

Abstract
Loss of telomeres stability is a hallmark of cancer cells. Exposed telomeres are prone to aberrant end-joining reactions leading to chromosomal fusions and translocations. Human telomeres contain repeated TTAGGG elements, in which the 3’ exposed strand may adopt a G-quadruplex (G4) structure. The guanine-rich regions of telomeres are hotspots for oxidation forming 8-oxoguanine, a lesion that is handled by the base excision repair (BER) pathway. One key player of this pathway is Ape1, the main human endonuclease processing abasic sites. Recent evidences showed an important role for Ape1 in telomeric physiology, but the molecular details regulating Ape1 enzymatic activities on G4-telomeric sequences are lacking. Through a combination of in vitro assays, we demonstrate that Ape1 can bind and process different G4 structures and that this interaction involves specific acetylatable lysine residues (i.e. K27/31/32/35) present in the unstructured N-terminal sequence of the protein. The cleavage of an abasic site located in a G4 structure by Ape1 depends on the DNA conformation or the position of the lesion and on electrostatic interactions between the protein and the nucleic acids. Moreover, Ape1 mutants mimicking the acetylated protein display increased cleavage activity for abasic sites. We found that nucleophosmin (NPM1), which binds the N-terminal sequence of Ape1, plays a role in modulating telomere length and Ape1 activity at abasic G4 structures. Thus, the Ape1 N-terminal sequence is an important relay site for regulating the enzyme’s activity on G4-telomeric sequences, and specific acetylatable lysine residues constitute key regulatory sites of Ape1 enzymatic activity dynamics at telomeres.
Keywords: telomeres, G4 structure, Ape1, abasic sites, acetylation, NPM1
Introduction
Genome instability is a central feature of most cancers, with effects ranging from numerous point mutations to changes in the size of repeated sequences, to gross chromosomal rearrangements [1,2]. These genetic changes are key elements of the etiology of cancer, through the activation of oncogenes or inactivation of tumor-suppressor genes [2]. Moreover, many tumors display continuing high-frequency mutagenesis, due to compromised checkpoint functions, loss of DNA repair activities and immortalization ending in a limitless replication [3,4]. In addition, the maintenance of chromosomal telomeres by special proteins and the replication by telomerase can enable for such limitless replication potential [5]. In the minor proportion of cell types that do not employ telomerase mechanism, the alternative lengthening of telomeres (ALT) pathway is active, which uses the homologous recombination pathway to maintain telomeric structures [6].
Various DNA maintenance and cell cycle-control pathways normally prevent genetic aberrations [3]. Exposed chromosomal termini pose another threat to genetic stability, namely a propensity to participate in aberrant end-joining reactions that can lead to chromosomal fusions and translocations [7]. Damage to DNA is a significant threat in this regard, with a well-known contribution of environmental (e.g., solar radiation) and lifestyle (e.g., tobacco smoking) exposures to the etiological role of DNA lesions in cancer development [7,8]. However, a considerable burden of DNA damage may arise from the intrinsic chemical instability of the molecule and from reactions with cellular metabolites [9]. Examples include the hydrolytic loss of purine bases, or the various lesions formed by reaction with free radical by-products of aerobic respiration [10]. Most of this endogenous burden is handled by the base excision DNA repair (BER) pathway [11,12]. BER involves several DNA N-glycosylases specific for removing a wide array of oxidized, alkylated or deaminated bases, and in some cases mispaired normal bases, which yields abasic (AP) sites [13,14]. AP sites can also be generated by hydrolytic base loss and some free-radical reactions [15], and these lesions enter the BER pathway. AP sites are avidly recognized by AP endonuclease(s) which, in mammalian cells, is uniquely represented by the Apurinic/apyrimidinic endonuclease 1 (Ape1) protein [16–19]. Ape1 incises the phosphodiesteric backbone at the 5’ side of the AP site allowing the excision of the abasic residue and DNA synthesis to replace the missing nucleotide and the ligation of the resulting nick to complete the repair. Ape1 in mammalian cells is essential for its DNA repair activity [20]. The portion of human Ape1 deputed to the endonuclease activity involves both the C-terminal domain and a part of the N-terminal domain (residues 61–127). The N-terminal sequence (first 35 amino acid residues) of the protein is crucial for the interaction with other proteins and contributes to the binding of nucleic acids [21,22]. Among the residues 35–127, the protein contains a redox activity (‘Ref-1’) to activate several transcription factors involved in DNA repair and stress responses, and other cellular functions [23]. Within the N-terminal sequence, several positively charged amino acids are important for modulating the binding to different DNA and RNA substrates [24], as well as for regulating protein-protein interactions [22,25–27]. Interestingly, some of these lysine residues (i.e. K27, K31, K32 and K35) have been shown to be acetylated in cancer cells [22,28], and cancer cells expressing this mutant protein display a reduced proliferation rate [25]. It is also intriguing that, under certain circumstances and in some human cell lines, the first 33–35 residues can be removed via specific cleavage sites that resides among lysine residues 27, 31, 32 and 35 [29,30].
It was found that Ape1 protein is required for proper telomere maintenance in both normal and in tumor cells [31] and that Ape1 deficiency promotes cell senescence and premature aging features [32]. Ape1 localization to telomeres is required for normal binding of the protective protein Telomeric repeat-binding factor 2 (TRF2) [31] whereas its deficiency leads to an increased telomeric binding of the Protection of telomeres protein 1 (POT1), which in turn stimulates Ape1’s AP-endonuclease activity in vitro [33,34]. Moreover, in telomerase-expressing cells, the telomeres become rapidly shorter upon Ape1 depletion and undergo frequent aberrations and fusions [31]. Telomeric defects are observed for Ape1 deficiency in cells utilizing either the telomerase pathway or the recombination-dependent Alternative lengthening of telomeres (ALT) pathway [30,34]. Surprisingly, the substitution of acetylatable lysine residues 6 and 7 (K6 and K7) with alanine in Ape1 N-terminal sequence, though not directly affecting its DNA repair activity, leads to aberrant telomeres formation, while abolition of the redox function does not [31]. Unfortunately, this first study did not reveal molecular explanation for the unexpected roles of lysine 6 and 7. Not only K6/K7 but also K27−35 can be acetylated and the acetylation status of K6 and K7 may modulate the modification of K27−35 [25]. However, the role of K27−35 and, more in general of its N-terminal sequence, on the ability of Ape1 to process telomeric sequences is completely unknown. We previously found that substitution of these lysine residues to mimic acetylation (in the mutant called Ape1K4pleA) increased the endonuclease activity at abasic sites in double-stranded (ds) DNA [22], but reduced the protein’s ability to recognize and process abasic sites in single-stranded (ss) nucleic acids [22,24]. Therefore, it is possible that charged status of lysine residues 27–35 may also modulate the Ape1 enzymatic activity on telomeric sequences. This work specifically addressed this issue.
It is well established that at least four repeats of the human telomeric motif TTAGGG fold into a variety of G-quadruplex (G4) conformations in vitro [35], in the presence of monovalent cations, such as Na+ or K+. Each G base serves as both donor and acceptor for hydrogen bond formation [36]. The factors that determine the conformation and stability of G-quadruplex forming sequences remain poorly understood.
Several types of G4 conformations exist; they are identified as parallel (with the guanine strands running in the same direction and double-chain reversal loops), antiparallel (with two strands running in each direction and either lateral or diagonal loops) or mixed ‘3 + 1’ hybrid, with three strands in one direction and one in the other [37]. Which of these structures is preferred depends on the sequence and length of the loops and on the cation type. In vitro, many G4 DNA structures, once formed, are thermodynamically more stable than dsDNA and their unfolding kinetics are much slower than those of DNA- or RNA-hairpin structures [38].
G-quadruplex structures may serve as DNA replication regulators: this structure makes the telomeric substrate non-accessible for the replication machinery [39]. The unfolding of the G4 is regulated by a phosphorylation of the telomere end binding protein (TEBPβ), whose expression is cell-cycle dependent, and by a telomerase associated RecQ-like helicase (RecQ protein-like 4) thus finely regulating the telomere synthesis process [40].
With G4s abundance throughout the genome, DNA damages also affects these structures. Specifically, oxidized and abasic sites might be generated in these structures under oxidative stress conditions. The absence of a purine resulted in a lower stability of the G4, since the tetrad is disrupted [41–43].
Ape1 is able to process the abasic lesions contained in the context of double-stranded telomeric repeats in vitro [44–46], including the G4 structure formed by the c-Myc promoter [44], but till now very little is known about its enzymatic activity on telomeric G4-folded structures. We have shown that the ability of Ape1 to cleave single stranded nucleic acids was strictly dependent on the propensity of the substrate to adopt secondary structures, and this effect was modulated by the N-terminal basic, unstructured portion of the protein [24].
Here, we highlight the importance of the N-terminal sequence of Ape1 for the stable recognition and enzymatic processing of telomeric substrates and we demonstrate the essential role exerted by acetylatable lysine residues of this unstructured protein portion.
Materials and methods
Protein expression and FPLC purification
BL21 cells were transformed with the proper pGEX-3X (GST-tag) or pET-15b (His-tag) plasmid following the manufacturer’s instructions. In order to produce the protein, the culture was harvested at 37°C at 250 rpm until the reaching of OD600 ~0,7. Finally, the culture was induced with 1 mM of IPTG (Sigma) for 4 hours and then was collected upon centrifugation. The pellet was lysed in the presence of Protease inhibitor 2.1 mg/ml (Sigma) and Lysozyme 0.3 mg/ml (Sigma) through sonication and the sample was centrifuged at 23,000 g for 20 minutes at 4° C, then the supernatants were purified through the appropriate column for affinity purification.
GST-tagged proteins (Ape1WT, Ape1N∆33 and Ape1K4pleA) were purified through a GSTrap column (GE Healthcare) and they were eluted through at increasing range of GSH (Sigma) concentration following the manufacturer’s instructions. Then the proteins were incubated with Factor X (Amersham) to remove the tag; the enzyme was separated from the protein through a benzamidine column (GE Healthcare) and finally a cation exchange purification was performed.
His-tagged NPM1 was purified through a His-Trap column (GE Healthcare) and was eluted through 3 steps at increasing imidazole (Sigma) concentrations. The protein was dialyzed and then purified through a cation exchange purification column.
All protein fractions were stored in a buffer containing 25 mM Tris pH 7.5, 100 mM NaCl, 1 mM DTT, 10% glycerol.
Oligonucleotides synthesis, purification and annealing
Oligonucleotides (ODN) reported in Table 1 were synthesized on a Millipore Cyclone Plus DNA synthesizer using standard solid phase β -cyanoethyl phosphoramidite chemistry at 1 µmol scale. The synthesis were performed by using Fast Deprotection DNA 3’-phosphoramidites, a 5’-dimethoxytrityl-3’-phosphoramidite-1’,2’-dideoxyribose (dSpacer, dS, Link Technologies) for the insertion of an abasic site mimic moiety and IRDye 800 phosphoramidite (LI-COR Biosciences) for the introduction of a Near IR fluorescent dye at the 5’-end of each ODN sequence. The oligomers were detached from the support and deprotected by treatment with concentrated aqueous ammonia in the dark for 1.5 hour at room temperature. The oligomers were purified by HPLC (Nucleosil C18 column Macherey–Nagel, 100–5; EC250/4.6) using standard methods. The fractions of the oligomers were collected and successively desalted by Sep-pak cartridges (C-18). The isolated oligomers proved to be >98% pure by HPLC. To perform in vitro experiments, some selected ODN sequences used previously [41] were employed and produced homemade as explained in the previous paragraph. A poly dT ODN of 23 bases, holding an abasic site (F) in 16th position (Poly dT-F) and an IRDye 800 at the 5’ was synthesized by Metabion, purified through HPLC and checked in Mass Check. This probe was used as negative control as it is unable to acquire secondary structures. All the ODN were resuspended in DNase-free water at 100 µM and annealing was performed at a final concentration of 5 µM in a solution containing 70 mM KCl, 20 mM KH2PO4 and 0.2 mM EDTA, at pH 7.0, heated at 70°C and cooling down over night. A double-stranded ODN, named ds-F, which is composed of 26 bases and it holds a tetrahydrofuran mimicking the abasic site in position 15, was also synthesized by Metabion, purified through HPLC and checked in Mass Check. This latter ODN was annealed in 10 mM Tris, 10 mM MgCl2, 1 mM EDTA pH 7.5, heated at 95°C and let to cool down. All ODN sequences are listed in Table 1.
Table 1-. Sequences of the ODN used in this study.
F: tetrahydrofuran abasic site analog. Nat, S4 and S8 substrates are single strand DNA folded in a G-quadruplex. Poly dT-F is an unstructured DNA single strand. ds-F is the only duplex DNA sequence. All the ODN are labeled with IRDye 800 at the 5’ end.
| ODN name | Sequences | Length of the product |
|---|---|---|
| Nat | 5’-TAG GGT TAGG GTT AGG GTT AGG G-3’ | - |
| S4 | 5’-TAG GGT TAFG GTT AGG GTT AGG G −3’ | 8 nt |
| S8 | 5’-TAG GGT TAGG GTT AGF GTT AGG G-3’ | 15 nt |
| Poly dT-F | 5’-TTT TTT TTTT TTT TTF TTT TTT T-3’ | 15 nt |
| ds-F | 5’-AAT TCA CCG GTA CCF TCT AGA ATT CG-3’ 3’-TTA AGT GGA CAT GGG AGA TCT TAA GC-5’ |
14 nt |
| Poly dT | 5’-TTT TTT TTTT TTT TTT TTT TTT T-3’ | - |
Circular dichroism (CD) spectroscopy
CD samples of the IRDye-labelled oligonucleotides reported in Table 1 were prepared at an ODN concentration of 25 µM by using a potassium phosphate buffer (20 mM KH2PO4/K2HPO4, 70 mM KCl, pH 7.0) and submitted to the annealing procedure (heating at 70°C and slowly cooling at RT). CD samples at 50 mM KCl were prepared by diluting the samples at 70 mM KCl and adding the suitable amounts of Tris and MgCl2 to final concentrations of 50 mM and 10 mM, respectively. CD samples at 5 mM KCl were prepared by dialyzing CD samples at 70 mM against a solution KCl 5 mM, Tris 5 mM and MgCl2 1 mM. CD spectra of all G-quadruplexes and CD melting curves were registered on a Jasco 715 CD spectrophotometer. For the CD spectra, the wavelength was varied from 220 to 320 nm at 100 nm min−1 scan rate, and the spectra recorded with a response of 4 s, at 1.0 nm bandwidth and normalized by subtraction of the background scan with buffer. The temperature was kept constant at 20°C with a thermoelectrically-controlled cell holder (Jasco PTC-348). CD melting curves were registered as a function of temperature from 20°C to 70°C for all G-quadruplexes at their maximum Cotton effect wavelengths. The CD data were recorded in a 0.1 cm pathlength cuvette with a scan rate of 0.5°C/min.
SDS-PAGE and Western blotting
All recombinant proteins were loaded onto a 10 (w/v) % sodium dodecyl sulphate-polyacrylamide (SDS-PAGE; acr:bis= 37.5:1) electrophoresis gel, which was subsequently stained using Coomassie Brilliant Blue stain (ThermoFisher). Each band, corresponding to the protein of interest, was quantified and normalized with BSA (bovine serum albumin) standardization curve. The image was finally developed by using NIR Fluorescence technology with an Odissey CLx scanner (LI-COR GmbH). Bands were quantified and analyzed using the ImageStudio software (LI-COR GmbH).
Cell extracts samples were loaded onto a 12 (w/v) % SDS-PAGE electrophoresis gel. Proteins were then transferred onto nitrocellulose membranes (Schleicher & Schuell). Monoclonal α -Ape1 was from Novus Biologicals (1:5,000 dilution). Membranes were incubated with secondary antibodies labeled with IRDye (1:10,000 dilution) in 5% milk, PBS and Tween 0.1% and finally developed by using NIR Fluorescence technology with an Odissey CLx scanner (LI-COR GmbH). Bands were quantified and analyzed using the ImageStudio software (LI-COR).
Electrophoretic mobility shift assay (EMSA)
EMSA assays were performed incubating 20 pmol of Ape1 protein (2 µM) with 250 fmol of the substrate (0.025 µM) for 1 hour at RT in 25 mM Tris, 100 mM KCl, 2 mM DTT, 10% glycerol, pH 7.5. Alternatively, EMSA assays were done with 3.5 pmol of NPM1 or with 7 pmol of Ape1WT or Ape1N∆33, co-incubated for 1 h at RT with increasing concentrations of NPM1 (varying between 3.5 and 17.5 pmol); 350 fmol of Nat substrate were added and left to incubate for 1 h at RT during the binding reactions. The mixtures were loaded on a native gel 6% polyacrylamide (acr: bis= 37.5: 1) then run in the cold apparatus (at 4°C) at 130 V for 4 hours using 0.5x TBE as buffer. Gels were scanned and band intensities were quantified using the Image Studio software (Odyssey CLx, LI-COR GmbH).
UV-crosslinking experiments
UV-crosslinking analysis were performed co-incubating 20 pmol of recombinant purified protein (Ape1WT, of Ape1N∆33 or Ape1K4pleA) for 1.5 h at 4°C with 250 fmol of S4 or S8 ODN. Alternatively, UV-crosslinking analysis were performed with Immunoprecipitated (IP) samples obtained from transfected U2OS cells employing 2 pmol of Ape1, co-incubated for 40 min at RT with 250 fmol of the G4-structured ODN in AP buffer containing 5 mM KCl and 1 mM MgCl2.
The mixtures were UV-crosslinked using a Vilber Lourmat UV-crosslinker BLX-254 at 0.2 J/cm2 and run onto SDS-PAGE 10% acrylamide.
Cell cultures, transfection and Co-immunoprecipitation
U2OS cells were grown in DMEM (EuroClone) medium supplemented with 10% fetal bovine serum (FBS, EuroClone), L-glutamine (2mM), penicillin (100 U/ml) and streptomycin (100 mg/ml) and cultured in a humidified incubator at 5% CO2 at 37°C. HeLa stable clones were grown as indicated in [26] and harvested 10 days after the addition of doxycycline in the medium [26]. OCI/AML2 and OCI/AML3 cells were grown in MEM-α (EuroClone) supplemented with 20% heat-inactivated FBS, L-glutamine (2mM), penicillin (100 U/ml) and streptomycin (100 mg/ml) and cultured in a humidified incubator at 5% CO2 at 37°C. CH12F3 cells were grown in advanced RPMI 1640 (EuroClone) supplemented with 10% FBS, Hepes (25 mM), β -mercaptoethanol (50 µM), L-glutamine (2mM), penicillin (100 U/ml) and streptomycin (100 mg/ml) and cultured in a humidified incubator at 5% CO2 at 37°C [47].
Co-immunoprecipitation studies were carried out with whole cell extracts from U2OS cells transfected with FLAG-tagged Ape1 mutants (Ape1WT-Flag, Ape1N∆33-Flag, Ape1K4pleA-Flag, Ape1K4pleQ-Flag and Ape1K4pleR-Flag). The cells were transiently transfected using 12 µg of DNA and 36 µL of Lipofectamine 3000 (Invitrogen). Cells were harvested 24 hours upon transfection, washed twice with PBS and re-suspended in lysis buffer (50 mM Tris HCl pH 7.4, 150 mM NaCl, 1 mM EDTA and 1 % Triton X-100) containing proteases inhibitor cocktail. After incubation for 20 minutes at 4°C under rotation, cell lysates were clarified by centrifugation at 12,000 × g for 10 minutes at 4°C and co-immunoprecipitation was performed with anti-FLAG M2 affinity gel (SIGMA-ALDRICH) at 4°C with gentle rocking for 3 hours. After washing three times with Tris-buffered saline (TBS), immunoprecipitates (IP) were then eluted by incubation with 0.15 mg/ml FLAG peptide in TBS and analyzed as indicated. Samples were then loaded onto a 12 w/vol % SDS-PAGE electrophoresis gel. Proteins were then transferred to nitrocellulose membranes (Schleicher & Schuell). Monoclonal α -Ape1 was from Novus Biologicals (1:5,000 dilution). Membranes were incubated with secondary antibodies labeled with IRDye (1:10,000 dilution) in 5% milk, PBS and Tween 0.1%. All gel images were captured with an Odissey CLx scanner (LI-COR GmbH) and analyzed using the ImageStudio software (LI-COR GmbH).
Chromatin immunoprecipitation and quantitative PCR
Chromatin immunoprecipitation (ChIP) was performed using the Diagenode High cell number ChIP kit, according to the manual. After colturing HeLa cells stably carrying p3XFLAG-CMV vectors in doxycycline 1 µg/ml, as previously described [26], the cells were crosslinked with 1% formaldehyde for 10 minutes at RT, and sonicated with the Diagenode bioruptor. Samples were immunoprecipitated overnight with anti-FLAG (Sigma-Aldrich). Telomeric DNA sequences were amplified by PCR using SensiFAST SYBR No-ROX Kit (Bioline).
The primer sequences were telomere forward primer (Tel 1) 5’-GGTTTTTGAGGG-TGAGGGTGAGG-GTGAGGGTGAGGGT-3’; telomere reverse primer (Tel 2) 5’-TCCCGACTA-TCCCTATCCCTATCCCTATCCCTATCCCTA-3’; 36B4 forward primer 5’-CAGCAAGTGGG-AAGGTGTAATCC-3’; 36B4 reverse primer 5’-CCCATTCTATCATCAACGGGTACAA-3’ [48,49]. Samples were run on a C1000 Thermal Cycler CFX96 Real-Time System (BIORAD). Each sample was analyzed in triplicate. 36B4, a single-copy gene that encodes the acidic ribosomal phosphoprotein P0, was used as a reference [49]. PCR reactions (15 µL) were set up as follows: 1 µL of recovered ChIP DNA, 2× SYBR Green master mix (Bioline), and the forward and reverse primers each at 100 nM final concentration. The thermal cycling conditions were as follows: 10 min at 95 °C, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min for both telomere and 36B4 amplification.
AP site incision assays
Cleavage assays were performed either at high ionic strength (Tris 50 mM pH 7.5, KCl 50 mM, MgCl2 10 mM, BSA 1 µg/µl, triton X-100 0.05%) or at low ionic strength (Tris 5 mM pH 7.5, KCl 5 mM, MgCl2 1 mM, BSA 0.1 µg/µl, triton X-100 0.005%). A fixed amount of substrate (250 fmol) was incubated with an increasing amount of Ape1 proteins for 40 minutes at 37°C. Alternatively, 10 pmol of Ape1WT were incubated for 1h at RT with increasing amounts of NPM1, varying between 5 and 200 pmol. Then the substrate was added for the reaction for 40 min at 37°C in a solution containing 50 mM KCl. The reactions were in any case stopped by adding an equal volume of stop solution containing formamide (10 mM EDTA, 0.5 % bromophenol blue, 80 % formamide), followed by heating for 5 minutes at 95°C. The mixtures were loaded on a 7 M urea 20% acrylamide gel and run in 0.5x TBE at 130 V for 50 minutes. The intensity of the obtained bands was determined using a fluorescence scanner (Odyssey CLx, LI-COR GmbH) and the quantification was obtained calculating the ratio between the product intensity over the sum between the intensities of the product and the substrate. The signals of the non-incised substrate (S) and the incision product (P) bands were quantified using Image Studio software.
Surface Plasmon Resonance (SPR) experiments
Real-time binding assays were performed at 25 °C on a Biacore 3000 Surface Plasmon Resonance (SPR) instrument (GE Healthcare). For immobilization, 5’-biotinylated Nat and Poly dT ODN were injected at a concentration of 20 µM on a SA Biacore sensor chip until the desired level of immobilization was achieved (averaged value of 700 RU) in an injection time of 7 min. Binding assays were carried out by injecting 90 µL of analyte, at 20 µL x min-1. Experiments were carried out at pH 7.4 using HBS (10 mM Hepes, 150 mM NaCl, 3 mM EDTA, pH 7.4). The association phase (kon) was followed for 270 s, whereas the dissociation phase (koff) was followed for 300 s. The reference chip sensorgrams were properly subtracted to sample sensorgrams. After each cycle, the sensor chip surface was regenerated with a 1.0 M NaCl solution for 30 s followed by multiple buffer injections to yield a stable baseline for the following cycles. Analyte concentrations were in the range 50–1000 nM, for all proteins. Experiments were carried out in duplicates. Kinetic parameters were estimated assuming a 1:1 binding model and using version 4.1 Evaluation Software (GE Healthcare).
Telomeric length assay
Telomere length analysis was performed employing TeloTAGGG Telomere Length Assay Kit (Roche). Genomic DNA was isolated form OCI/AML2, OCI/AML3, CH12F3 cells using the Blood & Cell Culture DNA Midi kit (Qiagen). OCI/AML2 were treated with #3 500 nM [50], or with DMSO as control, for 7 days. After digestion with Hinf I and Rsa I, DNA was separated on 0.8% agarose gel. Following denaturation, the DNA was transferred to a HybondN+ membrane (Boehringer Mannheim), and hybridized with a digoxigenin-labeled telomeric probe included in the kit. Non-radioactive telomeric signal intensity was scanned with high resolution chemiluminescence settings using a Molecular Imager Chemidoc XRS scanner (Bio Rad) with Image Lab™ Software (Bio Rad). The length of the fragments was quantified using the TeloTool software version 1.3 (Matlab) [51].
GST pull-down assay
300 pmol of NPM1 were added, together with 100 pmol of GST or Ape1-GST, to 15 µl of glutathione-sepharose 4B beads (GE healthcare). Binding was performed in AP buffer 1x at 4°C for 2h, under rotation. The beads were washed three times with PBS 1x supplemented with 0.1% w/v Igepal (CA-630 Sigma). Elution was performed with GSH 10 mM and the obtained samples were analyzed through western blot. Anti-GST (Sigma H9658) and anti-NPM1 (Invitrogen 32–5200) antibodies were employed.
Statistical analysis
Statistical analyses were performed by using the Student’s t test. P <0.05 was considered as statistically significant.
Results
Structural analysis of G4 sequences by circular dichroism (CD) spectroscopy
Previous studies, performed on dsDNA, demonstrated that Ape1 exhibits a substrate-dependent endonuclease activity [52,53] and may play a role in telomere maintenance [31]. The potential of Ape1 to process telomeric G-quadruplex structures containing abasic sites has been barely investigated [31,32,54,55]. In order to fill in this gap, we used three labelled oligonucleotides (ODN) (Table 1), including telomeric sequences already known to fold into G4 structures [41], some bearing abasic sites in different positions. Specifically, the ODN, indicated as S4 and S8 contain a tetrahydrofuran (F) residue resembling an abasic site [56], replacing the 4th or the 8th guanosine, respectively, while Nat represents the natural telomeric sequence containing undamaged DNA. In order to obtain structural insights, we first analyzed the ODN by CD spectroscopy, under several buffer conditions including those required for the enzymatic assays. The profile of the labeled Nat sequence (Fig. 1A) (annealed at 70 mM KCl) appears almost superimposable on that of its unlabeled counterpart [41], thus indicating that the presence of the IRDye moiety does not influence G-quadruplex conformation. This profile is characteristic of the hybrid 3+1 strand arrangement (Fig. S1)[57]. On the other hand, the CD spectrum of sequence S4 (Fig. 1B) shows a positive band around 260 nm, typical of parallel G-quadruplexes, and a further positive band at 295 nm. In turn, S8 exhibits a negative band at 243 nm and a positive band at 265 nm (Fig. 1C), that are characteristic of parallel G-quadruplex structures adopted by telomeric sequences in molecular crowding conditions [58]. These data would suggest the coexistence of parallel and other types of G-quadruplex conformations for S4 and S8, with a clear prevalence of the parallel conformation in the case of S8 [41]. Overall, the CD profiles of the three labeled ODN in the conditions tested appear very similar to those obtained in KCl 70 mM solution alone, thus indicating that the changes of buffer composition, due to addition of Tris and MgCl2, and KCl, do not affect the G-quadruplex folding topology adopted following the annealing procedure. Schematic representations of the hybrid 3+1 conformation, adopted by Nat, and that of the parallel G-quadruplex are reported in Fig. S1.
Fig. 1-. G4 structures analysis by CD spectroscopy.

Overlay of CD spectra of telomeric oligonucleotides Nat (A), S4 (B) and S8 (C) acquired at indicated buffer conditions.
CD-melting experiments were also carried out to investigate the thermal stability of the G-quadruplex structures adopted by the labeled ODN at the lowest salt concentration, namely the least favorable conditions for the G-quadruplex stability. The CD-melting profiles (Fig. S2) showed that all the G-quadruplex structures start to melt at around 45–50°C and thus the three ODN were completely folded at 37°C. These data clearly indicate the presence of suitable amount of G-quadruplex structures in all the conditions used for the following assays (see below).
The 33-residue Ape1 N-terminus is required to stably bind telomeric G4 structures
The 33 N-terminal residues of Ape1 play a role in the recognition of single stranded nucleic acids [24] Therefore, we analyzed the functional role of this N-terminal sequence of Ape1 for interaction with telomeric sequences. The recombinant proteins used here (unmodified Ape1 or Ape1WT, and a derivative lacking the first 33 N-terminal residues called Ape1N∆33) were expressed and purified in E. coli as previously described [22] and verified by SDS-PAGE analysis (Fig. S3A). The binding ability of purified recombinant Ape1 proteins to the ODN was tested through electrophoretic mobility shift assay (EMSA) analyses (Fig. 2A). These assays were performed by incubating the proteins with the telomeric substrates (i.e. Nat, S4 and S8) and the unstructured Poly dT-F, a poly dT oligonucleotide containing an abasic site, as a control. The analysis of native gels (Fig. 2A, left) revealed that Ape1WT bound the telomeric sequences whereas this ability was lost when the N-terminal sequence was missing. Specifically, three different shifted bands (indicated by asterisks in the figure) are clearly visible that correspond to protein-DNA complex formation between Ape1WT and the telomeric sequences (lanes 2, 4, 6). The bands present at the origin of the gel in all the DNA samples with APE1 in correspondence of the bottom of each well, are possibly due to large protein-DNA aggregates formed as a consequence of the high protein:DNA stechiometric ratio used in the experiment. The bands appearing at the origin of the gel, indicated as a mark (#) in Figure 2A, are possibly the result of formation of sovramolecular aggregates by the telomeric oligonucleotides. In fact, these low migrating bands are only present in native gel analyses (EMSA) but are absent when the samples are analysed under denaturing conditions (see below) demonstrating that they are not contaminant of the oligonucleotides used in the study. Lower bands, with weaker intensities, may have increased mobility due to conformational differences [59]. As expected, there was no indication of complex formation between Ape1 and the unstructured Poly dT-F (lane 8), confirming the importance of ODN secondary structure for stable binding [24]. Furthermore, the band pattern among the three ODN demonstrated that Ape1WT is proficient in binding the G-quadruplex sequences with no significant differences among the G4 substrates used. In addition, EMSA experiments with the Ape1N∆33 (Fig. 2A, right) highlight the requirement of the residues in the 33 N-terminal domain residues for binding to each G4 substrate (lanes 3–5-7). Again, no detectable complex was observed for the unstructured Poly dT-F (lane 9) incubated with Ape1N∆33. Notably, UV-crosslink experiments (Fig. S3B) showed that the more stable complex between Ape1 and the substrate has relative mobility corresponding to ~44 kDa in the case of the Ape1WT protein, and of about 42 kDa in the case of the Ape1N∆33 deletion mutant, consistent with a 1:1 stoichiometry in the complex. Moreover, the presence of a UV-crosslinked complex between Ape1N∆33 and the ODN (Fig. S3B) suggests that the absence of the 33 N-terminal sequence does not hamper the ability of the protein to interact with the DNA substrate, at least transiently.
Fig. 2-. Ape1 stably binds the G4 oligonucleotides.

A) Representative native EMSA polyacrylamide gel of recombinant Ape1WT (left) and Ape1N∆33 (right). Binding on the indicated ODN substrates (25 nM) is shown. Poly dT-F was used as negative control whereas Nat was used as positive control. The difference between this signal and the corresponding sample in the left panel relies only the intensity. *, ** and *** indicate the signals derived from metastable complexes that display different migration. # indicates the formation of sovramolecular aggregates of telomeric oligonucleotides. Reactions were performed as explained in Materials and methods section.
B) Overlay of sensorgrams relative to SPR experiments for the binding to immobilized Biot- Nat of Ape1WT (left) and Ape1N∆33 (right), respectively.
Altogether, these results demonstrated that: i) the molar ratio between the protein and the substrate is close to 1:1 as detected by the calculated molecular weight of the Protein-DNA complexes through UV-crosslink experiments; ii) the different mobility of the complexes and the weak signals of the retarded complexes, as seen from EMSA experiments, is possibly due to metastable complexes formation during the electrophoretic run [59] and iii) the absence of the 33 N-terminal sequence may probably affect the binding equilibrium of Ape1 with the substrate, possibly accelerating the dissociation step.
In order to quantitatively evaluate the affinity of both Ape1WT and for the telomeric Nat sequence, we used surface plasmon resonance (SPR) analyses. The overlays of the binding profiles are shown in Fig. 2B (and in Fig. S4). Both proteins exhibited concentration-dependent binding: whereas Ape1WT exhibited a low nanomolar value for its dissociation constant with the Nat sequence (Fig. 2B, left), the binding of Ape1N∆33 to the Nat sequence (Fig. 2B, right), exhibited a 9-fold reduced affinity (KD) compared to Ape1WT (Table 2). This was further corroborated by the SPR analysis of Poly dT-F/Ape1N∆33 interaction, that, in the range of concentrations tested, does not allow the evaluation of the KD value (Fig. S4); while Ape1WT shows an affinity toward this unstructured ligand five-fold reduced with respect to the G-quadruplex sequence (Table 2). The absence of a visible signal arising from the interaction between Ape1N∆33 and Nat substrate in EMSA assay, despite a five-fold difference in KD with respect to Ape1WT, may be due to the shifting of the equilibrium during the electrophoretic run toward the dissociated form of the complex. These data further confirm an important role exerted by the N-terminal sequence of Ape1 in the recognition of the G4 ODN, reflected in both dissociation and association phases shown by Ape1N∆33 with respect to full-length (Table 2).
Table 2-.
SPR based equilibrium dissociation constants (KD) and kinetic parameters for the interaction of Ape1WT and Ape1N∆33 and NPM1 with Nat and Poly dT ODN using the BIAevaluation v.4.1 software. Data reported were obtained through SPR analyses using proteins as analyte on the indicated biotinylated ODN ligands.
| Protein | ODN | kon(M−1 s −1× 104) | koff(s−1×10−3) | KD (nM) |
|---|---|---|---|---|
| Ape1WT | Nat | 8.23 | 2.6 | 31.6 |
| Poly dT | 2.82 | 4.79 | 170 | |
| NPM1 | Nat | 2.04 | 2.99 | 147 |
| Poly dT | 0.696 | 1.78 | 255 | |
| Ape1N Δ33 | Nat | 3.04 | 8.67 | 285 |
| Poly dT | Not measurable | |||
The enzymatic activity of Ape1 on G4 telomeric structures is influenced by ionic strength and the N-terminus
In order to test the endonuclease activity of Ape1 on telomeric substrates, we performed AP-site incision assays under different salt concentrations including, as substrate, a conventional dsDNA oligonucleotide containing an abasic site in the middle, named ds-F [60].
Cleavage assays were performed by incubating the indicated amounts of Ape1WT with a fixed amount of each G4 substrate in a high ionic strength solution (50 mM KCl). As Fig. 3A shows, Ape1WT exhibited different endonuclease activities on the three substrates: it did not significantly cleave the S4 ODN, generating a maximum of only about 4% of product. In contrast, Ape1 cleaved about one third of the S8 ODN substrate at the highest protein concentration (Fig. 3A). As expected, the Poly dT-F was cut less efficiently than S8, as it does not adopt secondary structures, and the Nat sequence was not processed (Fig. S5A). Moreover, as expected, the ds-F ODN was the best substrate, with a 1000-fold higher rate of cleavage by Ape1 than found for the G4 structures (Fig. S5A).
Fig. 3-. The position of the abasic site on the G4 influences Ape1 endonuclease activity.
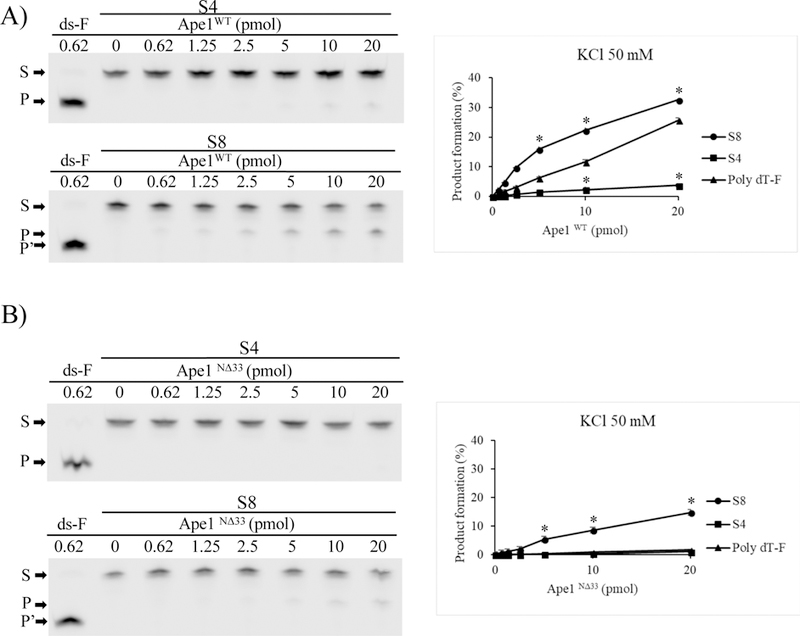
A) Representative denaturing polyacrylamide gels of AP site incision by Ape1WT on S4 and S8 incubated with the indicated amount of protein in a solution containing 50 mM KCl as described in Materials and methods section (left).
B) Representative denaturing polyacrylamide gels of AP site incision by Ape1N∆33 on S4 and S8 incubated with the indicated amount of protein in a solution containing 50 mM KCl as described in Materials and methods section (left).
Graphs describe the percentage of conversion of substrate (S) into product (P) as a function of the dose of Ape1 protein on the specified substrate (right). ds-F ODN was used as positive control. S denotes the substrate position; the length of the generated products is 1 nt different between S8 (P) and ds-F (P’). Average values are plotted with standard deviations of three loadings of the same experiment as a function of protein dosage. Standard deviation values were always less than 10% of the mean of experimental points. See also supplementary Fig. S5A and S5B for the gels.
Prompted by the importance of the Ape1 N-terminal sequence in binding G4 structures (Figs 1 and 2), we analyzed the endonuclease activity of the Ape1N∆33 on the same telomeric substrates (Fig. 3B). At high ionic strength (i.e. 50 mM KCl), Ape1N∆33 cleaved the G-quadruplex structures much less efficiently than Ape1WT protein. Cleavage of the S8 substrate reached a maximum of nearly 15%, while both the S4 and Poly dT-F ODN gave maxima of only about 1% product (Fig. 3B). The activity of Ape1N∆33 on ds-F, was weaker than that of the full-length protein but, at the highest concentration, Ape1N∆33 was able to process nearly all the substrate, with no activity on the Nat or the Poly dT-F ODN (Fig. S5B).
At low ionic strength, the Ape1 N-terminus affects the catalytic activity of the protein, via the product-release step of the reaction [22]. We therefore tested the effects of reduced salt concentration on Ape1-cleavage activity, by performing enzymatic experiments in 5 mM KCl (vs. 50 mM in the previous experiments). Under these conditions, Ape1WT displayed increased endonuclease activity on G4 structures (Figs 4A and S6A), then seen at higher KCl (Fig. 3A). In detail, the S4 resulted to be better processed under low ionic strength conditions (Fig. 4A) by Ape1WT, with an increase of about ten-fold with respect to high salt concentration (50 mM KCl). Similarly, the S8 appeared more efficiently processed at low ionic strength with a more than two-fold increase in product formation. Notably, also the Poly dT-F was processed under these conditions, with an efficiency similar to that displayed on the S8 (Fig. 4A). As expected for ds-F, the efficiency was significantly higher (about 100-fold), compared to the two G4 structures (Fig. S6B), and 4-fold higher with respect to the activity measured at 50 mM KCl. Therefore, the Ape1 enzymatic activity was significantly affected by the KCl concentration and it was modulated by an electrostatic effect possibly involving the contribution of the basic amino acids of the N-terminal sequence [22,24].
Fig. 4-. Ionic strength influences Ape1 endonuclease activity.

A) Graph depicting AP site incision activity of Ape1WT on the specified substrate in a solution containing 5 mM KCl. See also supplementary Fig. S6A for the gels.
B) Graph depicting AP site incision activity of Ape1N∆33 on the specified substrate in a solution containing 5 mM KCl. See also supplementary Fig. S7A for the gels. Graphs describe the percentage of conversion of substrate (S) into product (P) as a function of the dose of protein on the specified substrate. Average values are plotted with standard deviations of three loadings of the same experiment as a function of protein dosage. Standard deviation values were always less than 10% of the mean of experimental points.
C) AP site incision activity on S8 ODN decreases in raising ionic strength. Graph depicting endonuclease activity experiments performed with Ape1WT or Ape1N∆33 as described in Materials and methods section. Average values are plotted with standard deviations of three loadings of the same experiment as a function of KCl concentration used. Standard deviation values were always less than 10% of the mean of experimental points. See also supplementary Fig. S7C for the gels.
D) Histograms summarizing the different endonuclease activities of Ape1WT (left) and Ape1N∆33 (right) on the specified substrates at the indicated fmol of the enzyme in the stated KCl concentrations on the different substrates. Each bar was compared with the activity of the protein on ds-F in the corresponding saline condition.
Then, also the activity of Ape1N∆33 was tested under low ionic strength conditions. Both S4 and S8 substrates were processed more actively than the previous condition (Fig. 4B and S7A). Moreover, the activity of Ape1N∆33 on ds-F was even higher than the one displayed by Ape1WT protein (Fig. 5C and S7B), since the maximum product formation is reached at 1.5 fmol of Ape1N∆33. These data are in agreement with previous results and are due to an increase of the koff in the catalytic reaction [22].
Fig. 5-. Ape1 interacts with G4 oligonucleotides in a stoichiometric ratio.

Interaction between recombinant wild type and mutant Ape1 (A) with S4 (left) and S8 (right) was evaluated through crosslinking analysis.
The substrates were challenged with Ape1WT (lane 2 and 6), Ape1N∆33 (lane 3 and 7) or Ape1K4pleA (lane 4 and 8) as described in Materials and methods section. Reactions were resolved onto SDS-PAGE 10%. Lane 1 and 5: crosslinked ODN without proteins.
AP site incision activity of Ape1WT, Ape1N∆33, Ape1K4pleA in 50 mM KCl (B) and 5 mM KCl (C). ds-F was used as positive control.
Graphs describe the percentage of conversion of substrate into product as a function of the dose of protein on the indicated substrate. Average values are plotted with standard deviations of three loadings of the same experiment as a function of protein dosage. Standard deviation values were always less than 10% of the mean of experimental points. See also supplementary Figs S5, S6, S7 and S8 for the gels.
In order to evaluate the dependence of the Ape1 activity on ionic strength, we analyzed the variation of enzyme products formation by Ape1WT and Ape1N∆33 on the S8 substrate, as a function of KCl concentrations (Fig. 4C and S7C). Data obtained clearly indicated a linear dependence of the Ape1 endonuclease activity on KCl concentrations even though to a different extent for the two proteins since the trend line of Ape1N∆33 showed a greater steepness (m= −1.574) with respect to that of Ape1WT (m= −1.136). In Fig. 4D, the quantifications of the amount of products obtained under the different salt concentration conditions, as above, are summarized showing that the dependence of the enzymatic activities of Ape1WT and Ape1N∆33 proteins on salt concentrations appeared more pronounced for S4 and the S8 ODN with respect to the ds-F substrate.
Overall, these data demonstrated that the enzymatic activity of Ape1 on G4 telomeric structures strongly depends on the ionic strength and on the presence of the N-terminal sequence. The data show that the N terminus also modulates activity on ssDNA (the poly dT substrate). Therefore, these findings suggest that the charged residues present in the N-terminus of Ape1 may modulate its enzymatic activity on G4 and on non-canonical DNA secondary structures.
Acetylatable lysine residues in the Ape1 N-terminus are required for binding and enzymatic activity on G4 telomeric structures
In order to investigate whether the positive charges (i.e. acetylatable lysine residues) in the Ape1 N-terminal sequence might affect the processing of damaged telomeric substrates, we evaluated the endonuclease activity on the S4 and S8 substrates of Ape1K4pleA, in which replacement of the lysine by alanine residues (Fig. S8A) mimics acetylation by neutralizing the positive charges of the side chains [22,25].
Purified Ape1K4pleA (Fig. S8B) displayed an altered mobility with respect to Ape1WT, which was likely due to the alteration of the overall charge, as we previously observed [22]. First, we evaluated the ability of Ape1K4pleA to interact with the telomeric sequences. UV-crosslinking experiments (Fig. 5A) demonstrated that all the proteins form complexes with both S4 and S8, and that the complexes had the same relative mobility as the proteins. The enzymatic activities were assessed under different salt concentration conditions. At 50 mM KCl, none of the proteins had strong activity on S4 (<3% product formation). All the proteins had some activity on S8 at 50 mM KCl, with Ape1WT having a 2- to 3-fold higher rate of cleavage than Ape1N∆33 and Ape1K4pleA (Fig. 5B and Fig. S8C). In contrast, the activity of Ape1K4pleA on ds-F at 50 mM KCl resembled that of the Ape1WT, while Ape1N∆33 activity was somewhat lower (Figs 5B and S8C). At 5 mM KCl, the lower salt concentration strongly increased the activity of all the proteins on both the S4 and the S8 substrates (Fig. 5C, Fig. S8D). On the S4 substrate, Ape1K4pleA was more active (~44% product) than Ape1WT (Figs 5C and S8D). Notably, the activity of both modified Ape1 proteins on ds-F was similar and higher than that of Ape1WT (Figs 5C and S8D).
Overall, these data clearly demonstrate that lysine 27–35 of Ape1 contribute strongly to the binding and cleavage activity of the enzyme for a DNA lesion in telomeric sequences; acetylation to neutralize these residues in vivo may modulate Ape1 function at telomeres.
Activity of Ape1 protein variants from U2OS cells
The Ape1 enzymatic activity could be dynamically modulated by interacting proteins in cells [61,62]. With the aim to evaluate the contribution of such partners on the enzymatic activity of Ape1, we carried out enzymatic assays employing Ape1-protein complexes isolated from human cell lines. In these assays, in addition to the K-to-A protein, we introduced the alternative K-to-R protein (a non-acetylatable form - Ape1K4pleR) and the K-to-Q protein (mimicking a constitutive acetylated Ape1 form retaining the side chain with the charge neutralized - Ape1K4pleQ) substitutions [63]. We measured the activity of the Ape1 proteins on telomeric substrates using immunopurifed Ape1-protein complexes obtained from U2OS human cells (Fig. S9A and B). We used U2OS cells as a source of recombinant immunopurified Ape1 protein, because we could get a significant level of recombinant protein upon transfection. These complexes were affinity purified following transfection of vectors expressing the FLAG-tagged mutant proteins [63] and checked for the presence of Ape1’s partners such as nucleophosmin (NPM1) (Fig. S9B and [27]). The Ape1K4pleA and Ape1K4pleQ proteins immunopurifed from U2OS cells had different electrophoretic mobility than Ape1WT (Fig. S9A), consistent with the recombinant proteins obtained from E. coli (Fig. S3A). The separation of these IP samples on a urea-gel demonstrated that the altered migration obtained in SDS-PAGE gels was not due to a different apparent molecular weight of the mutants Ape1K4pleA and Ape1K4pleQ, but presumably to the change of the overall charge (Fig. S9C), as previously shown [22].
We then conducted UV-crosslinking experiments with these different Ape1 variants and Ape1WT, (Fig. 6A). The ability of each mutant protein to stably interact with the substrates was at least comparable to that of the immunocaptured Ape1WT. Interestingly, the Ape1K4pleR protein displayed an enhanced binding ability, relative to Ape1WT, with all the substrates used in the UV-crosslinking assay (Fig. 6A, lanes 7).
Fig. 6-. Acetylable lysine residues in Ape1 N-term are necessary for G4 oligonucleotide binding.

A) Crosslinking analysis of immunopurified (IP) recombinant Ape1-Flag proteins obtained from U2OS cells were performed as described in Materials and methods section and run onto an SDS-PAGE 10%. Recombinant protein, rApe1WT was used as control. Values reported at the bottom of the Figure, in correspondence of each lane, indicate the signals intensity of the retarded complex expressed as fold of change with respect to the wild-type protein. See also supplementary Figs S9A and B for the gels.
B) Graphs depicting AP site incision activity of IP for Ape1WT, Ape1N∆33, Ape1K4pleA, Ape1K4pleQ and Ape1K4pleR on the specified substrate in a solution containing 5 mM KCl. Graphs describe the percentage of conversion of substrate into product as a function of the dose of protein on the indicated substrate. Average values are plotted with standard deviations of three loadings of the same experiment as a function of protein dosage. Standard deviation values were always less than 10% of the mean of experimental points. See also supplementary Figs S9D for the gels.
C) ChIP analysis on telomeric sequences. U2O2 cells were transfected with pCMV plasmids for the expression of Flag-tagged Ape1 mutants as indicated in Materials and methods section. Empty vector used as control. Histogram shows the amount of telomeric sequence that was immunoprecipitated for the different Ape1 mutant. Data represent the binding of the telomeric sequences by each mutant of Ape1 resulting from ChIP experiments as percentage of input DNA. Error bars correspond to SDs of qPCR from one ChIP experiment.
We performed cleavage experiments, at low KCl concentration (5 mM), to compare with Ape1WT the activity of the two acetylation-mimicking proteins (Ape1K4pleA and Ape1K4pleQ) and the protein that mimicks non-acetylatable Ape1 (Ape1K4pleR). Endonuclease assays (Figs 6B and S9D) demonstrated that, particularly with the S8 substrate, the acetylation-mimicking mutants displayed a higher endonuclease activity than did the Ape1WT and Ape1K4pleR proteins, suggesting that the loss of positive charges of several Lys residues confers on the enzyme an increased ability to release the product upon cleavage, as we previously hypothesized [22]. The S4 substrate was significantly less processed by all the tested proteins, similarly to previous data obtained with recombinant proteins from E. coli with the activities of acetylation-mimicking proteins higher than those of the Ape1WT and Ape1K4pleR proteins. Similar experiments, performed at 50 mM KCl, indicated that the enzymatic activity of the tested proteins was significantly lower under those conditions, as found for the purified proteins (data not shown).
Overall, our data clearly indicate that the acetylation of lysine residues K27−35 is important for controlling the overall enzymatic activity of Ape1 on telomeric sequences, as in the case for ordinary abasic dsDNA substrates [22].
The telomeric-binding ability of Ape1 depends on K27, K31, K32 and K35 residues
Next, we checked the ability of Ape1 wild-type and mutated proteins to physiologically interact with telomeres by using Telomeric-Chromatin Immuno-Precipitation analysis (Telo-ChIP) analyses (see Materials and methods). Telo-ChIP analysis was performed on HeLa cells stably expressing FLAG-tagged Ape1 mutant proteins in place of the endogenous wild-type (which was down-regulated through a specific siRNA [25–27]). The expression levels of the ectopic forms of Ape1 in these cells was similar to that of the endogenous protein [27], which should minimize off-target and other confounding effects of overexpression. The DNA recovered from the ChIP assay was then analysed by quantitative PCR (Q-PCR). As shown in Fig. 6C, the telomeric binding ability of Ape1N∆33 was considerably reduced, compared to Ape1WT protein. In contrast, Ape1K4pleA showed a binding ability comparable to that of Ape1WT while Ape1K4pleR bound telomeric DNA with an activity 9-fold higher than that of Ape1WT. These data are consistent with the binding detected using UV-crosslinking (Figs 5A).
Interaction of NPM1 with Ape1 modulates its endonuclease activity on telomeric sequences
Since the N-terminal sequence of Ape1 is involved in modulating protein-protein interactions [24], we next checked whether the interaction with a known Ape1 partner NPM1 [26] may affect Ape1 enzymatic activity on telomeric DNA. We previously demonstrated that stable interaction between Ape1 and NPM1 requires charged K27−35 residue on Ape1 [25], and that this interaction modulates Ape1 endonuclease activity in vitro and in cancer cells [61]. Interestingly, NPM1 can bind G-quadruplex structures in the c-Myc promoter [64].
First, we first checked whether NPM1 affects telomere physiology by testing whether telomere length is affected in the OCI/AML3 cell line, which stably expresses the aberrant NPM1c+ protein, which is localized to the cytoplasm, compared to OCI/AML2 cells expressing wild-type NPM1 localized to the nucleus [65]. Moreover, the use of this cell model is strictly correlated with Ape1 function, since we previously demonstrated that a functional interaction between Ape1 and NPM1 is required to positively modulate the Ape1 endonuclease activity in vitro and in cancer cells [61]. Interestingly, OCI/AML3 cell line display an altered subcellular distribution of Ape1 as a function of NPM1c+ mislocalization and is associated with BER impairment [61]. Telomere-length assays clearly demonstrated an evident shortening of telomeres in OCI/AML3 cells with respect to control OCI/AML2 cells, confirming a role for nuclear NPM1 in telomere maintenance (Fig. 7A). To verify the importance of Ape1 role in the maintenance of telomere integrity, we inhibited the Ape1 endonuclease activity treating OCI/AML2 cells with sublethal doses of compound #3 for one week [50]. Upon the inhibition of Ape1 activity, we could detect a shortening of the telomeric signal, as the mean telomere length was 3.12 kbp compared with 3.28 kbp of the control sample (DMSO). This evidence suggests that the protein exerts its endonuclease activity at the telomeric level and its inhibition indeed affects telomere dimension. In order to confirm a role for Ape1 in telomere maintenance, we performed the telomere length analysis in CH12F3 cells, a mouse lymphoma cell line devoid of Ape1 (∆∆∆) or retaining one copy of Ape1 gene (∆++) [47]. The Ape1-proficient line had a telomere length averaging ~18 kbp, as expected for mouse telomeres. The Ape1 deficient cells showed a significantly shorter telomeres averaging 14 kbp (Fig. 7B). These results show a role for Ape1 in telomere maintenance in murine lymphocytes, consistent with a previous study on human cell lines [31].
Fig. 7-. NPM1 inhibits the endonuclease activity of Ape1 competing with the substrate for the interaction with the N-terminal sequence of the endonuclease.

A) Telomeric length assay as a function of NPM1 localization. Telomere length assay was performed as described in Materials and methods section on acute myeloid leukemia (AML) cells showing the different length of telomeres in cell types characterized by different NPM1 localization. AML2 cells were untreated (UNT) or treated with #3 or DMSO. The molecular weight marker (m) is provided by the kit; the weight of each band is indicated as kbp.
B) Telomeric length assay as a function of Ape1 expression level. Telomere length was assayed on mouse CH12F3 cells expressing (∆++) or depleted for Ape1 (∆∆∆) protein. Ctrl refers to a DNA control with a mean length of 7.4 kbp provided by the kit. The molecular weight marker (m) is provided by the kit; the weight of each band is indicated as kbp.
C) Representative native EMSA polyacrylamide gel of recombinant Ape1WT and Ape1N∆33 pre-incubated with increasing concentrations of NPM1. Binding on Nat substrate (25 nM) is shown. *, ** and *** indicate the signals derived from metastable complexes that display different migration. # indicates the formation of sovramolecular aggregates of telomeric oligonucleotides. Reactions were performed as explained in Materials and methods section.
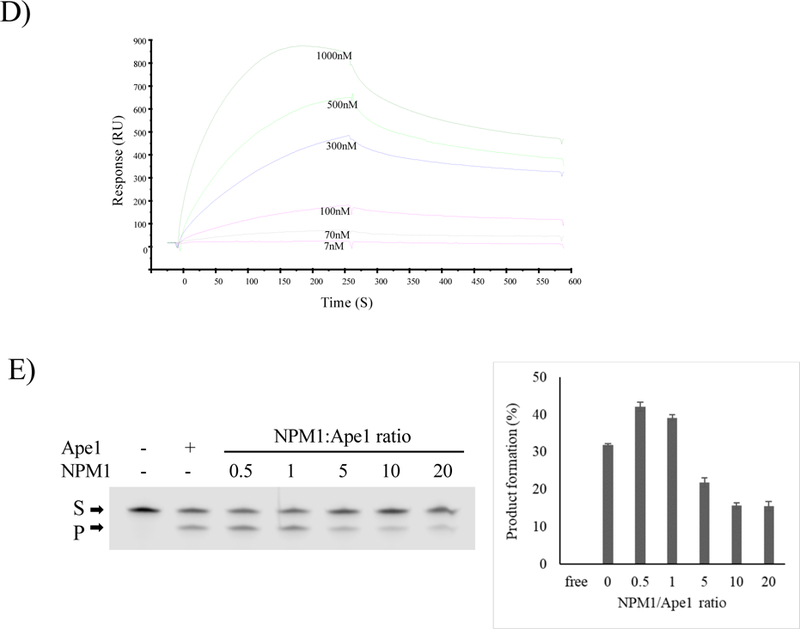
D) Overlay of sensorgrams relative to SPR experiments for the binding to immobilized Biot- Nat of NPM1.
E) Representative denaturing polyacrylamide gel of AP site incision by Ape1WT on S8 substrate in the presence of increasing amounts of NPM1 (left). The proteins were incubated in a solution containing 50 mM KCl as described in Materials and methods section. S denotes the substrate position and P denotes the product position. Histogram showing the level of substrate processing upon the increasing ratio between NPM1 and Ape1WT (right). Average values with standard deviations of three loadings of the same experiment are presented.
We then investigated the effects of NPM1 on Ape1 binding of the telomeric Nat substrate by EMSA assays. NPM1 did not form detectable complexes with the Nat, while there clearly was binding by Ape1 (Fig. 7C, lanes 2 and 3). When increasing amounts of NPM1 were added to reactions with a constant amount of Ape1, the extent of the protein-telomeric DNA complex was progressively decreased (Fig. 7C, lanes 4–7). As expected, Ape1N∆33 did not form detectable complexes with the Nat sequence, independently of NPM1 presence (Fig. 7C, lanes 8–12). GST-pulldown experiments (Fig. S10) confirmed the interaction of Ape1 with NPM1 recombinant proteins under these conditions.
The reported ability of NPM1 to bind G-quadruplex structures [64,66,67] prompted us to investigate telomere binding using SPR. As shown in Fig. 7D, NPM1 does interact with the Nat, although with a 4.6-fold lower affinity than Ape1WT (Table 2), consistent with the EMSA results (Fig. 7C).
We then analysed the effect of the interaction with NPM1 on the Ape1-AP endonuclease activity. When NPM1 was present in a sub-stoichiometric ratio with respect to Ape1, the endonuclease activity was modestly enhanced; in contrast when NPM1 was present in a supra-stoichiometric ratio with Ape1, the endonuclease activity was inhibited ~2-fold (Fig. 7E). Frome these data, we can conclude that interaction of NPM1 with Ape1 modulates its endonuclease activity on telomeric sequences, at least in vitro.
Discussion
This work was aimed at characterizing the ability of Ape1 to recognize and process telomeric DNA substrates containing abasic sites in different positions. Moreover, we wanted to evaluate the role of the unstructured N-terminal sequence and, in particular, that of the acetylatable Lys residues we previously identified [22,25]. To this end, we analyzed the ability of the wild type and several mutant forms of Ape1 to recognize and process different oligonucleotides that are able to fold into different G-quadruplex structures.
We found that Ape1WT was able to bind each G4 structured ODN, independently of the presence of the abasic site (Table 1 and Fig. 2). KD values obtained through SPR assays showed that the affinity of the protein toward the G4 structured substrates was considerably higher than that for an unstructured substrate, highlighting the importance of DNA secondary structure for recognition by Ape1, as we previously suggested [24].
While the G-quartet structure appears to be an important determinant, a possible effect of the abasic site position on Ape1 activity cannot be ruled out. In fact, the enzymatic activity on S4 was significantly lower than that on the S8 substrate, and the CD data indicate that the solution composition of G4 structures for S4 and S8 is different. There was a clear preference of Ape1 for a parallel G4 structure for S8, with this conformation the one preferred under molecular crowding conditions. Consistent with our results, other studies report a dependence of Ape1 activity on the type of G4 structure in telomeric sequences containing abasic sites [54]. Since the variation of ionic strength plays a crucial role in similar enzymes [68,69], we evaluated the enzymatic activity of Ape1WT under KCl concentrations of 5–50 mM. There was only a small effect of KCl for Ape1 acting on canonical abasic dsDNA (Fig. 5, panels C and D), as previously shown [22]; the KCl concentration does not affect the secondary structure of the G4-substrates (Fig. 1). Remarkably, Ape1N∆33 was not able to form any stable complexes with the substrates (Fig. 2) and exhibited a significantly lower efficiency in processing the abasic lesions in both the S4 and S8 structures (Fig. 3B), despite active Ape1 binding as demonstrated by UV-crosslinking experiments (Fig S3B). These findings further indicate that the unstructured N-terminal sequence of Ape1, though not directly participating in the enzymatic activity of the protein, can modulate stable DNA binding by Ape1, as we previously hypothesized [24] and as recently corroborated by other studies [59].
The Ape1 N-terminal sequence is rich in positively charged amino acids (8 Lys and 2 Arg residues); several of these lysine residues (K27, K31, K32, K35) can undergo acetylation in cells under genotoxic stress conditions [22,25]. Similarly to Ape1N∆33, the mutant Ape1K4pleA, which mimics the acetylated status by replacing all four Lys with Ala residues, displayed an endonuclease activity on S4 and S8 that was significantly lower than that of Ape1WT. Despite this difference, the Ape1K4pleA did not show a reduced ability to bind to G4-DNA (Fig. 5A). Interestingly, at lower ionic strength, the activity of the Ape1K4pleA and Ape1N∆33 mutants was higher than that of Ape1WT protein. This result indicates that additional charged amino acids outside the N-terminus may contribute to the catalytic activities of Ape1. In addition, the increased enzymatic activities at low ionic strength of Ape1K4pleA and Ape1K4pleQ on S4 and S8, relative to Ape1WT and Ape1K4pleR (Fig. 6B and Fig. S9D), suggest that charged K residues in the Ape1 N-terminus may regulate the product-release step, as we previously hypothesized [22]. These findings are in line with previous observations showing that release of the incision product may represent the limiting step in Ape1 turnover, particularly for dsDNA and structures such as the telomere G-quadruplex [24,53,70,71].
The observation that the Ape1K4pleR protein has increased telomere binding ability (Fig. 6A-6C), also supports the conclusion that the charged residues in the N-terminal sequence significantly contribute to the stability of Ape1-telomeric substrates complexes.
Recent studies pointed to the unstructured Ape1 N-terminus as a relay for regulating the different functions of the protein through modulation of the Ape1-protein interactome [22,27,61,72]. We previously demonstrated that NPM1 stimulates Ape1 endonuclease activity on abasic dsDNA in vitro and in cells through interaction with the unstructured Ape1 N-terminus [22,61]. Moreover, NPM1 by itself interacts with G-quadruplex-forming DNA [64], and mouse cells lacking NPM1 display general signs of genetic instability and activation of DNA damage responses [73]. In addition, Acute Myeloid Leukemia (AML)-associated mutations in the NPM1 gene, cause the re-localization of part of the protein to the cytoplasm (NPM1c+). This mis-localization hampers DNA binding of non-telomeric G4 structures by NPM1 [74,75] and may affect Ape1 nuclear BER function in cancer cells [61]. Therefore, the investigation of a potential effect of NPM1 in modulating the activity of Ape1 on G4-structures may represent an important model for understanding the role of mutual interactions between Ape1 and its interacting partners in the processing of damaged DNA. Using AML cell lines, we demonstrated that functional NPM1 is required for maintenance of appropriate telomeric length. Depending on the NPM1/Ape1 ratio, recombinant NPM1 may modulate the enzymatic activity of Ape1 on abasic telomeric substrates by having a stimulatory function under sub-stoichiometric ratio, and an inhibitory effect at supra-stoichiometric ratio. It must be noted that functional NPM1 may act as a pentameric molecule [76], so it is possible that, in vivo, a supra-stoichiometric ratio between NPM1 and Ape1 may represent a more physiologic condition, though further experiments are needed to better circumstantiate these findings in the telomeric context. Overall, the data show that, depending on the relative stoichiometric ratio between NPM1 and Ape1, the enzymatic activity of Ape1 on telomeric substrates may be strictly modulated by Ape1 protein interacting partners. Moreover, these findings suggest that an effort should be made to directly determine whether telomere maintenance is compromised, as a result of disrupting the normal Ape1-NPM1 interaction.
Since the interaction between NPM1 and Ape1 is also modulated by acetylation of the Lys residues at positions 27/31/32/35 [25], we may also infer that their acetylation may significantly affect the Ape1 role on telomere maintenance, not only through a direct effect on Ape1 binding on these substrates (Fig. S10), but also by means of protein-protein mediated effects. Interaction of NPM1 with Ape1 involves mainly the N-terminal sequence of Ape1 [24] and positive charges on Lys residues at positions 27/31/32/35 [25]. Thus, these data support the hypothesis that the Ape1 positively charged N-terminus is an important player in regulating the kinetics of the interaction of Ape1 with the telomeric substrates. Moreover, the endonuclease activity data support the conclusion that, depending on the relative stoichiometric ratio between NPM1 and Ape1, the enzymatic activity of Ape1 over telomeric substrates may be strictly modulated by Ape1 protein interacting partners. Therefore, these findings require an effort to directly determine whether telomere maintenance is compromised, as a result of disrupting the functional normal Ape1-NPM1 interaction.
Madlener et al. [31] showed that cells expressing an Ape1 mutant bearing substitution of K6/K7 with Ala residues underwent severe chromosomal instability. Though we did not evaluate the role of these residues in modulating the enzymatic activity and binding properties of Ape1 on telomeric G-quadruplex, we may speculate that our data might provide the molecular basis for those unexplained findings. In fact, we previously found that deacetylation of K6/K7 by Sirtuin 1 (SIRT1) is dependent on the charged status of the K27−35 residues [25]. Only when K27−35 are positively charged, K6/K7 may be de-acetylated by SIRT1, providing the positive charges essential for the normal function of Ape1 at telomeres. These observations support the hypothesis that dynamic cross-talk between the Lys residues in the Ape1 N-terminus may fine-tune the enzymatic activity of the protein and we speculate that there is a general role for acetylation of these residues in regulating Ape1 function at telomeres.
In another recent work [59], stimulation of the OGG1, MBD4 and ANPG glycosylases activities by Ape1 strictly depended on the presence of an intact Ape1 N-terminus. Through oligomerization of Ape1 along the DNA duplex dependent on its N-terminus, the protein was able to promote DNA-bridges formation and DNA aggregation, which, in turn, may cause structural deformation in the DNA double helix responsible for increasing the enzymatic rate of the glycosylases themselves. This new biochemical property of Ape1 may be particularly interesting in the context of the highly repetitive telomeric sequences, in which the secondary and tertiary structures of DNA play a fundamental regulatory function that influences genome stability.
Further studies along these lines are warranted and will deserve particular attention. A crystal structure of full-length Ape1 protein with its cognate substrate would provide physical evidence to our biochemical observations. To date, all the available crystal structures of Ape1 did not allow to visualize the N-terminal sequence of the protein as results of its disorder [77,78], thus limiting definitive conclusions on this unusual DNA repair enzyme.
An altered acetylation status of Ape1 K27−35 has been demonstrated in human cancers [28,29]. We may thus hypothesize that chromosomal instability in cancer development may result from defective coordination of Ape1 function at telomeres due to post-translational modifications on its N-terminal Lys residues. Specific experiments are needed to corroborate this hypothesis.
Supplementary Material
Acknowledgements
This work was supported by a grant from Associazione Italiana per la Ricerca sul Cancro (AIRC) (IG19862) to G. Tell and by a grant from the National Institutes of Health (NIH) (R21CA191856, NIH G22F16000880005) to B. Demple. The authors thank the Tell and Demple laboratories for constructive feedbacks during the development of this work.
Footnotes
The authors declare no conflict of interest.
Bibliography
- [1].Coleman WB, Tsongalis GJ, The role of genomic instability in human carcinogenesis, Anticancer Res 19 (1999) 4645–4664. [PubMed] [Google Scholar]
- [2].Grandér D, How do mutated oncogenes and tumor suppressor genes cause cancer?, Med. Oncol. Northwood Lond. Engl 15 (1998) 20–26. [DOI] [PubMed] [Google Scholar]
- [3].Ciccia A, Elledge SJ, The DNA Damage Response: Making It Safe to Play with Knives, Mol. Cell 40 (2010) 179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hanahan D, Weinberg RA, Hallmarks of Cancer: The Next Generation, Cell 144 (2011) 646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [5].Olovnikov AM, Telomeres, telomerase, and aging: Origin of the theory, Exp. Gerontol 31 (1996) 443–448. doi: 10.1016/0531-5565(96)00005-8. [DOI] [PubMed] [Google Scholar]
- [6].Nabetani A, Ishikawa F, Unusual telomeric DNAs in human telomerase-negative immortalized cells, Mol. Cell. Biol 29 (2009) 703–713. doi: 10.1128/MCB.00603-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bailey SM, Telomeres, chromosome instability and cancer, Nucleic Acids Res 34 (2006) 2408–2417. doi: 10.1093/nar/gkl303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wogan GN, Hecht SS, Felton JS, Conney AH, Loeb LA, Environmental and chemical carcinogenesis, Semin. Cancer Biol 14 (2004) 473–486. doi: 10.1016/j.semcancer.2004.06.010. [DOI] [PubMed] [Google Scholar]
- [9].Smolková B, Dušinská M, Rašlová K, McNeill G, Spustová V, Blažıček P, Horská A, Collins A, Seasonal changes in markers of oxidative damage to lipids and DNA; correlations with seasonal variation in diet, Mutat. Res. Mol. Mech. Mutagen 551 (2004) 135–144. doi: 10.1016/j.mrfmmm.2004.02.021. [DOI] [PubMed] [Google Scholar]
- [10].Lindahl T, Quality Control by DNA Repair, Science 286 (1999) 1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- [11].Demple B, Sung J-S, Molecular and biological roles of Ape1 protein in mammalian base excision repair, DNA Repair 4 (2005) 1442–1449. doi: 10.1016/j.dnarep.2005.09.004. [DOI] [PubMed] [Google Scholar]
- [12].Liu P, Demple B, DNA repair in mammalian mitochondria: Much more than we thought?, Environ. Mol. Mutagen 51 (2010) 417–426. doi: 10.1002/em.20576. [DOI] [PubMed] [Google Scholar]
- [13].Svilar D, Goellner EM, Almeida KH, Sobol RW, Base Excision Repair and Lesion-Dependent Subpathways for Repair of Oxidative DNA Damage, Antioxid. Redox Signal 14 (2011) 2491–2507. doi: 10.1089/ars.2010.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Friedman JI, Stivers JT, Detection of Damaged DNA Bases by DNA Glycosylase Enzymes, Biochemistry 49 (2010) 4957–4967. doi: 10.1021/bi100593a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wilson DM, Barsky D, The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA, Mutat. Res. Repair 485 (2001) 283–307. doi: 10.1016/S0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- [16].Wong D, Demple B, Modulation of the 5’-deoxyribose-5-phosphate lyase and DNA synthesis activities of mammalian DNA polymerase beta by apurinic/apyrimidinic endonuclease 1, J. Biol. Chem 279 (2004) 25268–25275. doi: 10.1074/jbc.M400804200. [DOI] [PubMed] [Google Scholar]
- [17].Tell G, Quadrifoglio F, Tiribelli C, Kelley MR, The many functions of APE1/Ref-1: not only a DNA repair enzyme, Antioxid. Redox Signal 11 (2009) 601–620. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tell G, Fantini D, Quadrifoglio F, Understanding different functions of mammalian AP endonuclease (APE1) as a promising tool for cancer treatment, Cell. Mol. Life Sci. CMLS 67 (2010) 3589–3608. doi: 10.1007/s00018-010-0486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Harrison L, Ascione G, Menninger JC, Ward DC, Demple B, Human apurinic endonuclease gene (APE): structure and genomic mapping (chromosome 14q11.2–12), Hum. Mol. Genet 1 (1992) 677–680. [DOI] [PubMed] [Google Scholar]
- [20].Mitra S, Izumi T, Boldogh I, Bhakat K, Chattopadhyay R, Szczesny B, Intracellular trafficking and regulation of mammalian AP-endonuclease 1 (APE1), an essential DNA repair protein, DNA Repair 6 (2007) 461–469. doi: 10.1016/j.dnarep.2006.10.010. [DOI] [PubMed] [Google Scholar]
- [21].Izumi T, Mitra S, Deletion analysis of human AP-endonuclease: minimum sequence required for the endonuclease activity, Carcinogenesis 19 (1998) 525–527. [DOI] [PubMed] [Google Scholar]
- [22].Fantini D, Vascotto C, Marasco D, D’Ambrosio C, Romanello M, Vitagliano L, Pedone C, Poletto M, Cesaratto L, Quadrifoglio F, Scaloni A, Radicella JP, Tell G, Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions, Nucleic Acids Res 38 (2010) 8239–8256. doi: 10.1093/nar/gkq691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kelley MR, Georgiadis MM, Fishel ML, APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1, Curr. Mol. Pharmacol 5 (2012) 36–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Poletto M, Vascotto C, Scognamiglio PL, Lirussi L, Marasco D, Tell G, Role of the unstructured N-terminal domain of the hAPE1 (human apurinic/apyrimidinic endonuclease 1) in the modulation of its interaction with nucleic acids and NPM1 (nucleophosmin), Biochem. J 452 (2013) 545–557. doi: 10.1042/BJ20121277. [DOI] [PubMed] [Google Scholar]
- [25].Lirussi L, Antoniali G, Vascotto C, D’Ambrosio C, Poletto M, Romanello M, Marasco D, Leone M, Quadrifoglio F, Bhakat KK, Scaloni A, Tell G, Nucleolar accumulation of APE1 depends on charged lysine residues that undergo acetylation upon genotoxic stress and modulate its BER activity in cells, Mol. Biol. Cell 23 (2012) 4079–4096. doi: 10.1091/mbc.E12-04-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vascotto C, Fantini D, Romanello M, Cesaratto L, Deganuto M, Leonardi A, Radicella JP, Kelley MR, D’Ambrosio C, Scaloni A, Quadrifoglio F, Tell G, APE1/Ref-1 Interacts with NPM1 within Nucleoli and Plays a Role in the rRNA Quality Control Process, Mol. Cell. Biol 29 (2009) 1834–1854. doi: 10.1128/MCB.01337-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Antoniali G, Serra F, Lirussi L, Tanaka M, D’Ambrosio C, Zhang S, Radovic S, Dalla E, Ciani Y, Scaloni A, Li M, Piazza S, Tell G, Mammalian APE1 controls miRNA processing and its interactome is linked to cancer RNA metabolism, Nat. Commun 8 (2017). doi: 10.1038/s41467-017-00842-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Poletto M, Loreto CD, Marasco D, Poletto E, Puglisi F, Damante G, Tell G, Acetylation on critical lysine residues of Apurinic/apyrimidinic endonuclease 1 (APE1) in triple negative breast cancers, Biochem. Biophys. Res. Commun 424 (2012) 34–39. doi: 10.1016/j.bbrc.2012.06.039. [DOI] [PubMed] [Google Scholar]
- [29].Bhakat KK, Sengupta S, Adeniyi VF, Roychoudhury S, Nath S, Bellot LJ, Feng D, Mantha AK, Sinha M, Qiu S, Luxon BA, Bhakat KK, Sengupta S, Adeniyi VF, Roychoudhury S, Nath S, Bellot LJ, Feng D, Mantha AK, Sinha M, Qiu S, Luxon BA, Regulation of limited N-terminal proteolysis of APE1 in tumor via acetylation and its role in cell proliferation, Oncotarget 7 (2016) 22590–22604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Timofeyeva NA, Koval VV, Knorre DG, Zharkov DO, Saparbaev MK, Ishchenko AA, Fedorova OS, Conformational dynamics of human AP endonuclease in base excision and nucleotide incision repair pathways, J. Biomol. Struct. Dyn 26 (2009) 637–652. doi: 10.1080/07391102.2009.10507278. [DOI] [PubMed] [Google Scholar]
- [31].Madlener S, Ströbel T, Vose S, Saydam O, Price BD, Demple B, Saydam N, Essential role for mammalian apurinic/apyrimidinic (AP) endonuclease Ape1/Ref-1 in telomere maintenance, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 17844–17849. doi: 10.1073/pnas.1304784110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li M, Yang X, Lu X, Dai N, Zhang S, Cheng Y, Zhang L, Yang Y, Liu Y, Yang Z, Wang D, Wilson DM, APE1 deficiency promotes cellular senescence and premature aging features, Nucleic Acids Res (2018). doi: 10.1093/nar/gky326. [DOI] [PMC free article] [PubMed]
- [33].Lee O-H, Kim H, He Q, Baek HJ, Yang D, Chen L-Y, Liang J, Chae HK, Safari A, Liu D, Songyang Z, Genome-wide YFP fluorescence complementation screen identifies new regulators for telomere signaling in human cells, Mol. Cell. Proteomics MCP 10 (2011) 10.1074/mcp.M110.001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Miller AS, Balakrishnan L, Buncher NA, Opresko PL, Bambara RA, Telomere proteins POT1, TRF1 and TRF2 augment long-patch base excision repair in vitro, Cell Cycle 11 (2012) 998–1007. doi: 10.4161/cc.11.5.19483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bugaut A, Alberti P, Understanding the stability of DNA G-quadruplex units in long human telomeric strands, Biochimie 113 (2015) 125–133. doi: 10.1016/j.biochi.2015.04.003. [DOI] [PubMed] [Google Scholar]
- [36].Miller MC, Buscaglia R, Chaires JB, Lane AN, Trent JO, Hydration Is a Major Determinant of the G-Quadruplex Stability and Conformation of the Human Telomere 3ʹ Sequence of d(AG3(TTAG3)3), J. Am. Chem. Soc 132 (2010) 17105–17107. doi: 10.1021/ja105259m. [DOI] [PubMed] [Google Scholar]
- [37].Dai J, Carver M, Punchihewa C, Jones RA, Yang D, Structure of the Hybrid-2 type intramolecular human telomeric G-quadruplex in K+ solution: insights into structure polymorphism of the human telomeric sequence, Nucleic Acids Res 35 (2007) 4927–4940. doi: 10.1093/nar/gkm522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lane AN, Chaires JB, Gray RD, Trent JO, Stability and kinetics of G-quadruplex structures, Nucleic Acids Res 36 (2008) 5482–5515. doi: 10.1093/nar/gkn517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bochman ML, Paeschke K, Zakian VA, DNA secondary structures: stability and function of G-quadruplex structures, Nat. Rev. Genet 13 (2012) 770–780. doi: 10.1038/nrg3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Postberg J, Tsytlonok M, Sparvoli D, Rhodes D, Lipps HJ, A telomerase-associated RecQ protein-like helicase resolves telomeric G-quadruplex structures during replication, Gene 497 (2012) 147–154. doi: 10.1016/j.gene.2012.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Virgilio A, Petraccone L, Esposito V, Citarella G, Giancola C, Galeone A, The abasic site lesions in the human telomeric sequence d[TA(G3T2A)3G3]: A thermodynamic point of view, Biochim. Biophys. Acta BBA - Gen. Subj 1820 (2012) 2037–2043. doi: 10.1016/j.bbagen.2012.09.011. [DOI] [PubMed] [Google Scholar]
- [42].Esposito V, Martino L, Citarella G, Virgilio A, Mayol L, Giancola C, Galeone A, Effects of abasic sites on structural, thermodynamic and kinetic properties of quadruplex structures, Nucleic Acids Res 38 (2010) 2069–2080. doi: 10.1093/nar/gkp1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Babinský M, Fiala R, Kejnovská I, Bednářová K, Marek R, Sagi J, Sklenář V, Vorlíčková M, Loss of loop adenines alters human telomere d[AG3(TTAG3)3] quadruplex folding, Nucleic Acids Res 42 (2014) 14031–14041. doi: 10.1093/nar/gku1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Broxson C, Hayner JN, Beckett J, Bloom LB, Tornaletti S, Human AP endonuclease inefficiently removes abasic sites within G4 structures compared to duplex DNA, Nucleic Acids Res 42 (2014) 7708–7719. doi: 10.1093/nar/gku417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Li M, Völker J, Breslauer KJ, Wilson DM III, APE1 Incision Activity at Abasic Sites in Tandem Repeat Sequences, J. Mol. Biol 426 (2014) 2183–2198. doi: 10.1016/j.jmb.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Theruvathu JA, Darwanto A, Hsu CW, Sowers LC, The effect of Pot1 binding on the repair of thymine analogs in a telomeric DNA sequence, Nucleic Acids Res 42 (2014) 9063–9073. doi: 10.1093/nar/gku602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Masani S, Han L, Yu K, Apurinic/Apyrimidinic Endonuclease 1 Is the Essential Nuclease during Immunoglobulin Class Switch Recombination, Mol. Cell. Biol 33 (2013) 1468–1473. doi: 10.1128/MCB.00026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gil ME, Coetzer TL, Real-Time Quantitative PCR of Telomere Length, Mol. Biotechnol 27 (2004) 169–172. doi: 10.1385/MB:27:2:169. [DOI] [PubMed] [Google Scholar]
- [49].O’Callaghan N, Dhillon V, Thomas P, Fenech M, A quantitative real-time PCR method for absolute telomere length, BioTechniques 44 (2008) 807–809. doi: 10.2144/000112761. [DOI] [PubMed] [Google Scholar]
- [50].Rai G, Vyjayanti VN, Dorjsuren D, Simeonov A, Jadhav A, Wilson DM, Maloney DJ, Small Molecule Inhibitors of the Human Apurinic/apyrimidinic Endonuclease 1 (APE1), in: Probe Rep. NIH Mol. Libr. Program, National Center for Biotechnology Information (US), Bethesda (MD), 2010. http://www.ncbi.nlm.nih.gov/books/NBK133448/ (accessed September 12, 2018). [PubMed] [Google Scholar]
- [51].Göhring J, Fulcher N, Jacak J, Riha K, TeloTool: a new tool for telomere length measurement from terminal restriction fragment analysis with improved probe intensity correction, Nucleic Acids Res 42 (2014) e21–e21. doi: 10.1093/nar/gkt1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Berquist BR, McNeill DR, Wilson DM III, Characterization of Abasic Endonuclease Activity of Human Ape1 on Alternative Substrates, as Well as Effects of ATP and Sequence Context on AP Site Incision, J. Mol. Biol 379 (2008) 17–27. doi: 10.1016/j.jmb.2008.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Marenstein DR, Wilson DM III, Teebor GW, Human AP endonuclease (APE1) demonstrates endonucleolytic activity against AP sites in single-stranded DNA, DNA Repair 3 (2004) 527–533. doi: 10.1016/j.dnarep.2004.01.010. [DOI] [PubMed] [Google Scholar]
- [54].Zhou J, Fleming AM, Averill AM, Burrows CJ, Wallace SS, The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures, Nucleic Acids Res 43 (2015) 4039–4054. doi: 10.1093/nar/gkv252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fleming AM, Ding Y, Burrows CJ, Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair, Proc. Natl. Acad. Sci. U. S. A 114 (2017) 2604–2609. doi: 10.1073/pnas.1619809114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Takeshita M, Chang CN, Johnson F, Will S, Grollman AP, Oligodeoxynucleotides containing synthetic abasic sites. Model substrates for DNA polymerases and apurinic/apyrimidinic endonucleases., J. Biol. Chem 262 (1987) 10171–10179. [PubMed] [Google Scholar]
- [57].Dai J, Carver M, Yang D, Polymorphism of human telomeric quadruplex structures, Biochimie 90 (2008) 1172–1183. doi: 10.1016/j.biochi.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Heddi B, Phan AT, Structure of human telomeric DNA in crowded solution, J. Am. Chem. Soc 133 (2011) 9824–9833. doi: 10.1021/ja200786q. [DOI] [PubMed] [Google Scholar]
- [59].Kladova OA, Bazlekowa-Karaban M, Baconnais S, Piétrement O, Ishchenko AA, Matkarimov BT, Iakovlev DA, Vasenko A, Fedorova OS, Le Cam E, Tudek B, Kuznetsov NA, Saparbaev M, The role of the N-terminal domain of human apurinic/apyrimidinic endonuclease 1, APE1, in DNA glycosylase stimulation, DNA Repair 64 (2018) 10–25. doi: 10.1016/j.dnarep.2018.02.001. [DOI] [PubMed] [Google Scholar]
- [60].Sanderson BJ, Chang CN, Grollman AP, Henner WD, Mechanism of DNA cleavage and substrate recognition by a bovine apurinic endonuclease, Biochemistry 28 (1989) 3894–3901. [DOI] [PubMed] [Google Scholar]
- [61].Vascotto C, Lirussi L, Poletto M, Tiribelli M, Damiani D, Fabbro D, Damante G, Demple B, Colombo E, Tell G, Functional regulation of the apurinic/apyrimidinic endonuclease 1 by nucleophosmin: impact on tumor biology, Oncogene 33 (2014) 2876–2887. doi: 10.1038/onc.2013.251. [DOI] [PubMed] [Google Scholar]
- [62].Moor NA, Vasil’eva IA, Anarbaev RO, Antson AA, Lavrik OI, Quantitative characterization of protein–protein complexes involved in base excision DNA repair, Nucleic Acids Res 43 (2015) 6009–6022. doi: 10.1093/nar/gkv569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Lirussi L, Antoniali G, D’Ambrosio C, Scaloni A, Nilsen H, Tell G, Lirussi L, Antoniali G, D’Ambrosio C, Scaloni A, Nilsen H, Tell G, APE1 polymorphic variants cause persistent genomic stress and affect cancer cell proliferation, Oncotarget 7 (2016) 26293–26306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Federici L, Arcovito A, Scaglione GL, Scaloni F, Lo Sterzo C, Di Matteo A, Falini B, Giardina B, Brunori M, Nucleophosmin C-terminal leukemia-associated domain interacts with G-rich quadruplex forming DNA, J. Biol. Chem 285 (2010) 37138–37149. doi: 10.1074/jbc.M110.166736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tiacci E, Spanhol-Rosseto A, Martelli MP, Pasqualucci L, Quentmeier H, Grossmann V, Drexler HG, Falini B, The NPM1 wild-type OCI-AML2 and the NPM1-mutated OCI-AML3 cell lines carry DNMT3A mutations, Leukemia 26 (2012) 554–557. doi: 10.1038/leu.2011.238. [DOI] [PubMed] [Google Scholar]
- [66].Gallo A, Lo Sterzo C, Mori M, Di Matteo A, Bertini I, Banci L, Brunori M, Federici L, Structure of Nucleophosmin DNA-binding Domain and Analysis of Its Complex with a G-quadruplex Sequence from the c-MYC Promoter, J. Biol. Chem 287 (2012) 26539–26548. doi: 10.1074/jbc.M112.371013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Scognamiglio PL, Di Natale C, Leone M, Poletto M, Vitagliano L, Tell G, Marasco D, G-quadruplex DNA recognition by nucleophosmin: New insights from protein dissection, Biochim. Biophys. Acta BBA - Gen. Subj 1840 (2014) 2050–2059. doi: 10.1016/j.bbagen.2014.02.017. [DOI] [PubMed] [Google Scholar]
- [68].Correa EME, De Tullio L, Vélez PS, Martina MA, Argaraña CE, Barra JL, Analysis of DNA structure and sequence requirements for Pseudomonas aeruginosa MutL endonuclease activity, J. Biochem. (Tokyo) 154 (2013) 505–511. doi: 10.1093/jb/mvt080. [DOI] [PubMed] [Google Scholar]
- [69].Abeldenov S, Talhaoui I, Zharkov DO, Ishchenko AA, Ramanculov E, Saparbaev M, Khassenov B, Characterization of DNA substrate specificities of apurinic/apyrimidinic endonucleases from Mycobacterium tuberculosis, DNA Repair 33 (2015) 1–16. doi: 10.1016/j.dnarep.2015.05.007. [DOI] [PubMed] [Google Scholar]
- [70].Mol CD, Hosfield DJ, Tainer JA, Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3’ ends justify the means, Mutat. Res 460 (2000) 211–229. [DOI] [PubMed] [Google Scholar]
- [71].Masuda Y, Bennett RA, Demple B, Dynamics of the interaction of human apurinic endonuclease (Ape1) with its substrate and product, J. Biol. Chem 273 (1998) 30352–30359. [DOI] [PubMed] [Google Scholar]
- [72].Antoniali G, Lirussi L, Poletto M, Tell G, Emerging roles of the nucleolus in regulating the DNA damage response: the noncanonical DNA repair enzyme APE1/Ref-1 as a paradigmatical example, Antioxid. Redox Signal 20 (2014) 621–639. doi: 10.1089/ars.2013.5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Colombo E, Bonetti P, Lazzerini Denchi E., Martinelli P, Zamponi R, Marine J-C, Helin K, Falini B, Pelicci PG, Nucleophosmin Is Required for DNA Integrity and p19Arf Protein Stability, Mol. Cell. Biol 25 (2005) 8874–8886. doi: 10.1128/MCB.25.20.8874-8886.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bañuelos S, Lectez B, Taneva SG, Ormaza G, Alonso-Mariño M, Calle X, Urbaneja MA, Recognition of intermolecular G-quadruplexes by full length nucleophosmin. Effect of a leukaemia-associated mutation, FEBS Lett 587 (2013) 2254–2259. doi: 10.1016/j.febslet.2013.05.055. [DOI] [PubMed] [Google Scholar]
- [75].Chiarella S, De Cola A, Scaglione GL, Carletti E, Graziano V, Barcaroli D, Lo Sterzo C, Di Matteo A, Di Ilio C, Falini B, Arcovito A, De Laurenzi V, Federici L, Nucleophosmin mutations alter its nucleolar localization by impairing G-quadruplex binding at ribosomal DNA, Nucleic Acids Res 41 (2013) 3228–3239. doi: 10.1093/nar/gkt001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Mitrea DM, Grace CR, Buljan M, Yun M-K, Pytel NJ, Satumba J, Nourse A, Park C-G, Madan Babu M., White SW, Kriwacki RW, Structural polymorphism in the N-terminal oligomerization domain of NPM1, Proc. Natl. Acad. Sci 111 (2014) 4466–4471. doi: 10.1073/pnas.1321007111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Mol CD, Izumi T, Mitra S, Tainer JA, DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected], Nature 403 (2000) 451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- [78].Freudenthal BD, Beard WA, Cuneo MJ, Dyrkheeva NS, Wilson SH, Capturing snapshots of APE1 processing DNA damage, Nat. Struct. Mol. Biol 22 (2015) 924–931. doi: 10.1038/nsmb.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.