In[In]-oxine showing that a change in the radiometal does not alter the biodistribution of this hydrophobic complex.
The analysis of the three different blood fractions, taken at time points from 1 to 24 h revealed, however, a low in vivo stability of the micelles. 70% of the total activity of the blood sample was found after 1 hour p.i. in the blood cell fraction, and it increased after 4 and 24 hours to ca. 79% and 85% respectively. These results indicate release of 111In-oxine from the micellar core.
The pure 111In[In]-oxine, given as control, accumulated to an even higher extend in the blood cell fraction (>95%) from 1 h to 24 hours. This fact could be explained as a consequence of the high hydrophobicity of the complex, which accumulate in the hydrophobic membrane structure of the blood cells [54]. 111In[In]-oxine is known to diffuse into red and white blood cells serving as an imaging agent of e.g. infection or inflammation in vivo [42,43]. Values below 2.5% in the water plasma indicate that the 111In[In]-oxine complex remained stable; there was almost no release of free 111In. So the hydrophobic oxine is probably lost from the micellar core in a similar way as recently reported for hydrophobic dyes in polymer coated colloids [8]. In this case the hydrophobic compounds move from the hydrophobic core to the hydrophobic lipid membrane on close contact. For this process no fusion or uptake is necessary [8]. The difference between the in vitro (only plasma proteins) and the in vivo situation may then be just the large amount of cellular membranes available during the in vivo experiments (both cellular membranes of blood cells and of endothelial cells), which act as perfect “sink” for the hydrophobic 111In-oxine.
These results are in concordance with the biodistribution analysis since we did not find major differences between the biodistribution of 111In[In]-oxine-micelles and 111In-oxine. Assuming that the micelles are not stable, the organ uptake values do not provide information on the micelles distribution, as around 70% of the activity measured belongs to released 111In[In]-oxine. Obviously, 111In[In]-oxine is increasingly removed from the micelles post injection. This gives also a new interpretation for the strong accumulation of 111In[In]-oxine in the lungs. As the thin blood capilllaries in the lungs are reached first after intraveneous injection, the hydrophobic 111In[In]-oxine might transfer to membranes of endothelial cells. Overall the polymeric micelles under investigation may thus be suitable for a solubilization of hydrophobic compounds, but they are not suitable for a controlled delivery since most of the hydrophobic cargo is left directly after injection.
68Ga[Ga]-oxine, additionally analyzed by in vivo PET in order to visualize its biodistribution, revealed uptake in liver, heart, lung and kidneys (Figure 9). The ex vivo biodistribution data 1 hour p.i. confirmed the PET data with highest values for liver, heart, lung and kidneys (Table 2), all comparable to the biodistribution data of 111In[In]-labeled oxine at the same time point.
Conclusions
Three different structures of amphiphilic HPMA-LMA based copolymers that self-assemble into micelles were studied to apply a novel radiolabeling technique. Spontaneous incorporation of hydrophobic 68Ga[Ga]-oxine or 111In[In]-oxine complexes in the micellar core was quantitative after one minute. The strategy does not require a previous modification of the polymer in order to be labeled with a radiometal and therefore the biological characteristics of the polymer will be preserved. In this context, the use of radiolabeled micelles allows for a systematic correlation between the chemical features of the polymers and the micelles formed thereof versus the stability and pharmacology of the final products in vitro and in particular in vivo. The incorporation efficiency as well as in vitro stability in human serum and saline clearly depends on the polymer morphology. The optimum results were achieved for the block-copolymers with a well-defined structure, in particular for the polymer that contains PEG moieties in the corona. Further ex vivo evaluation in healthy mice did not correlate with the in vitro stability values.
Further investigations are needed in order to ensure in vivo stability of each particular carrier system. For example, in a study using liposomal carriers, 18F-labeled fluoro-2-desoxy-D-glucose was used as a surrogate of a drug enclosed liposomal structures of different composition [55]. Subsequent to the i.p. injection of the 18F[F]-FDG-loaded liposomes, its release was quantified following the time-dependent 18F[F]-FDG accumulation in the brain of mice by in vivo small animal PET imaging.
Acknowledgements
The authors would like to thank Christian Herrmann for his support during the animal studies.
Disclosure of conflict of interest
None.
References
- 1.Simon WH, Joseph WS. Clinical imaging with indium 111 oxine-labeled leukocyte scan: review and case report. Clin Podiatr Med Surg. 1988;5:329–40. [PubMed] [Google Scholar]
- 2.Talelli M, Barz M, Rijcken CJ, Kiessling F, Hennink WE, Lammers T. Core-crosslinked polymeric micelles: principles, preparation, biomedical applications and clinical translation. Nano Today. 2015;1:93–117. doi: 10.1016/j.nantod.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cabral H, Kataoka K. Progress of drug-loaded polymeric micelles into clinical studies. J Control Release. 2014;190:465–76. doi: 10.1016/j.jconrel.2014.06.042. [DOI] [PubMed] [Google Scholar]
- 4.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–92. [PubMed] [Google Scholar]
- 5.Talelli M, Rijcken CJ, Hennink WE, Lammers T. Polymeric micelles for cancer therapy: 3 C’ s to enhance efficacy. Curr Opin Solid State Mater Sci. 2012;16:302–9. [Google Scholar]
- 6.Rijcken CJF, Talelli M, Van Nostrum CF, Storm G, Hennink WE. Crosslinked micelles with transiently linked drugs - a versatile drug delivery system. Ther Nanomedicine. 2010;3:19–24. [Google Scholar]
- 7.van Nostrum CF. Covalently cross-linked amphiphilic block copolymer micelles. Soft Matter. 2011;7:3246–59. [Google Scholar]
- 8.Tomcin S, Kelsch A, Staff RH, Landfester K, Zentel R, Mailänder V. Acta biomaterialia HPMA-based block copolymers promote differential drug delivery kinetics for hydrophobic and amphiphilic molecules. Acta Biomater. 2016;35:12–22. doi: 10.1016/j.actbio.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil - based paclitaxel in women with breast cancer. J. Clin. Oncol. 2005;23:7794–803. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- 10.Sparreboom A, Scripture CD, Trieu V, Williams PJ, De T, Yang A, Beals B, Figg WD, Hawkins M, Desai N. Cancer therapy: clinical comparative preclinical and clinical pharmacokinetics of a ( ABI-007) and paclitaxel formulated in cremophor (Taxol) Clin Cancer Res. 2005;11:4136–44. doi: 10.1158/1078-0432.CCR-04-2291. [DOI] [PubMed] [Google Scholar]
- 11.Gardner ER, Dahut WL, Scripture CD, Jones J, Aragon-ching JB, Desai N, Hawkins MJ, Sparreboom A, Figg WD. Cancer therapy: clinical randomized crossover pharmacokinetic study of solvent-based paclitaxel and nab-paclitaxel. Clin Cancer Res. 2008;14:4200–6. doi: 10.1158/1078-0432.CCR-07-4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kramer S, Kim KO, Zentel R. Size tunable core crosslinked micelles from HPMA-based amphiphilic block copolymers. Macromol Chem Phys. 2017;218:1700113. [Google Scholar]
- 13.Nuhn L, Barz M, Zentel R. New perspectives of HPMA-based copolymers derived by post-polymerization modification. Macromol Biosci. 2014;14:607–18. doi: 10.1002/mabi.201400028. [DOI] [PubMed] [Google Scholar]
- 14.Duncan R. Development of HPMA copolymer-anticancer conjugates: clinical experience and lessons learnt. Adv Drug Deliv Rev. 2009;61:1131–48. doi: 10.1016/j.addr.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 15.Weilbächer M, Allmeroth M, Hemmelmann M, Ritz S, Mailänder V, Bopp T, Barz M, Zentel R, Becker C. Interaction of N-(2-hydroxypropyl)methacrylamide based homo, random and block copolymers with primary immune cells. J Biomed Nanotechnol. 2013;9:1–11. doi: 10.1166/jbn.2014.1693. [DOI] [PubMed] [Google Scholar]
- 16.Hemmelmann M, Kurzbach D, Koynov K, Hinderberger D, Zentel R. Aggregation behavior of amphiphilic p(HPMA)-co-p(LMA) copolymers studied by FCS and EPR spectroscopy. Biomacromolecules. 2012;13:4065–74. doi: 10.1021/bm301364g. [DOI] [PubMed] [Google Scholar]
- 17.Mohr N, Kappel C, Kramer S, Bros M, Grabbe S, Zentel R. Targeting cells of the immune system: mannosylated HPMA-LMA block-copolymer micelles for targeting of dendritic cells. Nanomedicine (Lond) 2016;11:2679–2697. doi: 10.2217/nnm-2016-0167. [DOI] [PubMed] [Google Scholar]
- 18.Hemmelmann M, Mohr K, Fischer K, Zentel R, Schmidt M. Interaction of pHPMA-pLMA copolymers with human blood serum and its components. Mol Pharm. 2013;10:3769–75. doi: 10.1021/mp400254b. [DOI] [PubMed] [Google Scholar]
- 19.Barz M, Luxenhofer R, Zentel R, Kabanov AV. The uptake of hydroxypropyl methacrylamide based homo, random and block copolymers by human multi-drug resistant breast adenocarcinoma cells. Biomaterials. 2011;30:5682–90. doi: 10.1016/j.biomaterials.2009.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gong J, Chen M, Zheng Y, Wang S, Wang Y. Polymeric micelles drug delivery system in oncology. J Control Release. 2012;159:312–23. doi: 10.1016/j.jconrel.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 21.Hemmelmann M, Knoth C, Schmitt U, Allmeroth M, Moderegger D, Barz M, Koynov K, Hiemke C, Rösch F, Zentel R. HPMA based amphiphilic copolymers mediate central nervous effects of domperidone. Macromol Rapid Commun. 2011;32:712–7. doi: 10.1002/marc.201000810. [DOI] [PubMed] [Google Scholar]
- 22.Welch MJ, Thakur M, Coleman RE, Patel M, Siegel B, Ter-Pogossian M. Gallium-GB new labeled red cells and platelets agents for positron tomography. Radiochem Radiopharm. 1977;18:558–62. [PubMed] [Google Scholar]
- 23.Roca M, de Vries EF, Jamar F, Israel O, Signore A. Guidelines for the labeling of leucocytes with In-oxine. Eur J Nucl Med Mol Imaging. 2010;37:835–41. doi: 10.1007/s00259-010-1393-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson S, Rodnick ME, Stauff J, Arteaga J, Desmond TJ, Scott JH, Viglianti BL. Automated synthesis of [68Ga] oxine, improved preparation of 68Ga-labeled erythrocytes for blood-pool imaging, and preclinical evaluation in rodents. Medchemcomm. 2018;9:454–9. doi: 10.1039/c7md00607a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muhamad F, Mitry NR, Lewington VJ, Blower PJ, Terry SYA. Re-assessing gallium-67 as a therapeutic radionuclide. Nucl Med Biol. 2017;46:12–8. doi: 10.1016/j.nucmedbio.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yano Y, Budinger TF, Ebbe SN, Mathis CA, Singh M, Brennan KM, Moyer BR. Gallium-68 lipophilic complexes for labeling platelets. J Nucl Med. 1985;26:1429–37. [PubMed] [Google Scholar]
- 27.Sinzinger HB. Radioactive isotopes in clinical medicine and research. 1995:427–31. [Google Scholar]
- 28.Gogna R, Madan E, Keppler B, Pati U. Gallium compound GaQ(3) -induced Ca(2+) signalling triggers p53-dependent and -independent apoptosis in cancer cells. Br J Pharmacol. 2012;166:617–36. doi: 10.1111/j.1476-5381.2011.01780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bernstein LR, Tanner T, Godfrey C, Noll B, Road W, Park M. Chemistry and pharmacokinetics of gallium maltolate, a compound with high oral gallium bioavailability. Met Based Drugs. 2000;7:33–47. doi: 10.1155/MBD.2000.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Timerbaev AR. Advances in developing tris(8-quinolinolato)gallium (III) as an anticancer drug: critical appraisal and prospects of. Metallomics. 2009;1:193–8. doi: 10.1039/b902861g. [DOI] [PubMed] [Google Scholar]
- 31.Jungwirth U, Gojo J, Tuder T, Walko G, Holcmann M, Sch T, Nowikovsky K, Wil N, Schoonhoven S, Kowol CR, Lemmens-gruber R, Heffeter P, Keppler BK, Berger W. Calpain-mediated integrin deregulation as a novel mode of action for the anticancer gallium compound KP46. Mol Cancer Ther. 2014;13:2436–50. doi: 10.1158/1535-7163.MCT-14-0087. [DOI] [PubMed] [Google Scholar]
- 32.Kubista B, Schoefl T, Mayr L, Van Schoonhoven S, Heffeter P, Windhager R, Keppler BK, Berger W. Distinct activity of the bone-targeted gallium compound KP46 against osteosarcoma cells - synergism with autophagy inhibition. J Exp Clin Cancer Res. 2017;36:1–13. doi: 10.1186/s13046-017-0527-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willmann JK, van Bruggen N, Dinkelborg LM, Gambhir SS. Molecular imaging in drug development. Nat Rev Drug Discov. 2008;7:591–607. doi: 10.1038/nrd2290. [DOI] [PubMed] [Google Scholar]
- 34.van Dongen GA, Visser GW, Lub-de Hooge MN, de Vries EG, Perk LR. Immuno-PET: aNavigator in monoclonala ntibody development and applications. Oncologist. 2007;12:1379–89. doi: 10.1634/theoncologist.12-12-1379. [DOI] [PubMed] [Google Scholar]
- 35.Moderegger D. Radiolabeling of defined polymer architectures with fluorine-18 and iodine-131 for ex vivo and in vivo evaluation: visualization of structure property relationships. Johannes Gutenberg-University, Mainz. 2012 [Google Scholar]
- 36.Herth MM, Barz M, Moderegger D, Allmeroth M, Jahn M, Thews O, Zentel R, Rösch F. Radioactive labeling of defined HPMA-based polymeric structures using [18F]FETos for in vivo imaging by positron emission tomography. Biomacromolecules. 2009;10:1697–703. doi: 10.1021/bm8014736. [DOI] [PubMed] [Google Scholar]
- 37.Wagener K, Moderegger D, Allmeroth M, Reibel A, Kramer S, Biesalski B, Zentel R, Thews O, Rösch F. Long-term biodistribution study of HPMA-ran-LMA copolymers in vivo by means of 131I-labeling. Nucl Med Biol. 2018;58:59–66. doi: 10.1016/j.nucmedbio.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 38.Wadas TJ, Wong EH, Weisman GR, Anderson CJ. Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for PET and SPECT imaging of disease. Chem Rev. 2010;110:2858–902. doi: 10.1021/cr900325h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson CJ, Welch MJ. Radiometal-labeled agents (non-technetium) for diagnostic imaging. Chem Rev. 1999;99:2219–34. doi: 10.1021/cr980451q. [DOI] [PubMed] [Google Scholar]
- 40.Price EW, Orvig C. Matching chelators to radiometals for radiopharmaceuticals. Chem Soc Rev. 2014;43:260–290. doi: 10.1039/c3cs60304k. [DOI] [PubMed] [Google Scholar]
- 41.Yang DJ, Edmund EK, Inoue T. Targeted molecular imaging in oncology. Ann Nucl Med. 2006;20:1–11. doi: 10.1007/BF02985584. [DOI] [PubMed] [Google Scholar]
- 42.Heyns AD, Lotter MG, Kotze HF, Wessels P, Pieters H, Badenhorst PN. Kinetics, distribution, and sites of destruction of indium-111 oxine labeled red cells in haemolytic anaemia. J Clin Pathol. 1985;38:128–32. doi: 10.1136/jcp.38.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Becker W, Fischbach W, Jenett M, Reiners C, Börner W. 111In-oxine-labeled white blood cells in the diagnosis and follow-up of crohn’s disease. Klin Wochenschr. 1986;64:141–8. doi: 10.1007/BF01732640. [DOI] [PubMed] [Google Scholar]
- 44.Hemmelmann M, Metz VV, Koynov K, Blank K, Postina R, Zentel R. Amphiphilic HPMA-LMA copolymers increase the transport of Rhodamine 123 across a BBB model without harming its barrier integrity. J Control Release. 2012;163:170–7. doi: 10.1016/j.jconrel.2012.08.034. [DOI] [PubMed] [Google Scholar]
- 45.Chong YK, Le TPT, Moad G, Rizzardo E, Thang SH. A more versatile route to block copolymers and other polymers of complex architecture by living radical polymerization: The RAFT Process. Macromolecules. 1999;32:2071–4. [Google Scholar]
- 46.Eberhardt M, Mruk R, Zentel R, Théato P. Synthesis of pentafluorophenyl(meth)acrylate polymers: new precursor polymers for the synthesis of multifunctional materials. Eur Polym J. 2005;41:1569–75. [Google Scholar]
- 47.Zhernosekov KP, Filosofov DV, Baum RP, Aschoff P, Bihl H, Razbash AA, Jahn M, Jennewein M, Frank R. Processing of generator-produced medical application for medical application. Nucl Med. 2007;48:1741–9. doi: 10.2967/jnumed.107.040378. [DOI] [PubMed] [Google Scholar]
- 48.Mueller D, Klette I, Baum RP, Gottschaldt M, Schultz MK, Breeman WA. Simplified NaCl based 68Ga concentration and labeling procedure for rapid synthesis of 68Ga radiopharmaceuticals in high radiochemical purity. Bioconjugate Chem. 2012;23:1712–1717. doi: 10.1021/bc300103t. [DOI] [PubMed] [Google Scholar]
- 49.Fischer K, Schmidt M. Pitfalls and novel applications of particle sizing by dynamic light scattering. Biomaterials. 2016;98:79–91. doi: 10.1016/j.biomaterials.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 50.Bargeron CB. Measurement of a continuous distribution of spherical particles by intensity correlation spectroscopy: analysis by cumulants. J Chem Phys. 1974;61:10–5. [Google Scholar]
- 51.Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, Double JA, Everitt J. Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102:1555–77. doi: 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Allmeroth M, Moderegger D, Gündel D, Buchholz H, Mohr N, Koynov K, Rösch F, Thews O, Zentel R. PEGylation of HPMA-based block copolymers enhances tumor accumulation in vivo: a quantitative study using radiolabeling and positron emission tomography. J Control Release. 2013;172:77–85. doi: 10.1016/j.jconrel.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 53.Sant VP, Kang N, Maysinger D, Leroux J. Block copolymer micelles: preparation, characterization and application in drug delivery. J Control Release. 2005;109:169–88. doi: 10.1016/j.jconrel.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 54.Ebbe S, Taylor S, Maurer H, Kullgren B, Ebbe S, Taylor S, Maurer H, Kullgren B. Uptake of indium-111-labeled platelets and indium-111 oxine by murine kidneys after total-body irradiation. Radiat Res. 1996;146:216–22. [PubMed] [Google Scholar]
- 55.Hühn E, Buchholz H, Shazly G, Maus S, Thews O, Bausbacher N, Rösch F, Schreckenberger M, Langguth P. Predicting the in vivo release from a liposomal formulation by IVIVC and non-invasive positron emission tomography imaging. Eur J Pharm Sci. 2010;41:71–7. doi: 10.1016/j.ejps.2010.05.020. [DOI] [PubMed] [Google Scholar]