

Abstract
The dominantly inherited spinocerebellar ataxias (SCAs) are a large and diverse group of neurodegenerative diseases. The most prevalent SCAs (SCA1, SCA2, SCA3, SCA6 and SCA7) are caused by expansion of a glutamine-encoding CAG repeat in the affected gene. These SCAs represent a substantial portion of the polyglutamine neurodegenerative disorders and provide insight into this class of diseases as a whole. Recent years have seen considerable progress in deciphering the clinical, pathological, physiological and molecular aspects of the polyglutamine SCAs, with these advances establishing a solid base from which to pursue potential therapeutic approaches.
The spinocerebellar ataxias (SCAs) are a large and diverse group of autosomal-dominant, progressive neurodegenerative diseases. They share the clinical feature of progressive ataxia, reflecting degeneration of the cerebellum and, often, other connected regions of the nervous system. The prevalence of the SCAs varies markedly depending on geography and ethnicity but is estimated to be 1–3 per 100,000 among Europeans1. Although many of the SCAs result from point mutations, DNA rearrangements (SCA15, SCA16 and SCA20) or expansions of non-coding repeats (SCA8, SCA10, SCA31 and SCA36) (BOXES 1,2), the most common SCAs are caused by expansion of a CAG nucleotide repeat that encodes polyglutamine (polyQ) in the relevant disease proteins. These polyQ SCAs include SCA1–SCA3, SCA6, SCA7 and SCA17, which are caused by expanded polyQ sequences in ataxin 1 (ATXN1), ATXN2, ATXN3, subunit-α of the Cav2.1 voltage-gated calcium channel (CACNA1A), ATXN7 and TATA-box-binding protein (TBP), respectively. Disease severity tends to be greater, and survival shorter, for patients with a polyQ SCA than for those with a non-polyQ SCA2.
The genetic diversity of the SCAs, combined with the relative conservation of cerebellar structure among mammals, make these diseases a biologically robust experimental platform for discovering fundamental aspects of cerebellar function and dysfunction. In turn, these discoveries have implications for the development of potential therapeutic approaches and for the understanding of other neurodegenerative diseases — particularly other polyQ neurodegenerative diseases. In this article, we review the clinical, molecular and physiological aspects of the most common SCAs, the polyQ SCAs, and describe how molecular genetic approaches and model systems have advanced our understanding of essential features of their underlying pathogenesis. An emerging theme is that although the individual disease proteins that result in the polyQ SCAs are structurally and functionally different, the effects of the mutations on cellular processes and neuronal activity overlap considerably, leading to similar pathology and clinical features.
Clinical and pathological changes
The symptoms of polyQ SCAs typically start in adulthood and slowly progress over many years, culminating in death by brainstem failure, although this is not the case for SCA6, which typically spares cranial motor nerves3. Gait ataxia is most often the initial sign in patients; later signs include limb incoordination, speech disturbance and oculomotor abnormalities4, 5. There is considerable variability in disease features, however, partly owing to differences in the size of the polyQ repeat expansion; longer expansions are associated with earlier symptom onset and a broader range of neurological symptoms. Symptoms include cerebellar ataxia, pyramidal symptoms and extrapyramidal symptoms, as well as impaired control of eye movements sometimes culminating in ophthalmoplegia and peripheral neuropathy. Individuals with SCA1, SCA2 or SCA3 usually show signs of clinical sensorimotor or sensory neuropathy, consistent with peripheral nerve involvement. Brainstem dysfunction in the SCAs leads to abnormal visual and brainstem auditory evoked potentials6, 7.
The neuropathology of the polyQ ataxias is also complex and varied. In SCA1, SCA2 and SCA3, cerebellar and brainstem degeneration are nearly always prominent, whereas more anterior regions (such as the basal ganglia) and posterior regions (such as the spinal cord) exhibit a variable degree of damage8–10.
Perturbations in neuronal circuitry and eventual neuronal loss affect many aspects of the cerebellar motor system in the SCAs (FIG. 1). The affected afferent components include the mossy fibres that arise in the spinocerebellar tracts, the vestibular nuclei and basal pontine nuclei, as well as climbing fibres from the olivary nuclei. Other proprioceptive sensory pathways, such as those ascending from the posterior columns of the spinal cord, can also be impaired. The cerebellar cortex itself is a frequent target of the SCAs, with Purkinje cells being the neuronal population that is most frequently affected by these diseases. The efferent components that are affected include the deep nuclei of the cerebellum, as well as their targets in the red nucleus and thalamus. The pathological variability within each polyQ SCA makes it difficult to define a distinctive pattern for each type, but certain areas tend to be affected preferentially in each SCA.
Figure 1 |. Components of the cerebellar circuitry showing degeneration in the polyglutamine spinocerebellar ataxias.

The different spinocerebellar ataxias (SCAs) have similar but not identical patterns of pathological involvement, shown here for SCA1, SCA2, SCA3 and SCA6. The figure shows the areas that are consistently affected in the different SCAs (red), as well as those that are mildly or variably affected in the different SCAs (pink). The areas and cell types in the cerebellar circuitry that are affected include the cerebellar cortex (particularly Purkinje cells (PCs), the cell bodies of which are depicted by triangles here), the inferior olive and its climbing fibres, the pontine nuclei and their mossy fibres, the deep cerebellar nuclei, the red nucleus and the cranial nerve (CN) motor nuclei. GC, cerebellar granule cell.
SCA1.
The age of onset of SCA1 ranges from childhood to late adult life, with a mean onset in the fourth decade11, 12. Earlier age at onset and larger expanded alleles are associated with faster symptom progression11. Of the polyQ SCAs, SCA1 progresses the fastest (REF. 11) and is associated with concomitant progression of cerebellar and non-cerebellar symptoms.
The typical pathological pattern in SCA1 is olivopontocerebellar atrophy13, which also occurs in SCA2 and several other SCAs. In addition to the profound loss of cerebellar Purkinje cells and the neurons of the dentate, basal pontine and olivary nuclei, other brainstem areas, including the red nuclei, vestibular nuclei and motor cranial nerve nuclei4, are often affected, whereas the pars compacta of the substantia nigra is usually spared. Involvement of the spinal cord varies greatly, and the basal ganglia, thalamus, cerebral cortex and hippocampus are typically spared9, 13.
SCA2.
SCA2 typically presents with progressive ataxia, slowed eye movements and sensorimotor neuropathy. Extrapyramidal manifestations, including parkinsonism, motor weakness, ocular palsies and cognitive impairment, may also occur. The age of onset ranges from infancy to later adult life, with a mean onset in the mid-30s11, 12. As in SCA1, an earlier age at onset and larger expanded alleles are associated with faster symptom progression11.
As in SCA1 brains, most SCA2 brains at autopsy have olivopontocerebellar atrophy. Whereas the cerebellar cortical changes associated with SCA2 resemble those that occur in SCA1, the deep cerebellar nuclei are mostly spared in SCA2 (REFS 14–16). The striatonigral pathways are more severely affected in SCA2 than in SCA1, as dopaminergic neurons in the substantia nigra and neurons in the neostriatum and pallidum are lost14, 15. The red nucleus and thalamus are also usually affected in SCA215. Motor cranial nerve nuclei are variably involved16, and the spinal cord often shows a loss of neurons in the anterior horn and the dorsal nucleus of Clarke (also known as the posterior thoracic nucleus), along with atrophy of the spinocerebellar tracts and posterior columns14, 15.
Intermediate-range CAG repeat expansions (29–34 repeats) at the ATXN2 locus are also a risk factor for amyotrophic lateral sclerosis (ALS)17. ATXN2-associated ALS is classic ALS: it is associated with a combination of upper and lower motor neuron signs; disease onset typically occurs in the 50s; and the survival time after onset is less than 3 years18. However, selective loss of Purkinje cells in the cerebellar vermis has been reported in ATXN2-associated ALS, unlike in other types of ALS19.
SCA3.
Also known as Machado–Joseph disease, SCA3 most commonly presents with progressive ataxia and spasticity8. The clinical phenotype, however, is so variable that some have classified SCA3 into the following subtypes: early-onset disease with extrapyramidal signs and spasticity but minimal ataxia (Type 1); midlife progressive ataxia (Type 2); later-onset ataxia accompanied by neuropathy, amyotrophy and loss of reflexes (Type 3); and parkinsonism with or without ataxia (Type 4)8–10.
SCA3 neuropathology differs from that of SCA1 and SCA2 in that the cerebellar cortex and olivary nuclei are relatively less affected in SCA3, whereas the deep cerebellar nuclei and basis pontis are usually more severely affected20–22. Spinal, vestibular and pontine afferents to the cerebellum are also lost, along with their nuclei. The red nucleus is affected, but the thalamus is usually only minimally involved. Extrapyramidal pathology involves the substantia nigra, the subthalamic nuclei and the globus pallidus. There is often a loss of motor neurons in the cranial nerve nuclei and the anterior horns of the spinal cord.
SCA6.
SCA6 begins later than other polyQ ataxias, with the median age of onset in the early 50s23. Clinically, SCA6 is considered a relatively pure cerebellar syndrome with few extracerebellar signs1, and seems not to reduce lifespan23. Symptoms may be episodic in individuals with the shortest disease-causing repeat sequences4. Clinical dysfunction in SCA6 correlates best with the degree of cerebellar atrophy24. Macroscopically, atrophy is confined to the cerebellum. Microscopically, however, the neuropathology in SCA6 shares features with that of other polyQ SCAs, although involvement beyond the cerebellar Purkinje neurons is much less prominent22. Loss of the giant pyramidal Betz neurons of the cerebral cortex has been described in SCA6, and there is variable involvement of the brainstem and cerebellar nuclei25.
Functions and pathogenic mechanisms
Whereas it is widely recognized that expansion of the polyQ tract in each of the proteins associated with neurodegenerative diseases alters protein conformation and leads to protein aggregation26, there remains debate about the mechanisms underlying the neuronal vulnerability in each of the polyQ SCAs. As reviewed below, data suggest that protein context matters greatly and that for any given polyQ SCA, the perturbations of the native functions of the disease protein and the mechanisms responsible for toxic gain of function are probably interrelated (FIG. 2). A common refrain is that the disease proteins affected in polyQ SCAs participate in the molecular regulation of gene expression at the transcriptional and/or post-transcriptional level and that polyQ expansion perturbs this regulation. An actively pursued hypothesis is that the misfolding, accumulation and/or aggregation of the expanded polyQ protein disrupts the delicate balance of RNA and protein homeostasis in the nervous system and does so in a disease-specific manner that is dictated in part by the structure (FIG. 3) and function of the protein. In this section, we describe the normal functions of the proteins affected by polyQ expansion in the polyQ SCAs and examine how a loss of these functions and/or a gain of toxic functions may be induced by expanded polyQ tracts.
Figure 2 |. Cellular processes affected by mutant polyglutamine proteins in spinocerebellar ataxias.

Each panel illustrates the cellular processes affected by the polyglutamine (polyQ) proteins in spinocerebellar ataxia 1 (SCA1), SCA2, SCA3, SCA6 and SCA7. In all of the SCAs depicted, except SCA3, the cerebellar Purkinje cells are prominently affected, whereas in SCA3, the Purkinje cells are more mildly affected. Top panel (SCA1): in the ataxin 1 (ATXN1) protein, it is believed that the AXTN1 HBP1 (AXH) domain and the U2AF homology (UHM) domain are interaction motifs for the transcriptional regulator capicua (CIC) and the RNA splicing factor RBM17, respectively, and a shift in these interactions is critical for mutant, polyglutamine-expanded ATXN1 to cause disease in Purkinje cells. PolyQ expansion is depicted by ‘exp[Q]’ in all panels. Phosphorylation of Ser776 in ATXN1 (pSer 776) also shifts the balance between these interactions. Second panel (SCA2): ATXN2 is known to interact directly or indirectly with numerous proteins implicated in RNA metabolism, as well as RNA itself, to regulate translation, stress granule formation and P-body formation. Of particular interest are the interactions of ATXN2 with polyadenylate-binding protein (PABP) and TAR DNA-binding protein 43 (TDP43), each of which also binds directly to RNA. One hypothesis is that the polyQ tract length in ATXN2 impairs interactions with PABP and TDP43 and thereby contributes to SCA2 pathogenesis, as well as the risk for ALS (not shown). Third panel (SCA3): as a deubiguitinase (DUB), ATXN3 binds and cleaves polyubiguitin chains and has been implicated in a variety of ubiguitin (Ub)-dependent protein quality control pathways. Although expanded ATXN3 retains DUB activity in vitro, changes in polyQ-repeat length may alter its function in the complex cellular environment, with deleterious consequences. As with many polyQ disease proteins, mutant ATXN3 becomes concentrated in the nucleus. Fourth panel (SCA6): the CACNA1A gene encodes a bicistronic mRNA that, on translation, yields the following two proteins: the membrane-localized α1A subunit of the Cav2.1 channel and the transcription factor α1ACT. Expansion of the polyQ-encoding repeat in CACNA1A leads to toxicity through altered α1ACT-mediated regulation of transcription, as well as through nuclear translocation of a peptide cleaved from the carboxyl terminus of the mutant Cav2.1 channel subunit. Fifth panel(SCA7): ATXN7 is a component of the SPT-ADA-GCN5 acetyltransferase (SAGA) complex. SAGA regulates transcription through its dual histone-modifying enzymes, the histone acetyltransferase GCN5 and the DUB ubiguitin C-terminal hydrolase 22 (USP22). PolyQ-expanded ATXN7 forms insoluble complexes that are thought to sequester other components of the DUB module such that the SAGA complex can no longer remove ubiquitin from its substrates. Pol II, polymerase II.
Figure 3 |. Functional motifs in ATXN1, ATXN2 and ATXN3.

Functional motifs are diagrammed for each polyglutamine (polyQ) spinocerebellar ataxia (SCA)-associated ataxin (ATXN) protein. The diagrams also show the polyQ regions (denoted as ‘Q’), as well as the phosphorylation sites (denoted as ‘P’) and ubiquitylation sites (denoted as ‘Ub’ to represent ubiquitin). a | ATXN1 with a 30Q polyQ region is shown. The ATXN1 HBP1 (AXH) domain and the U2AF homology motif (UHM) of ATXN1 are interaction motifs for capicua (CIC) and RBM17, respectively; the AXH domain itself forms an oligonucleotide/oligosaccharide-binding (OB) fold. ATXN1 also features a nuclear-localization signal (NLS) near the carboxyl terminus of the protein that facilitates its localization to the nucleus and a phosphorylation-dependent binding motif for the chaperone 14-3-3 (14-3-3 LM). b | ATXN2 interacts directly or indirectly with numerous proteins implicated in RNA metabolism. Its poly(A)-binding protein (PABP)-interacting motif PAM2 enables ATXN2 to interact with PABP and TAR DNA-binding protein 43 (TDP43). ATXN2 also features a like-Sm(LSm) motif and an LSm-associated domain (LSmAD). c | The deubiquitinase (DUB) ATXN3 has an N-terminal catalytic (Josephin) domain, which contains two Ub-binding sites and two nuclear export sites (NES), and a C-terminal Ub-binding domain bearing three Ub-interacting motifs (UIMs) and an NLS.
ATXN1 and SCA1.
Wild-type ATXN1 includes a polyQ tract that normally contains 6–34 glutamines, with repeats longer than 20 glutamines containing 1–3 histidine interruptions. In addition, several other functional motifs and domains have been identified in ATXN1 (FIG. 3). A monopartite nuclear-localization signal (NLS) motif near the carboxyl terminus directs the localization of the protein to the nucleus27. ATXN1 containing a wild-type polyQ tract shuttles between the cytoplasm and nucleus. By contrast, ATXN1 containing an expanded polyQ tract is transported to the nucleus but cannot be exported from the nucleus28. A single amino-acid substitution within its NLS prevents expanded ATXN1 from entering the nuclei of Purkinje cells and eliminates its toxicity27, indicating that at least in these cells, disease pathogenesis may be linked to a function of ATXN1 in the nucleus.
Wild-type and expanded ATXN1 interact with various nuclear components, including RNA28,29, several regulators of transcription, SMRT30 (silencing mediator of retinoic acid and thyroid hormone receptor; also known as NCOR2), capicua (CIC)31, the zinc finger protein GFI1 (homologue of senseless in Drosophila)32 and the nuclear receptor-α (RORα)-60 kDa Tat-interactive protein (TIP60; also known as KAT5) complex33,34. The interaction with the transcriptional repressor CIC is subserved by the 130-amino-acid region of ATXN1 known as the ATXN1 HBP1 (AXH) domain, which independently forms an oligonucleotide/oligosaccharide-binding (OB) fold35,36. The importance of ATXN1–CIC complexes in SCA1 pathogenesis is suggested by the observation that reducing CIC levels — either genetically or through exercise — dampens the deficit in motor performance and the premature lethality of the SCA1-like phenotypes seen in Atxn1 154Q/2Q–knock-in mice37 (see BOX 3 on modelling SCAs in mice).
Box 3 |. Mouse models of polyglutamine spinocerebellar ataxias.
An important approach to understanding spinocerebellar ataxia (SCA) pathogenesis has been to model disease features in mice. An issue in using mice to model SCAs is the extent to which slowly progressive, mid-life diseases, such as the SCAs, can be recapitulated in the short lifespan of a mouse, which is typically 2 years. Two general approaches to creating a discernible clinical phenotype in mice are to overexpress an SCA-associated transgene or to express a disease-associated transgene with a CAG-repeat expansion that is longer than the maximum repeat length seen in humans. These manipulations create the potential to produce results unrelated to the human disease and thus require careful comparison to controls, such as transgenic mouse lines that overexpress normal alleles to the same degree.
Another strategic issue is whether to target the SCA-associated transgene such that it is selectively expressed in particularly relevant populations, such as Purkinje cells118, 159, 160, or such that it is expressed in all neurons or cells of the CNS that normally express the gene. The former approach can be advantageous when applied to Purkinje cells, for example, as these cells are crucial for cerebellar function and are affected in nearly all forms of SCA, and when these cells are damaged, a common result in mouse models is an easily discernible motor deficit. The latter approach uses either knock-in strategies to insert an expanded polyglutamine-encoding region into the mouse disease gene with its endogenous transcriptional regulator elements161, 162 or transgenic strategies using vectors capable of carrying an entire human gene, including its regulatory elements135. One advantage of such a strategy is that the resulting model is arguably more genetically precise and more fully captures the molecular genetic features of the human disease.
Last, there are transgenic approaches to generating conditional mouse models that allow the disease phenotype to be measured after stopping the expression of a mutant SCA protein136, 163. For example, a model of SCA1 pathogenesis in which mutant ataxin 1 (ATXN1) was conditionally expressed in Purkinje cells revealed that the disease is dependent on the continuous expression of mutant ATXN1. The extent of recovery when mutant ATXN1 was no longer expressed was directly related to how long the ATXN1 transgene had already been expressed. When mutant ATXN1 expression was discontinued at an early stage, the disease was completely reversed; by contrast, discontinuation of ATXN1 expression at a later stage led to only partial recovery. Notably, Purkinje cells were at least partly able to recover from mutant ATXN1 expression even at a late stage of the disease.
Near the C terminus of ATXN1, just downstream of the NLS, is Ser776, one of seven phosphorylation sites in ATXN1 (REFS 38,39). Replacement of S776 with an alanine, which cannot be phosphorylated, mitigates the ability of ATXN1 [82Q] to induce Purkinje cell disease in transgenic mice38. Substituting an aspartic acid at position 776, to mimic phosphorylation, enhances the ability of ATXN1[82Q] to induce Purkinje cell disease and converts ATXN1[30Q] into a protein that can induce ataxia in mice when expressed in Purkinje cells40. Ser776 phosphorylation leads to a reduction in the proteolytic clearance of ATXN1 (REF. 41), activation of a binding motif in ATXN1 for the chaperone 14-3-3 (REE 42), an increase in ATXN1 binding to the splicing factor RBM17 (REF. 43) and a decrease in ATXN1 interactions with the splicing factor U2AF65 (also known as U2AF2)44.
FIGURE 2 presents a model of SCA1 pathogenesis that incorporates the crucial nuclear interactions and functions of ATXN1. An important aspect underlying this model is the concept that transcription and RNA processing, including splicing, are coupled spatially and kinetically45. On entering the nuclei of Purkinje cells, ATXN1 readily binds to CIC. Although ATXN1 alone does not bind to DNA29, CIC can bind DNA; thus, the ATXN1-CIC complex is targeted to chromatin and transcription sites. Notably, as transcription proceeds, ATXN1 is hypothesized to shuttle between complexes with CIC and those with RBM17. This idea is supported by analyses of cerebellar extracts revealing that ATXN1 can be found in two high-molecular-weight soluble complexes: one that includes CIC and another, distinct complex that includes RBM17 (REF. 43). These two ATXN1 complexes are envisioned to be in a dynamic equilibrium — perhaps determined by the length of the polyQ tract and the phosphorylation of Ser776 — and the dynamics of the complexes affect gene expression. Importantly, expansion of the polyQ tract in ATXN1 favours ATXN1-RBM17 complex formation43, implicating this complex in SCA1 pathogenesis.
Cerebellar RNA-sequencing studies using transgenic mice expressing ATXN1 [82Q] identified a gene network whose expression correlated with disease progression46. Pathway analysis indicated that genes involved in synaptic long-term depression (LTD) and glutamate receptor signalling were particularly affected in transgenic mice compared with controls; both of these pathways have been linked to the pathogenesis of SCA1 and other ataxias47–49. Proteomic approaches indicate that alterations in mechanistic target of rapamycin (mTOR) signalling contribute to SCA1 cerebellar pathogenesis50,51. In the cerebellar proteomes of transgenic mice with ATXN1 [82Q]-expressing Purkinje cells, mTOR levels were decreased before the onset of symptoms51. Proteomic analysis of synaptic proteins in disease-susceptible Purkinje cells in early-stage Seal154Q/2Q-knock-in mice showed that the levels of HOMER3, a synaptic scaffold protein enriched in Purkinje cells, were dramatically reduced50. Importantly, either pharmacological or genetic reductions in Purkinje cell expression of mTOR complex 1 (mTORC1) worsened cerebellar-linked behaviour and pathology in the Sca1154Q/2Q mouse model, whereas increased Purkinje cell expression of HOMER3 attenuated these disease features.
Interestingly, mTORC1 signalling is also altered in Huntington disease (HD) and in mouse HD models52. Restoring mTORC1 activity in the N171-82Q mouse model of HD by expressing a constitutively active form of the GTP-binding proteins RHEB (an mTORC1 regulator) or RHES (an mTORC1 activator enriched in the striatum) alleviated HD-like motor deficits and pathology. Thus, a decrease in mTOR signalling — particularly mTORC1 signalling — is observed in SCA1 and HD and probably in several other neurodegenerative disorders as well.
Changes in DNA-damage and DNA-repair pathways have also been implicated in the pathogenesis of neurodegenerative disorders53, including many polyQ diseases54. In SCA1, enhanced expression of replication factor A1 (RPA1), a protein that functions in several DNA-repair pathways, was shown to improve motor performance and Purkinje cell morphology, as well as molecular functions, in Sca1154Q/2Q mice55. In addition, restoration of mitochondrial DNA repair by expressing the DNA-repair protein high mobility group protein B1 (HMGB1) attenuated SCA1-like phenotypes in Sca1154Q/2Q mice56.
Thus, several cellular pathways are implicated in SCA1 pathogenesis. The multi-pathway picture of SCA1 pathogenesis creates challenges for the development of new therapies, as discussed later in this Review.
ATXN2 and SCA2.
ATXN2 is a 145 kDa protein that is highly conserved across species57. Considerable evidence suggests that ATXN2 functions in cytoplasmic aspects of RNA metabolism. Initial evidence for this came from the finding that ATXN2 bears like-Sm (LSm) motifs, which are associated with mRNA decay in the cytoplasm58. Many identified ATXN2-interacting proteins were subsequently shown to function in RNA metabolism. For example, the polyadenylate-binding protein (PABP)-interacting motif of ATXN2, PAM2 (REF. 59), enables the protein to interact with PABP and a putative RNA-binding protein called ATXN2-binding protein60. One way in which ATXN2 functions in RNA metabolism is in the assembly of stress granules and processing bodies (P-bodies): both ATXN2 and PABP are components of stress granules and P-bodies61. Moreover, the Drosophila homologue of ATXN2, Atx2, is needed for microRNA (miRNA)-mediated repression of the translation of several mRNAs, raising the possibility that ATXN2 might also act in miRNA pathways in mammals62.
Further studies of ATXN2 homologues in different model organisms provide evidence linking ATXN2 to the regulation of translation. Yeast Pab1 (a homologue of PABP1), which promotes translation, genetically interacts with the ATXN2 homologue, Pbp1 (REF. 63). Furthermore, overexpression of Pbp1 in yeast causes effects similar to those observed when translation is inhibited by cycloheximide. Direct evidence linking ATXN2 to translation comes from studies showing that ATXN2 or its homologue forms complexes with polyribosomes. Atx2 in Drosophila (and ATXN2 in transfected human cells) co-sediments with polyribosomes, and this association is mediated by the LSm and PAM2 motifs64. The association of ATXN2 with components of translationally active polyribosomes and translationally silent RNA granules suggests that it may regulate the trafficking of mRNA between these structures, allowing a cell to tailor translation to its needs in response to changes in the environment. For example, ATXN2 regulates the cellular growth response to nutrient signals: studies in yeast65, nematode66 and mammalian cells67 show that ATXN2 inhibits the mTOR pathway in part by sequestering mTORC in stress granules.
Another intriguing functional link between ATXN2 and RNA metabolism relates to the observation that intermediate-length CAG-repeat tracts (encoding 27–33 glutamines) in ATXN2 are an important risk factor for sporadic ALS. This finding fits with the emerging view that many ALS risk genes, including the gene encoding TAR DNA-binding protein 43 (TDP43; also known as TARDBP), regulate RNA metabolism68; moreover, several findings functionally link TDP43 and ATXN2. TDP43 is a heterogeneous nuclear ribonucleoprotein that, similar to ATXN2, is involved in several aspects of RNA metabolism, including transcription, alternative splicing and RNA stability. In a yeast screen for modifiers of TDP43 toxicity, the yeast ATXN2 homologue, Pbp1, increased TDP43-mediated toxicity. Moreover, Atx2 also increases TDP43 toxicity in Drosophila. In addition, in yeast and mammalian cells, the interaction between TDP43 and ATXN2 is RNA-dependent17. Perhaps most intriguing is that, similar to TDP43, ATXN2 mislocalizes to cytoplasmic puncta in ALS motor neurons. Intermediate-length CAG-repeat expansions in ATXN2 (27–33 repeats) are considerably more prevalent in individuals with ALS than in unaffected individuals17,18, suggesting that variation in polyQ tract length — even in the wild-type range — directly affects function, almost certainly through an aspect of RNA metabolism involving ATXN2 and TDP43.
How polyQ expansion in ATXN2 causes SCA2 remains unknown. The role of ATXN2 in stress granule formation and translational regulation, however, raises the intriguing possibility that polyQ expansion in ATXN2 alters its interactions with key proteins and complexes (such as PABP, TDP43 and mTORC), thereby perturbing neuronal RNA and protein homeostasis. Evidence from a mouse model expressing ATXN2 [Q42] supports this view; in this model, the mutant ATXN2 was observed to sequester, in insoluble form, its known interactor PABPC1 (REF. 69). Recent expression profiling in a different SCA2 mouse model expressing ATXN2[Q127] revealed dysregulated expression of cerebellar gene clusters in several pathways, including GTPase signalling pathways, calcium signalling pathways and cell death pathways70. These changes were largely absent in Atxn2−/− mice, suggesting that toxic gain-of-function mutations rather than loss-of-function mutations are a key driver of disease70.
ATXN3 and SCA3.
The SCA3 disease protein ATXN3 is a specialized deubiquitinase (DUB) involved in ubiquitin-dependent protein quality control (reviewed in REFS 71,72). This 42 kDa protein has an amino-terminal catalytic domain (termed the Josephin domain) and a flexible C-terminal tail that includes three ubiquitin-interacting motifs (UIMs) that flank the polyQ tract. In addition to the UIMs, which bind ubiquitin chains with high affinity (FIG. 3), the Josephin domain of ATXN3 contains other ubiquitin-binding sites that regulate catalytic activity and protein stability73,74. As a DUB, ATXN3 preferentially acts on longer chains of at least four ubiquitins in length and may cleave K63-linked chains and mixed-linkage chains more readily than K48-linked chains. The UIMs probably specify the types of ubiquitin chains (in terms of both length and linkage) that are bound and cleaved by ATXN3, and mono-ubiquitylation of ATXN3 itself on a specific lysine residue, K117, enhances its activity75. This post-translational modification may accelerate ATXN3 function under conditions of ubiquitin stress.
Evidence suggests that ATXN3 participates in various ubiquitin-dependent pathways to maintain protein homeostasis. A primary function of ATXN3 may be to partner with ubiquitin ligases to regulate, or edit, the lengths and linkage types of ubiquitin chains on substrates. Two important E3 ligase partners are parkin and CHIP (also known as STUB1), both of which have crucial roles in ubiquitin-dependent protein homeostasis76,77. ATXN3 has also been implicated in endoplasmic reticulum-associated degradation78, aggresome production75 and DNA repair80–83, among other pathways. In all these processes, ATXN3 acts through ubiquitin-dependent functions. ATXN3 has been reported to interact with more than 100 proteins, many of which are directly linked to ubiquitin-dependent protein quality control. These include valosin-containing protein (VCP), UV excision repair protein RAD23 and many molecular chaperones. Strikingly, when overexpressed, non-expanded ATXN3 actually suppresses expanded polyQ-mediated neurotoxicity in Drosophila models84, supporting the idea that ATXN3 has a role in maintaining ubiquitin-dependent protein homeostasis.
It is intriguing that a protein whose very function counters perturbations in protein homeostasis in neurodegeneration also causes neurodegeneration when harbouring a polyQ expansion. Although data indicate that polyQ expansion confers a toxic gain-of-function to the disease proteins, is it possible that, in the case of ATXN3 a loss of this vital quality-control protein contributes in some way to disease development? In vitro, at least, expanded ATXN3 still binds and cleaves polyubiquitin chains, suggesting that it retains activity. However, polyQ expansion could conceivably alter the ability of ATXN3 to bind to or cleave ubiquitin chains in the cellular milieu. For example, the ability of expanded ATXN3 to interact with parkin or CHIP is indeed altered; expanded ATXN3 binds CHIP more tightly, reducing the levels of this important quality-control ubiquitin ligase77. Further investigation of the DUB function of ATXN3 in neurons is needed, however, to support the notion that a partial loss of function contributes to disease. Nevertheless, it seems clear that a profound loss of ATXN3 function is not the primary cause of SCA3: mice lacking ATXN3 are essentially normal except for increased levels of polyubiquitin in the brain, and neurodegeneration in a knock-in mouse model of HD is not accelerated when ATXN3 is eliminated85.
One attractive hypothesis for the pathogenesis of SCA3 is that conformational changes induced by polyQ expansion could alter ATXN3 DUB function in multiple ubiquitin-dependent pathways. The roles of ubiquitin in the nervous system extend far beyond proteasomal degradation, and mutant ATXN3 could precipitate myriad subtle changes in ubiquitin-dependent cellular processes that collectively prove deleterious (‘death by a thousand cuts’). A normal-sized polyQ domain in ATXN3 may permit conformational flexibility in the C-terminal domain that facilitates binding to diverse polyubiquitin chains and ubiquitylated substrates, whereas expansion would favour a kinetically ‘trapped’ conformation that perturbs ATXN3 interactions with its normal binding partners, with many deleterious consequences for the cell.
The well-described aggregation of mutant ATXN3 in SCA3 occurs both in the form of large intranuclear inclusions in neurons and as smaller cytoplasmic and axonal puncta. In SCA3, or when cells are stressed, ATXN3 also moves from the cytoplasm, its primary site of residence, into the nuclei of neurons. Thus, its nuclear relocation in disease may reflect an indirect response to stress or may directly result from changes to the mutant protein itself. Its presence in the nucleus contributes to its toxicity86, but precisely how its nuclear localization contributes to such toxicity remains unclear.
Although the downstream consequences of polyQ expansion in ATXN3 that are principally responsible for its toxicity are currently unknown, the mutant gene product is clearly somehow neurotoxic. As ATXN3 is a nonessential gene, reducing levels of the mutant protein — that is, to intervene proximally in the neurotoxic cascade — is an attractive therapeutic option. Nucleotidebased gene-silencing approaches, including RNA interference and antisense oligonucleotides, have established that suppressing expression of the mutant protein in mouse models does not have deleterious consequences for the nervous system87–90. Moreover, small-molecule screens have begun to identify existing US-approved drugs that can reduce the toxicity, or steady-state levels, of mutant ATXN3. For example, citalopram and other selective serotonin-reuptake inhibitors reduce ATXN3 toxicity in nematode and mouse models of SCA3 through a serotonin receptor-dependent pathway91, and aripiprazole, an atypical antipsychotic with complex pharmacology, recently surfaced from a cell-based drug screen as a compound that reduces the levels of mutant ATXN3 (REF. 92). Studies of these and other compounds will be required to determine their potential as disease-modifying agents and their mechanisms of action. One of many potential mechanisms of action is through the boosting of one or more protein quality-control pathways, such as molecular chaperone-mediated clearance or autophagy93,94, the efficacy of which has been shown in animal models. In addition to strategies intended to lower mutant ATXN3 levels, strategies intended to alter its nuclear relocation, phosphorylation and proteolysis — all of which can modulate toxicity — are under investigation95–97.
CACNA1A and SCA6.
With the discovery of CACNA1A as the disease gene affected in SCA6 (REFS 98,99), the basis of the disease was attributed to an expanded C-terminal polyQ tract in the gene product, the α1A transmembrane subunit of the Cav2.1 calcium channel. The Cav2.1 channel is abundantly expressed by cerebellar Purkinje cells, and CACNA1A is mutated in several other inherited neurological disorders, including episodic ataxia type 2 and familial hemiplegic migraine type 1 (REF. 100). The possibility that the function of the channel is altered, however, continues to be debated, and recent data suggest that the pathogenesis of the disease is driven by an alternative mechanism.
In wild-type mice and in individuals without SCA6, the polyQ-containing cytoplasmic C terminus of the α1A subunit of the Cav2.1 channel is cleaved, generating a stable C-terminal peptide that transits to the nuclei of Purkinje cells. In culture, the nuclear localization of the expanded polyQ-containing C-terminal peptide is toxic to cells 101. In addition, CACNA1A encodes a bicistronic mRNA (with an internal ribosomal entry site (IRES)) that can be translated into two proteins 102(FIG. 2): the α1A subunit of the Cav2.1 channel and the transcription factor α1ACT, which enhances the expression of numerous genes implicated in neuronal differentiation and Purkinje cell development, including GRN (which encodes granulin), PMCA2 (which encodes a calcium ATPase) and BTG1 (which encodes B cell translocation gene 1 protein). α1ACT containing an expanded polyQ is less able to enhance the expression of these genes, and blocking the IRES-driven translation of polyQ-expanded α1ACT rescues SCA6-like phenotypes in a mouse model of SCA6. These results suggest that an expanded polyQ-containing C-terminal domain of the gene product — whether by cleavage of the α1A subunit of the Cav2.1 channel or by IRES-mediated translation of α1ACT — and its subsequent translocation to the nucleus contribute to disease.
Compared with other polyQ SCAs, SCA6 contains several unique features, including a much lower repeat-length threshold for disease (disease-causing tracts are only 20–33 repeats long) and the fact that the Cav2.1 protein is embedded in the plasma membrane (all other polyQ disease proteins are soluble). Perhaps linked to these distinctive features, there is much less evidence to suggest that disease-protein accumulation and aggregation occur in SCA6 compared with what is known about the other polyQ SCAs. Arguably, the best evidence is found in mouse models expressing hyper-expanded forms of CACNA1A. For example, knock-in mice expressing the major Cav2.1 subunit splice isoform with a 118Q expansion show selective degeneration of Purkinje cells and accumulation of the disease protein in what seem to be lysosomes103. Moreover, inclusion formation and Purkinje cell degeneration in this mouse model were accelerated by the absence of the key lysosomal protease cathepsin B. Although these findings suggest that the endolysosomal pathway has a role in SCA6, further studies exploring the behaviour of the Cav2.1 channel subunit and α1ACT with polyQ-repeat lengths in the actual disease range are needed.
Other polyQ proteins implicated in cerebellar ataxia.
Three other polyQ diseases can present primarily with ataxia: SCA7, SCA17 (REF. 104] and dentatorubral-pallidoluysian atrophy (DRPLA)105. SCA7 resembles SCA1, SCA2 and SCA3 clinically and pathologically but is unique among the SCAs in that it is typically accompanied by profound retinal degeneration. SCA17 and DRPLA are rather rare, show more widespread degeneration than most SCAs and are markedly heterogeneous in clinical features; sometimes, they resemble HD more than an SCA. The disease proteins in all three diseases are closely linked to transcription, supporting that alterations in transcription-associated complexes are a common feature of polyQ disease pathogenesis, as already illustrated in SCA1.
The SCA7 protein ATXN7 is a component of the SPT-ADA-GCN5 acetyltransferase complex (SAGA complex)106. SAGA broadly regulates transcription through its dual histone-modifying enzymes, the histone acetyltransferase GCN5 (also known as KAT2A) and the DUB ubiquitin C-terminal hydrolase 22 (USP22). Although ATXN7 itself has no enzymatic activity, it anchors USP22 within the DUB module of the SAGA complex, enhancing the DUB activity of SAGA. In vitro, normal and polyQ-expanded ATXN7 enhance DUB activity equally; however, in vivo, expanded ATXN7 forms insoluble complexes that sequester other components of the DUB module such that the SAGA complex can no longer remove ubiquitin from its substrates, including histone H2B107. This dominant-negative effect of expanded ATXN7 probably disrupts the careful balance between the acetylation and de-ubiquitylation mediated by SAGA, leading to deleterious changes in gene expression that help to precipitate retinal and neurodegeneration in SCA7 (REFS 107–109.).
Studies of mice in which the expression of mutant, expanded ATXN7 could be conditionally inactivated in Bergmann glia or in Purkinje cells and inferior olivary neurons revealed that cerebellar disease involves the action of mutant ATXN7 in both neurons and glia110 and implicated the dysfunction of inferior olivary neurons, whose climbing fibre projections are one of the two major excitatory Purkinje cell inputs, in disease pathogenesis. These findings nicely illustrate the complexity of SCA7 — and, presumably, other SCAs — and remind us that much more must be discovered about the contributions of cell types besides neurons to SCA disease pathogenesis.
Among the rarest polyQ SCAs is SCA17, which is caused by a repeat expansion in TBP104. TBP is an essential general transcription factor that forms dimers and binds the TATAAA sequence just upstream of the transcription start site in most genes; here, it recruits the many other factors needed to initiate transcription. Expansion of the polyQ tract in TBP reduces dimer formation and DNA binding by TBP; alters TBP interactions with various transcription factors, including the general transcription initiation factor IIB; and promotes aggregation of TBP in neurons.
Given that TBP is such an important transcription factor throughout the body, its involvement in selective neurodegeneration in SCA17 is particularly intriguing. It has been speculated, but not yet experimentally proven, that polyQ-expanded TBP may disrupt neuron-specific transcriptional events. Similar to TBP, the DRPLA protein atrophin is a transcription co-regulator that may, when harbouring a polyQ expansion, elicit cell-specific transcriptional changes that are deleterious in the nervous system105. Although less common than the other polyQ SCAs, both SCA17 and DRPLA warrant further investigation, as such investigation may reveal that these diseases share pathways of relevance to the broader field of polyQ diseases.
Changes in physiology
In the course of a neurodegenerative disorder, alterations in neuronal structure and function are believed to result in part from perturbations to intracellular homeostatic processes. These alterations may represent relatively nonspecific changes in the ability of neurons to maintain the normal trafficking, assembly and energetics of membrane proteins required for regulating excitability (FIG. 4). Nevertheless, some ofthese changes may contribute to behavioural dysfunction in ataxia and may thus represent potential targets for symptom improvement in SCA. Recent work suggests that early changes in the expression of specific receptors and ion channels that are important for regulating membrane excitability contribute not only to motor dysfunction but also to structural changes in neurons that consistently precede cell death. Thus, targeting these changes might not only improve symptoms but also protect neurons from degeneration.
Figure 4 |. Alterations in Purkinje cell electrophysiology in spinocerebellar ataxias.

Concurrent with the motor dysfunction that occurs in the spinocerebellar ataxias (SCAs), and before the onset of substantial cellular morphological alterations, the expression levels and functions of ion channels and receptors are altered in SCA. a | Expression and function of ion channels in an unaffected Purkinje cell. The function of excitatory amino acid transporters (EAATs), which carry glutamate, and metabotropic glutamate receptors (mGluRs) yield slow excitatory postsynaptic currents (EPSCs), whereas normal large-conductance calcium-activated potassium (BK) channel function keeps the cell membrane hyperpolarized, maintaining spiking, b | The reduction in mGluRs and EAATs results in reductions in the amplitude of slow EPSCs (normal currents are shown in blue, and the currents in SCA are shown in red). The reduction in glutamate transporters prolongs the effect of glutamate at the synapse and also prolongs the mGluR-mediated slow EPSCs (red). In addition to alterations in synaptic signalling, the intrinsic excitability of the neuron is altered secondary to a loss of potassium channels. A reduction in BK channel expression and function results in unopposed calcium entry through voltage-gated calcium channels, with impairments in Purkinje neuron spiking (normal spiking indicated in blue; altered spiking indicated in red).
Some components of cerebellar circuitry seem to be preferentially vulnerable early in the disease. Climbing fibre, but not parallel fibre, innervation of Purkinje cells undergoes structural and functional alterations in a transgenic mouse model of SCA1, and these alterations are concurrent with the onset of mot or symptoms. A failure of climbing fibres to extend fully onto Purkinje cell dendrites is associated with impaired Purkinje neuron responsiveness to climbing fibre activation111. Although the degeneration of Purkinje cell dendrites that is associated with SCA1 would inevitably lead to synaptic changes, recent work suggests that early involvement of specific synaptic signalling pathways contributes to motor dysfunction. Specifically, before substantial dendritic degeneration has occurred and while fast synaptic glutamatergic transmission is still intact, metabotropic glutamate receptor (mGluR) signalling and the amplitude of mGluR-mediated excitatory postsynaptic currents (EPSCs) in Purkinje cells are reduced112. This impairment in mGluR-mediated signalling also impairs LTD of Purkinje cell-parallel fibre synapses in transgenic and lentivirus-mediated models of mutant ATXN1 expression. In a lentivirus-mediated model of SCA1, baclofen, a GABA type B receptor agonist that also potentiates mGluR1 responses in Purkinje neurons, partially rescued impairment in Purkinje neuron–parallel fibre LTD and motor dysfunction. Surprisingly, despite a reduction in the maximum amplitude of mGluR1-mediated responses observed in this study, mGluR1-mediated slow EPSCs were observed to be prolonged in a different study employing ATXN1 transgenic mice, possibly secondary to loss of glutamate transporter activity112. Systemic administration of a negative allosteric modulator of mGluR1, JNJ 16259685, improved the motor phenotype in mutant ATXN1-expressing mice. Hence, modulating mGluR signalling in Purkinje neurons may be an attractive therapeutic strategy for SCA1.
Purkinje neurons exhibit remarkably regular autonomous pacemaker spiking with little variability in interspike intervals, even in the absence of synaptic input. Altered Purkinje cell spiking occurs in many mouse models of ataxia113,114, including models of SCA1 (REFS 115,116), SCA2 (REFS 117–119), SCA3 (REF. 120.) and SCA6 (REF. 121). In mouse models of SCA1 and SCA2, a progressive reduction in firing frequency precedes cell loss115–117. In SCA1 Purkinje cells, this reduction in firing frequency may reflect increased A-type voltage-gated potassium channel currents115. In one mouse model of SCA1, the pacemaker firing of Purkinje cells was found to become disrupted with the onset of motor dysfunction, and these cells showed abnormal depolarization116. The inability of SCA1 Purkinje cells to support repetitive spiking reflected reduced levels of large-conductance calcium-activated potassium (BK) channels and subthreshold-activated potassium channels. Virus-mediated upregulation of BK channel expression in SCA1 Purkinje cells improved motor function and partly restored Purkinje cell morphology116. In a transgenic model of SCA2, Purkinje cell spiking in cerebellar slices and in vivo is irregular, with considerable variability among interspike intervals98,117. Although the basis for these alterations in spiking is still unclear, a positive modulator of small-conductance calcium-activated potassium channels improved the regularity of spiking to control levels, preventing motor dysfunction and reducing the number of darkly stained Purkinje cells on electron microscopy (so-called dark cell degeneration)117. In a different transgenic model of SCA2, reduced BK channel transcripts, and the associated impaired function of BK channels and other potassium channels, are responsible for impaired Purkinje neuron spiking164.
These studies suggest that changes in the intrinsic excitability of Purkinje neurons occur early in both SCA1 and SCA2 and are due in part to dysregulation of calcium-activated potassium channel physiology. These channels may be important targets for the treatment of motor dysfunction and neurodegeneration.
Therapeutic perspectives
Emerging insights into the mechanisms underlying polyQ SCAs suggest that these diseases may be treated through at least two principal therapeutic routes. The first is to pharmacologically modulate the perturbed cerebellar circuitry, as a symptomatic or possibly diseasemodifying therapy. The second is to reduce levels of toxic disease-gene products.
Given that mutant polyQ disease proteins can perturb gene expression, RNA homeostasis and protein homeostasis through diverse pathways with myriad downstream consequences, acting proximally in the disease cascade to lower levels of the toxic protein is currently the most compelling strategy towards developing a disease-modifying therapy. To date, efforts to target the expression of polyQ SCA proteins have used antisense oligonucleotides (ASOs) or virus-mediated delivery of short hairpin RNAs or artificial miRNAs to interfere with translation. Preclinical tests of ASOs or miRNA-based drugs for SCA1, SCA2, SCA3 and SCA6 (reviewed else-where122; also see REFS 90,123) show considerable promise, making clinical trials in the near future very likely. Notably, ATXN2-targeting ASOs ameliorated disease not only in SCA2 model mice124 but also in a TDP43 transgenic mouse model of ALS125. Certainly, the recent clinical trial successes with an ASO-based drug for spinal muscular atrophy126,127, as well as a phase I clinical trial for ASOs in superoxide dismutase 1 (SOD1)-variant-mediated ALS128, demonstrate that CNS delivery of ASOs by lumbar puncture is a viable therapeutic approach. The future looks promising for gene suppression as disease-altering therapy for the polyQ SCAs.
Well-designed interventional studies of SCA are crucial for a better understanding of the natural history of disease progression and to identify biomarkers for assessing disease progression and therapeutic target engagement. The rates of progression in SCA1, SCA2, SCA3 and SCA6 have been assessed by the European and US consortia of ataxia investigators (the European Integrated Project on SCAs (EUROSCA) and the Clinical Research Consortium for SCAs, respectively), permitting calculation of the sample sizes needed in SCA clinical trials129,130. Moreover, structural MRI and proton magnetic resonance spectroscopy detect disease-relevant changes with higher sensitivity than do clinical scales and thus have potential as noninvasive surrogate markers in clinical trials130,131. Transcranial magnetic stimulation is showing promise for detecting alterations in corticospinal and corticomuscular coherence in presymptomatic and symptomatic individuals harbouring the ATXN2 mutation132,133. As recently shown for another polyQ protein, huntingtin134, detection of the SCA polyQ proteins in the cerebrospinal fluid of patients may also prove to be an effective biomarker of target engagement for the polyQ SCAs.
Conclusions
The polyQ SCAs are a prime example of the power of genetics to advance the understanding of the cellular pathways crucial for neuronal function and dysfunction. One example highlighting this point is the recent elucidation of the role of CACNA1A bicistronic mRNA and the effects of a cleaved C-terminal peptide on nuclear transcription in SCA6102; this study identified an exciting new signal transduction pathway in neurons. As a group, the polyQ SCA proteins function in diverse fundamental processes affecting neuronal function and survival. With this in mind, the relationship between ATXN2 polyQ tract length and the risk of developing ALS17,18 probably portends the existence of similar effects of polyQ tract length in other SCA proteins.
As in other dominant neurodegenerative disorders, several modes and pathways have been identified as potential contributors to disease pathogenesis in the polyQ SCAs46,70,135. Understanding the relative contribution of each to pathogenesis in a given disease, however, remains a work in progress. Regardless of the precise molecular underpinnings of the toxic gain of function in polyQ SCAs, targeting the expression of the mutant disease proteins is a particularly compelling therapeutic strategy. Two therapeutically relevant observations from SCA mouse models are that neurological dysfunction precedes neuronal loss and that mouse models in which the mutant gene can be conditionally inactivated demonstrate an age-dependent recovery, with earlier silencing of mutant SCA gene expression conferring more complete recovery. Importantly, even at a relatively late stage in disease progression, but before neuronal loss, neurons may retain some ability to recover136, a finding that emphasizes the importance of intervening early in the disease. It remains unclear, however, how early in polyQ SCA a therapeutic intervention must be administered to have a disease-modifying effect.
Pyramidal symptoms
Spasticity, increased reflexes and weakness caused by pathology in cerebral cortical primary motor neurons or their axonal projections.
Extrapyramidal symptoms
Rigidity, tremor, difficulty initiating movement and gait disturbances typical of parkinsonism or, occasionally, increased involuntary movement, such as chorea. These symptoms are secondary to pathology in pathways involving the basal ganglia and substantia nigra.
Ophthalmoplegia.
Inability to move the eyes, secondary to pathology in the oculomotor cranial nerve nuclei (cranial nerves III, IV and VI) or the nerves themselves. In some cases, the pathology may be above the cranial nerve nuclei (supranuclear), in the voluntary gaze centres.
Oligonucleotide/oligosaccharide-binding fold.
(OB fold). A structural feature found in many oligonucleotide-binding proteins.
Stress granules
Subcellular organelles that are major sites for the regulation of mRNA translation. These are non-membrane-bound aggregates composed of proteins and mRNA molecules that form in a reversible manner during cellular stress.
Processing bodies
(P-bodies). Cytoplasmic domains that contain collections of proteins involved in diverse processes, such as mRNA degradation, nonsense-mediated mRNA decay, translational repression and inhibitory-RNA-mediated gene silencing.
Cycloheximide
An antibiotic that interferes with protein synthesis by blocking translational elongation. The drug is used experimentally as a rapidly reversible means to block protein synthesis in vitro.
Heterogeneous nuclear ribonucleoprotein
Protein that forms nuclear complexes with RNA during gene transcription and post-translational modification of pre-mRNA.
Aggresome
A cytoplasmic collection of misfolded proteins that forms when the normal protein degradation system is overtaxed. This possibly protective mechanism may result in formation of the intracellular inclusions observed in neurodegenerative diseases.
Bicistronic mRNA
mRNA that has two open-reading frames, both of which are translated into a polypeptide. These polypeptides often are functionally related and may be regulated together.
Internal ribosomal entry site
(IRES). RNA structures that allow initiation of translation independent of the usual 5’-cap mechanism, thus enabling translation of protein in the middle of an mRNA.
SPT-ADA-GCN5 acetyltransferase complex
(SAGA complex). A histone acetyltransferase complex that regulates gene transcription by altering chromatin structure via histone modification. The complex is also involved in the biogenesis and nuclear export of mRNA.
Bergmann glia.
A type of astrocyte found only in the cerebellar cortex. These cells have intimate contacts with Purkinje cell dendrites in the superficial layer of the Cortex and are involved in the re-uptake of glutamate.
Negative allosteric modulator.
A molecule that decreases the function of a receptor or an enzyme by binding to a locus that is not the active enzymatic site, typically by effecting a conformational change.
Box 1 |. Mutational basis for some non-polyglutamine spinocerebellar ataxias.
Genetics has played a seminal role in revealing the varied biology of the spinocerebellar ataxias (SCAs). In addition to the polyglutamine (polyQ) SCAs, which are the most common SCAs and the focus of this Review, many other less common forms of SCA are caused by other repeat expansions, DNA deletions or classical point mutations. Together, they demonstrate the many cellular and molecular pathways that, if disrupted, can cause spinocerebellar degeneration. The table lists non-polyQ SCAs that are associated with point mutations or DNA deletions, as well as the proteins affected in the diseases and their physiological functions.
| Disorder | Mutant protein (mutated gene) | Function of protein | Refs |
|---|---|---|---|
| SCA5 | β2-spectrin (SPTBN2) | Helps to form neuronal membrane skeleton | 137 |
| SCA11 | Tau tubulin kinase 2 (TTBK2) | Tau phosphorylation | 138 |
| SCA13 | Voltage-gated potassium channel subunit Kv3.3 (KCNC3) | Regulates membrane potential | 139 |
| SCA14 | Protein kinase Cγ (PRKCG) | Protein phosphorylation | 140 |
| SCA15/16 | Inositol 1,4,5-triphosphate receptor type 1 (ITPR1) | Ca2+ signalling | 141–143 |
| SCA23 | Prodynorphin (PDYN) | Synaptic transmission | 144 |
| SCA26 | Eukaryotic translation elongation factor 2 (EEF2) | Translation | 145 |
| SCA27 | Fibroblast growth factor 14 (FGF14) | Cell growth and survival | 146 |
| SCA28 | AFG3-like protein 2 (AFG3L2) | ATP-dependent protease | 147 |
| SCA34 | Elongation of very long chain fatty acids 4 (ELOVL4) | Lipid metabolism | 148 |
| SCA35 | Transglutaminase 6 (TGM6) | Protein crosslinking | 149 |
| SCA38 | Elongation of very long chain fatty acids 5 (ELOVL 5) | Lipid metabolism | 150 |
| SCA40 | Coiled-coil domain-containing protein 88C (CCDC88C) | WNT signalling | 151 |
| SCA41 | Transient receptor potential 3 (TRPC3) | Regulates membrane potential, Ca2+ signalling | 152 |
| SCA42 | Voltage-gated calcium channel subunit-α1G Cav3.1 (CACNA1G) | Ca2+ signalling | 153 |
| SCA43 | Neprilysin (MME) | Zinc-dependent metalloprotease | 154 |
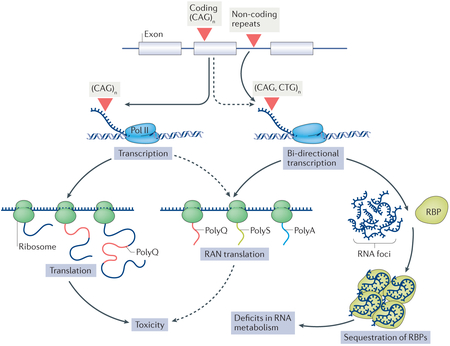
Box 2 |. Toxic RNA and RAN peptides in spinocerebellar ataxias.
Considerable evidence suggests that the toxicity of RNA that contains non-coding expanded repeat seguences can underlie the pathogenesis in spinocerebellar ataxias (SCAs). This notion is underlined by the toxicity of RNA containing the large non-coding ATTCT repeat expansion in SCA10. RNA molecules that contain the expanded repeat form foci that bind and sequester RNA-binding proteins (RBPs)155, resulting in a loss of RBP function and thus impairing aspects of RNA metabolism such as splicing and translation, depending on the normal role of the sequestered RBP (see the figure).
In addition, expanded repeats can cause protein-mediated effects through repeat-associated non-ATG initiated (RAN) translation, as first demonstrated in SCA8 (REF. 156). RAN translation, which may contribute to disease pathogenesis in many repeat expansion diseases157, including polyglutamine (polyQ) diseases, can occur across the expanded repeat in all three frames and possibly in both the sense and antisense directions, producing putatively toxic RAN peptides. Experimental evidence supports the presence of RAN-translated peptides in Huntington disease and their toxicity when overexpressed in tissue culture cells158. Although it remains unclear whether RAN-translated peptides are generated in the polyQ SCAs and contribute to toxicity in vivo (depicted as dashed arrows in the figure), the role of such peptides is an active topic of investigation in the polyQ SCAs. Pol II, polymerase II; polyA, polyalanine; polyS, polyserine.
Acknowledgements
The authors thank J. Friedrich for designing the artwork that formed the foundation for the figures in this Review. Financial support for the authors’ research was provided by the US National Institute of Neurological Disorders and Stroke, the US National Institutes of Health and the US National Ataxia Foundation.
Footnotes
Competing interests statement
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Durr A Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 9, 885–894 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Monin M-L et al. Survival and severity in dominant cerebellar ataxias. Ann. Clin. Transl. Neurol. 2, 202–207 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klockgether T et al. The natural history of degenerative ataxia: a retrospective study in 466 patients. Brain 121,589–600 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Globas C et al. Early symptoms in spinocerebellar ataxia type 1, 2, 3, and 6. Mov. Disord. 23, 2232–2238 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Luo L et al. The initial symptom and motor progression in spinocerebellar ataxias. Cerebellum 16, 616–622 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schols L, Linnemann C & Globas C Electrophysiology in spinocerebellar ataxias: spread of disease and characteristic findings. Cerebellum 7, 198–203 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Liang L, Chen T & Wu Y The electrophysiology of spinocerebellar ataxias. Neurophysiol. Clin. 46, 27–34 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Paulson H Machado–Joseph disease/spinocerebellar ataxia type 3. Handb. Clin. Neurol. 103, 437–449 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seidel K et al. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 124, 1–21 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Riess O et al. SCA3: neurological features, pathogenesis and animal models. Cerebellum 7, 125–137 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Jacobi H et al. The natural history of spinocerebellar ataxia type 1, 2, 3, and 6: a 2-year follow-up study. Neurology 77, 1035–1041 (2011).One of several recent natural history studies performed by the EUROSCA that will inform future clinical prevention trials in polyQ SCAs (also see ref. 130).
- 12.Ashizawa T et al. Clinical characteristics of patients with spinocerebellar ataxias 1, 2, 3 and 6 in the US; a prospective observational study. Orphanet. J. Rare Dis. 8, 177 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robitaille Y, Schut L & Kish SJ Structural and immunocytochemical features of olivopontocerebellar atrophy caused by the spinocerebellar ataxia type 1 (SCA-1) mutation define a unique phenotype. Acta Neuropathol. 90, 572–581 (1995). [DOI] [PubMed] [Google Scholar]
- 14.Orozco G et al. Dominantly inherited olivopontocerebellar atrophy from eastern Cuba. Clinical, neuropathological, and biochemical findings. J. Neurol. Sci. 93, 37–50 (1989). [DOI] [PubMed] [Google Scholar]
- 15.Estrada R et al. Spinocerebellar ataxia 2 (SCA2): morphometric analyses in 11 autopsies. Acta Neuropathol. 97, 306–310 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Adams C, Starkman S & Pulst SM Clinical and molecular analysis of a pedigree of southern Italian ancestry with spinocerebellar ataxia type 2. Neurology 49, 1163–1166 (1997). [DOI] [PubMed] [Google Scholar]
- 17.Elden AC et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075 (2010).This study demonstrates that intermediate-length alleles are a risk factor for ALS, indicating that even non-pathogenic changes in repeat length can have profound effects on neuronal function.
- 18.Van Damme P et al. Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology 76, 2066–2072 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Tan RH et al. Cerebellar neuronal loss in amyotrophic lateral sclerosis cases with ATXN2 intermediate repeat expansions. Ann. Neurol. 79, 295–305 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Sesqueiros J & Coutinho P Epidemiology and clinical aspects of Machado–Joseph disease. Adv. Neurol. 61, 139–153 (1993). [PubMed] [Google Scholar]
- 21.Rüb U, Brunt ER & Deller T New insights into the pathoanatomy of spinocerebellar ataxia type 3 (Machado–Joseph disease). Curr. Opin. Neurol. 21, 111–116 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Rüb U et al. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol. 104, 38–66 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Stevanin G et al. Clinical and molecular features of spinocerebellar ataxia type 6. Neurology 49, 1243–1246(1997). [DOI] [PubMed] [Google Scholar]
- 24.Schulz JB et al. Visualization, quantification and correlation of brain atrophy with clinical symptoms in spinocerebellar ataxia types 1, 3 and 6. Neuroimage 49, 158–168 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Gierga K et al. Spinocerebellar ataxia type 6 (SCA6): neurodegeneration goes beyond the known brain predilection sites. Neuropathol. Appl. Neurobiol. 35, 515–527 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Shao J & Diamond MI Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum. Mol. Genet. 16, R115–R123 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Klement IA et al. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95, 41–53 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Irwin S et al. RNA association and nucleocytoplasmic shuttling by ataxin-1. J. Cell Sci. 118, 233–242 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Yue S et al. The spinocerebellar ataxia type 1 protein, ataxin-1, has RNA-binding activity that is inversely affected by the length of its polyglutamine tract. Hum. Mol. Genet. 10, 25–30 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Tsai C-C et al. Ataxin-1, a SCA1 neurodegenerative disorder protein, is functionally linked to the silencing mediator of retinoid and thyroid hormone receptors. Proc. Natl Acad. Sci. USA 101,4047–4052 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lam YC et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 127, 1335–1347 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Tsuda H et al. The AXH domain in mammalian/ Drosophila Ataxin-1 mediates neurodegeneration in spinocerebellar ataxia 1 through its interaction with Gfi-1/Senseless proteins. Cell 122, 633–644 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Serra HG et al. RORα-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell 127, 697–708 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Gehrking KM et al. Partial loss of Tip60 slows midstage neurodegeneration in a spinocerebellar ataxia type 1 (SCA1) mouse model. Hum. Mol. Genet. 20,2204–2212 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Chiara C et al. The AXH module: an independently folded domain common to ataxin-1 and HBP1. FEBS Lett. 551, 107–112 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Chen YW et al. The structure of the AXH domain of spinocerebellar ataxin-1. J. Biol. Chem. 279, 3758–3765 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Fryer JD et al. Exercise and genetic rescue of SCA1 via the transcriptional repressor Capicua. Science 334, 690–693 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emamian ES et al. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 38, 375–387 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Huttlin EL et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duvick L et al. SCA1-like disease in mice expressing wild type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron 67, 929–935 (2010).The disease-like phenotypes elicited by the engineered ATXN1 in this study, despite a normal repeat length, indicate that specific altered protein interactions play a key part in SCA1.
- 41.Jorgensen ND et al. Phosphorylation of ATXN1 at Ser776 in the cerebellum. J. Neurochem. 110, 675–686 (2009) (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen H-K et al. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell 113,457–468 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Lim J et al. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 452, 713–719 (2008).The study presents data supporting the concept that an altered balance in the interaction of expanded ATXN1 with CIC and RBM17 drives pathogenesis in SCA1.
- 44.de Chiara C et al. Phosphorylation of S776 and 14–3-3 binding modulate Ataxin-1 interaction with splicing factors. PLoS ONE 4, e8372 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bentley DL Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 15, 163–175 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ingram M et al. Cerebellar transcriptome profiles of ATXN1 transgenic mice reveal SCA1 disease progression and protection pathways. Neuron 89, 1194–1207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Serra HG et al. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum. Mol. Genet. 13, 2535–2543 (2004).One of several reports suggesting that altered glutamate signalling in the cerebellum contributes to disease in polyQ SCAs.
- 48.Carlson KM, Andresen MJ & Orr HT Emerging pathogenic pathways in the spinocerebellar ataxias. Curr. Opin. Genet. Dev. 19, 247–253 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schorge S et al. Human ataxias: a genetic dissection of inositol triphosphate receptor (ITPR1)-dependent signaling. Trends Neurosci. 33, 209–211 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruegsegger C et al. Impaired mTORC1 -dependent expression of Homer-3 influences SCA1 pathophysiology. Neuron 89, 129–146 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Sánchez I, Balagué E & Matilla-Dueñas A Ataxin-1 regulates the cerebellar bioenergetics proteome through the GSK3β–mTOR pathway which is altered in spinocerebellar ataxia type 1 (SCA1). Hum. Mol. Genet 25, 4021–4040 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Lee JH et al. Reinstating aberrant mTORC1 activity in Huntington’s disease mice improves disease phenotypes. Neuron 85, 303–315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ross CA & Truant R A unifying mechanism in neurodegeneration. Nature 541,34–35 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Bettencourt C et al. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann. Neurol. 79, 983–990 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taniguchi JB et al. RpA1 ameliorates symptoms of mutant ataxin-1 knock-in mice and enhances DNA damage repair. Hum. Mol. Genet. 25, 4432–4447 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Ito H et al. HMGB1 facilitates repair of mitochondrial DNA damage and extends the lifespan of mutant ataxin-1 knock-in mice. EMBO Mol. Med. 7, 78–101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nechiporuk T et al. The mouse SC42 gene: cDNA sequence, alternative splicing and protein expression. Hum. Mol. Genet. 7, 1301–1309 (1998). [DOI] [PubMed] [Google Scholar]
- 58.Neuwald AF & Koonin EV Ataxin-2, global regulators of bacterial gene expression and spliceosomal snRNP proteins share a conserved domain. J. Mol. Med. 76, 3–5 (1998). [DOI] [PubMed] [Google Scholar]
- 59.Kozlov G et al. Structure and function of the C-terminal PABC domain of human poly(A)-binding protein. Proc. Natl Acad. Sci. USA 98, 4409–4413 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shibata H, Huynh DP & Pulst SM A novel protein with RNA-binding motifs interacts with ataxin-2. Hum. Mol. Genet. 9, 1303–1313 (2000). [DOI] [PubMed] [Google Scholar]
- 61.Nonhoff U et al. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol. Biol. Cell 18, 1385–1396 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCann C et al. The Ataxin-2 protein is required for microRNA function and synapse-specific long-term olfactory habituation. Proc. Natl Acad. Sci. USA 108, E655–E662 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mangue A, Amrani N & Jacobson A Pbp 1 p, a factor interacting with Saccharomyces cerevisiae poly(A)-binding protein, regulates polyadenylation. Mol. Cell. Biol. 18, 7383–7396 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Satterfield TF & Pallanck LJ Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum. Mol. Genet. 15, 2523–2532 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Takahara T & Maeda T Transient sequestration of TORC1 into stress granules during heat stress. Mol. Cell AT, 242–252 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Bar DZ et al. Cell size and fat content of dietary- restricted Caenorhabditis elegans are regulated by ATX-2, an mTOR repressor. Proc. Natl Acad. Sci. USA 113, E4620–E4629 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lastres-Beckei I et al. Mammalian ataxin-2 modulates translation control at the pre-initiation complex via PI3K/mTOR and is induced by starvation. Biochim. Biophys. Acta 1862, 1558–1569 (2016).One of several recent studies showing that ATXN2 has a complex role in regulating translation in cells.
- 68.Lagier-Tourenne C & Cleveland DC Rethinking ALS: the FUS about TDP-43. Cell 136, 1001–1004 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Damrath E, et al. ATXN2-CAG42 sequesters PABPC 1 into insolubility and induces FBXW8 in cerebellum of old ataxic knock-in mice. PLoS Genet. 8, el002920 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pflieger LT et al. Gene co-expression network analysis for identifying modules and functionally enriched pathways in SCA2. Hum. Mol. Genet, 10.1093/hmg/ddxl91 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Costa Mdo C & Paulson HL Toward understanding Machado–Joseph disease. Prog. Neurobiol. 97, 239–257 (2012).A thorough review describing the molecular features of SCA3, also known as Machado–Joseph disease.
- 72.Li X et al. Toward therapeutic targets for SCA3: Insight into the role of Machado–Joseph disease protein ataxin-3 in misfolded proteins clearance. Prog. Neurobiol. 132, 34–58 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Faggiano S et al. Allosteric regulation of deubiquitylase activity through ubiquitination. Front. Mol. Biosci. 2, 2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blount JR et al. Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23. Nat. Commun. 5, 4638 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsou WL et al. Ubiquitination regulates the neuroprotective function of the deubiquitinase ataxin-3 in vivo. J. Biol. Chem. 288, 34460–34469 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Durcan TM & Fon EA Ataxin-3 and its E3 partners: implications for Machado–Joseph disease. Front. Neurol. 4, 46 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Scaglione KM et al. Ube2w and ataxin-3 coordinately regulate the ubiquitin ligase CHIP Mol. Cell 43,599–612 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang Q, Li L & Ye Y Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J. Cell. Biol. 174, 963–971(2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang H, Ying Z & Wang G Ataxin-3 regulates aggresome formation of copper-zinc superoxide dismutase (SOD 1) by editing K63-linked polyubiquitin chains. J. Biol. Chem. 287, 28576–28585 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chatterjee A et al. The role of the mammalian DNA end-processing enzyme polynucleotide kinase 3’-phosphatase in spinocerebellar ataxia type 3 pathogenesis. PLoS Genet. 11, el004749 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gao R et al. Inactivation of PNKP by mutant ATXN3 triggers apoptosis by activating the DNA damage-response pathway in SCA3. PLoS Genet. 11, el004834 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pfeiffer A et al. Ataxin-3 consolidates the MDC 1 -dependent DNA double-strand break response by counteracting the SUMO-targeted ubiquitin ligase RNF4. EMBO J. 36, 1066–1083 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ashkenazi A et al. Polyglutamine tracts regulate beclin 1 -dependent autophagy. Nature 545, 108–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Warrick JM et al. Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol. Cell. 18, 37–48 (2005). [DOI] [PubMed] [Google Scholar]
- 85.Zeng L et al. The de-ubiquitinating enzyme ataxin-3 does not modulate disease progression in a knock-in mouse model of Huntington disease. J. Huntingtons Dis. 2,201–215 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bichelmeier U et al. Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: in vivo evidence. J. Neurosci. 27, 7418–7428 (2007).An important study demonstrating that expanded ATXN3 concentrates in the nucleus, where it may be most toxic to neurons.
- 87.Nobrega C et al. RNA interference mitigates motor and neuropathological deficits in a cerebellar mouse model of Machado–Joseph disease. PLoS ONE. 9, el00086 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Costa Mdo C et al. Toward RNAi therapy for the polyglutamine disease Machado–Joseph disease. Mol. Ther. 21, 1898–1908 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aiba Y et al. Allele-selective inhibition of expression of huntingtin and ataxin-3 by RNA duplexes containing unlocked nucleic acid substitutions. Biochemistry 52, 9329–9338 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Moore LR et al. Evaluation of antisense oligonucleotides targeting ATXN3 in SCA3 mouse models. Mol. Ther. Nucleic Acids 7, 200–210 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Teixeira-Castro A et al. Serotonergic signalling suppresses ataxin 3 aggregation and neurotoxicity in animal models of Machado–Joseph disease. Brain 138,3221–3237 (2015).In this study, an unbiased screen suggested that antidepressants in the serotonin reuptake inhibitor class could be protective against the SCA3 disease protein.
- 92.Costa MD et al. Unbiased screen identifies aripiprazole as a modulator of abundance of the polyglutamine disease protein, ataxin-3. Brain 139, 2891–2908 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pavel M et al. CCT complex restricts neuropathogenic protein aggregation via autophagy. Nat. Commun. 7, 13821 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cushman-Nick M, Βοηίηί NM & Shorter J Hspl04 suppresses polyglutamine-induced degeneration post onset in a drosophila MJD/SCA3 model. PLoS Genet. 9, el003781 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Simoes AT et al. Calpain inhibition reduces ataxin-3 cleavage alleviating neuropathology and motor impairments in mouse models of Machado–Joseph disease. Hum. Mol. Genet. 23, 4932–4944 (2014). [DOI] [PubMed] [Google Scholar]
- 96.Liman J et al. CDK5 protects from caspase-induced Ataxin-3 cleavage and neurodegeneration. J. Neurochem. 129, 1013–1023 (2014). [DOI] [PubMed] [Google Scholar]
- 97.Matos CA et al. Ataxin-3 phosphorylation decreases neuronal defects in spinocerebellar ataxia type 3 models. J. Cell Biol. 212, 465–480 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Riess O et al. SCA6 is caused by moderate CAG expansion in the a, A-voltage-dependent calcium channel gene. Hum. Mol. Genet. 6, 1289–1293 (1997). [DOI] [PubMed] [Google Scholar]
- 99.Zhuchenko O et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the a, A-voltage-dependent calcium channel. Nat. Genet. 15, 62–69 (1997). [DOI] [PubMed] [Google Scholar]
- 100.Pietrobon D Calcium channels and channelopathies of the central nervous system. Mol. Neurobiol. 25, 31–50 (2002). [DOI] [PubMed] [Google Scholar]
- 101.Kordasiewicz ΗB et al. C-termini of P/Q-type Ca2 + channel al A subunits translocate to nuclei and promote polyglutamine-mediated toxicity. Hum. Mol. Genet. 15, 1587–1599 (2006). [DOI] [PubMed] [Google Scholar]
- 102.Du X et al. Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell 154, 118–133 (2013).This study reshaped thoughts about SCA6 pathogenesis with the discovery that the SCA6 disease gene is complex and encodes both the α 1A subunit of the Cav2.1 calcium channel and a much smaller polyOcontaining transcription factor.
- 103.Unno T et al. Development of Purkinje cell degeneration in a knockin mouse model reveals lysosomal involvement in the pathogenesis of SCA6. Proc. Natl Acad. Sci. USA 109, 17693–17698 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang S, Li XJ & Li S Molecular mechanisms underlying spinocerebellar ataxia 17 (SCA17) pathogenesis. Rare Dis. 4, el223580 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsuji S Dentatorubral-pallidoluysian atrophy. Handb. Clin. Neurol. 103, 587–594 (2012). [DOI] [PubMed] [Google Scholar]
- 106.Helmlinger D et al. Both normal and polyglutamine- expanded ataxin-7 are components of TFTC-type GCN5 histone acetyltransferase-containing complexes. Biochem. Soc. Symp. 73, 155–163 (2006). [DOI] [PubMed] [Google Scholar]
- 107.Lan X et al. Poly(Q) expansions in ATXN7 affect solubility but not activity of the SAGA deubiquitinating module. Mol. Cell. Biol. 35, 1777–1787 (2015).An important study showing that although expanded ATXN7 still functions in deubiquitination, its propensity to aggregate ultimately impairs DUB activity in cells.
- 108.McCullough SD & Grant PA Histone acetylation, acetyltransferases, and ataxia — alteration of histone acetylation and chromatin dynamics is implicated in the pathogenesis of polyglutamine- expansion disorders. Adv. Protein Chem. Struct. Biol. 79, 165–203 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Palhan VB et al. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc. Natl Acad. Sci. USA 102, 8472–8477 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Furrer SA et al. Spinocerebellar ataxia type 7 cerebellar disease requires the coordinated action of mutant ataxin-7 in neurons and glia, and displays non- cell-autonomous Bergmann glia degeneration. J. Neurosci. 31, 16269–16278 (2011).One of the more compelling studies that illustrates the complex interrelationship between neurons and non-neuronal cells in the precipitation of polyQ-mediated cerebellar degeneration.
- 111.Barnes JA et al. Abnormalities in the climbing fiber-Purkinje cell circuitry contribute to neuronal dysfunction in ATXN1 [82 Q] mice. J. Neurosci. 31, 12778–12789 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Power EM, Morales A & Empson RM Prolonged type 1 metabotropic glutamate receptor dependent synaptic signaling contributes to spino-cerebellar ataxia type 1. J. Neurosci. 36,4910–4916(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.De Zeeuw CI et al. Spatiotemporal firing patterns in the cerebellum. Nat. Rev. Neurosci. 12, 327–344 (2011). [DOI] [PubMed] [Google Scholar]
- 114.Chopra R & Shakkottai VG Translating cerebellar Purkinje neuron physiology to progress in dominantly inherited ataxia. Future Neurol. 9, 187–196 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hourez R et al. Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J. Neurosci. 31, 11795–11807 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dell’Orco JM et al. Neuronal atrophy early in degenerative ataxia is a compensatory mechanism to regulate membrane excitability. J. Neurosci. 35, 11292–11307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kasumu AW et al. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem. Biol. 19, 1340–1353 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hansen ST et al. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum. Mol. Genet. 22, 271–283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Egorova PA et al. In vivo analysis of cerebellar Purkinje cell activity in SCA2 transgenic mouse model. J. Neurophysiol. 115, 2840–2851 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shakkottai VG et al. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J. Neurosci. 31, 13002–13014 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jayabal S et al. 4-Aminopyridine reverses ataxia and cerebellar firing deficiency in a mouse model of spinocerebellar ataxia type 6. Sci. Rep. 6, 29489 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Keisei MS, Kordasiewicz ΗB & McBride JL Gene suppression strategies for dominantly inherited neurodegenerative diseases: lessons from Huntington’s disease and spinocerebellar ataxia. Hum. Mol. Genet. 25, R53–R64 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Miyazaki Y et al. An miRNA-mediated therapy for SCA6 blocks IRES-driven translation of the CACNAIA second cistron. Sci. Transl Med 8, 347ra94 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Scoles DR et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 544, 362–366 (2017).One of several recent reports showing the promise of ASO-based disease gene silencing as a treatment for the polyQ SCAs. In this reference, the target is ATXN2, which the next reference shows is a therapeutic target in a TDP43 mouse model of ALS, providing further support for the concept that ATXN2 is an ALS risk factor.
- 125.Becker LA et al. Therapeutic reduction of ataxin 2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544, 367–371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chriboga CA et al. Results from a Phase 1 study of ISIS-SMNRX in children spinal muscular atrophy. Neurology 12, 435–442 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Finkel RS et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open- label, dose-escalation study. Lancet 388, 3017–3026 (2016). [DOI] [PubMed] [Google Scholar]
- 128.Miller TM et al. An antisense oligonucleotide against SOD 1 delivered intrathecally for patients with SOD 1 familial amytrophic lateral sclerosis: a phase 1, randomized, first-in man study. Lancet Neurol. 12, 435–442 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jacobi H et al. Long-term disease progression in spinocerebellar ataxia types 1, 2, 3, and 6: a longitudinal cohort study. Lancet Neurol. 14, 1101–1108 (2015). [DOI] [PubMed] [Google Scholar]
- 130.Reetz K et al. Genotype-specific patterns of atrophy progression are more sensitive than clinical decline in SCA1, SCA3, and SCA6. Brain 136, 905–917 (2013). [DOI] [PubMed] [Google Scholar]
- 131.Adanyeguh IM et al. In vivo neurometabolic profiling in patients with spinocerebellar ataxia types 1, 2, 3, and 7. Mov. Disord 30, 662–670 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Velazquez-Perez L et al. Abnormal corticospinal tract function and motor cortex excitability in non-ataxic SCA2 mutation carriers: a TMS study. Clin. Neurophysiol. 127, 2713–2719 (2016). [DOI] [PubMed] [Google Scholar]
- 133.Velazquez-Perez L et al. Corticomuscular coherence: a novel tool to assess the pyramidal tract dysfunction in spinocerebellar ataxia type 2. Cerebellum 16, 602–606 (2017). [DOI] [PubMed] [Google Scholar]
- 134.Southwell AL et al. Ultrasensitive measurement of huntingtin protein in cerebrospinal fluid demonstrates increase with Huntington disease stage and decrease following brain huntingtin suppression. Sci. Rep. 5, 12166 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dansithong W et al. Ataxin-2 regulates RGS8 translation in a new BAC-SCA2 transgenic mouse model. PLoS Genet. 11, el005182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zu T et al. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J. Neurosci. 24, 8853–8861 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ikeda Y et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat. Genet. 38, 184–190 (2006). [DOI] [PubMed] [Google Scholar]
- 138.Houlden H et al. Mutations in TTBK2, encoding a kinase implicated in tau phosphorylation, segregate with spinocerebellar ataxia type 11. Nat. Genet. 39, 1434–1436(2007). [DOI] [PubMed] [Google Scholar]
- 139.Waters MF & Pulst SM SCA13. Cerebellum 7, 165–169 (2008). [DOI] [PubMed] [Google Scholar]
- 140.Chen DH et al. Missense mutations in the regulatory domain of PKCy: a new mechanism for dominant nonepisodic cerebellar ataxia. Am. J. Hum. Genet. 72, 839–849 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Storey E & Gardner RJ Spinocerebellar ataxia type 15.Handb. Clin. Neurol. 103, 561–565 (2012). [DOI] [PubMed] [Google Scholar]
- 142.Iwaki A et al. Heterozygous deletion of ITPRI, but not SUMFI, in spinocerebellar ataxia type 16. J. Med Genet. 45, 32–35 (2008). [DOI] [PubMed] [Google Scholar]
- 143.van de Leemput J et al. Deletion at ITPRI underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 3, e108 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Verbeek DS et al. Mapping of the SCA23 locus involved in autosomal dominant cerebellar ataxia to chromosome region 2Op 13–12.3. Brain 127, 2551–2557 (2004). [DOI] [PubMed] [Google Scholar]
- 145.Hekman KE et al. A conserved eEF2 coding variant in SCA26 leads to loss of translational fidelity and increased susceptibility to proteostatic insult. Hum. Mol. Genet. 21,5472–5483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Brusse E et al. Spinocerebellar ataxia associated with a mutation in the fibroblast growth factor 14 gene (SCA27): a new phenotype. Mov. Disord. 21, 396–401 (2006). [DOI] [PubMed] [Google Scholar]
- 147.Mariotti C et al. Spinocerebellar ataxia type 28. Handb. Clin. Neurol. 103, 575–579 (2012). [DOI] [PubMed] [Google Scholar]
- 148.Turcotte-Gauthier M et al. Expanding the clinical phenotype associated with ELOVL4 mutation: study of a large French-Canadian family with autosomal dominant spinocerebellar ataxia and erythrokeratodermia. JAMA Neurol. 71,470–475 (2014). [DOI] [PubMed] [Google Scholar]
- 149.Guo Y-C et al. Spinocerebellar ataxia 35: novel mutations in TGM6 with clinical and genetic characterization. Neurology 83, 1554–1561 (2014). [DOI] [PubMed] [Google Scholar]
- 150.Di Gregorio E et al. ELOVL5 mutations cause spinocerebellar ataxia 38. Am. J. Hum. Genet. 95, 209–217 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tsoi H et al. A novel missense mutation in CCDC88C activates the JNK pathway and causes a dominant form of spinocerebellar ataxia. J. Med. Genet. 51, 590–595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fogel BL, Hanson SM & Becker EBE Do mutations in the murine ataxia gene TRPC3 cause cerebellar ataxia in humans? Mov. Disord. 30, 284–286 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Coutelier M et al. A recurrent mutation in CACNAIG alters Cav3.1 T-type calcium-channel conduction and causes autosomal-dominant cerebellar ataxia. Am. J. Hum. Genet. 97, 726–737 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Depondt C et al. MME mutation in dominant spinocerebellar ataxia with neuropathy (SCA43). Neurol. Genet. 2, e94 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.White M et al. Transgenic mice with SCA10 pentanucleotide repeats show motor phenotypes and susceptibility to seizure: a toxic RNA gain-of-function model. J. Neurosci. Res. 90, 706–714 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zu T et al. Non-ATG-initiated translation directed by miciOsatellite expansions. Proc. Natl Acad. Sci. USA 108, 260–265 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cleary JD & Ranum LP Repeat associated non- ATG (RAN) translation: new starts in miciOsatellite expansion disorders. Curr. Opin. Genet. Dev. 26, 6–15 (2014).A comprehensive review of emerging evidence supporting the idea that RAN translation-generated peptides play a part in repeat- expansion-associated neurological disease.
- 158.Banez-Coronel M et al. RAN translation in Huntington disease. Neuron 88, 667–677 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Burright EN et al. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cel! 82, 937–948 (1995). [DOI] [PubMed] [Google Scholar]
- 160.Huynh DP et al. Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat. Genet. 26, 44–50 (2000). [DOI] [PubMed] [Google Scholar]
- 161.Lorenzetti D et al. Repeat instability and motor incoordination in mice with a targeted expanded CAG repeat in the Seal locus. Hum. Mol. Genet. 9, 779–785 (2000). [DOI] [PubMed] [Google Scholar]
- 162.Watase K et al. A long CAG repeat in the mouse Sea I locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 34, 905–919 (2002). [DOI] [PubMed] [Google Scholar]
- 163.Boy J et al. Reversibility of symptoms in a conditional mouse model of spinocerebellar ataxia type 3. Hum. Mol. Genet. 18, 4282–4295 (2009). [DOI] [PubMed] [Google Scholar]
- 164.DelTOrco JM, Pulst SM & Shakkottai VG Potassium channel dysfunction underlies Purkinje neuron spiking abnormalities in spinocerebellar ataxia type 2. Hum. Mol. Genet, 10.1093/hmg/ddx281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]