

Abstract
Targeted molecular imaging with positron emission tomography (PET) constitutes a successful technique for detecting and diagnosing disease conditions promptly and accurately, and for effectively prognosticating outcomes and treating patients with a tailored and more individualized intervention. In order to expand the success of PET in nuclear medicine, it is important to assure access to radiotracers of desired quantities and qualities. In this context, the benefit of accessing PET radiotracers through a radionuclide generator (RNG) cannot be overstated, as generators offer the potential of enriching the PET radiotracer arsenal at the medical centers both with and without onsite cyclotrons. While RNG technology to avail PET tracers is in its infancy, their use is expected to revitalize current PET practices and seems poised to broaden the palette of PET in nuclear medicine in the foreseeable future. In this review, we discuss the principles of RNGs, assess major parent/daughter pairs of interest for PET, RNGs currently in use in clinical PET, and identify the potentially useful RNGs which have made substantial progress or are likely to be used in daily clinical practices in the near future. Availability of the parent radionuclides required for PET RNGs is an important criterion and hence their production will also be reviewed. This overview outlines a critical assessment of RNGs to avail PET tracers, the contemporary status of RNGs, and key challenges and apertures to the near future.
Keywords: Bifunctional chelator (BFC), coronary artery disease (CAD), parent/daughter radionuclide, positron emission tomography (PET), radionuclide generator (RNG), radiopharmaceuticals
Introduction
The role of PET to enable in vivo visualization of physiological processes on the molecular level in real time and quantify them by measuring regional concentration of the radiotracer for diagnosing disease, monitoring disease progression, and tracking therapeutic response, hardly needs to be reiterated [1-12]. This momentous molecular imaging paradigm, straddling the disciplines of molecular biology and medical imaging technology, has not only brought a perpetual shift in healthcare practice but also heralds a significant leap forward in treatment outcomes [13-15]. Growth in the field of PET has been phenomenal and will continue in the foreseeable future. The expanding role of PET in nuclear medicine (NM) procedures has led to an exponential growth of the research and development (R&D) efforts around the world. The driving force behind the success has been largely due to the rapid ascent of the positron emission tomography-computed tomography (PET/CT) system which offers anatomic (CT) as well as metabolic (PET) information, in addition to providing data for attenuation correction [16,17]. While the utility of PET/CT lives at the interface between many scientific disciplines, cost effective availability of PET tracers of required quality and quantity is a key determinant that underpins survival, strength, and success of the modality. Currently, 18F, 15O, 13N and 68Ga are the most commonly used PET tracers, among which 18F has dominated significantly and has been regarded as the workhorse of PET in clinics [3,6,12,18-22]. Reflecting on the last decade of development on PET, one can clearly see the impact of 18F which was blended into a number of formulations to improve current imaging practices and overall performance. Although the use of these tracers in clinical PET constitutes successful advancement in the field and heralds a new era of molecular imaging, the requirement of an onsite cyclotron to produce these tracers, due to their short half-life, has emerged as the major impediment that continue to thwart efforts for their widespread use in daily NM routines. In this context, the prospect of accessing PET tracers through an RNG seems to be a promising proposition as it enables PET examinations at remote hospitals and at the same time offers the prospect of enriching the PET radiotracer arsenal at the medical centers both with and without onsite cyclotrons. The tremendous prospects associated with the use of RNGs, along with the challenge of providing PET tracers of requisite quality for a variety of NM diagnostic procedures, have led to a considerable amount of fascinating research and innovative strategies, the flow of which shows no sign of diminishing. With to the aim of obtaining PET tracers in an acceptable chemical form amenable for the formulation radiopharmaceuticals through the use of an RNG, essentially every conceivable strategy has been exploited.
Rapid and burgeoning research interests in the use of RNGs in PET have been the driving force to provide a detailed review on this subject, in an attempt to stimulate interest in this exiting field. In order to improve the utility of RNGs in PET, it is of utmost importance to nurture emerging RNG technologies in an appropriate manner to facilitate their transition from laboratory research to the clinical settings. The aim of this article is thus to provide an overview of RNGs to avail PET radiotracers which are currently in use in clinical practice, or which have made substantial progress or are likely to be materialized in the foreseeable future. In the following sections, we provide a detailed coverage on the different types of RNGs available to obtain PET tracers, their contemporary status, and their aperture to the near future. Given the multi-disciplinary field, speculative options reported mainly of academic interest are not included and the authors apologize for possible oversights of important contributions. This review is intended to serve as a resource for the researchers and to offer an impetus for further development, as well as to become familiar with the expectations, capabilities, constraints, and gratification involved in the development of RNGs for today and tomorrow.
Radionuclide generator
A RNG is a self-contained system housing an equilibrium mixture of a parent/daughter radionuclide pair and designed to provide the daughter radionuclide formed by the decay of the parent radionuclide [23-29]. The parent/daughter nuclear relationships offer the possibility of separating the daughter radionuclide at repeated time intervals [30]. The inherent determinant for the success of RNGs resides with the selection of parent/daughter pairs and appropriate radiochemical separation strategies, which are based on a number of considerations [31]. Overviews of the principle, criteria for selection of parent/daughter pairs, and growth and equilibrium of the daughter radionuclide with parent radionuclide have been elaborately discussed in detail in recent reviews [26,30-32]. The striking diffusion and the exciting perspective of RNGs in NM are mainly attributed to the following causes:
RNGs ensure onsite availability of PET tracers on demand and without reliance on an onsite accelerator, thereby, representing a cost-effective proposition for the onsite formulation of radiopharmaceuticals.
Offer the prospect of availing PET tracers in no-carrier-added (NCA) form.
Provides the flexibility to perform multiple studies, due to ready availability of multiple doses of PET tracer on-demand.
Constitute the only on-site option of availing certain short-lived radionuclides (82Rb) which cannot be shipped as with commercial sources.
Several requirements need to be fulfilled for effective separation of the daughter radionuclide, and in general the process should be fast, reproducible and provide the daughter radionuclide of required radionuclidic, radiochemical and chemical purity with a high radiochemical yield [23-27,29-32]. A wide range of separation procedures, each with different characteristics, are currently being used or can potentially be used for RNG technology. Overviews of radiochemical separation processes with respect to RNGs are elaborated in recent reviews [26,27,31,32]. In-growth of the daughter radionuclide is continuous and depends on its half-life as well as the frequency of separation. For practical considerations, RNGs are eluted at periodic intervals, depending on the daughter activity requirements.
Table 1 presents examples of RNG systems capable of providing positron-emitting daughter nuclides relevant for quantitative PET. Generator produced, short half-life PET radionuclides (on the order of minutes) do not allow for radiochemical synthesis and are mainly used for characterizing the faster kinetics of smaller tracer molecules. The longer-lived PET radionuclides are better suited to studying the slower kinetics of labeled peptides, antibodies and cells.
Table 1.
Key examples of RNGs to provide positron emitting radionuclides with potential for PET imaging
| Generator | Half life | β+ branch (%) | E β+/MeV | Application | |
|---|---|---|---|---|---|
|
| |||||
| Parent | Daughter | ||||
| 68Ge/68Ga | 270.8 d | 1.14 h | 89.0 | 0.74 | Labelling Perfusion |
| 82Sr/82Rb | 25.6 d | 1.27 min | 95.0 | 1.41 | Perfusion |
| 44Ti/44Sc | 60.3 y | 3.927 h | 94.0 | 0.597 | Labelling |
| 62Zn/62Cu | 9.26 h | 9.74 min | 97.0 | 1.28 | Labelling, perfusion |
| 110Sn/110mIn | 4.1 h | 1.15 h | 62.0 | 0.623 | Labelling |
| 72Se/72As | 8.4 d | 1.083 d | 88.0 | 1.02 | Labelling |
| 140Nd/140Pr | 3.37 d | 3.39 min | 51.0 | 0.544 | Perfusion |
| 118Te/118Sb | 6.00 d | 3.6 min | 74.0 | 0.882 | Perfusion |
| 122Xe/122I | 20.1 h | 3.6 min | 77.0 | 1.09 | Perfusion |
| 128Ba/128Cs | 2.43 d | 3.62 min | 69.0 | 0.869 | Perfusion |
| 134Ce/134La | 3.16 d | 6.4 min | 63.0 | 0.756 | Perfusion |
| 52Fe/52mMn | 8.28 d | 21.1 min | 97.0 | 1.13 | Perfusion |
Potentially useful PET RNGs for biomedical applications
The interest in RNGs will vary according to the scenarios considered. The following section provides an overview of the potential RNGs that could be used to avail PET tracers for clinical and research needs.
68Ge/68Ga generator
In recent years, the 68Ge/68Ga generator has evoked excitement among radiopharmaceutical researchers and captured the imagination of NM physicians, thanks to recent progress in PET instrumentation, hybrid imaging modalities, and advances in molecular and cellular biology [33-40]. The parent 68Ge (t½ = 270 d) radionuclide decays by EC to 68Ga (t½ = 68 min) which subsequently decays to stable 68Zn via positron emission and electron capture (branching ratios: β+ = 89%, EC = 11%). The nuclear transitions are also accompanied by low intensity gamma emission [Eg = 1077 keV (3%)]. Decay characteristics of the 68Ge/68Ga generator are depicted in Figure 1.
Figure 1.
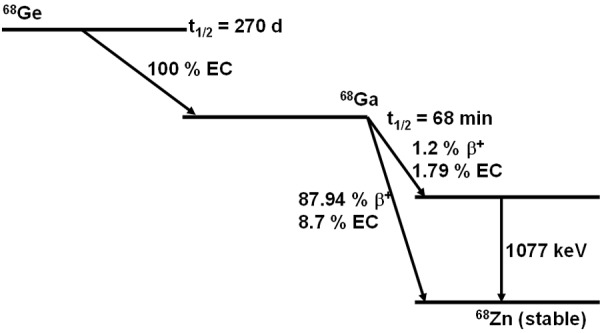
Decay characteristics of 68Ge/68Ga.
The use of 68Ge/68Ga generators in PET is very attractive for the following reasons:
The 270-day half-life of 68Ge ensures the ability to use the generator for extended periods, potentially up to 1 year or longer.
The decay characteristics of the short-lived daughter 68Ga (t½ = 68 min, 89% β+ and 11% EC, Eβ+, max= 1.92 MeV) are convenient for PET imaging.
With a physical half-life of 68 min, 68Ga also matches the biological half-life of many peptides used for imaging, due to their rapid diffusion, localization at the target and fast blood clearance.
The ability of 68Ga3+ to form stable complexes with many ligands containing oxygen and nitrogen as donor atoms offers the prospect for complexation with a wide variety of chelators, as well as some macromolecules having significant clinical potential.
Commercial 68Ge/68Ga generators have a longer history than many people would believe. Although 68Ge/68Ga generators have been investigated since 1960s, the chemical forms of generator derived 68Ga and the unacceptable level of 68Ge breakthrough emerged as the major obstacles that continued to thwart efforts for the development of gallium radiochemistry and the practical application. The first 68Ge/68Ga RNG based on liquid-liquid extraction technology, using acetyl-acetone in cyclohexane, was described in 1960 [41]. Due to the inherent of limitations of liquid-liquid extraction, and the requirement of three to four separation steps in order to obtain the requisite purity amenable for the preparation of 68Ga compounds for radiopharmaceuticals applications, subsequent developmental efforts were directed toward the use of column chromatography techniques. The first 68Ge/68Ga generator based on a column chromatography technique [42] consisted of an Al2O3 matrix from which 68Ga was eluted as a strong 68Ga-EDTA complex using a 5-mM EDTA solution at pH 7.0. While the use of alumina based 68Ge/68Ga generators is prolific in providing 68Ga-EDTA complex to measure the increased blood flow of brain tumors, it requires decomposition of the EDTA for the preparation of other 68Ga-radiopharmaceuticals because of the high stability of the complex (log K = 21.7). This extra step is not only cumbersome but also results in the loss of useful 68Ga radioactivity. In an attempt to mitigate this shortcoming, Kopecky et al. [43] eluted the column with 0.2 M HCl with a 48% elution yield. Malyshev et al. [44] used ZrO2, SnO2, and TiO2 matrices and eluted 68Ga with either HCl, HNO3, or CH3COOH as eluents. Of the three-column matrices used, ZrO2 was found to be the best, in which 68Ga was eluted with 0.1 M HCl with 35% elution yield. Subsequently Arino et al. [45] used a polyantimonic acid column matrix and eluted 68Ga with 2% sodium oxalate. The poor elution yield of 68Ga, the toxicity of the eluent, and the presence of oxalic acid emerged as the major impediments that prevented clinical use without further chemical manipulation. A generator based on the tin dioxide/HCl couple has also been tried by Loc’h et al. [46]. In all these metal oxide matrix-based systems, the release of column matrix due to limited solubility in the eluent constituted a major limitation which ruled out all possibility of clinical use. With the aim of circumventing this limitation, use of organic adsorbents and elution of 68Ga with dilute HCl or HF has also been tried, but met with limited success [47,48]. McElvany et al., in 1984 [49], evaluated three 68Ge/68Ga generators based on different metal oxide matrices over a period of 1 year and compared their performances with respect to 68Ga elution profiles and yields, parent 68Ge breakthrough levels, amounts of column matrix contaminants present in the generator eluate, and the ease of 68Ga-radiopharmaceutical production. The ionic 68Ga/68Ga generator utilizing a tin dioxide column eluted with 1 M HCl was found to be the most suitable for preparation of 68Ga-labeled compounds for use in conjunction with PET. In another embodiment, α-Fe2O3, activated carbon, and graphite were tried [50], and it was found that α-Fe2O3, having a 68Ga elution yield of 50-70% with a HCl solution of pH 2.0, was the most suitable. In spite of the great publicity received by 68Ga imaging, its impact in NM started to fade in the late 1970s due to the commercial non-availability of 68Ge/68Ga generators adaptable to the syntheses of a wide range of 68Ga radiopharmaceuticals, in comparison to the parallel and rapid developments of several new classes of radiopharmaceuticals based on 99mTc and l8F. While the utility of 68Ge/68Ga generators for clinical 68Ga imaging would be in hibernation for decades, the foundation needed to build to the next level has been well established and separation procedures developed for the isolation of 68Ga from 68Ge with HCl solutions proved to be fertile ground for the realization of a generator for direct application in a clinical context.
The first commercial 68Ge/68Ga generator based on a modified TiO2 solid phase support is the product of years of hard work by scientists of Cyclotron Ltd, Obninsk, Russian Federation [51]. Each generator contains 68Ge activities of up to 3.7 GBq from which “ionic” 68Ga3+ can be availed in 0.1 N HCl solution with ~80% initial 68Ga elution yields and with a 68Ge breakthrough of about 1 × 10-3%.
Commercial success of this generator, together with the advancement in imaging techniques, drew new players to enter the market. 68Ge/68Ga generator development during the last few decades is briefly summarized in Table 2. Currently, four different 68Ge/68Ga generators are commercially available and only a few of them (Eckert & Ziegler; iThemba Labs) hold license for good manufacturing practice (GMP) production. It is pertinent to mention that a draft monograph, “Gallium (68Ga) chloride solution for radiolabeling”, commensurate with the regulatory requirements for quality and patient safety, is available [52]. Characteristics of major commercial 68Ge/68Ga generators available today for use in NM centers are tabulated in Table 3 and shown in Figure 2.
Table 2.
Types of 68Ge/68Ga generators
| Years | Generator type | Remarks |
|---|---|---|
| 1960-1970 | liquid-liquid extraction | Tedious time consuming separation; not amenable for hospital radiopharmacy. |
| 1970-2000 | Column chromatography system based on Al2O3 and EDTA as eluent | This system served as a convenient and economical source of 68Ga-EDTA; clinical use was limited to measure increased blood flow of brain tumors, in particular. |
| After 2000 | Column chromatography system based on TiO2, ZrO2, CeO2, SnO2 or an organic resin | 68Ga is availed in an ionic form, with elution yields 70% to 80%; 68Ge breakthrough still in the range, 0.01-0.001%. |
Table 3.
Commercial 68Ge/68Ga generators
| Manufactures | Type | Year | Column matrix | Eluent | Elution yield | 68Ge breakthrough |
|---|---|---|---|---|---|---|
| Eckert & Ziegler AG, Berlin, Germany | Obninsk | 1996 | TiO2 | 0.1 M HCl | 60-75% | < 0.01% |
| IGG100 | 2008 | TiO2 | 0.1 M HCl | 70-75% | < 0.001% | |
| iThemba, South Africa | 2008 | SnO2 | 0.6 M HCl | 80% | < 0.002% | |
| ITG Garching, Germany | 2010 | Silica based organic matrix | 0.05 M HCl | > 80% | < 0.005% | |
Figure 2.

Commercially available 68Ge/68Ga generators according to column matrix, manufacturers and the year of introduction to the market.
Despite remarkable advancements, the low radioactive concentration, high [H+] burden, 68Ge breakthrough, and the presence of potential metal ions such as Al, Fe, Cu, Zn, Ti or Sn from generator eluate have emerged as the major deterrents in the path of direct labeling of 68Ga to make radiotracers from generator availed 68Ga. Although the concentrations of these metallic impurities are low (~ ppm level), their concentration can still be much higher than that of NCA 68Ga3+, impeding efforts for the preparation of 68Ga-labeled radiopharmaceuticals. Most of the commercially available 68Ge/68Ga generator systems demonstrate deteriorating performance in terms of increased 68Ge breakthrough and reduced 68Ga elution yields on repeated elutions over prolonged periods of time [53,54]. In order to circumvent such limitations, a variety of post-elution purification and/or concentration procedures based on (1) anion-exchange chromatography, (2) cation-exchange chromatography and (3) solvent extraction have been developed to obtain 68Ga of acceptable radionuclidic purity and radioactive concentration [53-59]. Among them, the chromatographic procedure reported by Zhernosekov et al. [54], based on selective trapping of 68Ga eluate on a cation-exchanger, followed by elution in a small volume of acetone-HCl mixture, was a major step and has reached the stage of commercial exploitation. The trace level of acetone present in the final reaction mixture could be averted by making use of the column-based radiochemical procedure reported by Loktionova et al. [57]. Even though there is no technical impediment to adapt this efficient and robust procedure, the requirement of an automated module is viewed as a necessary one not only to minimize the decay loss of 68Ga but also to reduce the radiation exposure. Nevertheless, the scope of using this technique is restricted to TiO2 and organic-resin based generators due to the explicit need of primary 68Ga eluate in HCl concentration ≤ 0.1 N for effective sorption in the cation-exchange column. In the case of SnO2 based generators, where 68Ga is eluted in 0.6-1 N HCl, the aforementioned post-elution processing can be effectively adapted after appropriate dilution. Commercial availability of 68Ge/68Ga generators yielding the ionic form of 68Ga, along with a post-elution processing system, has led to the exploration of a broad spectrum of 68Ga-labeled products for use as radiopharmaceuticals for PET imaging of cancer [60].
The availability of 68Ge/68Ga generators providing 68Ga3+ of requisite quality amenable for direct radiolabeling is a much more desirable option. In this regard, the potential of the BARC-developed 68Ge/68Ga generator, based on the nanoceria-polyacrylonitrile (CeO2-PAN) composite [61,62] sorbent, is capable of yielding 68Ga of requisite quality (free from metal ion impurities such as Al, Fe, Cu, Zn, Ti or Sn ions and very low 68Ge breakthrough) and without the need for any post-elution processing, and is poised to significantly expand the scope of 68Ga products formulation in radiopharmacy. Based on a similar strategy, a nano zirconia based 68Ge/68Ga generator has also been developed, which demonstrates comparable performance [63]. A schematic diagram of the ‘BARC’ 68Ge/68Ga generator is shown in Figure 3. Over the years, the ‘BARC’ 68Ge/68Ga generator has been effectively developed and refined for use in the preparation of 68Ga tracers, both for research and limited clinical use. A comprehensive quality assurance system is necessary to ensure that the quality of 68Ga availed from the generator meets the standards for clinical use.
Figure 3.
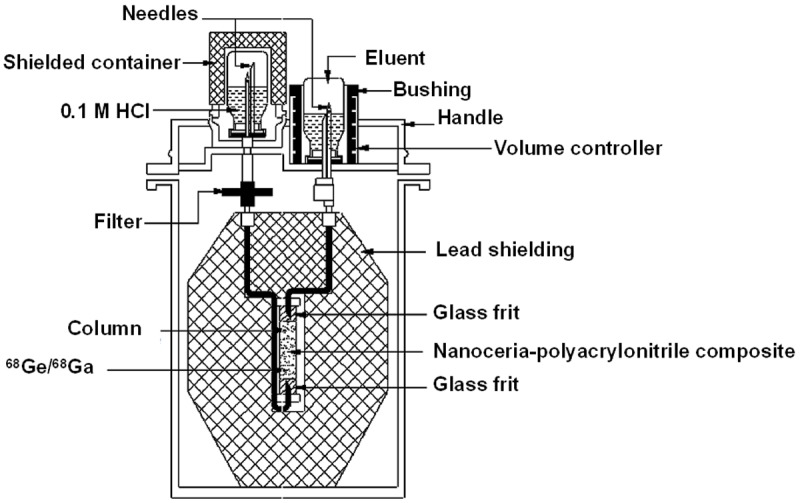
‘BARC’ 68Ge/68Ga generator based on nanoceria-polyacrylonitrile composite.
68Ga is exclusively in the oxidation state III in aqueous solution as well as at physiological pHs. In light of the perceived need to preclude the formation of insoluble Ga(OH)3 and soluble Ga(OH)4-, synthesis of 68Ga-labeled radiopharmaceuticals is carried out in the presence of weakly coordinating ligands such as citrate, acetate, or oxalate, which dramatically reduce the kinetics of complex formation [64]. Gallium (III) is classified as hard Lewis acidic, and therefore binds to hard Lewis base donor atoms such as nitrogen and oxygen, generally to form six coordinate bonds in close to octahedral geometry. Several promising 68Ga tracers ccomprising of small molecules, large biomolecules, and particles, targeting biological activities such as proliferation, angiogenesis, and apoptosis are currently under preclinical and clinical investigation [37,60,65-73]. Given the very broad field and long history of 68Ga tracers, agents that are being tested in preclinical and clinical trials, which promise an exciting future in clinical PET, are included in this review.
While 68Ga has made significant inroads into the field of clinical PET and undergone phenomenal expansion and growth, one of the most rapidly expanding areas is the development of 68Ga labeled peptide-based agents for targeted imaging, of tumors in particular [60]. The use of 68Ga labeled small tumor-affine peptides targeting somatostatin receptors (SSTR) has not only changed the diagnostic approach to neuroendocrine tumors (NETs) such as pituitary adenoma, pancreatic islet cell tumor, carcinoid, pheochromocytoma, paraganglioma, medullary thyroid cancer, and small cell lung carcinoma, but also has unlocked the potential of these agents for molecular imaging applications [37,38,72,74-78]. Among the SSTR agonists, 68Ga-labelled DOTA-conjugated somatostatin analogues have emerged as the breakthrough vector molecules owing to in vivo stability, favorable pharmacokinetics, and high and specific receptor-mediated tumor uptake [60]. They have proven to be increasingly useful due to several technical and biological advantages, such as fast clearance, rapid tissue penetration, and low antigenicity. The possibility of using 68Ga-labelled DOTA-conjugated somatostatin analogues for diagnosis is enticing, because the same compound labeled with 90Y or 177Lu can be used for therapy as a theranostic paradigm. This not only provides the prospect for better planning of therapy but also offers the opportunity to evaluate the therapeutic outcome, as in personalized medicine. This will allow for dosimetry before therapy to ensure the optimum balance between risk and benefit by enabling prediction and avoidance of potential radiotoxicity. The Society of Nuclear Medicine and Molecular Imaging (SNMMI) announced on November 18, 2013 that, 68Ga-DOTATOC has been officially designated an orphan drug by the U.S. Food and Drug Administration for the management of neuroendocrine tumors (NET) [79]. This is considered as a potential step in right direction that will contribute not only to potentially faster regulatory approval but to streamlined clinical trials.
The currently available data for potential use of other 68Ga-labeled peptide analogs as PET tracers are mainly preclinical and limited human studies have been carried out. In this context, 68Ga-labeled DOTA-4-amino-1-carboxymethyl-piperidine-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2 peptide (BAY86-7548), having a high affinity for the bombesin receptor subtype II for the detection of primary and metastatic prostate carcinoma, merits attention [80,81]. Integrin αvβ3 is an important member of receptor family and expressed preferentially on regenerative vascular endothelial cells and some tumor cells. Preliminary clinical studies indicate that integrin is an effective target for detecting intra-prostatic prostate cancer [82]. 68Ga-labeled αvβ3 integrin-targeting 68Ga-c[Lys-(NOTA)-Arg-Gly-Asp-D-Phe] is one of the radiolabeled arginine-glycin-aspartic acid (RGD) peptides for which initial clinical data are available. A biodistribution and radiation dosimetry study containing 10 patients having lung cancer or lymphoma showed that the excretion route with the highest activity was found to be the renal pathway. A clinical trial on 68Ga-BNOTA-PRGD2 [68Ga-p-SCN-Bn-NOTA-PEG3-RGD2] through intravenous injection of a single dose of nearly 111 MBq (≤ 40 µg BNOTA-PRGD2), is in progress to investigate post-myocardial infarction and post-stroke repair. Preliminary clinical studies indicate that 68Ga-PRGD2 uptake was found at or around the ischemic region in both MI and stroke patients, and significantly correlated with the disease phase and severity [83].
Urea-based low-molecular-weight peptidomimetic inhibitors of the prostate-specific membrane antigen (PSMA), a which is a cell surface protein and is expressed at higher levels in prostate cancer compared to other tissues, constitute a promising target for specific imaging due to the protein’s transmembrane location and internalization after ligand binding [84-86]. 68Ga-Glu-NH-CO-NH-Lys-(Ahx) [68Ga-PSMA-11] has emerged as an attractive agent currently used in clinical studies for the detection of recurrent prostate cancer and metastatic spread [86].
The enormous success of 68Ga-labelled peptides served as a springboard to spur the development of other 68Ga-labelled radiopharmaceuticals, which is primarily based on two main trends. First is the technetium “shake and shoot” concept, which primarily relied on the substitution of 68Ga in lieu of 99mTc in the preparation of radiopharmaceuticals. Some of the 68Ga labeled radiopharmaceuticals which can be substituted for 99mTc are shown in Table 4. A few examples of this type include the use of commercially available phosphonate kits for bone scintigraphy of lesions, MAA (macroaggregated human serum albumin) particulate for perfusion studies, 68Ga-NOTA-MSA (mannosylated human serum albumin) for immune system imaging, and 68Ga-BAPEN myocardial uptake as substitute for MIBI [87]. The second trend is the development of 68Ga labeled radiopharmaceuticals in place of 18F and 11C labeled compounds with an aim to achieve on-line cyclotron independence. These are elaborated in Table 5.
Table 4.
68Ga labeled radiopharmaceuticals substituting 99mTc
| Diagnostics modality | Established 99mTc labeled agents | Potential 68Ga labeled agents |
|---|---|---|
| Peptide receptors | 99mTc-HYNIC-peptide | 68Ga-DOTA-peptide |
| Bone metastases | 99mTc-MDP | 68Ga-phosphonates |
| Renal function | 99mTc-DTPA/MAG3/DMSA | 68Ga-EDTA |
| Cardiac function | 99mTc-RBC/MIBI | 68Ga-BAPEN |
| Lung function | 99mTc-MAA | 68Ga-MAA |
| Hepatobiliary | 99mTc-IDA | 68Ga-IDA |
| Infection | 99mTc-WBC | 68Ga-citrate |
| Brain imaging (perfusion) | 99mTc-ECD | 68Ga-ECD |
Table 5.
68Ga labeled radiopharmaceuticals substituting 18F, 11C labeled compounds
| Diagnostics modality | Established 11C/18F labeled agents | Potential 68Ga labeled agents |
|---|---|---|
| Angiogenesis | 18F-galacto-RGD | 68Ga-DOTA-RGD, 68Ga-VEGF |
| General cancer imaging | 18FDG | 68Ga-CXCR4 biomarker, 68Ga-uPAR biomarker, 68Ga-SCN-NOTA-BZA |
| Hypoxia | 18F-nitroimidazloes (FAZA, FMISO, FETNIM) | 68Ga-DOTA-imidazoles |
| Proliferation | 18FLT | 68Ga-DO3A-thymidine |
| Glioma | 18FET, 11C-methionine | 68Ga-glutamine, 68Ga-DO3A-alanine, 68Ga-DO2A-tyrosine |
| Prostate cancer | 18FDG, 11C-acetate, 18F-choline, 11C-choline | 68Ga-DOTA-PSMA |
Several innovative radiopharmaceuticals labeled with 68Ga are under active investigation and it is anticipated that quite a few novel agents labeled with this positron-emitting radionuclide will be available in the near future [88,89]. With expanding areas of application and growing interest in the use of 68Ga labeled radiopharmaceuticals, it is simply a matter of time until imaging of bone, sentinel lymph node, and/or lung ventilation/perfusion will be carried out routinely by 68Ga/PET-CT. Conspicuous harnessing of the 68Ga tracers in conjunction with their convergence with theranostics seems poised to bridge the quantitative diagnosis with subsequent therapeutic management. This bridging will benefit from therapeutic strategies aimed at the same or closely related processes and may be of great value for supporting and strengthening the concept of personalized medicine. It is time to bring this innovative paradigm changing concept out of the shadows and into the light.
82Sr/82Rb generator
There is a great deal of interest on the use of 82Sr/82Rb generators to avail 82Rb for clinical myocardial perfusion PET investigations. 82Rb is the first of the generator-produced positron emitters that made its entry into clinical NM. 82Rb, with a half-life of 76 s, decays by positron emission into stable 82Kr, which is a noble element and is therefore non-reactive and safe for biological use. Decay characteristics of the 82Sr/82Rb generator are depicted in Figure 4.
Figure 4.
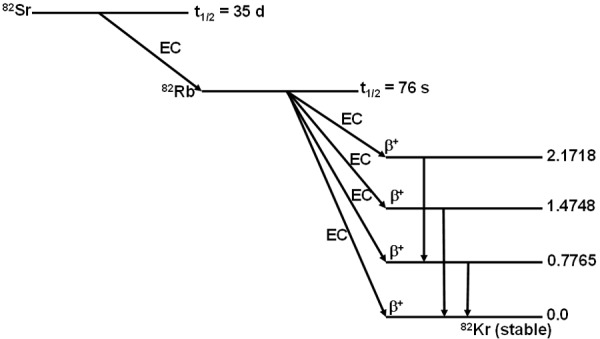
Decay characteristics of 82Sr/82Rb.
Being a cationic analogue of potassium with similar chemical and biological properties, 82Rb accumulates, as a function of blood flow, in cells of myocardium and other tissues in a manner similar to potassium. The myocardial uptake of 82Rb reflects blood flow through the myocardium. Compared with normal myocardium, areas of ischemia or infarction exhibit low 82Rb uptake because of diminished blood flow and/or viability, and this is useful for qualitative infarct imaging and for the detection of coronary artery stenosis and characterization of the severity. Synthesis of 82Rb radiopharmaceuticals for diverse imaging applications is precluded not only by the relatively limited chemistry of the alkali metals, but also due to the short physical half-life of 82Rb [90]. The exciting perspective of 82Sr/82Rb generators in clinical PET is attributed mainly due to the following:
The short half-life of 82Rb (t½ = 76 s) enables rapid rest/stress paired studies within a very short time (30-45 min), allowing for rest and stress imaging under virtually identical conditions and decreasing the total time required to scan each patient. This is convenient for the patient and permits high-throughput imaging and efficient use of the technology.
The ultra-short half-life of 82Rb presents limited radiation exposure to the patients (whole body dose of 5.5 mSv for 2.22 GBq) during investigation. The low-dose (10 MBq/kg) protocol allows simultaneous quantification of absolute flow, on 3D PET systems with adequate dynamic range to permit accurate measurement of the bolus first-pass activity, and measurement of ventricular function close to peak hyperemia with pharmacologic stress.
The suitably short physical half-life of 82Rb not only allows administration of high doses and short imaging times, but also offers the prospect of performing repeated and sequential perfusion studies within 20-minute intervals.
The 25.36 d half-life of parent radionuclide 82Sr is convenient for shipping and provides useful radioactivity in a generator for at least a month of frequent elutions.
Use of an RNG provides the scope to perform PET investigations in medical institutes without the necessity to maintain expensive cyclotron facilities. 82Rb has significant clinical potential and can be a considered as a low-cost alternative to short-lived, cyclotron-produced PET isotopes.
82Rb is extracted from the plasma with high efficiency by myocardial cells via the Na+/K+ ATPase pump [91]. While the myocardial extraction of 82Rb is similar to that of 201Tl, it is slightly less than 13NH3. The uptake of 82Rb is a function of blood flow, metabolism, and myocardial cell integrity. Cardiac PET using 82Sr possesses several advantages over traditional SPECT, with direct patient centric benefits including the accurate, diagnosis of coronary artery disease in asymptomatic or symptomatic patients, assessment of coronary stenosis severity, myocardial infarct imaging, evaluation of myocardial viability, collateral function, and cardiomyopathy.
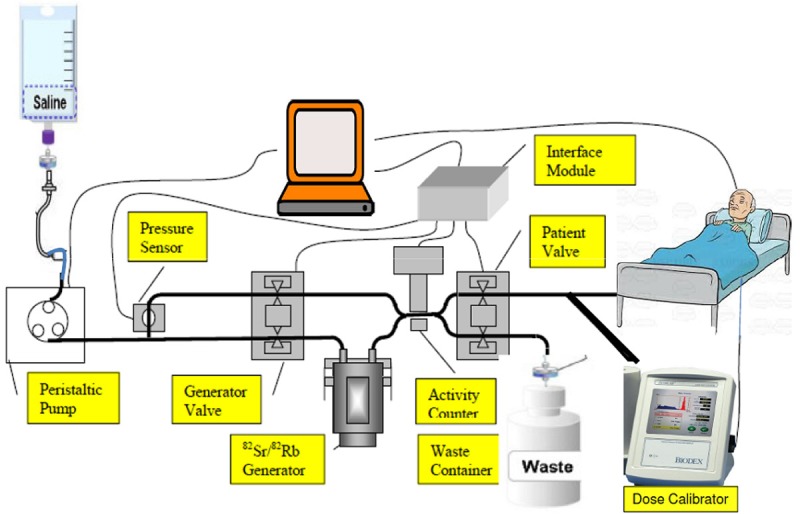
In 1989 the United States Food and Drug Administration (FDA) approved the use of 82RbCl availed from a commercial 82Sr/82Rb generator system trade name CardioGen-82, which has been available from Bracco Diagnostics Inc. for some years. This generator is composed of a small chromatography column [~4 cm (l) × 0.5 cm (Φ)] containing hydrous SnO2 housed in a lead shielding container. A schematic diagram of the 82Sr/82Rb generator system is depicted in Figure 5. When the column is flushed with a solution, such as 0.9% NaCl saline, the 82Rb+ is displaced by Na+ and is eluted. Quality control of the generator eluate includes 82Sr breakthrough measurements using a dose calibrator and pyrogen tests of the eluate.
Figure 5.
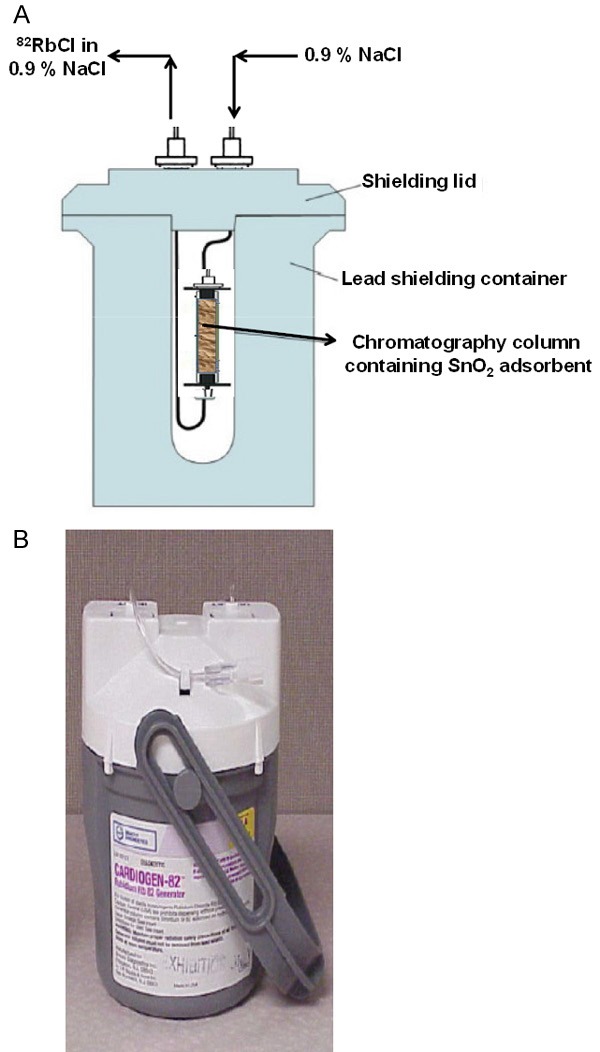
82Sr/82Rb generator system (A) Schematic diagram with shielding, (B) Commercial generator with shielding.
As the 82Rb supply is 90% replenished within 5 to 10 minutes of the previous elution, serial studies can be performed in rapid succession, maximizing patient throughput. There appeared to be enticing interest in the use of an automated infusion system to administer the 82Rb eluted from the 82Sr/82Rb generator directly to patients, in light of the short half-life of 82Rb and the decreasing amount of available 82Rb as the generator ages [92]. In the quest for an effective strategy to avail consistent levels of 82Rb activity from the 82Sr/82Rb generator, Klein et al. have developed an automatic infusion system [93]. A schematic representation of such a conceptual 82Rb infusion system that can control the concentration of 82Rb activity administered to the patient, perform quality assessment, and flush the activity from the patient line at the end of the infusion is shown in Figure 6.
Figure 6.
Schematic representation of 82Rb infusion system that can control concentration of 82Rb activity administered to the patient, perform quality assessment and flush the activity from the patient line at the end of the infusion.
Automated 82Rb infusion systems capable of accurate measurement and delivery of adequate doses of 82RbCl from a 82Sr/82Rb generator have been developed by Bracco Diagnostics, NJ, USA (CardioGen-82 Infusion System) and Jubilant DraxImage Inc, Canada (Ruby-Fill™ Infusion System). 82Rb is eluted from the generator by a computer-regulated elution pump and infused directly into patients using commercially available IV infusion system. Use of these systems substantially reduces the radiation dose, ensures optimum performance of the 82Sr/82Rb generator, and provides a log of the 82Rb activity infused into the patients. These developments are considered as successful steps forward in the promotion of widespread clinical use of 82Rb. A photograph of the CardioGen-82 Infusion System, along with various part of the system, is shown in Figure 7. Such 82Rb infusion systems clearly outperform conventional infusion systems or semi-automated systems, and their usefulness has been convincingly proven. Commercial availability of automated 82Rb infusion systems has a far-reaching effect on realizing the full promise and potential of 82Rb in the diagnosis of coronary artery disease (CAD) and is poised to bring functional imaging to the forefront in cardiac PET imaging.
Figure 7.

Photograph of the CardioGen-82 Infusion System along with various part of the system.
82Rb permits clinical imaging with short protocols (20-30 min in total) and a high patient throughput, providing better image quality and overall sensitivity in the diagnosis of coronary artery disease (CAD) as compared to myocardial scintigraphy with 201Tl or 99mTc-based radiotracers [94-96]. It also offers the potential for deriving absolute quantification of myocardial blood flow (MBF) [97]. New approaches to kinetic modeling and software tools for analysis of clinical 82Rb studies are being developed and evaluated, which could enable quantification of absolute myocardial blood flow and flow reserve.
Use of generator availed 82Rb in PET myocardial perfusion imaging has become far more accessible in daily practices, offers clinical feasibility of positron-based cardiac imaging without a cyclotron [98], and is poised to take a major leap forward to an exciting new stage. While the use of 82Rb availed from 82Sr/82Rb generators is a successful paradigm for the diagnosis of obstructive coronary artery disease (CAD) and has made considerable progress, the primary impediment for its widespread clinical use is the expense associated with 82Sr production and the need for generator replacement at 3-5 week intervals. Conscientious utilization of the generator in high throughput will not only offset this cost but can foster sustainability. The improved accuracy of PET leads to total cost savings in CAD management in clinical practice by getting rid of unnecessary diagnostic and therapeutic procedures such as coronary arteriography and coronary artery bypass grafting (CABG). A number of studies have demonstrated the routine use of 82Rb PET for myocardial perfusion imaging (MPI) at a cost similar to that for 99mTc SPECT [99]. With the advent of hybrid PET/CT driven by technological advances in medical imaging, there are a growing number of centers using cardiac PET with 82Rb [100] and this has greatly contributed to increasing acceptance of PET for clinical practice. This trend is expected to continue in the foreseeable future.
44Ti/44Sc generator
The 44Ti radionuclide (t½ = 60.6 y) decays by electron capture (EC) to 44Sc (t½ = 3.97 h) which subsequently decays to stable 44Ca. 44Sc is also a positron emitter (β+ branch = 94.3 %) and is a valuable alternative to 68Ga as a matched pair for radiotherapy. The use of 44Sc with a half-life more than 3 times longer than that of 68Ga (t½ = 68 min) makes it a valuable alternative for diagnostic and dosimetry purposes. Decay characteristic of 44Ti/44Sc system is shown in Figure 8.
Figure 8.
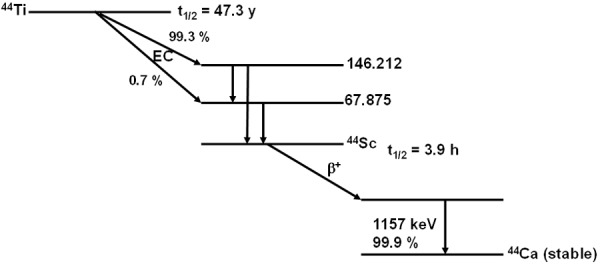
Decay characteristics of 44Ti/44Sc.
The 3.97-hour half-life of 44Sc is 3 times longer than that of 68Ga and therefore it may be a useful alternative not only for diagnostic purposes but also for dosimetry studies and further therapy planning with the use peptides labeled with the β-emitting radionuclide such as 177Lu or 90Y as radiotherapeutic agents.
The 3.92-hour half-life of 44Sc provides the ability to elute at 4 h interval with about a 50% elution yield. Hence, a 370 MBq (10 mCi) generator should be able to give 148-185 MBq (4-5 mCi) dose every four hours.
The 60.6 years half-life of 44Ti offers long-term application without generator replacement.
Despite its high β+-branching (94.3%), 44Sc shows an additional 99.9% photon emission of 1157 keV which is expected to be useful for nuclear medicine imaging using β-γ coincidences.
The chemistry of 44Sc is similar to that of the lanthanides, and the “lanthanide like” elements. Hence chelators developed for the complexation of 177Lu and 90Y such as DOTA and DTPA analogs can also be used for the complexation of 44Sc.
While 68Ga labeled compounds have significant clinical potential and present a convenient, low-cost alternative to cyclotron-produced PET radiopharmaceuticals, the 68 min physical half-life of 68Ga limits the spectrum of clinical applications of 68Ga-labelled radiodiagnostics. In addition, 68Ga-labelled analogues of endoradiotherapeuticals with longer biological half-lives, such as 90Y- or 177Lu-labeled peptides and proteins, cannot be used to determine individual radiation dosimetry directly. In this context, the notion of using generator-derived positron emitters with longer physical half-lives, such as 44Sc (t½ = 3.97 h), is deemed worthy of consideration.
While the outlook of 44Ti/44Sc generator is promising, development of a generator amenable for clinical use will not be a trivial process and poses formidable scientific and technical challenges, such as effective separation strategies of availing optimum 44Sc elution yields and low 44Ti breakthrough, and selection of an appropriate eluent to obtain 44Sc in a chemical form suitable for subsequent radiolabeling (i.e. high radioactive concentration (RAC), low pH, free from metallic impurities etc.). With an aim to tap the potential of 44Ti/44Sc generator to avail 44Sc of requisite purity, several strategies have been explored [101] among which the method reported by Filosofov et al. [102] merits attention. This 185 MBq (5 mCi) 44Ti/44Sc generator system has been studied in detail and has been successfully exploited for preclinical investigations.
The generator consists of a chromatography column containing Bio-Rad AG1 × 8 (200-400 mesh, Br --form), an anion-exchange resin, on to which purified 44Ti dissolved in 20 mL of 0.1 M H2C2O4 was loaded. 44Sc could be eluted with 20 mL of 0.005M H2C2O4/0.07 M HCl solution with > 97% elution yield and low 44Ti breakthrough (5 × 10-5%). The low RAC and the chemical composition of the generator eluate emerged as the major hindrances that thwarted efforts for fast, reliable and quantitative radiolabeling of nanomolar concentration of precursors. In order to enhance the RAC, fractionation of the elution of 44Sc was resorted to, but met with limited success, as the concentration of hydrochloric acid was too high for realizing direct radiolabeling.
With to the goal of enhancing RAC, reducing HCl concentration, and removing the oxalate anions, the prospect of using post elution processing was considered to be reliable and subsequently pursued [103]. In this procedure 0.005 M H2C2O4 / 0.07 M HCl solution was passed through the 44Ti/44Sc generator to elute 44Sc adsorbs, which was again passed online through a small cation exchange column to retain 44Sc. The cation-exchange column was washed with 2-4 mL of H2O to remove the remaining traces of the initial eluate solution. 44Sc retained by the cation exchange column was then eluted with 3 mL of 0.25 M ammonium acetate buffer, pH = 4.0. Reconditioning of the cation exchange cartridge was performed by passing 1 mL of 4 M HCl and finally by 1 mL H2O. A schematic representation of the procedure is depicted in Figure 9. A 185 MBq (5 mCi) generator provides about 170 MBq of 44Sc after direct elution and about 150 MBq following online post-processing.
Figure 9.
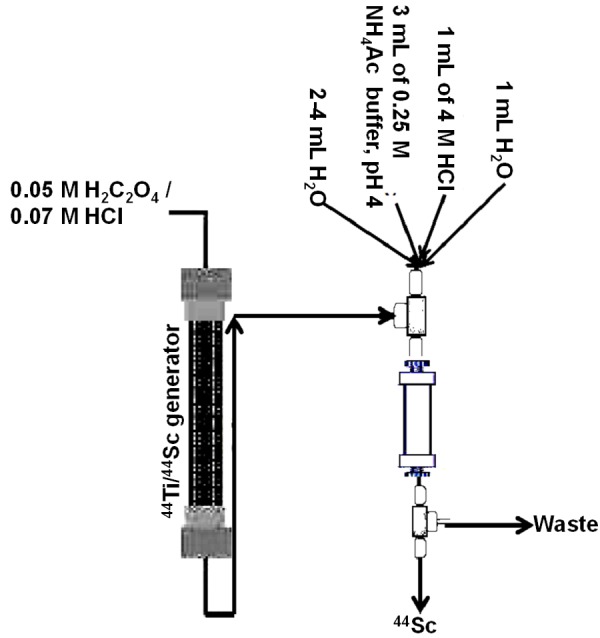
Post elution processing of 44Sc obtained from a 44Ti/44Sc generator.
Scandium is chemically similar to Y3+ and lanthanides. While the ionic radius of Sc3+ (74.5 pm) is smaller than that of lanthanides, it is larger than any of the M3+ cations formed by the 3d transition metals. It has been established that Sc3+ in solution has a tendency to forms DOTA complexes with coordination number 8 and 9 similar to that Y3+ and Lu3+, whereas Ga3+ forms 6 coordinated octahedral complexes [104]. 44Sc3+ as a metallic cation is suitable for complexation with chelators alone or with BFCs (DOTA, DTPA, NOTA, etc) conjugated to peptides or other molecular targeting vectors similar to the currently-used, trivalent radionuclides such as 68Ga ,111In or 90Y and 177Lu [105].
Recent studies on a series of polyamino-polycarboxylate ligands has shown that Sc-8-dentate ethylene glycol-bis (2-aminoethylether)-N,N,N’,N’-tetraacetic acid (EGTA) is a promising moiety for coupling 47Sc to proteins [106]. It has been demonstrated that DOTA is the most preferred chelator for 44Sc(III) among a series of macrocyclic ligands [52]. All 4 amine and carboxyl groups of a DOTA chelator contribute to the complexation of 44Sc(III), resulting in a stability constant comparable to those of Y(III) and lanthanides [52]. Also, 44Sc-DOTATATE has similar lipophilicity to that of 177Lu- and 90Y-DOTATATE [52]. Due to chemical similarity of Sc3+ with Lu3+ and Y3+ cations, it is anticipated that 44Sc-DOTA bioconjugates will have similar in vivo characteristics (i.e. receptor affinity, kidney clearance) to the 177Lu- and 90Y-conjugates currently used in therapy.
The 44Sc-DOTA-conjugated tumor targeting vectors such as somatostatin analogs investigated so far are stable in vitro and in vivo [107,108], and the data obtained reveal pharmacological parameters which are adequate for performing molecular imaging. Thus, 44Sc seems poised to become an alternative to 68Ga for imaging and dosimetry before 177Lu- or 90Y-radionuclide tumor therapy [101].
62Zn/62Cu generator
62Cu is a PET radionuclide with a half-life of 9.74 minutes, and decays 97.8% by β+ and 2.2% by EC to the ground state of stable 62Ni, with 99.6% of the β+ emissions having a 2.9 MeV positron Emax [109].
The 9.7 min half-life of 62Cu is long enough to facilitate radiopharmaceutical synthesis procedures and for image acquisition, while at the same time, it is short enough to afford favorable dosimetry.
The 9.7 min half-life of 62Cu enables serial imaging procedures in the same clinical visit without interference of 62Cu background activity from previous injections.
The versatile chemistry of copper provides the ability to prepare a wide range of 62Cu-labeled PET-radiopharmaceuticals for a variety of imaging applications.
The short half-life and high positron decay fraction (99.6%) of 62Cu make it suitable for characterizing the faster kinetics of smaller tracer molecules, such as Cu-PTSM or Cu-ATSM.
62Cu could be used as a PET-surrogate to therapeutic isotope 67Cu.
While the 9.7 min half-life of 62Cu restricts imaging to less than an hour following injection, this has the advantage of allowing several repeat scans in quick succession, which can be used to evaluate the response to a number of physiological or pharmacological stimuli.
The availability of 62Zn/62Cu generators is often the ultimate limiting factor in realizing the benefit of 62Cu-radiopharmaceuticals. Evolution and continued success of 62Cu in PET has been, in large part, due to development of the 62Zn/62Cu generator (Figure 10). Advances in the development of 62Zn/62Cu generator have contributed substantially to the deployment of 62Cu-radiopharmaceuticals in clinical PET.
Figure 10.
Schematic diagram of 62Zn/62Cu generator to avail 62Cu for radiopharmaceuticals formulation.
Robinson et al. [110] prepared a 62Zn/62Cu generator system based on “Dowex 1-X10” resin of 200-400 mesh [0.7 cm (Φ) × 8.0 cm (l)] from which 62Cu could be eluted with 3.5 mL of 0.1 N HCl containing 100 mg/mL NaCl and 1 μg/mL CuCl2, with 85% elution yield and with 62Zn breakthrough less than 0.001%. Fujibayashi et al. [111] has developed a generator based on a cation exchanger (CG-120, Amberlite), from which 62Cu is eluted in a glycine complex. While this system offers 70% elution efficiency, the 62Zn breakthrough was significantly high (2.2%). Zweit et al. [112] have developed a 62Zn/62Cu generator based on anion-exchange resin from which 62Cu could be eluted in a solution of 0.3 M HCl/40% ethanol with greater than 90% elution yield in a 3-mL volume and with low 62Zn breakthrough (< 3 × 10-7%). Bormans et al. [113] have developed an improved 62Cu-generator system based on Dowex 1 × 16, 200-400 mesh, from which 62Cu is eluted using a solution containing 1.7 M in NaCl/0.1 M in HCl. Fukumura et al. prepared an improved 62Zn/62Cu using Sep-Pak plus CM cartridge, from which 62Cu was eluted with high elution efficiency (approximately 96%) using a small volume (ca. 3 mL) of a 200-mM glycine solution with a very low breakthrough of 62Zn (< 0.1%) [114]. El-Azony has reported a method based on a De-Acidite FF anion exchanger [115], from which 62Cu is eluted using a solution of 0.2 M HCl-60% acetone with an elution efficiency of 92.5%. As the 0.2 M HCl-60% acetone medium is unsuitable for medical use, it was evaporated and reconstituted in a chemical form suitable for radiolabeling.
The chemistry of copper is restricted to two principle oxidation states (I and II), and the relatively simple coordination and redox chemistry of copper is well documented [116]. Copper salts generally exists as [Cu(OH)6]2+ in aqueous solution. Compounds of the Cu(I) oxidation state are unstable in aqueous solution and readily oxidize to Cu(II) to form 4, 5, or 6 coordination bonds with ligands. The Cu(II) oxidation state has a pronounced tendency to form coordination complexes with ligands that contain electron donor atoms, such as, N and S. Complex formation with chelating agents occurs at pH < 7 since formation of insoluble Cu(OH)2 at higher pH is a major concern [117]. The in vivo stability of the radio-copper complex is a critical factor for designing 62Cu-radiopharmaceuticals. Ligands that can form kinetically inert Cu(II) complexes are ideal since this is more significant than thermodynamic stability.
62Cu(II) diacetyl-bis(N4-methylthiosemicarbazone), or 62Cu-ATSM, has emerged as an effective agent to image tissue hypoxia [118-120]. 62Cu-ATSM is a neutral, lipophilic compound that can be readily diffuses into all the cells with perfusion. However, its structure changes due to the reduction of Cu2+ cation to Cu+ cation in hypoxic conditions resulting in accumulation in the hypoxic cell. The mechanism of 62Cu-ATSM retention in hypoxic tissues is ascribed mainly due to the low oxygen tensions and the subsequent altered redox environment of hypoxic tumors. Results from in vivo studies inferred that tissue Cu-ATSM uptake is dependent on oxygen concentration [121]. 62Cu-ATSM has shown heterogeneous uptake in tumors with homogeneous perfusion images, strongly suggesting uptake reflecting hypoxic heterogeneity. Results from several studies demonstrated the utility of Cu-ATSM PET hypoxic imaging with improved prognosis and effectiveness of radiotherapy [122-128].
62Cu(II) pyruvaldehyde bis(N4-methylthiosemicarbazone), or 62Cu-PTSM, is a promising tracer for myocardial, cerebral, renal, and tumor perfusion agents, and has attracted considerable interest [128-132]. 62Cu-PTSM demonstrated a high, first-pass extraction along with prolonged tissue retention due to intracellular reductive decomposition of the lipophilc Cu-PTSM complex to liberate the 62Cu ion, with negligible washout from the myocardium cell, suggesting the ability of 62Cu-PTSM to quantify blood flow in organs such as the heart, brain, and kidneys [130,133,134]. 62Cu-PTSM is reduced readily by the mitochondria, and is retained in most tissues, and thus can be used as a marker of tissue perfusion [135]. While Cu-ATSM and Cu-PTSM share the same molecular structural backbone, they have significantly different properties when utilized in PET imaging due to the presence of an additional methyl group on the ligand backbone of Cu-ATSM [136].
Another agent in the bis (thiosemicarbazone) family which merits attention is the 62Cu ethylglyoxal bis (thiosemicarbazone), or Cu-ETS, which is under investigation in human studies. The 62Cu-ETS radiopharmaceutical exhibits properties similar to 62Cu-PTSM in vivo due to its structural similarity with 62Cu-PTSM, while avoiding limitations imposed by human albumin binding. This agent has shown more linear uptake at high blood flow rates and thus may provide a superior PET perfusion tracer for applications such as myocardial perfusion and renal blood flow measurements [137-139].
As blood flow and hypoxia are complex, inter-related factors of physiology, the prospect of using dual-tracer studies with 62Cu-PTSM (blood flow) and 62Cu-ATSM (hypoxia) affords the potential for characterizing both tumor blood flow and hypoxia in a single scan [120,140,141]. In this modality, dynamic signals from overlapping tracers with staggered injections are recovered using differences in tracer kinetics and decay. The multi-tracer PET imaging paradigm has great potential to avail complementary information that would help for treatment selection, planning, and early response monitoring.
Proportional Technologies, Inc. USA has developed a micro 62Zn/62Cu generator to avail 62Cu tracer at very high isotonic concentration amenable for clinical use. Proportional Technologies, Inc. has also developed include H2PTSM, H2ETS and H2TASM kits available for both non-human studies and clinical investigation. Components in each injectable solution are described in Table 6.
Table 6.
Components of H2PTSM, H2ETS and H2TASM kits
| Component | H2PTSM kit | H2ETS kit | H2ATSM kit |
|---|---|---|---|
| Sodium acetate | 4.3 mg | 4.3 mg | 4.3 mg |
| Acetic acid | 3.1 mg | 3.1 mg | 3.1 mg |
| Sodium chloride | 30 mg | 30 mg | 30 mg |
| Excipient | 20 mg | 20 mg | 20 mg |
| Free ligand | 2 µg | 2 µg | 0.4 µg |
| 62Cu labeled ligand (per 555 MBq (15 mCi) dose) | 2.3 × 10-7 mg | 2.2 × 10-7 mg | 2.4 × 10-7 mg |
Despite the many advantages, the utility of 62Zn/62Cu generators comes with a few limitations. Currently, 62Zn/62Cu generators are procured on an as-needed basis as they are relatively expensive and have a shelf life of 1-2 days, and patients must be scheduled several days in advance of imaging. The cost of 62Zn/62Cu generator can be significantly off set if demand for the 62Cu compounds increases to the point at which generators can be produced for routine clinical use and the scheduling of several patients for 62Cu PET studies on the day of generator delivery. One generator will support 20 or more dose preparations during 1 day of use, and these doses can be produced at frequent intervals of 30-45 minutes between elution. In this context use, of a lyophilized kit formulation seems poised to provide a practical means of producing 62Cu-PTSM, 62Cu-ATSM and 62Cu-ETS on-site and bring substantial benefits at lower cost, as is well known in radiopharmaceutical practice. Additionally, one of two distribution models for the 62Zn/62Cu generator can be considered. Production of 62Zn can be carried out either in or near the 18F radiopharmacies using a 19 MeV cyclotron and 62Zn/62Cu generator can then be delivered using the same local established delivery network already in place for 18F. Alternatively, the 62Zn parent can be produced using > 25 MeV cyclotrons and loaded into generators which can be shipped to the local radiopharmacies or hospital radiopharmacies.
72Se/72As generator
72As is a positron-emitting arsenic isotope with a half life of 1.08 d, with branching ratio of 88% and Eβ+, max = 2.49 MeV. The chemical properties of 72As are amenable for the preparation of wide range of 72As-labelled PET radiopharmaceuticals. Interest in the use of a 72Se/72As generator is primarily attributed to the following:
Its relatively high β+ abundance and hours-long half-life allows for imaging of slower biological processes.
The relatively long half-life of the parent 72Se (t½ = 8.4 d) allows usage of the 72Se/72As generator for 2-3 weeks.
The long physical half-life of 1.08 d may render 72As a PET radionuclide of choice for the quantitative imaging of biochemical and physiological processes with longer biological half-lives, e.g. immunoimaging and receptor mapping.
The longer half-life allows for more elaborate chemical modification and labeling methodologies.
The versatile chemistry of arsenic would permit the radiolabelling of a broad spectrum of potentially valuable pharmaceuticals.
72Se/72As generators offer the prospect of availing 72As at the hospital radiopharmacies without dependence on an accelerator facility in close proximity.
Recognizing the potentially important role of the 72Se/72As generator in NM, a wide range of adaptive separation strategies with an aim to obtain 72As in a suitable chemical form and of requisite purity have been reported. An ion- exchange chromatography 72Se/72As generator based on Dowex 50 was developed by Al-Kouraishi and Boswell [142]. In this process, cold selenous acid was added to 72Se solution containing sulfur to reduce Se which was obtained as a precipitate. This was then centrifuged, washed with deionized water, loaded onto the chromatography column containing cation exchanger column and held for 90 hours for the in-growth of the 72As daughter. It is possible to elute 72As from the column using deionized water with a 70% elution yield. Another generator system developed at Los Alamos National Laboratory was based on the addition of selenium carrier in the form of selenic acid and hydrazine for cyclic reduction of selenium to Se(0) followed by separation of 72As by filtration with subsequent oxidative dissolution of Se(0) using H2O2 prior to each separation cycle [143]. A 72Se/72As generator based on an electrochemical process with selective deposition of 72Se on Pt electrodes as Cu72Se were also reported [144]. Jennewein et al. reported a 72Se/72As generator based on distillation technique [145]. In this work, 72As produced by radioactive decay of 72Se was distilled off as the volatile AsCl3 at 105°C for 10 minutes in a gaseous HCl stream, and collected on a charcoal filter with a radiochemical yield > 99% [145]. The same group of authors have reported a system based on a polystyrene solid support [Varian ENV solid phase extraction cartridge] to adsorb elemental 72Se (0) [146]. The in-grown 72As was then eluted using 2 mL of concentrated HF with a 50% elution yield [146]. Chajduk et al. have proposed a method based on extraction chromatography for isolation of arsenic from selenium [147]. In this method aromatic o-diamine extractant was impregnated into a polystyrene column matrix. The extractant selectively retains 72Se from which 72As could be selective eluted using 0.9% NaCl solution. The proposed separation procedure assures > 95 % elution yield with low (< 0.01%) 72Se breakthrough [147]. A 72Se/72As RNG system utilizing chelation and liquid-liquid extraction has been reported [148]. Despite the impressive progress, the promise to develop a commercial, clinical scale 72Se/72As generator amenable for use in a hospital radiopharmacy has not been fulfilled.
Arsenic forms stable covalent bonds with carbon and sulphur and can substitute phosphorus in certain compounds with minimal alteration of the biologically activities of the parent molecule. Jennewein et al. has reported a method to radiolabel 74As with N-succimidyl Sacetylthiacetate (SATA) derivitized bavituximab [149]. Arsenic has a high affinity to sulfur and As is able to bind covalently to sulfhydryl groups. In antibodies, the sulfur moieties are mainly associated with dithiol bridges. To increase the number of free thiols, conscious modification of antibodies with SATA (N-succinimidyl S-acetylthioacetate) seemed sagacious and was pursued [149]. It has been demonstrated that 74As labeled bavituximab-SATA-conjugate retained its immunoreactivity and exhibits favorable in vitro stability in fetal bovine serum. Biodistribution studies carried out with Dunning prostate R3327-AT1 tumor bearing rats reveal that tumors were clearly visible after 48 hours, exhibiting 8-fold higher uptake at 72 hours as compared to the control antibody [149,150].
It is clear that 72As based radiopharmaceuticals for clinical PET are still in their infancy and their utility is limited by the commercial unavailability of 72Se/72As generators as well as end-user radiolabeling techniques. Additional preclinical and clinical studies are warranted to exploit the full potential of 72As. Having established their importance in NM, the exploration of 72Aslabeled PET tracers will continue to rise.
110Sn/110mIn generator
110mIn is a positron-emitting isotope of In, with a half-life of 69.1 min and branching ratio of 62% with Eβ+, max = 2.26 MeV. This radioisotope is of interest as it would provide the scope of preparation of radiolabeled agents (analogous to SPECT tracers) to be quantified with PET and allow for sequential studies to be performed within a relatively short time. Potential use includes preparation of 110mIn labeled leukocytes for infection/inflammation imaging [151] and 110mIn-octreotide in clinical imaging of neuroendocrine tumors (NET) [152]. The prospect of using 110mIn-labeled octreotide seemed to be an interesting proposition as it can better detect small tumors and might be able to more accurately quantify tumor uptake with PET than can 111In-labelled octreotide and SPECT. Due to the short half-life of the 110mIn, use of the Octreoscan kit has been embraced by investigators [152]. Since octreotide is commercially available in the Octreoscan kit (Mallinckrodt Medical, St. Louis), an optimized procedure for labeling with 110mIn could be routinely used, solving the problems associated with GMP production of the peptide-chelator conjugate. The 69-min half-life of 110mIn matches the rapid kinetics of the octreotide well. Apart from enabling detection of smaller tumors, 110mIn-octreotide-PET can also provide quantitative information about receptor kinetics and concentrations with better temporal and spatial resolution than 111In-octreotide-SPECT.
This generator was prepared using Kieselgel 40 (silica gel) conditioned with 0.02 M HCl [153]. 110Sn was loaded in the column at 0.02 M HCl and elution of 110mIn was performed using 0.02 M HCl [153]. Owing to the relatively short half-life of the parent, 110Sn (t½ = 4.9 h), this generator has a limited shelf-life of 10-12 hours. The generator was eluted at 2 h interval with > 90% elution yield of 110mIn and the level of 110Sn breakthrough was < 0.003%.
52Fe/52mMn generator
52mMn is a positron-emitting isotope which decays with a branching ratio of 98.3% with a 21.1 min half-life and with Eβ+, max = 2.631 MeV. In addition to the annihilation radiation, 52mMn also emits a l434-keV gamma ray (98.3%). The remainder of the decay is by isomeric transition to 52Mn, which has a 5.59-day half-life. This PET isotope has been considered as a as a potential agent for myocardial imaging studies owing to the possibility to perform a number of studies in the course of a day [154-156]. The tremendous prospect associated with the use of 52Fe/52mMn generators in NM has led to the development of a number of strategies [155-159]. Due to the short half life of 52Fe (t½ = 8.27 h) breakthrough up to 1% would not significantly alter the efficacy of the 52mMn eluted. This generator has a very limited shelf life due to the short half life of 52Fe, the parent radioisotope.
Despite the favorable physical properties of 52mMn2+ as an agent for myocardial imaging studies, results from pig model studies concluded that 52mMn allows the qualitative assessment of myocardial perfusion but does not meet the requirements of a quantitative myocardial perfusion agent [160]. The 52Fe/52mMn generator presents no chemical or practical advantages over the 62Zn/62Cu generator, which has similar parent and daughter half-lives.
122Xe/122I generator
The use of 122Xe (t½ = 20.1 h)/122I (t½ = 3.6 min; β+ = 77%, EC = 23%, Eβ+, max = 3.1 MeV) generator system as a means of availing 122I for PET studies has caught the attention of NM clinicians and evoked excitement among radiopharmaceutical scientists. l22I is unique among all generator produced PET radionuclides in that it is not a metallic element and it is availed from a parent nuclide that is an inert gas. Due to the gaseous nature of the parent radionuclide, the customary use of column chromatography technique for the separation of daughter radionuclide will not be appropriate for this generator. In view of this, suitable alternative means for this separation have been described [161,162].
While the 3.6-min half-life of 122I, as well as the versatile chemistry of iodine, are appealing for many PET applications, the chemistry of iodine is not particularly amenable to the rapid synthetic chemistry which is dictated by the physical half-life of this label. Nevertheless, a few attempts have already been made. Potential utility of 122I labeled Iodoperidol (IP), an iodinated analogue of the antipsychotic drug haloperiodol, as a cerebral blood flow radiopharmaceutical for PET has been evaluated and showed promising results [163]. This study indicates that this class of compounds holds promise for development as perfusion radiopharmaceuticals. 122I has been successfully incorporated into an amphetamine analog, 2,4-dimethoxy-N,N-dimethyl-5-[122I]iodophenylisopropylamine (5-[122I]-2,4-DNNA), using a remote synthesis protocol and studied in dog models as a quantitative cerebral blood flow agent for PET [164].
In vivo generators
This concept essentially consists of labeling of molecular carriers (complexes, peptides, monoclonal antibodies and their fragments, etc.) with intermediate half-life generator parents, which continuously decay and generate shorter half-life daughter radionuclides much more than the parent [165-167]. It is pertinent to note that the chemical binding of the daughter nuclide must be analogous to the parent one to preclude the release of daughter radionuclide from the original position. Owing to the decay kinetics, the daughter nucleus experiences some recoil. However, this is assumed to be negligible. This is the reverse of the usual use of a generator, where the daughter is initially separated from the parent prior to use. By contrast, with this strategy the daughter is removed from the parent using a chemical separation technique and the parent is then attached to tissue-specific therapeutic agents (complexes, chelate, peptides, monoclonal antibodies and their fragments, etc.) to be administered. While this innovative paradigm has been practiced in radionuclide therapy to minimize the radiation exposure to non-target tissues, this could be extended to imaging agents [168]. Such a strategy seemed appealing, as it has the potential for quantitative PET to inform on a more personalized treatment strategy, since the same carrier moiety can be radiolabeled with a suitable parent/daughter pair to deliver therapeutic or diagnostic imaging radionuclides. In this context two generators that merit attention for the “Theranostics” concept are: 140Nd/140Pr and 134Ce/134La generators [168].
140Nd/140Pr generator
140Nd decays 100% by EC with a half-life of 3.37 days to produce a short-lived positron emitter 140Pr which decays to stable 140Ce by positron emission with a branching ratio of 49 %, with Eβ+, max = 2.4 MeV and with a 3.4 min half-life. This system shows potential as an RNG or as an in vivo generator system for PET [168].
A 140Nd/140Pr RNG system based on physico-chemical transitions (hot-atom effects) of the daughter 140Pr following the electron capture process of 140Nd has been developed [169]. In this process 140Nd(III), in the form of 140Nd-DOTA-conjugated complexes, was quantitatively retained on a cation-exchange resin (Bio-Rad AG 50W-X8, 200-400 mesh, in hydrogen form). 140Pr was eluted using 10-3 M DTPA in > 93% yield with negligible levels of 140Nd breakthrough.
134Ce/134La generator
134Ce is an Auger electron-emitting radionuclide which decays to 134La with a half-life of 3.16 d and the daughter nuclide 134La (t½ = 6.45 min) is a positron and Auger-electron emitter. While 134La was proposed as PET perfusion imaging agent [170], real practical applications have not yet been described. Both the low-energy Auger electrons, emitted in the 134Ce and 134La decays, as well as the high energy positrons emitted in the 134La decay, can be conscientiously exploited for radionuclide therapy. The kinetics of the positron-emitting daughter nuclide can be measured with PET and used for dosimetry. 134Ce/134La absorbed doses to single cells were reported to be higher than absorbed doses from 90Y and 111In [170]. Further investigations are warranted to realize the full potential of this system.
Production of parent radionuclide
Any advancement in PET RNGs will be largely dependent upon the availability of required quantity and quality of parent radionuclides. The major challenge for the production of parent radionuclide requires selection of the most economical processes from the pool of available options. Most of the parent radionuclides used in RNGs are produced using solid targets of metal-based materials (e.g. elemental metal or metal oxide) with the target either mounted to the cyclotron (under vacuum) or external to the cyclotron/accelerator. In each case, the target system needs to tolerate heat generated from the particle bombardment. Depending on the energy and current of the beam, the cooling systems can be quite elaborate. As the number of nuclear reactions in a given target material can be potentially large, one needs to design the target so that the preferred nuclear reaction dominates. This may be achieved in a number of ways:
Choice of beam incident particle [i.e. p, d, or α] energy of the beam, target material (i.e. natural or enriched isotope of target element).
Routine production of parent radionuclides for RNGs should meet the following requirements:
The production method, involving the bombardment as well as the radiochemical separation procedure for the isolation radionuclide of interest, must be economically viable.
Ability to isolate the radionuclide of interest in high specific activity, with acceptable radionuclidic, radiochemical and chemical impurities.
The radiochemical separation method selected must be as simple as possible to facilitate remote operation/automation within a hot cell to minimize radiation exposure to the operator.
The following section provides an overview of the issues associated with production of parent radionuclides which should be considered to identify recent advances in this field.
68Ge
Two nuclear reactions have been utilized for the routine production of 68Ge, namely, by 66Zn(α,2n)68Ge (66Zn natural abundance being 27.8%) giving a yield of up to 2 μCi.(μA.h)-1 (yield per µA beam current calculated for 1 h irradiation) at 35 MeV beam energy, or by 69Ga(p,2n)68Ge (69Ga natural abundance being 60%) giving a yield of up to 20 μCi.(μA.h)-1 at 23 MeV beam energy [171]. Of the two reactions, 69Ga(p,2n)68Ge has been regarded as the reaction of choice due to the higher yields obtained and ease of chemical separation. In this method, only two elements need to be separated from each other, where with the other reaction, a third element (Zn) needs to be taken into consideration [172].
When selecting the target, chemical, mechanical and thermal properties, and corrosion and radiation resistance need to be considered since the target will be exposed to high current irradiations and the power dissipated in the targets reaches values of about 300-1000 W or more. Target materials used for the production of 68Ge include Ga2O3, Ga4Ni, GaAg and Ga metal. The use of encapsulated Ga2O3 with high-current proton beams is precluded as the oxide changes from a hexagonal α-form to a monoclinic β-form at about 600°C accompanied by a volume increase, which leads to capsule rupture. While the use of alloys as target material (Ga4Ni and GaAg) has the advantage of attaining good thermal conductivity, the requirement of an elaborated chemical separation procedure to make 68Ge free from coproduced impurities discourage their use. The prospect of using Ga2O3 is prohibited by the difficulties encountered during the dissolution of irradiated target. In this regard, the idea of using Ga metal seemed attractive. As Ga metal is corrosive to Al, it is essential that it be encapsulated, using Nb for example, as Ga would not react with this encapsulation.
In order to avail satisfactory batch yields, proton energy > 20 MeV, high-current accelerators on the order of the mA’s and long irradiation periods of several days duration are required. Hence there are very few suppliers of 68Ge, despite huge demands. Four major centers which produce 68Ge currently are: iThemba laboratories (South Africa), Brookhaven National Laboratory (USA), Los Alamos National Laboratory (USA) and Cyclotron Co Ltd (Obninsk, Russia) [171]. These centers report on production capacities of about 18.5 to 74 GBq (0.5 to 2 Ci) of 68Ge per batch. Separation techniques used to isolate micro quantities of no-carried-added (NCA) 68Ge from macro amount of irradiated target range from solvent extraction [173-176] and ion exchange chromatography, to using organic [177] and inorganic [172,178,179] materials, have been employed.
At BNL, production is carried out using natGa targets with ~45 MeV protons. For a typical batch production, 81 g of natGa metal encapsulated in a Nb container is used and irradiation was carried for period of 4 weeks (0.52 MBq. (µAh)-1. 68Ge is recovered from the target by extraction into 4 N HCl and 30% H2O2 after two weeks cooling. Further purification is achieved by solvent extraction using carbon tetrachloride and back-extraction of 68Ge into H2O. An overall recovery yields > 85% having a batch yields of 33.3-51.8 GBq (900-1400 mCi) with radionuclidic purity > 99.9%, and activity concentrations > 3.15 GBq/mL (85 mCi/mL) have been reported [180]. At the Los Alamos National Laboratory (LANL), 100 MeV protons are used [181]. For a typical batch production, 4 g of natGa metal encapsulated in a Nb container is used and irradiation was carried for period 16-20 days (1.18 MBq /µAh). The batch yield at end of bombardment (EOB) is about 55-70 GBq. Chemical processing is carried out after two weeks of cooling using solvent extraction with CCl4 and re-extraction of 68Ge into water followed by an ion exchange purification step using alumina [182]. At the iThemba Laboratories, a 66 MeV proton beam is used. For a typical batch production, 5 g of target with a production rate 1.18 MBq (0.032 mCi). (µAh)-1 is used. The radionuclidic purity of the processed 68Ge is > 99.9% and the final product contains < 1 µg of Ga per 37 MBq (1 mCi) of 68Ge [183]. At the Cyclotron Co. Ltd. in Obninsk, Russian Federation, Ga-Ni alloy prepared on Cu backings is used as the target material. Irradiations are performed at a high proton beam intensity of several hundred microamperes of 23 MeV protons. 68Ge of high specific activity [> 74 GBq (> 2 Ci)/mg] and 99.8% radionuclidic purity is obtained [184].
82Sr
82Sr has been produced via several different reaction routes, as shown in Table 7. Each method presents different challenges for targetry, process chemistry, practical yield, radionuclidic purity, and availability. While the use of high energy spallation reactions with Mo or Y metal as the target constitutes a method for the production of 82Sr [185-187], low reaction cross-sections and the requirement of an elaborate radiochemical procedure to isolate 82Sr from other spallation products, as well as the bulk target material, emerged as the major impediments which continue to discourage this method’s wide scale adaptability. In an attempt to produce 82Sr, high energy 3He and alpha particle irradiations of natural Kr have also been tried [188-190], but met with limited success due to low yields and the scarcity of operating accelerators capable of accelerating 3He ions to the energies required. In this light, the natRb(p,xn)82Sr reaction seemed most attractive and thus stands as the technique of choice for large scale production [191-193].
Table 7.
Possible routes for the production of 82Sr
| Reaction | Target material | Projectile energy (MeV) | Comment |
|---|---|---|---|
| 89Y (p, spallation) 82Sr | Yttrium oxide | 60-240 | Low radiopurity & yield |
| NatMo (p, spallation) 82Sr | Molybdenum metal | 500-700 | Low radiopurity, high cost |
| NatRb (p, xn) 82Sr | RbCl or Rb metal | 40-90 | Preferred |
| NatKr (α, pxn) 82Sr | Kr gas | 20-120 | Low radiopurity, low yield, little availability |
| NatKr (3He, xn) | Kr gas | 20-90 | Low radiopurity, low yield, very little availability |
The requirement to use high energy proton reactions leads to relatively large government owned accelerator facilities around the world, and poses challenges associated with irradiation scheduling (as these facilities have other missions and do not operate continuously year round) and timely transport of large quantities of radioactivity to the generator production site; these are some of the major road blocks that need to be surmounted for widespread use of 82Sr for medical applications. As the potential for 82Sr/82Rb generators is evident, several commercial companies have started producing 82Sr to meet the demand on a routine basis. Production details adopted by the current suppliers of 82Sr are depicted in Table 8.
Table 8.
Production details from the current suppliers of 82Sr
| Laboratory | Target | Irradiation conditions | Typical batch yield at EOB (GBq) |
|---|---|---|---|
| Brookhaven National Lab (BNL), NY, USA | RbCl pressed pellet in inconel | 2 targets, 93-70 & 64-41 MeV | 220-300 (5.9-8.1Ci) |
| Los Alamos National Lab (LANL), NM, USA | RbCl cast puck in inconel | 2 targets, 97-71 & 65-45 MeV | 300-450 |
| Institute for Nuclear Research (INR), Troitsk Russia | Rb metal in stainless steel | 100-40 MeV | 120-220 |
| iThemba Labs, Faure, S. Africa | Rb metal in stainless steel | 66-44 MeV | 100 |
| Arronax, Nantes, France | RbCl pressed pellet in stainless steel | 8 thin targets, 69-44 MeV | 80-90 |
| Nordion/TRIUMF, Vancouver Canada | Rb metal in stainless | 60-48 MeV | 60-100 |
With the goal of undertaking a large scale production process using the natRb(p,xn)82Sr reaction, both RbCl and Rb metals have been used. The advantages of using RbCl are its commercial availability in high chemical and isotopic purity, resistance to corrosion, ability to be pressed into a pellet at near theoretical density, or melted and cast into a capsule, and easy dissolution of irradiated targets in water as well as common mineral acid. Limitations on the use of RbCl include hygroscopic behavior of the material and low thermal conductivity. The poor thermal conductivity requires careful target design and limits target thickness in order to withstand high beam current.
The use of Rb metal permit permits the use of very thick targets in order to increase batch quantities because of high thermal conductivity. Disadvantages of using Rb metal include commercial non-availability of very high purity material and the serious safety issues in handling metallic Rb, due to its susceptibility to ignite or explode upon exposure to air or moisture, and the subsequent requirement to perform initial irradiated target dissolution within an inert atmosphere (e.g. dry argon) using an anhydrous higher alcohol, typically propan-2-ol.
At BNL and LANL, processing of the irradiated RbCl target was carried out by removing the exterior of the target capsules with acid, followed by dissolution of irradiated RbCl pellet in water. 82Sr2+ was isolated using an ion exchange chromatography separation process using Chelex 100, wherein 82Sr2+ is chelated and retained on the resin while Rb+ gets eluted. 82Sr2+ was eluted from the column using 6 M HCl and further purification of 82Sr2+ was carried out using ion exchange chromatography with AG 50W-X8 and Chelex 100 [171,194]. At iThemba Labs, the irradiated target is dissolved in dilute ammonium chloride solution and 82Sr is separated from the target material using column chromatographic methods with Purolite S950, a macroporous aminophosphonic acid chelating resin. Further purification of 82Sr is carried out following column chromatography technique using AG MP-50 macroporous cation exchange resin. The final 82Sr possesses high radionuclidic purity with negligible Rb and Fe chemical impurities [194].
Proton irradiated metallic Rb targets from Institute for Nuclear Research (INR) of the Russian Academy of Sciences, Russian Federation, are also processed at Los Alamos National Laboratory (USA) [195], as well as in Institute for Physics and Power Engineering (IPPE), Obninsk, Russia, using a patented method [196]. Pure 82Sr obtained in IPPE is then transported to the Russian Research Center of Radiology and Surgical Technologies (RRCRST), St. Petersburg, for loading the 82Rb-generator. Positron Corporation, USA has entered into a license agreement with INR to produce 82Sr in its proposed 70 MeV cyclotron and using the patented process technology of INR [197]. The rights to this patented technology have been awarded by INR to Positron Corporation.
Over the past decade, the growth of cardiac PET imaging has driven a significant increase in the use of 82Sr/82Rb generators. Currently, the US Department of Energy (DOE) is the only supplier of 82Sr in the US. Positron Corp, U.S., is working with the US FDA for certification so it can begin its own production of 82Sr, through its subsidiary, Manhattan Isotope Technology (MIT), to provide a second source for 82Sr in the USA. In August 2012, MIT submitted its drug master file (DMF) with the US FDA and has begun production of active pharmaceutical ingredient (API) grade 82Sr at its Lubbock, Texas, facility with strontium received from iThemba South Africa.
44Ti
44Ti/44Sc RNGs described in the literature thus far are all based on the 44Ti produced by proton irradiation of scandium, by the 45Sc(p,2n)44Ti reaction. The long half-life of 44Ti (60 years) and a low cross-section necessitates the use of high proton flux and long-duration irradiations. The targets are prepared by melting Sc in 150-220 µm thick layers onto copper backings, which themselves are covered by intermediate layers of Ag in order to reduce 65Zn coproduction. About 1.5 g Sc targets are subjected to proton currents of ~200 µA for ~10 h at ~30 MeV proton energy. Irradiated Sc target processing was carried out by dissolving the target in 18 mL of 2 M HCl. Separation of 44Ti from silver as well as co-produced 65Zn and 109Cd have been achieved following a multi-step ion-exchange chromatography separation, and purification procedures using AG 50W-X8, 200-400 mesh in H+-form, as reported [101,102]. Using the reported procedure, about 185 MBq (5 mCi) of 44Ti was successfully recovered.
62Zn
The common method for the production of the 62Zn parent is natCu(p,xn)62Zn using intermediate energy (30-60 MeV) proton accelerators [110,112]. Using low-energy protons (5-14 MeV), 62Cu can be produced following 62Ni (p,n) 62Cu reaction [198,199]. As reported, the natZn (p,x)62Zn process can be adopted as a production route if protons of energies higher than 50 MeV are available [200]. The radiochemical processing step involves dissolution of the irradiated target in concentrated HNO3, reconstitution in 3 M HCl and separation from Cu using ion exchange chromatography (AG1-X8, Cl- form) [114].
72Se
Spurred by the perceived need to avail 72Se of requisite purity for the development of 72Se/72As generators, a number of production strategies have been described in the literature [142,146,201-203] and are elaborated in Table 9. When preparing for the production of 72Se, following one of the different production pathways, a thorough assessment of the production yield, the ease of the required separation chemistry, and the radiochemical purity of the 72Se product needs to be considered. It is pertinent to point out that highest production yield for 72Se is achieved following the proton induced reaction paths using RbBr salt targets, although this production approach is associated with challenges to isolate 72Se owing to the complexity of the product mixture in dissolved solution. While the irradiation of As targets with intermediate energy protons offers the next best yields for 72Se, chemical processing of multi-gram production targets is not only challenging but also associated with radioactive waste disposal issues because of co-production of long lived 75Se (t½ = 120 d).
Table 9.
Possible ways for the production of 72Se
| Reaction | Energy Range (MeV) | Yield [mCi (μA.h)-1] | ||
|---|---|---|---|---|
|
| ||||
| 72Se | 73Se | 75Se | ||
| 75As (p, 4n) | 45-24 | 0.21 | 57 | 0.03 |
| 75As (d, 5n) | 56-26 | 0.15 | 32 | 0.08 |
| natGe (3He, xn) | 36 | 0.08 | NR | NR |
| 70Ge (3He, xn) | 36 | 0.22 | NR | NR |
| Cu3 natGe (4He, xn) | 28-10 | 0.0005 | 0.09 | 0.0004 |
| Cu3 70Ge (4He, xn) | 28-10 | 0.002 | 0.4 | 0.0003 |
| KnatBr (p, x) | 100-69 | 0.10 | 7.18 | 0.12 |
| KnatBr (p, x) | 62-42 | 0.05 | 2.36 | 0.03 |
| RbnatBr (p, x) | 800 | 3.03 | 30.3 | 3.03 |
NR: Not reported.
Among the various possible routes to be considered for the production of 72Se, the use of alkaline bromide targets seems attractive, as they balance a relatively inexpensive and biologically benign target material with sufficiently high yields for the production of 72Se [148]. Other advantages include ease of dissolution of the irradiated target due to high solubility in water, and a reasonably high thermal conductivity. The target processing step involves dissolution of the irradiated target in dilute HCl. Quantitative removal of bromide is achieved by oxidation to elemental bromine, followed by distillation. A liquid-liquid extraction system using dithiocarbamate/ethyl acetate is used to purify Se [148].
52Fe
A number of nuclear reactions using proton, 3He and α particles have been studied for the 52Fe production: 52Cr(3He,3n)52Fe [204], 50Cr(4He,2n)52Fe [205], 55Mn(p,4n)52Fe [157,206,207], natNi(p,x)52Fe [207]. While the 55Mn (p,4n)52Fe [157,206-208] route of production using a 65-70 MeV proton beam has been appealing, the beams available from most medical accelerators are below this energy. Therefore, attempts to prepare sufficient quantities of 52Fe following this path have met with limited success. The production of 52Fe by spallation suffers from the requirement of a high energy cyclotron and the coproduction of 59Fe, a troublesome contaminant with a 45 day half-life.
The irradiation of Mn with 65-70 MeV protons is expected to produce isotopes of Fe and Mn, and possibly of Cr with reasonable yield, while V and Ti radionuclides will be formed in very small quantities [206]. The irradiated target is dissolved in 6 N HCl containing H2O2. In order to avail 52Fe of required quality, these radionuclides were separated using an ion-exchange chromatography method with Dowex-1 × 8, wherein the Fe gets trapped and the Mn, Na, Cr, and Al pass through the resin column with the 6 N HCl loading solution. Fe retained on the column is eluted with 0.5 N HCl. This step is repeated 2-3 times to obtain Fe of required purity [206]. One of the major limitations of this production route is that 52Fe produced is contaminated with 55Fe impurity.
The use of the 50Cr(4He,2n)52Fe nuclear reaction with an enriched 50Cr target and an irradiation with an ~40 MeV 4He beam seemed to be appealing option [209] as it precluded the production of the long-lived 59Fe isotope. However, it requires the recycling of precious enriched 50Cr target in order to make the production cost effective. The irradiated target contains 52Mn, 54Mn, 56Mn, 49Cr, 51Cr, 48V and 55Fe radionuclides in addition to the 52Fe. Separation of 52Fe from radioactive contaminants, as well as 50Cr, is achieved by solvent extraction of Fe(III) from 8 M HCI using di-isopropyl ether (DIPE) [209].
122Xe
The nuclear reactions used for the production of 122Xe are: 120Te(α, 2n)122Xe, 122Te(α, 4n)122Xe, 124Xe(p, 3n)122Cs which decays to 122Xe, 124Xe(p, p2n)122Xe and 127I(p,6n)122Xe [210]. The proton induced reaction using 127I is most abundantly used because the product and reactants are chemically different. The maximum cross-section for the 127I(p,6n)122Xe reaction is expected to be in the 72-74 MeV energy range [162,211]. While the use of a higher energy proton beam results in significantly higher 122Xe yields, it also leads to the production of other radioisotopes of iodine such as 121I, 123I, 125I due to the decay of 121Xe, 123Xe, 125Xe coproduced in the same energy region. With the aim to reduce the radiation dose hazard and to minimize the level of radioiodine isotopic impurities, proton energy in the range of 72-74 MeV energy is desirable. However, 123Xe is also obtained as a byproduct of irradiation of NaI targets due to l27I(p, 5n)123Xe reaction with 67.5 MeV proton beams and this is undesirable [211]. 124Xe(p, 3n)122Cs which decays to 122Xe, and 124Xe(p, p2n)122Xe are not used for production for economic reasons due to the requirement of enriched and expensive target material which cannot be recovered for recycling as the separation of 124Xe from the 122Xe is not feasible.
While the use of 120Te(α, 2n)122Xe and 122Te(α, 4n)122Xe reactions offer the potential of availing 122Xe, the requirement of 30-40-MeV α beams, expeditious as well as continuous removal of the radioxenons formed using solid/gas separation technique, and low natural abundances of 120Te (0.096%) and 122Te (2.60%) have emerged as the major impediments that discourage their large scale adaptability. Use of expensive enriched target material, the need to recover the target, and restricted availability of an accelerator to produce intense 30-40 MeV α beams are other major hurdles that must be tackled while following this route [211].
The production of 122Xe by spallation using CsI, La2O3 or BaCO3 targets and protons of energies from 320 to 590 MeV has also been reported by Peek and Hegedus [212]. Production of many xenon isotopes, such as 127Xe, 125Xe, 123Xe, 121Xe, during spallation and the requirement of > 200-MeV proton beams have emerged as the major deterrents that limit the utility of this production pathway.
Radiation safety
Radiation safety is another important part of the RNG development process. Because of the recent advancements in cyclotron targetry, radiochemistry, and separation techniques, new RNGs capable of providing PET radionuclides with different decay characteristics are entering biomedical research and seem poised to enter into clinical practices. Therefore, there is a clear need to reconsider the radiation safety aspects in the development of PET RNGs. Most of the RNGs are appropriately placed in a shielded container, designed to enable elution of the daughter radionuclide. Meeting the regulatory requirements for safe operation is an expensive proposition, but constitutes a necessity. Careful planning with the design engineer, generator architect, and a qualified medical physicist is necessary to produce a cost-effective design while maintaining the radiation safety standard.
With the aim to minimize both manufacturing and transportation expenses, and to make the system portable, it is essential to design an RNG such that the amount of shielding can be kept as small as possible. Adequate shielding is essential to reduce the risk of radiation exposure to workers. The purpose of the RNG shielding is to attenuate radiation by absorption (and to some extent scattering). This protects radiation workers by reducing their exposure. The design of proper shielding requires the use of accurate γ-ray dose (C) constants and tenth value layers (TVLs). In order to achieve the greatest possible radiation safety, the amount of radiation exposure from RNGs should be kept below a safe limit following the “ALARA” (As Low As Reasonably Achievable) principle. Based on the type of PET radionuclide, the design and radiation safety features of RNGs need to be adjusted appropriately.
Regulatory need
While the use of PET RNGs is a successful paradigm for ensuring sustained growth and future expansion of PET, their development and the possibility of their clinical translation have been stymied by a number of factors, such as: the complexity of pharmaceutical legislation and regulations, the lengthy process of obtaining a marketing authorization, and, in some cases, a limited return on investment, as well as lack of consensus. Despite the encouraging prospects and the favorable results of many PET RNGs, there is still quite a long way to go before any one of them become standard for widespread use in daily NM routine.
Among many other requirements, such as those relating to establishment of chemical and radionuclidic purity of the PET radionuclide obtained from an RNG, clinical use of RNGs is dependent on the condition that they are manufactured under conditions of good manufacturing practice (GMP). Radionuclides obtained from RNGs are considered as approved pharmaceutical ingredients (API) as they are used as a starting material for the preparation of radiopharmaceuticals for human use [213], and therefore subjected to regulatory approval to ensure both quality and safety. The emphasis on quality is most prominently manifested by the fact that not only do radionuclides obtained from the generators have to meet strict specifications but also the separation processes and associated accessories must fulfill the preset criteria.
Directive 2001/83 of European Union [214] states that all medicinal products must comply with the current standards of GMP. In Europe, these GMP rules have legal character as they are issued by the European Medicines Agency (EMEA) and the DG Enterprise and Industry of the European Commission. They are published online in the Eudralex, a web-based compendium of European pharmaceutical legislation [215]. However, Directive 2001/83, the basis for European GMP as outlined in Eudralex, only has a mandate for the industrial manufacture of medicinal products and does not legally cover the small-scale, patient-specific preparation, e.g., in pharmacies. Similarly, the US FDA approved a set of regulations describing production of PET radiopharmaceuticals according to current good manufacturing practice (cGMP), outlined in the Code of Federal Regulations (21CFR212), [216]. Because of the differences and complexities of the regulatory machinery in different countries, it would be expected that each country is different, and the introduction of an RNG to avail PET radionuclides for routine PET imaging would, of course, depend on regional and country regulations. Use of PET RNGs in radiopharmacies requires either dedicated radionuclide specific radio-synthesizers or kit-based, micro-scale radio-synthesizers that offer fully integrated radiochemistry solutions on the bench top to synthesize the radiotracers, and quality control (QC) testing equipment [e.g. gas chromatography (GC), analytical radio-high-performance liquid chromatography (radio-HPLC), dose calibrators, radio-thin layer chromatography (radio-TLC), etc.] to ensure the safety of the patient. Additionally, it also requires the availability specially trained and skilled personnel to operate the entire process, from generator elution to the aseptic dose preparation.
In light of the perceived need to address regulatory requirements and to achieve Current Good Manufacturing Practices (cGMP) compliance, the field of RNGs is increasingly migrating toward the use of automated systems. Automation offers several advantages, including providing robust, repeatable processes, reducing the radiation dose to operators, offering consistent performance of the generator system, ability to handle and distribute multiple doses daily, and providing a log of the steps performed in the elution of the medical radionuclide [217]. Automation also facilitates regulatory compliance through manufacturer design qualification/operational qualification/performance qualifications and scheduled maintenance protocols performed on automated RNGs by trained service engineers.
Due to the short half-life of most of the generator produced radionuclides, the prospect of using kit-type formulation strategies seemed sagacious, as it would reduce reagent preparation and setup time, curtail human errors, eliminate the possibility of contamination, facilitate cleanup, and simplify operation. A further advantage of kits is the simplification of reagent handling. Rather than individually managing multiple reagents, the radiopharmacy need only to manage the kit as a single unit, greatly simplifying compliance with FDA regulations concerning production of PET tracers for injection into humans. Kit formulation strategies are beginning to put the capability to make PET tracers directly into the hands of the scientists and clinicians who need them.
Summary and outlook
A review of the current use and potential applications of PET RNGs indicates that this dynamic field is evolving rapidly from being an “art rather than a science”, to a radionuclide delivery system. This multidisciplinary technological paradigm, which resides at the intersection of radiochemistry and chemical separation techniques, has undergone a tremendous metamorphosis and is destined to bring perpetual changes in clinical PET practices. New PET generators are emerging persistently, taking shape progressively and after maturation, steadily migrating towards clinical practice. The relentless parade of new PET generators, destined to unfold on many new fronts in clinical PET, harbor boundless possibilities and a wellspring to spur myriad applications in clinical PET. Almost every advance in PET RNGs is billed as a breakthrough, and the list of “next generation generators” grows longer every day. While the potential benefits of the new PET generators are tremendous, so are the challenges of bringing them from laboratory research to clinical practice for their impact.
RNGs have their roots in NM, and progress is inextricably linked to the continued research and advancements of PET imaging in NM. As PET is moving to the forefront of NM, demands for new radionuclides are emerging far more quickly than they did over the past decade. Because of the pace at which the PET scene is evolving, RNG strategies need a vision for today and tomorrow. Given the importance of PET in molecular imaging, paradigm-changing PET RNGs are quick to grab the headlines.
The advances made so far are exciting and efforts to develop new generators for use in clinical practices are evolving persistently. While the clinical realization of any of these paradigm-changing new PET RNGs would be expected to revitalize current PET practices and provide myriad opportunities to instill new possibilities to reshape the clinical PET of today and tomorrow, it requires effective harnessing of emerging technology and inspired vision from clinical as well as industrial partners to provide functional systems in appropriate volumes at the right cost. In this premise, each new generator and associated daughter PET radionuclide must be evaluated on the basis of their performance, affordability, cost effectiveness, and propitious clinical outcome. “Synergy, understanding, and collaboration” have emerged as the three most common descriptors that underpin their survival and growth.
Much has been written about the 82Sr/82Rb generator due to its dominance in the field of cardiac PET, but this example should really be viewed largely as a model that can be copied for several alternative PET RNGs. They deserve greater attention not only because a greater range of PET radionuclides will be needed, but also for the adaptability to use different radionuclides for different applications. With a better understanding of the properties, including the physical and chemical properties of each of the radionuclides, a wide range of PET radiopharmaceuticals can be prepared which can be effectively used for molecular imaging of targets for a broad range of biological or disease processes. Despite being a late entrant to PET, the 68Ge/68Ga generator has not only consolidated its presence but also established a strong foothold at the forefront of PET. In a relatively short time-span, 68Ga has virtually pervaded all areas of PET and has become a key PET radionuclide of choice for the foreseeable future. It may be surmised that diffusion of 68Ga tracers seem poised to bring spectacular developments and serve as the springboard to spur new breakthroughs in PET imaging.
The restricted clinical utility of PET in diagnostic NM is mainly due to limited availability of this technology, its cost, and limited published data supporting its use. In recent years, PET imaging has moved from an exotic diagnostic modality for very few patients to a mainstream modality. The future of generator derived PET radionuclides is, of course, difficult to predict, and there will be surprising inventions, as in the past, in which PET radionuclides availed from a generator may have an unexpected application that will continue to fuel development the field. Healthcare service providers should seize the opportunity by keeping their organizational strategies updated in the face of continually evolving PET radiotracer technologies and ensure that their organizations continue to pursue optimum PET radiotracers as well as innovative technologies in an effort to reduce the gap between requirements and capabilities.
In spite of the huge potential of PET RNGs, cost-effective availability of parent radionuclides is one major challenge which currently obstructs the path for their widespread progress. To traverse these obstacles, a constant and reliable supply of parent radionuclides of the required quality in the desired quantities needs to be assured. Owing to the inherent requirement of capital intensive installation of cyclotrons, significant expertise, skilled manpower and adequate resources to undertake regular production of radionuclides, the number of commercial radioisotope suppliers remains finite and their current production capabilities remain limited. Additional research efforts and large resources are warranted to undertake their production on a regular basis to meet the current and foreseeable demands.
There are several reasons why marketing authorization of PET RNGs are not attractive. First, the economic investment costs for a PET RNG with a very small potential market cannot be recouped. Second, an RNG housing a short-lived parent radionuclide is unattractive and the investment required for its large-scale production would usually require the availability of targets as well as high energy accelerators. Furthermore, such short-lived parent radionuclides would have to be produced regularly to replenish the generators. This demands extremely efficient logistics for the overall process of availing the proven benefit of PET tracers.
The clinical success of a new RNG largely depends on its application, cost-effective availability of the parent radionuclide, the overall cost associated with its GMP production, and the process of obtaining regulatory approval, which is gradually increasing. The process of converting a promising RNG for the clinical application is rife with scientific and regulatory obstacles. In order to surmount the regulatory barrier and become accepted in daily clinical practice, PET RNGs will likely need a strong corporate sponsor willing to invest. Industry sponsorship is possible in this era because academic inventors now routinely secure strong intellectual property rights on RNGs. If strong intellectual property rights are secured and industry makes a substantial investment, the cost of new PET RNGs will be significantly high, which may lead to few early users and consequently inhibit their widespread use as well as convergence into standard clinical practice. In order to mitigate such unforeseen situations, a more holistic, pragmatic and outcome-oriented regulatory approach is required. NM needs leaders who are capable of managing both technical and non-technical issues, whether in academia, business, government or the medical sector. It is the responsibility of all the stake holders, including research scientists, clinicians, radiopharmacists, hospitals and industries to share a common platform to convince regulatory authorities of the need for change to address the challenges in an ever-demanding regulatory environment. While the utility of generator derived PET tracers has passed many milestones and made considerable inroads into the arena of PET imaging, but without a doubt this is just the tip of the iceberg and further excitement in this field is promised in the foreseeable future.
Acknowledgements
Research at the Bhabha Atomic Research Centre is part of the ongoing activities of the Department of Atomic Energy, India and is fully supported by government funding. Figures of commercial generators were taken from the product brochure of respective manufacturers. Figures of 82Rb infusion system have been derived from the product brochures of Bracco Diagnostics Inc., United States. The author gratefully acknowledges the manufacturers while reproducing the figures and information in this article.
Disclosure of conflict of interest
None.
References
- 1.Castello A, Olivari L, Lopci E. Adenoid cystic carcinoma: focus on heavy ion therapy and molecular imaging. Am J Nucl Med Mol Imaging. 2018;8:1–14. [PMC free article] [PubMed] [Google Scholar]
- 2.Davidson CQ, Phenix CP, Tai TC, Khaper N, Lees SJ. Searching for novel PET radiotracers: imaging cardiac perfusion, metabolism and inflammation. Am J Nucl Med Mol Imaging. 2018;8:200–227. [PMC free article] [PubMed] [Google Scholar]
- 3.Filippi L, Chiaravalloti A, Bagni O, Schillaci O. 18F-labeled radiopharmaceuticals for the molecular neuroimaging of amyloid plaques in Alzheimer’s disease. Am J Nucl Med Mol Imaging. 2018;8:268–281. [PMC free article] [PubMed] [Google Scholar]
- 4.Hagemann CE, Ghotbi AA, Kjaer A, Hasbak P. Quantitative myocardial blood flow with Rubidium-82 PET: a clinical perspective. Am J Nucl Med Mol Imaging. 2015;5:457–468. [PMC free article] [PubMed] [Google Scholar]
- 5.Houshmand S, Salavati A, Hess S, Ravina M, Alavi A. The role of molecular imaging in diagnosis of deep vein thrombosis. Am J Nucl Med Mol Imaging. 2014;4:406–425. [PMC free article] [PubMed] [Google Scholar]
- 6.Jensen MM, Kjaer A. Monitoring of anti-cancer treatment with 18F-FDG and 18F-FLT PET: a comprehensive review of pre-clinical studies. Am J Nucl Med Mol Imaging. 2015;5:431–456. [PMC free article] [PubMed] [Google Scholar]
- 7.Karakatsanis NA, Fokou E, Tsoumpas C. Dosage optimization in positron emission tomography: state-of-the-art methods and future prospects. Am J Nucl Med Mol Imaging. 2015;5:527–547. [PMC free article] [PubMed] [Google Scholar]
- 8.Keiding S, Sorensen M, Frisch K, Gormsen LC, Munk OL. Quantitative PET of liver functions. Am J Nucl Med Mol Imaging. 2018;8:73–85. [PMC free article] [PubMed] [Google Scholar]
- 9.Li F, Joergensen JT, Hansen AE, Kjaer A. Kinetic modeling in PET imaging of hypoxia. Am J Nucl Med Mol Imaging. 2014;4:490–506. [PMC free article] [PubMed] [Google Scholar]
- 10.Lopci E, Grassi I, Chiti A, Nanni C, Cicoria G, Toschi L, Fonti C, Lodi F, Mattioli S, Fanti S. PET radiopharmaceuticals for imaging of tumor hypoxia: a review of the evidence. Am J Nucl Med Mol Imaging. 2014;4:365–384. [PMC free article] [PubMed] [Google Scholar]
- 11.Sarikaya I. PET studies in epilepsy. Am J Nucl Med Mol Imaging. 2015;5:416–430. [PMC free article] [PubMed] [Google Scholar]
- 12.Vali R, Loidl W, Pirich C, Langesteger W, Beheshti M. Imaging of prostate cancer with PET/CT using 18F-Fluorocholine. Am J Nucl Med Mol Imaging. 2015;5:96–108. [PMC free article] [PubMed] [Google Scholar]
- 13.Chakravarty R, Goel S, Hong H, Chen F, Valdovinos HF, Hernandez R, Barnhart TE, Cai W. Hollow mesoporous silica nanoparticles for tumor vasculature targeting and PET image-guided drug delivery. Nanomedicine (Lond) 2015;10:1233–1246. doi: 10.2217/nnm.14.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakravarty R, Hong H, Cai W. Positron emission tomography image-guided drug delivery: current status and future perspectives. Mol Pharm. 2014;11:3777–3797. doi: 10.1021/mp500173s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quarles van Ufford HM, Quekel LG, van Waes PF, de Haas PJ, de Klerk JM. Diagnostic evaluation of separately acquired PET and CT images by nuclear medicine physicians and radiologists in cancer patients. Hell J Nucl Med. 2007;10:105–108. [PubMed] [Google Scholar]
- 16.Grammaticos P, Zerva C, Asteriadis I, Trontzos C, Hatziioannou K. Is hybridic positron emission tomography/computerized tomography the only option? The future of nuclear medicine and molecular imaging. Hell J Nucl Med. 2007;10:74–76. [PubMed] [Google Scholar]
- 17.Townsend DW. Positron emission tomography/computed tomography. Semin Nucl Med. 2008;38:152–166. doi: 10.1053/j.semnuclmed.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Cho MW, Chin BB. 18F-FDG PET/CT findings in hepatosplenic Gamma-Delta T-cell lymphoma: case reports and review of the literature. Am J Nucl Med Mol Imaging. 2018;8:137–142. [PMC free article] [PubMed] [Google Scholar]
- 19.Eary JF, Hawkins DS, Rodler ET, Conrad EU. 18F-FDG PET in sarcoma treatment response imaging. Am J Nucl Med Mol Imaging. 2011;1:47–53. [PMC free article] [PubMed] [Google Scholar]
- 20.Usmani S, Sit C, Gnanasegaran G, den Wyngaert TV, Marafi F. Pictorial atlas of symptomatic accessory ossicles by 18F-Sodium Fluoride (NaF) PET-CT. Am J Nucl Med Mol Imaging. 2017;7:275–282. [PMC free article] [PubMed] [Google Scholar]
- 21.Vallabhajosula S, Solnes L, Vallabhajosula B. A broad overview of positron emission tomography radiopharmaceuticals and clinical applications: what is new? Semin Nucl Med. 2011;41:246–264. doi: 10.1053/j.semnuclmed.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Mankoff DA, Katz SI. PET imaging for assessing tumor response to therapy. J Surg Oncol. 2018;118:362–373. doi: 10.1002/jso.25114. [DOI] [PubMed] [Google Scholar]
- 23.Chakraborty S, Chakravarty R, Sarma HD, Dash A, Pillai MR. The practicality of nanoceria-PAN-based 68Ge/68Ga generator toward preparation of 68Ga-labeled cyclic RGD dimer as a potential PET radiotracer for tumor imaging. Cancer Biother Radiopharm. 2013;28:77–83. doi: 10.1089/cbr.2012.1252. [DOI] [PubMed] [Google Scholar]
- 24.Chakravarty R, Chakraborty S, Ram R, Vatsa R, Bhusari P, Shukla J, Mittal BR, Dash A. Detailed evaluation of different 68Ge/68Ga generators: an attempt toward achieving efficient 68Ga radiopharmacy. J Labelled Comp Radiopharm. 2016;59:87–94. doi: 10.1002/jlcr.3371. [DOI] [PubMed] [Google Scholar]
- 25.Chakravarty R, Chakraborty S, Shukla R, Bahadur J, Ram R, Mazumder S, Dev Sarma H, Tyagi AK, Dash A. Mechanochemical synthesis of mesoporous tin oxide: a new generation nanosorbent for 68Ge/68Ga generator technology. Dalton Trans. 2016;45:13361–13372. doi: 10.1039/c6dt01921h. [DOI] [PubMed] [Google Scholar]
- 26.Chakravarty R, Dash A, Pillai MR. Availability of yttrium-90 from strontium-90: a nuclear medicine perspective. Cancer Biother Radiopharm. 2012;27:621–641. doi: 10.1089/cbr.2012.1285. [DOI] [PubMed] [Google Scholar]
- 27.Chakravarty R, Dash A, Pillai MR. Electrochemical separation is an attractive strategy for development of radionuclide generators for medical applications. Curr Radiopharm. 2012;5:271–287. doi: 10.2174/1874471011205030271. [DOI] [PubMed] [Google Scholar]
- 28.Chakravarty R, Ram R, Pillai KT, Pamale Y, Kamat RV, Dash A. Ammonium molybdophosphate impregnated alumina microspheres as a new generation sorbent for chromatographic 137Cs/137mBa generator. J Chromatogr A. 2012;1220:82–91. doi: 10.1016/j.chroma.2011.11.059. [DOI] [PubMed] [Google Scholar]
- 29.Chakravarty R, Shukla R, Ram R, Venkatesh M, Tyagi AK, Dash A. Exploitation of nano alumina for the chromatographic separation of clinical grade 188Re from 188W: a renaissance of the 188W/188Re generator technology. Anal Chem. 2011;83:6342–6348. doi: 10.1021/ac201232m. [DOI] [PubMed] [Google Scholar]
- 30.Chakravarty R, Dash A. Nanomaterial-based adsorbents: the prospect of developing new generation radionuclide generators to meet future research and clinical demands. J Radioanal Nucl Chem. 2014;299:741–757. [Google Scholar]
- 31.Dash A, Chakravarty R. Pivotal role of separation chemistry in the development of radionuclide generators to meet clinical demands. RSC Adv. 2014;4:42779–42803. [Google Scholar]
- 32.Dash A, Knapp FF Jr, Pillai MR. 99Mo/99mTc separation: an assessment of technology options. Nucl Med Biol. 2013;40:167–176. doi: 10.1016/j.nucmedbio.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 33.Amor-Coarasa A, Schoendorf M, Meckel M, Vallabhajosula S, Babich JW. Comprehensive Quality control of the ITG 68Ge/68Ga generator and synthesis of 68Ga-DOTATOC and 68Ga-PSMA-HBED-CC for clinical imaging. J Nucl Med. 2016;57:1402–1405. doi: 10.2967/jnumed.115.171249. [DOI] [PubMed] [Google Scholar]
- 34.Maecke HR, André JP. 68Ga-PET radiopharmacy: a generator-based alternative to 18F-radiopharmacy. Ernst Schering Res Found Workshop. 2007:215–242. doi: 10.1007/978-3-540-49527-7_8. [DOI] [PubMed] [Google Scholar]
- 35.Martiniova L, Palatis L, Etchebehere E, Ravizzini G. Gallium-68 in medical imaging. Curr Radiopharm. 2016;9:187–207. doi: 10.2174/1874471009666161028150654. [DOI] [PubMed] [Google Scholar]
- 36.Prata MI. Gallium-68: a new trend in PET radiopharmacy. Curr Radiopharm. 2012;5:142–149. doi: 10.2174/1874471011205020142. [DOI] [PubMed] [Google Scholar]
- 37.Velikyan I, Lindhe O. Preparation and evaluation of a 68Ga-labeled RGD-containing octapeptide for noninvasive imaging of angiogenesis: biodistribution in non-human primate. Am J Nucl Med Mol Imaging. 2018;8:15–31. [PMC free article] [PubMed] [Google Scholar]
- 38.Velikyan I, Rosenstrom U, Eriksson O. Fully automated GMP production of [68Ga]Ga-DO3A-VS-Cys(40)-Exendin-4 for clinical use. Am J Nucl Med Mol Imaging. 2017;7:111–125. [PMC free article] [PubMed] [Google Scholar]
- 39.Rosch F. Past, present and future of 68Ge/68Ga generators. Appl Radiat Isot. 2013;76:24–30. doi: 10.1016/j.apradiso.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 40.Roesch F. Maturation of a key resource - the germanium-68/gallium-68 generator: development and new insights. Curr Radiopharm. 2012;5:202–211. doi: 10.2174/1874471011205030202. [DOI] [PubMed] [Google Scholar]
- 41.Gleason GI. A positron cow. Int J Appl Radiat Isot. 1960;8:90–94. doi: 10.1016/0020-708x(60)90052-1. [DOI] [PubMed] [Google Scholar]
- 42.Greene MW, Tucker WD. An improved gallium-68 cow. Appl Radiat Isot. 1961;12:62–63. [Google Scholar]
- 43.Kopecký P, Mudrová B. 68Ge/68Ga generator for the production of 68Ga in an ionic form. Appl Radiat Isot. 1974;25:263–268. [Google Scholar]
- 44.Malyshev KV, Smirnov VV. A generator of galium-68 based on zirconium hydroxide. Radiokhimiya. 1975;17:137–140. [Google Scholar]
- 45.Arino H, Skraba WJ, Kramer HH. A new 68Ge/68Ga radioisotope generator system. Appl Radiat Isot. 1978;29:117–120. [Google Scholar]
- 46.Loc’h C, Maziere B, Comar D. A new generator for ionic gallium-68. J Nucl Med. 1980;21:171–173. [PubMed] [Google Scholar]
- 47.Neirinckx RD, Davis MA. Potential column chromatography generators for ionic Ga-68. I. Inorganic substrates. J Nucl Med. 1979;20:1075–1079. [PubMed] [Google Scholar]
- 48.Neirinckx RD, Davis MA. Potential column chromatography for ionic Ga-68. II: organic ion exchangers as chromatographic supports. J Nucl Med. 1980;21:81–83. [PubMed] [Google Scholar]
- 49.McElvany KD, Hopkins KT, Welch MJ. Comparison of 68Ge/68Ga generator systems for radiopharmaceutical production. Appl Radiat Isot. 1984;35:521–524. [Google Scholar]
- 50.Ambe S. 68Ge/68Ga generator with alpha-ferric oxide support. Appl Radiat Isot. 1988;39:49–51. [Google Scholar]
- 51.Razbash AA, Sevastianov YG, Krasnov NN, Leonov AI, Panickin VE. Germanium-68 row of products; Proceedings of the 5th International Conference on Isotopes 5ICI; April 25-29; Brussels, Belgium. 2005. pp. 147–151. [Google Scholar]
- 52.Majkowska-Pilip A, Bilewicz A. Macrocyclic complexes of scandium radionuclides as precursors for diagnostic and therapeutic radiopharmaceuticals. J Inorg Biochem. 2011;105:313–320. doi: 10.1016/j.jinorgbio.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 53.Eppard E, Wuttke M, Nicodemus PL, Rosch F. Ethanol-based post-processing of generator-derived 68Ga toward kit-type preparation of 68Ga-radiopharmaceuticals. J Nucl Med. 2014;55:1023–1028. doi: 10.2967/jnumed.113.133041. [DOI] [PubMed] [Google Scholar]
- 54.Zhernosekov KP, Filosofov DV, Baum RP, Aschoff P, Bihl H, Razbash AA, Jahn M, Jennewein M, Rosch F. Processing of generator-produced 68Ga for medical application. J Nucl Med. 2007;48:1741–1748. doi: 10.2967/jnumed.107.040378. [DOI] [PubMed] [Google Scholar]
- 55.Asti M, De Pietri G, Fraternali A, Grassi E, Sghedoni R, Fioroni F, Roesch F, Versari A, Salvo D. Validation of 68Ge/68Ga generator processing by chemical purification for routine clinical application of 68Ga-DOTATOC. Nucl Med Biol. 2008;35:721–724. doi: 10.1016/j.nucmedbio.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 56.Mueller D, Klette I, Baum RP, Gottschaldt M, Schultz MK, Breeman WA. Simplified NaCl based 68Ga concentration and labeling procedure for rapid synthesis of 68Ga radiopharmaceuticals in high radiochemical purity. Bioconjug Chem. 2012;23:1712–1717. doi: 10.1021/bc300103t. [DOI] [PubMed] [Google Scholar]
- 57.Loktionova NS, Belozub AN, Filosofov DV, Zhernosekov KP, Wagner T, Turler A, Rosch F. Improved column-based radiochemical processing of the generator produced 68Ga. Appl Radiat Isot. 2011;69:942–946. doi: 10.1016/j.apradiso.2011.02.035. [DOI] [PubMed] [Google Scholar]
- 58.Rosch F. Post-processing via cation exchange cartridges: versatile options. Recent Results Cancer Res. 2013;194:33–42. doi: 10.1007/978-3-642-27994-2_3. [DOI] [PubMed] [Google Scholar]
- 59.Mueller D, Klette I, Baum RP. Purification and labeling strategies for 68Ga from 68Ge/ 68Ga generator eluate. Recent Results Cancer Res. 2013;194:77–87. doi: 10.1007/978-3-642-27994-2_5. [DOI] [PubMed] [Google Scholar]
- 60.Velikyan I. Prospective of 68Ga-radiopharmaceutical development. Theranostics. 2013;4:47–80. doi: 10.7150/thno.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chakravarty R, Shukla R, Ram R, Venkatesh M, Dash A, Tyagi AK. Nanoceria-PAN composite-based advanced sorbent material: a major step forward in the field of clinical-grade 68Ge/68Ga generator. ACS Appl Mater Interfaces. 2010;2:2069–2075. doi: 10.1021/am100325s. [DOI] [PubMed] [Google Scholar]
- 62.Chakravarty R, Chakraborty S, Ram R, Dash A, Pillai MR. Long-term evaluation of ‘BARC 68Ge/68Ga generator’ based on the nanoceria-polyacrylonitrile composite sorbent. Cancer Biother Radiopharm. 2013;28:631–637. doi: 10.1089/cbr.2012.1470. [DOI] [PubMed] [Google Scholar]
- 63.Chakravarty R, Shukla R, Ram R, Tyagi AK, Dash A, Venkatesh M. Development of a nano-zirconia based 68Ge/68Ga generator for biomedical applications. Nucl Med Biol. 2011;38:575–583. doi: 10.1016/j.nucmedbio.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 64.Bartholomä MD. Recent developments in the design of bifunctional chelators for metal-based radiopharmaceuticals used in Positron Emission Tomography. Inorganica Chimica Acta. 2012;389:36–51. [Google Scholar]
- 65.Chakravarty R, Chakraborty S, Radhakrishnan ER, Kamaleshwaran K, Shinto A, Dash A. Clinical 68Ga-PET: is radiosynthesis module an absolute necessity? Nucl Med Biol. 2017;46:1–11. doi: 10.1016/j.nucmedbio.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 66.Abongwa C, Mott S, Schafer B, McNeely P, Abusin G, O’Dorisio T, Zamba G, O’Dorisio MS, Menda Y. Safety and accuracy of 68Ga-DOTATOC PET/CT in children and young adults with solid tumors. Am J Nucl Med Mol Imaging. 2017;7:228–235. [PMC free article] [PubMed] [Google Scholar]
- 67.Afzelius P, Nielsen OL, Alstrup AK, Bender D, Leifsson PS, Jensen SB, Schonheyder HC. Biodistribution of the radionuclides 18F-FDG, 11C-methionine, 11C-PK11195, and 68Ga-citrate in domestic juvenile female pigs and morphological and molecular imaging of the tracers in hematogenously disseminated Staphylococcus aureus lesions. Am J Nucl Med Mol Imaging. 2016;6:42–58. [PMC free article] [PubMed] [Google Scholar]
- 68.Esfahani SA, Salcedo S, Heidari P, Catalano OA, Pauplis R, Hesterman J, Kronauge JF, Mahmood U. A phase one, single-dose, open-label, clinical safety and PET/MR imaging study of 68Ga-DOTATOC in healthy volunteers. Am J Nucl Med Mol Imaging. 2017;7:53–62. [PMC free article] [PubMed] [Google Scholar]
- 69.Madru R, Tran TA, Axelsson J, Ingvar C, Bibic A, Stahlberg F, Knutsson L, Strand SE. 68Ga-labeled superparamagnetic iron oxide nanoparticles (SPIONs) for multi-modality PET/MR/Cherenkov luminescence imaging of sentinel lymph nodes. Am J Nucl Med Mol Imaging. 2013;4:60–69. [PMC free article] [PubMed] [Google Scholar]
- 70.Mojtahedi A, Alavi A, Thamake S, Amerinia R, Ranganathan D, Tworowska I, Delpassand ES. Assessment of vulnerable atherosclerotic and fibrotic plaques in coronary arteries using 68Ga-DOTATATE PET/CT. Am J Nucl Med Mol Imaging. 2015;5:65–71. [PMC free article] [PubMed] [Google Scholar]
- 71.Olsen IH, Langer SW, Federspiel BH, Oxbol J, Loft A, Berthelsen AK, Mortensen J, Oturai P, Knigge U, Kjaer A. 68Ga-DOTATOC PET and gene expression profile in patients with neuroendocrine carcinomas: strong correlation between PET tracer uptake and gene expression of somatostatin receptor subtype 2. Am J Nucl Med Mol Imaging. 2016;6:59–72. [PMC free article] [PubMed] [Google Scholar]
- 72.Retamal J, Sorensen J, Lubberink M, Suarez-Sipmann F, Borges JB, Feinstein R, Jalkanen S, Antoni G, Hedenstierna G, Roivainen A, Larsson A, Velikyan I. Feasibility of 68Ga-labeled Siglec-9 peptide for the imaging of acute lung inflammation: a pilot study in a porcine model of acute respiratory distress syndrome. Am J Nucl Med Mol Imaging. 2016;6:18–31. [PMC free article] [PubMed] [Google Scholar]
- 73.Selvaraju RK, Bulenga TN, Espes D, Lubberink M, Sorensen J, Eriksson B, Estrada S, Velikyan I, Eriksson O. Dosimetry of [68Ga]Ga-DO3A-VS-Cys(40)-Exendin-4 in rodents, pigs, non-human primates and human - repeated scanning in human is possible. Am J Nucl Med Mol Imaging. 2015;5:259–269. [PMC free article] [PubMed] [Google Scholar]
- 74.Decristoforo C, Pickett RD, Verbruggen A. Feasibility and availability of 68Ga-labelled peptides. Eur J Nucl Med Mol Imaging. 2012;39(Suppl 1):S31–40. doi: 10.1007/s00259-011-1988-5. [DOI] [PubMed] [Google Scholar]
- 75.Win Z, Al-Nahhas A, Rubello D, Gross MD. Somatostatin receptor PET imaging with Gallium-68 labeled peptides. Q J Nucl Med Mol Imaging. 2007;51:244–250. [PubMed] [Google Scholar]
- 76.Persson M, Madsen J, Ostergaard S, Ploug M, Kjaer A. 68Ga-labeling and in vivo evaluation of a uPAR binding DOTA- and NODAGA-conjugated peptide for PET imaging of invasive cancers. Nucl Med Biol. 2012;39:560–569. doi: 10.1016/j.nucmedbio.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 77.Mojtahedi A, Thamake S, Tworowska I, Ranganathan D, Delpassand ES. The value of 68Ga-DOTATATE PET/CT in diagnosis and management of neuroendocrine tumors compared to current FDA approved imaging modalities: a review of literature. Am J Nucl Med Mol Imaging. 2014;4:426–434. [PMC free article] [PubMed] [Google Scholar]
- 78.Wild D, Bomanji JB, Benkert P, Maecke H, Ell PJ, Reubi JC, Caplin ME. Comparison of 68Ga-DOTANOC and 68Ga-DOTATATE PET/CT within patients with gastroenteropancreatic neuroendocrine tumors. J Nucl Med. 2013;54:364–372. doi: 10.2967/jnumed.112.111724. [DOI] [PubMed] [Google Scholar]
- 79.FDA grants orphan drug designation for 68Ga-DOTATOC. J Nucl Med. 2014;55:10N. [Google Scholar]
- 80.Jensen RT, Battey JF, Spindel ER, Benya RV. International Union of Pharmacology. LXVIII. Mammalian bombesin receptors: nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacol Rev. 2008;60:1–42. doi: 10.1124/pr.107.07108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kahkonen E, Jambor I, Kemppainen J, Lehtio K, Gronroos TJ, Kuisma A, Luoto P, Sipila HJ, Tolvanen T, Alanen K, Silen J, Kallajoki M, Roivainen A, Schafer N, Schibli R, Dragic M, Johayem A, Valencia R, Borkowski S, Minn H. In vivo imaging of prostate cancer using [68Ga]-labeled bombesin analog BAY86-7548. Clin Cancer Res. 2013;19:5434–5443. doi: 10.1158/1078-0432.CCR-12-3490. [DOI] [PubMed] [Google Scholar]
- 82.Tateishi U, Oka T, Inoue T. Radiolabeled RGD peptides as integrin alphavbeta3-targeted PET tracers. Curr Med Chem. 2012;19:3301–3309. doi: 10.2174/092986712801215937. [DOI] [PubMed] [Google Scholar]
- 83.Sun Y, Zeng Y, Zhu Y, Feng F, Xu W, Wu C, Xing B, Zhang W, Wu P, Cui L, Wang R, Li F, Chen X, Zhu Z. Application of 68Ga-PRGD2 PET/CT for alphavbeta3-integrin imaging of myocardial infarction and stroke. Theranostics. 2014;4:778–786. doi: 10.7150/thno.8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Banerjee SR, Pullambhatla M, Byun Y, Nimmagadda S, Green G, Fox JJ, Horti A, Mease RC, Pomper MG. 68Ga-labeled inhibitors of prostate-specific membrane antigen (PSMA) for imaging prostate cancer. J Med Chem. 2010;53:5333–5341. doi: 10.1021/jm100623e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eder M, Schafer M, Bauder-Wust U, Hull WE, Wangler C, Mier W, Haberkorn U, Eisenhut M. 68Ga-complex lipophilicity and the targeting property of a urea-based PSMA inhibitor for PET imaging. Bioconjug Chem. 2012;23:688–697. doi: 10.1021/bc200279b. [DOI] [PubMed] [Google Scholar]
- 86.Afshar-Oromieh A, Zechmann CM, Malcher A, Eder M, Eisenhut M, Linhart HG, Holland-Letz T, Hadaschik BA, Giesel FL, Debus J, Haberkorn U. Comparison of PET imaging with a 68Ga-labelled PSMA ligand and 18F-choline-based PET/CT for the diagnosis of recurrent prostate cancer. Eur J Nucl Med Mol Imaging. 2014;41:11–20. doi: 10.1007/s00259-013-2525-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kilian K. 68Ga-DOTA and analogs: current status and future perspectives. Rep Pract Oncol Radiother. 2014;19:S13–S21. doi: 10.1016/j.rpor.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Doughton JA, Hofman MS, Eu P, Hicks RJ, Williams SG. A first-in-man study of 68Ga-nanocolloid PET-CT sentinel lymph node imaging in prostate cancer demonstrates aberrant lymphatic drainage pathways. J Nucl Med. 2018;59:1837–1842. doi: 10.2967/jnumed.118.209171. [DOI] [PubMed] [Google Scholar]
- 89.Evertsson M, Kjellman P, Cinthio M, Andersson R, Tran TA, In’t Zandt R, Grafstrom G, Toftevall H, Fredriksson S, Ingvar C, Strand SE, Jansson T. Combined magnetomotive ultrasound, PET/CT, and MR imaging of 68Ga-labelled superparamagnetic iron oxide nanoparticles in rat sentinel lymph nodes in vivo. Sci Rep. 2017;7:4824. doi: 10.1038/s41598-017-04396-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yoshinaga K, Klein R, Tamaki N. Generator-produced rubidium-82 positron emission tomography myocardial perfusion imaging-from basic aspects to clinical applications. J Cardiol. 2010;55:163–173. doi: 10.1016/j.jjcc.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 91.Alvarez-Diez TM, deKemp R, Beanlands R, Vincent J. Manufacture of strontium-82/rubidium-82 generators and quality control of rubidium-82 chloride for myocardial perfusion imaging in patients using positron emission tomography. Appl Radiat Isot. 1999;50:1015–1023. doi: 10.1016/s0969-8043(98)00170-5. [DOI] [PubMed] [Google Scholar]
- 92.J Epstein N, Benelfassi A, S B Beanlands R, DeKemp RA. An 82Rb infusion system for quantitative perfusion imaging with 3D PET. Appl Radiat Isot. 2004;60:921–927. doi: 10.1016/j.apradiso.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 93.Klein R, Adler A, Beanlands RS, Dekemp RA. Precision-controlled elution of a 82Sr/82Rb generator for cardiac perfusion imaging with positron emission tomography. Phys Med Biol. 2007;52:659–673. doi: 10.1088/0031-9155/52/3/009. [DOI] [PubMed] [Google Scholar]
- 94.Machac J. Cardiac positron emission tomography imaging. Semin Nucl Med. 2005;35:17–36. doi: 10.1053/j.semnuclmed.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 95.Bateman TM, Heller GV, McGhie AI, Friedman JD, Case JA, Bryngelson JR, Hertenstein GK, Moutray KL, Reid K, Cullom SJ. Diagnostic accuracy of rest/stress ECG-gated Rb-82 myocardial perfusion PET: comparison with ECG-gated Tc-99m sestamibi SPECT. J Nucl Cardiol. 2006;13:24–33. doi: 10.1016/j.nuclcard.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 96.Sampson UK, Dorbala S, Limaye A, Kwong R, Di Carli MF. Diagnostic accuracy of rubidium-82 myocardial perfusion imaging with hybrid positron emission tomography/computed tomography in the detection of coronary artery disease. J Am Coll Cardiol. 2007;49:1052–1058. doi: 10.1016/j.jacc.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 97.Lortie M, Beanlands RS, Yoshinaga K, Klein R, Dasilva JN, DeKemp RA. Quantification of myocardial blood flow with 82Rb dynamic PET imaging. Eur J Nucl Med Mol Imaging. 2007;34:1765–1774. doi: 10.1007/s00259-007-0478-2. [DOI] [PubMed] [Google Scholar]
- 98.Gould KL, Goldstein RA, Mullani NA, Kirkeeide RL, Wong WH, Tewson TJ, Berridge MS, Bolomey LA, Hartz RK, Smalling RW, Fuentes F, Nishikawa A. Noninvasive assessment of coronary stenoses by myocardial perfusion imaging during pharmacologic coronary vasodilation. VIII. Clinical feasibility of positron cardiac imaging without a cyclotron using generator-produced rubidium-82. J Am Coll Cardiol. 1986;7:775–789. doi: 10.1016/s0735-1097(86)80336-9. [DOI] [PubMed] [Google Scholar]
- 99.Merhige ME, Breen WJ, Shelton V, Houston T, D’Arcy BJ, Perna AF. Impact of myocardial perfusion imaging with PET and 82Rb on downstream invasive procedure utilization, costs, and outcomes in coronary disease management. J Nucl Med. 2007;48:1069–1076. doi: 10.2967/jnumed.106.038323. [DOI] [PubMed] [Google Scholar]
- 100.Groves AM, Speechly-Dick ME, Dickson JC, Kayani I, Endozo R, Blanchard P, Shastry M, Prvulovich E, Waddington WA, Ben-Haim S, Bomanji JB, McEwan JR, Ell PJ. Cardiac 82Rubidium PET/CT: initial European experience. Eur J Nucl Med Mol Imaging. 2007;34:1965–1972. doi: 10.1007/s00259-007-0537-8. [DOI] [PubMed] [Google Scholar]
- 101.Roesch F. Scandium-44: benefits of a long-lived PET radionuclide available from the 44Ti/44Sc generator system. Curr Radiopharm. 2012;5:187–201. doi: 10.2174/1874471011205030187. [DOI] [PubMed] [Google Scholar]
- 102.Filosofov DV, Loktionova NS, Rosch F. A 44Ti/44Sc radionuclide generator for potential nuclear-medical application of 44Sc-based PET-radiopharmaceuticals. Radiochim Acta. 2010;98:149–156. [Google Scholar]
- 103.Pruszynski M, Loktionova NS, Filosofov DV, Rosch F. Post-elution processing of 44Ti/44Sc generator-derived 44Sc for clinical application. Appl Radiat Isot. 2010;68:1636–1641. doi: 10.1016/j.apradiso.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 104.Viola-Villegas N, Doyle RP. The coordination chemistry of 1,4,7,10-tetraazacyclododecane-N,N’,N’’,N’’’-tetraacetic acid (H4DOTA): structural overview and analyses on structure-stability relationships. Coord Chem Rev. 2009;253:1906–1925. [Google Scholar]
- 105.Mausner LF, Joshi V, Kolsky KL, Meinken GE, Mease RC, Sweet MP, Srivastava SC. Evaluation of chelating agents for radioimmunotherapy with scandium-47. J Nucl Med. 1995;36:104. [Google Scholar]
- 106.Polosak M, Piotrowska A, Krajewski S, Bilewicz A. Stability of 47Sc-complexes with acyclic polyamino-polycarboxylate ligands. J Radioanal Nucl Chem. 2013;295:1867–1872. doi: 10.1007/s10967-012-2188-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pruszynski M, Majkowska-Pilip A, Loktionova NS, Eppard E, Roesch F. Radiolabeling of DOTATOC with the long-lived positron emitter 44Sc. Appl Radiat Isot. 2012;70:974–979. doi: 10.1016/j.apradiso.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 108.Muller C, Bunka M, Reber J, Fischer C, Zhernosekov K, Turler A, Schibli R. Promises of cyclotron-produced 44Sc as a diagnostic match for trivalent beta--emitters: in vitro and in vivo study of a 44Sc-DOTA-folate conjugate. J Nucl Med. 2013;54:2168–2174. doi: 10.2967/jnumed.113.123810. [DOI] [PubMed] [Google Scholar]
- 109.Laforest R, Dehdashti F, Lewis JS, Schwarz SW. Dosimetry of 60/61/62/64Cu-ATSM: a hypoxia imaging agent for PET. Eur J Nucl Med Mol Imaging. 2005;32:764–770. doi: 10.1007/s00259-004-1756-x. [DOI] [PubMed] [Google Scholar]
- 110.Robinson GD Jr, Zielinski FW, Lee AW. The zinc-62/copper-62 generator: a convenient source of copper-62 for radiopharmaceuticals. Int J Appl Radiat Isot. 1980;31:111–116. doi: 10.1016/0020-708x(80)90053-8. [DOI] [PubMed] [Google Scholar]
- 111.Fujibayashi Y, Matsumoto K, Yonekura Y, Konishi J, Yokoyama A. A new zinc-62/copper-62 generator as a copper-62 source for PET radiopharmaceuticals. J Nucl Med. 1989;30:1838–1842. [PubMed] [Google Scholar]
- 112.Zweit J, Goodall R, Cox M, Babich JW, Potter GA, Sharma HL, Ott RJ. Development of a high performance zinc-62/copper-62 radionuclide generator for positron emission tomography. Eur J Nucl Med. 1992;19:418–425. doi: 10.1007/BF00177368. [DOI] [PubMed] [Google Scholar]
- 113.Bormans G, Janssen A, Adriaens P, Crombez D, Witsenboer A, de Goeij J, Mortelmans L, Verbruggen A. A 62Zn/62Cu generator for the routine production of 62Cu-PTSM. Appl Radiat Isot. 1992;43:1437–1441. [Google Scholar]
- 114.Fukumura T, Okada K, Suzuki H, Nakao R, Mukai K, Szelecsenyi F, Kovacs Z, Suzuki K. An improved 62Zn/62Cu generator based on a cation exchanger and its fully remote-controlled preparation for clinical use. Nucl Med Biol. 2006;33:821–827. doi: 10.1016/j.nucmedbio.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 115.El-Azony KM. Improving the separation of Cu(II) from Zn(II) based on an anion exchanger for the preparation a 62Zn/62Cu generator. Appl Radiat Isot. 2011;69:1176–1180. doi: 10.1016/j.apradiso.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 116.Wadas TJ, Wong EH, Weisman GR, Anderson CJ. Copper chelation chemistry and its role in copper radiopharmaceuticals. Curr Pharm Des. 2007;13:3–16. doi: 10.2174/138161207779313768. [DOI] [PubMed] [Google Scholar]
- 117.Szymanski P, Fraczek T, Markowicz M, Mikiciuk-Olasik E. Development of copper based drugs, radiopharmaceuticals and medical materials. Biometals. 2012;25:1089–1112. doi: 10.1007/s10534-012-9578-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lewis JS, Sharp TL, Laforest R, Fujibayashi Y, Welch MJ. Tumor uptake of copper-diacetyl-bis(N(4)-methylthiosemicarbazone): effect of changes in tissue oxygenation. J Nucl Med. 2001;42:655–661. [PubMed] [Google Scholar]
- 119.Obata A, Yoshimi E, Waki A, Lewis JS, Oyama N, Welch MJ, Saji H, Yonekura Y, Fujibayashi Y. Retention mechanism of hypoxia selective nuclear imaging/radiotherapeutic agent cu-diacetyl-bis(N4-methylthiosemicarbazone) (Cu-ATSM) in tumor cells. Ann Nucl Med. 2001;15:499–504. doi: 10.1007/BF02988502. [DOI] [PubMed] [Google Scholar]
- 120.Wong TZ, Lacy JL, Petry NA, Hawk TC, Sporn TA, Dewhirst MW, Vlahovic G. PET of hypoxia and perfusion with 62Cu-ATSM and 62Cu-PTSM using a 62Zn/62Cu generator. AJR Am J Roentgenol. 2008;190:427–432. doi: 10.2214/AJR.07.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.O’Donoghue JA, Zanzonico P, Pugachev A, Wen B, Smith-Jones P, Cai S, Burnazi E, Finn RD, Burgman P, Ruan S, Lewis JS, Welch MJ, Ling CC, Humm JL. Assessment of regional tumor hypoxia using 18F-fluoromisonidazole and 64Cu(II)-diacetyl-bis(N4-methylthiosemicarbazone) positron emission tomography: comparative study featuring microPET imaging, Po2 probe measurement, autoradiography, and fluorescent microscopy in the R3327-AT and FaDu rat tumor models. Int J Radiat Oncol Biol Phys. 2005;61:1493–1502. doi: 10.1016/j.ijrobp.2004.12.057. [DOI] [PubMed] [Google Scholar]
- 122.Dehdashti F, Grigsby PW, Lewis JS, Laforest R, Siegel BA, Welch MJ. Assessing tumor hypoxia in cervical cancer by PET with 60Cu-labeled diacetyl-bis(N4-methylthiosemicarbazone) J Nucl Med. 2008;49:201–205. doi: 10.2967/jnumed.107.048520. [DOI] [PubMed] [Google Scholar]
- 123.Dehdashti F, Grigsby PW, Mintun MA, Lewis JS, Siegel BA, Welch MJ. Assessing tumor hypoxia in cervical cancer by positron emission tomography with 60Cu-ATSM: relationship to therapeutic response-a preliminary report. Int J Radiat Oncol Biol Phys. 2003;55:1233–1238. doi: 10.1016/s0360-3016(02)04477-2. [DOI] [PubMed] [Google Scholar]
- 124.Lewis JS, Laforest R, Dehdashti F, Grigsby PW, Welch MJ, Siegel BA. An imaging comparison of 64Cu-ATSM and 60Cu-ATSM in cancer of the uterine cervix. J Nucl Med. 2008;49:1177–1182. doi: 10.2967/jnumed.108.051326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lohith TG, Kudo T, Demura Y, Umeda Y, Kiyono Y, Fujibayashi Y, Okazawa H. Pathophysiologic correlation between 62Cu-ATSM and 18F-FDG in lung cancer. J Nucl Med. 2009;50:1948–1953. doi: 10.2967/jnumed.109.069021. [DOI] [PubMed] [Google Scholar]
- 126.Minagawa Y, Shizukuishi K, Koike I, Horiuchi C, Watanuki K, Hata M, Omura M, Odagiri K, Tohnai I, Inoue T, Tateishi U. Assessment of tumor hypoxia by 62Cu-ATSM PET/CT as a predictor of response in head and neck cancer: a pilot study. Ann Nucl Med. 2011;25:339–345. doi: 10.1007/s12149-011-0471-5. [DOI] [PubMed] [Google Scholar]
- 127.Tateishi K, Tateishi U, Nakanowatari S, Ohtake M, Minamimoto R, Suenaga J, Murata H, Kubota K, Inoue T, Kawahara N. 62Cu-diacetyl-bis (N(4)-methylthiosemicarbazone) PET in human gliomas: comparative study with [18F]fluorodeoxyglucose and L-methyl-[11C]methionine PET. AJNR Am J Neuroradiol. 2014;35:278–284. doi: 10.3174/ajnr.A3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Herrero P, Hartman JJ, Green MA, Anderson CJ, Welch MJ, Markham J, Bergmann SR. Regional myocardial perfusion assessed with generator-produced copper-62-PTSM and PET. J Nucl Med. 1996;37:1294–1300. [PubMed] [Google Scholar]
- 129.Haynes NG, Lacy JL, Nayak N, Martin CS, Dai D, Mathias CJ, Green MA. Performance of a 62Zn/62Cu generator in clinical trials of PET perfusion agent 62Cu-PTSM. J Nucl Med. 2000;41:309–314. [PubMed] [Google Scholar]
- 130.Herrero P, Markham J, Weinheimer CJ, Anderson CJ, Welch MJ, Green MA, Bergmann SR. Quantification of regional myocardial perfusion with generator-produced 62Cu-PTSM and positron emission tomography. Circulation. 1993;87:173–183. doi: 10.1161/01.cir.87.1.173. [DOI] [PubMed] [Google Scholar]
- 131.Wallhaus TR, Lacy J, Whang J, Green MA, Nickles RJ, Stone CK. Human biodistribution and dosimetry of the PET perfusion agent copper-62-PTSM. J Nucl Med. 1998;39:1958–1964. [PubMed] [Google Scholar]
- 132.Tadamura E, Tamaki N, Okazawa H, Fujibayashi Y, Kudoh T, Yonekura Y, Magata Y, Nohara R, Sasayama S, Konishi J. Generator-produced copper-62-PTSM as a myocardial PET perfusion tracer compared with nitrogen-13-ammonia. J Nucl Med. 1996;37:729–735. [PubMed] [Google Scholar]
- 133.Shelton ME, Green MA, Mathias CJ, Welch MJ, Bergmann SR. Assessment of regional myocardial and renal blood flow with copper-PTSM and positron emission tomography. Circulation. 1990;82:990–997. doi: 10.1161/01.cir.82.3.990. [DOI] [PubMed] [Google Scholar]
- 134.Mathias CJ, Welch MJ, Raichle ME, Mintun MA, Lich LL, McGuire AH, Zinn KR, John EK, Green MA. Evaluation of a potential generator-produced PET tracer for cerebral perfusion imaging: single-pass cerebral extraction measurements and imaging with radiolabeled Cu-PTSM. J Nucl Med. 1990;31:351–359. [PubMed] [Google Scholar]
- 135.Green MA, Mathias CJ, Welch MJ, McGuire AH, Perry D, Fernandez-Rubio F, Perlmutter JS, Raichle ME, Bergmann SR. Copper-62-labeled pyruvaldehyde bis(N4-methylthiosemicarbazonato)copper(II): synthesis and evaluation as a positron emission tomography tracer for cerebral and myocardial perfusion. J Nucl Med. 1990;31:1989–1996. [PubMed] [Google Scholar]
- 136.Tateishi K, Tateishi U, Sato M, Yamanaka S, Kanno H, Murata H, Inoue T, Kawahara N. Application of 62Cu-diacetyl-bis (N4-methylthiosemicarbazone) PET imaging to predict highly malignant tumor grades and hypoxia-inducible factor-1alpha expression in patients with glioma. AJNR Am J Neuroradiol. 2013;34:92–99. doi: 10.3174/ajnr.A3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fletcher JW, Logan TF, Eitel JA, Mathias CJ, Ng Y, Lacy JL, Hutchins GD, Green MA. Whole-body PET/CT evaluation of tumor perfusion using generator-based 62Cu-ethylglyoxal bis (thiosemicarbazonato) copper (II): validation by direct comparison to 15O-water in metastatic renal cell carcinoma. J Nucl Med. 2015;56:56–62. doi: 10.2967/jnumed.114.148106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Green MA, Mathias CJ, Willis LR, Handa RK, Lacy JL, Miller MA, Hutchins GD. Assessment of Cu-ETS as a PET radiopharmaceutical for evaluation of regional renal perfusion. Nucl Med Biol. 2007;34:247–255. doi: 10.1016/j.nucmedbio.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 139.Ng Y, Lacy JL, Fletcher JW, Green MA. Performance of a 62Zn/62Cu microgenerator in kit-based synthesis and delivery of [62Cu]Cu-ETS for PET perfusion imaging. Appl Radiat Isot. 2014;91:38–43. doi: 10.1016/j.apradiso.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Black NF, McJames S, Rust TC, Kadrmas DJ. Evaluation of rapid dual-tracer 62sup>Cu-PTSM+ 62Cu-ATSM PET in dogs with spontaneously occurring tumors. Phys Med Biol. 2008;53:217–232. doi: 10.1088/0031-9155/53/1/015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rust TC, Kadrmas DJ. Rapid dual-tracer PTSM+ ATSM PET imaging of tumour blood flow and hypoxia: a simulation study. Phys Med Biol. 2006;51:61–75. doi: 10.1088/0031-9155/51/1/005. [DOI] [PubMed] [Google Scholar]
- 142.Al-Kouraishi SH, Boswell GGJ. An isotope generator for 72As. Int J Appl Radiat Isot. 1978;29:607–609. [Google Scholar]
- 143.Phillips DR, inventor. Chemistry and concept for an automated 72Se/72As generator. No. 5,371,372. United States Patent. 1994
- 144.Phillips DR, Hamilton VT, Taylor MD, Farnham JE, Emran AM, Rowe RW, Pattel D. Generator-produced arsenic-72 in positron emission tomography. Radioact Radiochem. 1992;3:53–58. [Google Scholar]
- 145.Jennewein M, Schmidt A, Novgorodov AF, Qaim SM, Rösch F. A no-carrier-added 72Se/72As radionuclide generator based on distillation. Radiochim Acta. 2004;92:245–249. [Google Scholar]
- 146.Jennewein M, Qaim SM, Kulkarni PV, Mason RP, Hermanne A, Roesch F. A no-carrier-added 72Se/72As radionuclide generator based on solid phase extraction. Radiochim Acta. 2005;93:579–583. [Google Scholar]
- 147.Chajduk E, Doner K, Polkowska-Motrenko H, Bilewicz A. Novel radiochemical separation of arsenic from selenium for 72Se/72As generator. Appl Radiat Isot. 2012;70:819–822. doi: 10.1016/j.apradiso.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 148.Ballard B, Wycoff D, Birnbaum ER, John KD, Lenz JW, Jurisson SS, Cutler CS, Nortier FM, Taylor WA, Fassbender ME. Selenium-72 formation via nat Br(p,x) induced by 100 MeV protons: steps towards a novel 72Se/72As generator system. Appl Radiat Isot. 2012;70:595–601. doi: 10.1016/j.apradiso.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 149.Jennewein M, Hermanne A, Mason RP, Thorpe PE, Rösch F. A new method for the labelling of proteins with radioactive arsenic isotopes. Nucl Instrum Methods Phys Res A. 2006;569:512–517. [Google Scholar]
- 150.Jennewein M, Lewis MA, Zhao D, Tsyganov E, Slavine N, He J, Watkins L, Kodibagkar VD, O’Kelly S, Kulkarni P, Antich PP, Hermanne A, Rosch F, Mason RP, Thorpe PE. Vascular imaging of solid tumors in rats with a radioactive arsenic-labeled antibody that binds exposed phosphatidylserine. Clin Cancer Res. 2008;14:1377–1385. doi: 10.1158/1078-0432.CCR-07-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lundqvist H, Scott-Robson S, Einarsson L, Malmborg P. 110Sn/110In--a new generator system for positron emission tomography. Int J Rad Appl Instrum A. 1991;42:447–450. doi: 10.1016/0883-2889(91)90104-9. [DOI] [PubMed] [Google Scholar]
- 152.Lubberink M, Tolmachev V, Widstrom C, Bruskin A, Lundqvist H, Westlin JE. 110mIn-DTPA-D-Phe1-octreotide for imaging of neuroendocrine tumors with PET. J Nucl Med. 2002;43:1391–1397. [PubMed] [Google Scholar]
- 153.Rösch F, Qaim SM, Novgorodov AF, Ying-Ming T. Production of positron-emitting 110mIn via the 110Cd(3He, 3n)110Sn→110mIn process. Appl Radiat Isot. 1997;48:19–26. [Google Scholar]
- 154.Atkins HL, Som P, Fairchild RG, Hui J, Schachner E, Goldman A, Ku T. Myocardial positron tomography with manganese-52m. Radiology. 1979;133:769–774. doi: 10.1148/133.3.769. [DOI] [PubMed] [Google Scholar]
- 155.Atcher RW, Friedman AM, Huizenga JR, Rayudu GV, Silverstein EA, Turner DA. Manganese-52m, a new short-lived, generator-produced radionuclide: a potential tracer for positron tomography. J Nucl Med. 1980;21:565–569. [PubMed] [Google Scholar]
- 156.Herscheid JD, Vos CM, Hoekstra A. Manganese-52m for direct application: a new 52Fe/52mMn generator based on a hydroxamate resin. Int J Appl Radiat Isot. 1983;34:883–886. doi: 10.1016/0020-708x(83)90147-3. [DOI] [PubMed] [Google Scholar]
- 157.Ku TH, Richards P, Stang LG Jr, Prach T. Preparation of 52Fe and its use in a 52Fe/52mMn generator. Radiology. 1979;132:475–477. doi: 10.1148/132.2.475. [DOI] [PubMed] [Google Scholar]
- 158.Atcher RW, Friedman AM, Huizenga JR. Production of 52Fe for use in a radionuclide generator system. Int J Nucl Med Biol. 1980;7:75–78. doi: 10.1016/0047-0740(80)90018-2. [DOI] [PubMed] [Google Scholar]
- 159.Blauenstein P, Pellikka R, Schubiger PA. Reinvestigation of a physiological eluate of the 52Fe/52mMn generator. Appl Radiat Isot. 1997;48:1097–1101. doi: 10.1016/s0969-8043(97)00106-1. [DOI] [PubMed] [Google Scholar]
- 160.Buck A, Nguyen N, Burger C, Ziegler S, Frey L, Weigand G, Erhardt W, Senekowitsch-Schmidtke R, Pellikka R, Blauenstein P, Locher JT, Schwaiger M. Quantitative evaluation of manganese-52m as a myocardial perfusion tracer in pigs using positron emission tomography. Eur J Nucl Med. 1996;23:1619–1627. doi: 10.1007/BF01249625. [DOI] [PubMed] [Google Scholar]
- 161.Richards P, Ku TH. The 122Xe/122I system: a generator for the 3.62-min positron emitter, 122I. Int J Appl Radiat Isot. 1979;30:250–252. [Google Scholar]
- 162.Mathis CA, Lagunas-Solar MC, Sargent T 3rd, Yano Y, Vuletich A, Harris LJ. A 122Xe-122I generator for remote radio-iodinations. Int J Rad Appl Instrum A. 1986;37:258–260. doi: 10.1016/0883-2889(86)90183-8. [DOI] [PubMed] [Google Scholar]
- 163.Moerlein SM, Mathis CA, Brennan KM, Budinger TF. Synthesis and in vivo evaluation of 122I- and 131I-labelled iodoperidol, a potential agent for the tomographic assessment of cerebral perfusion. Int J Rad Appl Instrum B. 1987;14:91–98. doi: 10.1016/0883-2897(87)90137-1. [DOI] [PubMed] [Google Scholar]
- 164.Mathis CA, Sargent T 3rd, Shulgin AT. Iodine-122-labeled amphetamine derivative with potential for PET brain blood-flow studies. J Nucl Med. 1985;26:1295–1301. [PubMed] [Google Scholar]
- 165.Borchardt PE, Yuan RR, Miederer M, McDevitt MR, Scheinberg DA. Targeted actinium-225 in vivo generators for therapy of ovarian cancer. Cancer Res. 2003;63:5084–5090. [PubMed] [Google Scholar]
- 166.Knapp FF Jr, Mirzadeh S. The continuing important role of radionuclide generator systems for nuclear medicine. Eur J Nucl Med. 1994;21:1151–1165. doi: 10.1007/BF00181073. [DOI] [PubMed] [Google Scholar]
- 167.Edem PE, Fonslet J, Kjaer A, Herth M, Severin G. In Vivo Radionuclide generators for diagnostics and therapy. Bioinorg Chem Appl. 2016;2016:6148357. doi: 10.1155/2016/6148357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rosch F, Baum RP. Generator-based PET radiopharmaceuticals for molecular imaging of tumours: on the way to THERANOSTICS. Dalton Trans. 2011;40:6104–6111. doi: 10.1039/c0dt01504k. [DOI] [PubMed] [Google Scholar]
- 169.Zhernosekov KP, Filosofov DV, Qaim SM, Rösch F. A 140Nd/140Pr radionuclide generator based on physico-chemical transitions in 140Pr complexes after electron capture decay of 140Nd-DOTA. Radiochim Acta. 2007;95:319–327. [Google Scholar]
- 170.Lubberink M, Lundqvist H, Tolmachev V. Production, PET performance and dosimetric considerations of 134Ce/134La, an Auger electron and positron-emitting generator for radionuclide therapy. Phys Med Biol. 2002;47:615–629. doi: 10.1088/0031-9155/47/4/305. [DOI] [PubMed] [Google Scholar]
- 171.International Atomic Energy Agency (IAEA) publication 10-00628. Vienna, Austria: 2010. Production of long lived parent radionuclides for generators: 68Ge, 82Sr, 90Sr, and 188W. [Google Scholar]
- 172.Pao PJ, Silvester DJ, Waters SL. A new method for the preparation of 68Ga-generators following proton bombardment of gallium oxide targets. J Radioanal Nucl Chem. 1981;64:267–272. [Google Scholar]
- 173.Loc’h C, Maziere B, Comar D, Knipper R. A new preparation of germanium 68. Int J Appl Radiat Isot. 1982;33:267–270. [Google Scholar]
- 174.van der Walt TN, Vermeulen C. Thick targets for the production of some radionuclides and the chemical processing of these targets at iThemba LABS. Nucl Instrum Methods Phys Res A. 2004;521:171–175. [Google Scholar]
- 175.Fassbender M, Nortier FM, Phillips DR, Hamilton VT, Heaton C, Jamriska DJ, Kitten JJ, Pitt LR, Salazar LL, Valdez FO, Peterson EJ. Some nuclear chemical aspects of medical generator nuclide production at the Los Alamos hot cell facility. Radiochim Acta. 2004;92:237–243. [Google Scholar]
- 176.Ehrhardt GJ, Welch MJ. A new germanium-63/gallium-68 generator. J Nucl Med. 1978;19:925–929. [PubMed] [Google Scholar]
- 177.Naidoo C, van der Walt TN, Raubenheimer HG. Cyclotron production of 68Ge with a Ga2O target. J Radioanal Nucl Chem. 2002;253:221–225. [Google Scholar]
- 178.Cheng WL, Jao Y, Lee CS, Lo AR. Preparation of 68Ge/68Ga generator with a binary Ga/Ag electrodepositions as solid target. J Radioanal Nucl Chem. 2000;245:25–30. [Google Scholar]
- 179.Schuhmacher J, Maier-Borst W. A new 68Ge/68Ga radioisotope generator system for production of 68Ga in dilute HCl. Int J Appl Radiat Isot. 1981;32:31–36. [Google Scholar]
- 180.Meinken GE, Kurczak S, Mausner LF, Kolsky KL, Srivastava SC. Production of high specific activity 68Ge at Brookhaven National Laboratory. J Radioanal Nucl Chem. 2005;263:553–557. [Google Scholar]
- 181.Peterson EJ, editor. Accelerator radioisotopes save lives: the isotope production facility at Los Alamos. Los Alamos Science No. 30. 2006. Available at: http://lansce.lanl.gov/news/docs/LA%20Science.pdf.
- 182.Philips DR, editor. Radioisotope production at Los Alamos National Laboratory, presented at Specialization School on Health Physics, Milan. 2002. Available at: www.mi.infn.it/conferences/phillips/Lanl.pdf.
- 183.The National Isotope Development Center (NIDC) Product Catalog. Available at http://www.isotopes.gov/catalog/product.php?element=Titanium&type=rad&rad_product_index=61.
- 184.Alenitzky YG, Novgorodov AF, Skripnik AV, Filosofov DV, Skripnik AV, Kaplun VG, Suzikov AG, Eliseev IA, Rösch F, editors. Annual report. Institute of Nuclear Chemistry, University of Mainz; 2005. 44Ti: investigation of target preparation, irradiation and yields in the 45Sc(p,2n) process. Available at http://www.kernchemie.uni-mainz.de/Dateien/b3_05.pdf. [Google Scholar]
- 185.Caretto AA, Wiig EO. Interaction of yttrium with protons of energy between 60 and 240 Mev. Phy Rev. 1959;115:1238–1242. [Google Scholar]
- 186.Grant PM, Erdal BR, O’Brien HA. Medium-energy spallation cross sections. 2. Mo irradiation with 800-MeV protons. Int J Appl Radiat Isot. 1983;34:1631–1632. [Google Scholar]
- 187.Thomas KE. Isotope production from molybdenum targets. Radiochim Acta. 1984;37:137–141. [Google Scholar]
- 188.Tárkányi F, Qaim SM, Stöcklin G. Excitation functions of high-energy 3He- and alpha-particle induced nuclear reactions on natural krypton with special reference to the production of 82Sr. Appl Radiat Isot. 1990;41:91–95. [Google Scholar]
- 189.Tarkanyi F, Qaim SM, Stoecklin G. Excitation-functions of He-3-particle and alpha-particle induced nuclear-reactions on enriched Kr-82 and Kr-83. Radiochim Acta. 1988;43:185–189. [Google Scholar]
- 190.Tárkányi F, Qaim SM, Stöcklin G. Production of 82Sr via the 82Kr(3He,3n)-process at a compact cyclotron. J Label Compd Radiopharm. 1989;26:153–154. [Google Scholar]
- 191.Buthelezi EZ, Nortier FM, Schroeder IW. Excitation functions for the production of 82Sr by proton bombardment of natRb at energies up to 100 MeV. Appl Radiat Isot. 2006;64:915–924. doi: 10.1016/j.apradiso.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 192.Qaim SM, Steyn GF, Spahn I, Spellerberg S, van der Walt TN, Coenen HH. Yield and purity of 82Sr produced via the natRb(p,xn) 82Sr process. Appl Radiat Isot. 2007;65:247–252. doi: 10.1016/j.apradiso.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 193.Cackette MRC, Ruth TJ, Vincent JS. 82Sr production from metallic Rb targets and development of an 82Rb generator system. Appl Radiat Isot. 1993;44:917–922. [Google Scholar]
- 194.Meulen NP, Walt TN, Steyn GF, Raubenheimer HG. The production of 82Sr using larger format RbCl targets. Appl Radiat Isot. 2013;72:96–99. doi: 10.1016/j.apradiso.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 195.Phillips DR, Peterson EJ, Taylor WA, Jamriska DJ, Hamilton VT, Kitten JJ, Valdez FO, Salazar LL, Pitt LR, Heaton RC, Kolsky KL, Mausner LF. Production of strontium-82 for the Cardiogen® PET generator: a project of the Department of Energy Virtual Isotope Center. Radiochim Acta. 2000;88:149–155. [Google Scholar]
- 196.Zhuikov BL, Ermolaev SV, Kokhanyuk VM, inventors. Method for producing radiostrontium. 2356113. Russian Patent. 2009
- 197.Institute for Nuclear Research of the Russian Academy of Sciences granted U.S. Patent for radiostrontium production technology included in licensing agreement with Positron Corporation. Available at: http://money.cnn.com/news/newsfeeds/articles/prnewswire/AQ11623.htm.
- 198.Piel H, Qaim SM, Stöcklin G. Excitation functions of (p,xn)-reactions on natNi and highly enriched 62Ni: possibility of production of medically important radioisotope 62Cu at a small cyclotron. Radiochim Acta. 1992;57:1–5. [Google Scholar]
- 199.Szelecsényi F, Suzuki K, Kovács Z, Takei M, Okada K. Production possibility of 60,61,62Cu radioisotopes by alpha induced reactions on cobalt for PET studies. Nucl Instrum Methods Phys Res B. 2002;187:153–163. [Google Scholar]
- 200.Szelecsényi F, Kovács Z, van der Walt TN, Steyn GF, Suzuki K, Okada K. Investigation of the natZn(p,x)62Zn nuclear process up to 70MeV: a new 62Zn/62Cu generator. Appl Radiat Isot. 2003;58:377–384. [Google Scholar]
- 201.Mushtaq A, Qaim SM, Stöcklin G. Production of 73Se via (p, 3n) and (d, 4n) reactions on arsenic. Appl Radiat Isot. 1988;39:1085–1091. [Google Scholar]
- 202.Fassbender M, de Villiers D, Nortier M, van der Walt N. The natBr(p,x) 73,75Se nuclear processes: a convenient route for the production of radioselenium tracers relevant to amino acid labelling. Appl Radiat Isot. 2001;54:905–913. doi: 10.1016/s0969-8043(00)00359-6. [DOI] [PubMed] [Google Scholar]
- 203.Phillips DR, Moody DC, Taylor WA, Segura NJ, Pate BD. Electrolytic separation of selenium isotopes from proton irradiated RbBr targets. Appl Radiat Isot. 1987;38:521–525. [Google Scholar]
- 204.Dahl JR, Tilbury RS. The use of a compact, multi-particle cyclotron for the production of 52Fe, 67Ga, 111In and 123I for medical purposes. Int J Appl Radiat Isot. 1972;23:431–437. doi: 10.1016/0020-708x(72)90110-x. [DOI] [PubMed] [Google Scholar]
- 205.Thakur ML, Nunn AD, Waters SL. Iron-52: Improving its recovery from cyclotron targets. Int J Appl Radiat Isot. 1971;22:481–483. [Google Scholar]
- 206.Saha GB, Farrer PA. Production of 52Fe by the 55Mn(p,4n)52Fe reaction for medical use. Int J Appl Radiat Isot. 1971;22:495–498. doi: 10.1016/0020-708x(71)90174-8. [DOI] [PubMed] [Google Scholar]
- 207.Steyn GF, Mills SJ, Nortier FM, Simpson BR, Meyer BR. Production of 52Fe via proton-induced reactions on manganese and nickel. Int J Rad Appl Instrum A. 1990;41:315–325. doi: 10.1016/0883-2889(90)90197-o. [DOI] [PubMed] [Google Scholar]
- 208.Suzuki K. Production of 52Fe by the 55Mn(p,4n)52Fe reaction and milking of 52mMn from 52Fe. Radioisotopes. 1985;34:537–542. doi: 10.3769/radioisotopes.34.10_537. [DOI] [PubMed] [Google Scholar]
- 209.Zweit J, Downey S, Sharma H. A method for the production of iron-52 with a very low iron-55 contamination. Int J Rad Appl Instrum A. 1988;39:1197–1201. doi: 10.1016/0883-2889(88)90099-8. [DOI] [PubMed] [Google Scholar]
- 210.Tárkányi F, Qaim SM, Stöcklin G, Sajjad M, Lambrecht RM. Nuclear reaction cross sections relevant to the production of the 122Xe → 122I generator system using highly enriched 124Xe and a medium-sized cyclotron. Appl Radiat Isot. 1991;42:229–233. [Google Scholar]
- 211.Lagunas-Solar MC, Carvacho OF, Harris LJ, Mathis CA. Cyclotron production of 122Xe(20.1 h)→122I (β+ 77%; EC 23%; 3.6 min) for positron emission tomography. Current methods and potential developments. Appl Radiat Isot. 1986;37:835–842. [Google Scholar]
- 212.Peek NF, Hegedüs F. The production of xenon isotopes with protons of energies from 320 to 590 MeV. Int J Appl Radiat Isot. 1979;30:631–635. [Google Scholar]
- 213.Guideline on Radiopharmaceuticals, CHMP, EMEA/CHMP/QWP/306970/2007 (draft released for consultation) Available at: http://www.emea.europa.eu/pdfs/human/qwp/30697007en.pdf.
- 214.Directive 2001/83/EC of the European Parliament and the Council of November 6, 2001 on the community code relating to medicinal products for human use. Available at: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol/dir_2001_83_cons2009/2001_83_cons2009_en.pdf.
- 215.EudraLex. The Rules Governing Medicinal Products in the European Union. Available at: http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/eudralex_en.htm.
- 216.Current good manufacturing practice for positron emission tomography drugs. Final rule. Available at: http://www.gpo.gov/fdsys/pkg/FR-2009-12-10/pdf/E9-29285.pdf. [PubMed]
- 217.Shimchuk G, Gr S, Pakhomov G, Avalishvili G, Zavrazhnov G, Polonsky-Byslaev I, Fedotov A, Polozov P. Generators and automated generator systems for production and on-line injections of PET radiopharmaceuticals. J Phys Conf Ser. 2017;784:012059. [Google Scholar]