

Abstract
Purpose:
Glioblastoma (GBM) is the most common primary malignant tumor in the central nervous system. Our recent pre-clinical work has suggested that PD-1/PD-L1 plays an important immunoregulatory role to limit effective anti-tumor T cell responses induced by active immunotherapy. However, little is known about the functional role that PD-1 plays on human T lymphocytes in malignant glioma patients.
Experimental Design:
In this study, we examined the immune landscape and function of PD-1 expression by T cells from tumor and peripheral blood in malignant glioma patients.
Results:
We found several differences between PD-1+ tumor-infiltrating lymphocytes (TILs) and patient-matched PD-1+ peripheral blood T lymphocytes. Phenotypically, PD-1+ TILs exhibited higher expression of markers of activation and exhaustion than peripheral blood PD-1+ T cells, which instead had increased markers of memory. A comparison of the T cell receptor variable chain populations revealed decreased diversity in T cells that expressed PD-1, regardless of the location obtained. Functionally, peripheral blood PD-1+ T cells had a significantly increased proliferative capacity upon activation compared with PD-1− T cells.
Conclusion:
Our evidence suggests that PD-1 expression in glioma patients reflects chronically-activated effector T cells that display hallmarks of memory and exhaustion depending on its anatomical location. The decreased diversity in PD-1+ T cells suggests that the PD-1 expressing population has a narrower range of cognate antigen targets compared to the PD-1 non-expression population. This information can be used to inform how we interpret immune responses to PD-1 blocking therapies or other immunotherapies.
Keywords: cancer, immunotherapy, glioblastoma, T cell exhaustion, PD-1, checkpoint inhibitors
INTRODUCTION
Glioblastoma (GBM) is the most common primary brain tumor in adults and remains one of the most lethal of all human cancers (1). Current standard of care includes surgical resection, chemotherapy, and radiation therapy and is associated with a median survival of 12–18 months (2–4). The dismal outcome of this disease clearly highlights the need for new approaches in therapy. Novel treatment strategies, such as GBM-targeted immunotherapy and vaccination therapy, have shown promise in both pre-clinical and clinical trials (5–9). However, the survival benefit has been indeterminate. Antitumor immune responses induced by immunotherapy and, more specifically active vaccination in GBM, may in turn induce an adaptive immune resistance mechanism mediated by immune checkpoints such as signaling of programmed death 1 and its ligand (PD-1/PD-L1) (10–12).
PD-1 is a cell surface co-inhibitory receptor that is expressed on T cells after activation and limits the immune response, resulting in what is traditionally considered functionally “exhausted” T cells (13,14). In normal physiologic states, the function of this immune checkpoint protein is to inhibit or reduce autoimmunity and to maintain homeostasis during chronic infections (15,16). In the setting of cancer, the PD-1/PD-L1 pathway may be an adaptive mechanism by the tumor to evade anti-tumor immune attack. Although PD-1 has previously been considered to be a marker of terminally differentiated exhausted T cells, PD-1 expression may in fact be a marker of continued effector or memory function needed to maintain pathogen or tumor control while minimizing tissue damage in a chronic state (17). In fact, a number of recent studies have shown that PD-1 expression of CD8+ T cells early after activation may temporally coincide with rapid proliferation and production of signature effector cytokines and be an important signaling pathway in programming of memory CD8+ T cells (18–20).
In this study, we set out to define and characterize PD-1 expression in tumor-infiltrating lymphocytes (TILs) and peripheral blood from patients diagnosed with malignant glioma. Numerous studies have shown PD-L1 expression in GBM as well as PD-1 expression in a subset of GBM TIL (10,12,13). PD-1 expression on malignant glioma TILs in comparison to peripheral blood has not been investigated, nor has the function of these malignant glioma PD-1+ TILs been examined in this detail. We found that the proportion of TILs expressing PD-1 was significantly elevated when compared to T cells in patient peripheral blood and T cells from healthy controls. Furthermore, we found increased levels of activation and exhaustion markers on PD-1+ TILs when compared to PD-1+ peripheral blood T cells, while we found increased markers of memory/antigen experience on the peripheral blood PD-1+ T cells. Our results highlight the complex relationship between PD-1 expression on T cells and its relation to exhaustion and chronic activation within human malignant brain tumors.
MATERIALS & METHODS
Patients
We collected 52 patient tumor and blood specimen samples and an additional 14 peripheral blood samples from healthy donors at UCLA. In parallel, matched TILs and peripheral blood were obtained from six consented glioblastoma patients who presented to the University of Heidelberg neurosurgery service for surgical removal of their tumors. All patients provided written informed consent and this study was conducted in accordance with recognized ethical guidelines and under Institutional Review Board (IRB) approved protocols from both institutions.
Collection of PBMC for Immune Monitoring
Peripheral blood was drawn from patients prior to surgical resection of their gliomas. Once drawn, we diluted peripheral blood at 1:1 dilution of RPMI media (Fisher Scientific Cat. MT10041CV); PBMCs were harvested through subsequent extraction with Ficoll (Fisher Scientific Cat. 45–001-750) gradient. After a series of two washes in RPMI media, we placed cells in freezing media made of 90% Human AB serum (Fisher Scientific Cat. MT35060CI) and 10% DMSO (Sigma Cat. C6295–50ML) and stored in liquid nitrogen.
Collection of Tumor Infiltrating Lymphocytes
Tumor infiltrating lymphocytes were collected in two ways. In the first method, we collected tumor and digested the tissue via incubation with collagenase (1mg/mL) (Advance Biofactures Corporation) and DNAse (0.02mg/mL) (Sigma Cat. D4263–5VL) in RPMI media overnight on the day of resection. TILs were isolated using Percoll (Sigma Cat. P4937) gradient and washed in RPMI twice. In the second method, patient-resected tumors were first digested using Miltenyi’s Brain Tumor Dissociation Kit (Miltenyi Biotec Cat. 130–095-42) and gentleMACS Dissociator (Cat. 130–093-235) and labeled with CD45+ microbeads (Cat. 130–045-801). CD45+ cells were positively selected for using Miltenyi LS columns (Cat. 130–042-401) and MidiMACS Separator. TILs were frozen in the same freezing media as the PBMCs.
Flow Cytometric Analysis of PBMCs and TILs
We thawed donor PBMCs and patient PBMCs and TILs at 37 °C for three minutes in a water bath and immediately washed with RPMI media to remove DMSO. We resuspended cells at one million cells/100 µl PBS.
In the characterization of PD-1-expression of TILs and PBMCs, we first stained cells with live-dead Zombie Yellow stain (BioLegend, Cat. 423104). In two separate experimental reactions, we stained cells with a 7-antibody and an 8-antibody panel. Two antibody panels were necessary to investigate all of the surface markers of interest due to the spectral limitations of the flow cytometer. The first antibody cocktail included seven antibodies: CD3 Pe-Cy7 (eBioscience, Cat. 25–0038-42), CD4 Alexa Fluor 700 (BD Biosciences, Cat. 557922), CD8 Pacific Blue (BD Biosystems, Cat. 558207), CD16 FITC (BD Biosystems, Cat. 555406), CTLA-4 PE (BD Biosystems, Cat. 555853), PD-1 AF647 (eBioscience, Cat. 51–9969-73), and CD69 PerCP-CY5.5 (BD Biosystems, Cat. 560738) antibodies where each antibody was diluted to a 1:40 antibody to buffer ratio. The second 8-antibody cocktail contained CD3 Pe-Cy7 (eBioscience, Cat. 25–0038-42), CD4 Alexa Fluor 700 (BD Biosystems, Cat. 557922), CD8 Pacific Blue (BD Biosystems, Cat. 558207), CD16 FITC (BD Biosystems, Cat. 555406), CD19 PE (BD Biosystems, Cat. 555413), CD25 APC-Cy7 (BD Biosystems, Cat. 557753), CD127 AF647 (BD Biosystems, Cat. 558598), and CD69 PE-Cy5 (BD Biosystems, Cat. 555532) antibodies at the same dilution as above.
We used a BD LSR II flow cytometer and the BD FACS diva software to analyze all cell-samples. Cytometer settings were restricted to the nine colors used. We used unstained controls to adjust FSC and SSC voltages such that the lymphocyte population became visible and then acquired single color compensation controls, adjusting voltages such that the stained cells did not exceed 105. We set gates on stained controls in comparison to the negative control and recorded data from all samples, while subsequently applying compensation controls. We recorded staining expression at 80,000 – 100,000 events per sample, as quantity of T cells were variable from specimen to specimen. Thus, we analyzed the data acquired from the flow cytometer by using frequency measurements rather than absolute numbers to compare sample expression profiles using FlowJo.
CyTOF
Patient PBMCs and TILs were collected as described above. Between 1–2 million patient PBMCs and TILs were labeled with Cell-ID Cisplatin (Fluidigm, Cat. 201064) to assess for live/dead and then labeled with a variety of metal-conjugated antibodies. From the patient PBMCs, 1 million cells were set aside and labeled with a smaller panel containing only cell lineage markers to aid in setting the proper gates of other markers. After labeling, cells were fixed using MaxPar Fix and Perm buffer (Fluidigm, Cat. 201067) and labelled for single cell discrimination with Cell-ID Intercalator-Ir (Fluidigm, CA 201192A) and run on the Helios CyTOF system.
The collected mass cytometry data was analyzed as previously described (21). Briefly, samples were normalized utilizing a bead standard, then manually gated to remove debris and dead cells. Raw marker intensities were transformed using hyperbolic inverse sine with cofactor of 5. Cell population identification was carried out utilizing the FlowSOM (22) and ConsensusClusterPlus packages in R using 11 lineage markers, with the number of clusters determined by elbow criterion. For differential analysis of cell population abundance and functional marker expression, we fit fixed and mixed linear models to our data and performed testing using the general linear hypothesis function glht in R. To account for multiple testing, we applied the Benjamini-Hochberg adjustment with a false discovery rate of 5%.
Fluorescence-activated Cell Sorting of CD3+ PD-1+ and CD3+ PD-1− T Cell Populations
As described above, we thawed frozen donor PBMCs and patient PBMCs and TILs, stained samples with CD3 Pe-Cy7 (eBioscience, Cat. 25–0038-42) and PD-1 AF647 (BD Biosystems, Cat. 558598) and sorted samples with the Aria II cell sorter (Becton Dickinson) into PD-1+ and PD-1− T cell populations. We defined PD-1+ T cells as CD3+ and PD1+ and PD-1− T cells as CD3+ and PD1−.
RNA Gene Expression
We used the NanoString nCounter Dx Analysis System which measures the expression of 511 human immune function genes through digital readouts of the abundance of mRNA transcripts in order to analyze cell lysates of PD-1-expressing and PD-1 void cells (23). The nCounter Dx Analysis System uses gene-specific probe pairs that hybridize directly to mRNA samples. A Reporter Probe carries the fluorescent signal; a Capture Probe allows the complex to be immobilized for data collection. We included, in each assay, six positive quality controls comprised of a linear titration of in vitro transcribed RNA transcripts and corresponding probes, and eight negative quality controls consisting of probes with no sequence homology to human RNA sequences. Each Human Immunology assay run includes a reference sample consisting of in vitro transcribed RNAs of the 13 targets that are used for normalization purposes.
TCR V-β Sequencing
We extracted genomic DNA from sorted CD3+ PD-1+ and CD3+ PD-1− T cells from malignant glioma patient TILs and PBMCs and from healthy donor PBMCs using DNeasy Blood & Tissue Kit (Qiagen Cat. 69504) and sequenced DNA at the TCR V-beta CDR3 region using high throughput sequencing in collaboration with Adaptive Biotechnologies, as we have recently published (24).
IFN-γ Activation Assay
CD3+ PD-1− and CD3+ PD-1+ cells were sorted in a similar manner described above. In a 96-well plate, we added 200,000 cells into each well at a concentration of 1×106 cells/mL. For the cells that were to be activated, we added 50 ng/mL anti-CD3 Ab (BioLegend, Cat. 317304) and 1 ug/mL anti-CD28 Ab (BD Biosciences, Cat. 555725). In all wells, we added 50 IU/mL IL-2. After 24 hours, the supernatant was collected and fresh media with 50 IU/mL IL-2 was added in each well, and the supernatant was collected 24hrs later (total of 48hrs after activation). To measure IFN-γ levels, we used the Human IFN-γ Uncoated ELISA Kit (Fisher, Cat. 88–7613-86).
Statistics
The percentage of T lymphocytes expressing the various T cell markers studied were compared between patient PBMCs, TILs, and donor PBMCs. Unpaired Welch’s t-test was applied to compare the means of the frequencies of expression with two-sided p-values (*p<0.05, **p<0.005, ***p<0.0005).
RESULTS
Glioma-infiltrating T cells have elevated expression of PD-1 and other markers of T cell exhaustion and activation compared to peripheral T cells.
We recently demonstrated a critical role for PD-L1 on T cells infiltrating murine gliomas, as well as analogous human tumors (10,12). To examine the role of PD-1 in human glioma, we first analyzed the PD-1 expression profile using traditional flow cytometric approaches in TILs and matched peripheral blood from 14 malignant glioma patients undergoing surgical resection at UCLA Medical Center (Fig. 1A). The clinical and pathologic characteristics of our patient population are outlined in Table 1. Similarly, we simultaneously analyzed the expression of PD-1 in the peripheral blood T cells of 14 healthy donors (Suppl Fig. 1A).
Figure 1: CD3+ T cells from malignant glioma patients demonstrate significantly elevated expression of PD-1 compared to healthy donor PBMCs and glioma patient PBMCs.

A) Percent of PD-1 expression on CD3+ T cell from all the patient samples as measured by flow cytometry (*P<0.05, **P≤0.005, ***P≤0.0005).
B) The percentage of PD-1 expressing CD3+ T cells from a validation cohort of malignant glioma patients from the University of Heidelberg (*P≤0.05).
C) Histogram of the expression levels of the different exhaustion markers (PD-1, TIM3, LAG3, CTLA-4).
D) Percentage of CD3+ T cells that express one or more markers of exhaustion (PD-1, TIM3, LAG3, CTLA-4).
E) The percentage of cells expressing different combinations of the various exhaustion markers (**P≤0.005, ***P≤0.0005). Blue bars represent healthy donor PBMC samples, red bars represent glioma patient PBMC samples, and green bars represent glioma patient TIL samples.
Table 1:
The clinical and pathological characterization of our patient population
| Flow Cytometry/Gene Expression/TCR sequencing | Cytof | Activation IFN-γ Studies | All Studies | |
|---|---|---|---|---|
| Total Number | 20 | 28 | 4 | 52 |
| Mean age at diagnosis | 51.3 years | 51.4 years | 43.1 years | 50.7 years |
| Diagnosis | Number (percent) | Number (percent) | Number (percent) | Number (percent) |
| Glioblastoma | 18 (90.0%) | 20 (71.4%) | 3 (75.0%) | 41 (78.8%) |
| Gliosarcoma | 0 (0%) | 1 (3.6%) | 0 (0%) | 1 (1.9%) |
| Anaplastic Oligodendroglioma | 0 (0%) | 3 (10.7%) | 0 (0%) | 3 (5.8%) |
| Anaplastic Astrocytoma | 2 (10.0%) | 4 (14.3%) | 1 (25.0%) | 7 (13.5%) |
| Sex | ||||
| Male | 13(65.0%) | 17 (60.7%) | 4 (100%) | 34 (65.4%) |
| Female | 7 (35.0%) | 11 (39.3%) | 9(0%) | 27 (51.9%) |
| IDH | ||||
| Mutated | 4 (20.0%) | 7 (25.0%) | 2 (50.0%) | 13 (25.0%) |
| Wildtype | 15 (75.0%) | 21 (75.0%) | 2 (50.0%) | 38 (73.1%) |
| Unknown | 1(5.0%) | 1 (1.9%) | ||
| MGMT | ||||
| Methylated | 10 (50.0%) | 11 (39.3%) | 0 (0%) | 21 (40.4%) |
| Non-methylated | 10 (50.0%) | 16 (57.1%) | 3 (75.0%) | 29 (55.8%) |
| Unknown | 1 (3.6%) | 1 (25.0%) | 2 (3.8%) | |
| Presentation | ||||
| New diagnosis | 14 (70.0%) | 16 (57.1%) | 0 (0%) | 30 (57.7%) |
| Recurrent | 6(40.0%) | 12 (42.9%) | 4(100%) | 22 (42.3%) |
PD-1 was significantly elevated in the CD3+ TIL population (mean ± SE: 51.7% ± 7.2% compared to the CD3+ glioma patient PBMC population (19.7% ± 1.8%, p=0.0005) and healthy donor PBMCs (17.0% ± 2.4%, p=0.02) (Fig. 1A). The proportion of TILs expressing PD-1 was significantly elevated in the CD3+CD4+ T cell population (mean ± SE: 53.2% ± 6.8%) as compared to patient peripheral blood (17.6% ± 2.6%, p<0.0004) or donor peripheral blood (17.3% ± 1.9%, p<0.0001; Suppl Fig. 1B). Similarly, the percentage of CD3+CD8+ cells expressing PD-1 was also significantly higher in TILs (51.9% ± 7.39%) than on patient peripheral blood T cells (22.5% ± 3.13%; p<0.0023) or donor peripheral blood T cells (15.9% ± 2.24%; p<0.0005; Suppl Fig. 1C). In order to validate these findings independently, PD-1 expression was analyzed on GBM TILs, matched patient peripheral blood T cells, and normal controls from a separate cohort of six GBM patients and six healthy controls at the University of Heidelberg (Germany). Similarly, PD-1 expression was significantly increased in the GBM TIL population when compared to matched patient peripheral blood T cells and healthy patient’s T cells (Fig. 1B).
Because PD-1 has typically been associated with an exhausted phenotype, in addition to PD-1, the expression of three other exhaustion markers (CTLA-4, TIM-3, and LAG-3) was measured in the patients from our German validation cohort (Fig. 1C). The GBM TIL population showed greater expression of TIM-3 and LAG-3 and marginal increase in expression of CTLA-4 compared to both healthy donor PBMC and patient PBMCs. To examine this further, we noted the percentage of cells in each population that showed one more of the exhaustion markers (Fig. 1D). The GBM TIL population showed the largest subpopulation of cells that expressed at least one of the exhaustion markers and the only population that contained cells that expressed more than two of the exhaustion markers. In contrast, peripheral blood T cells from matched patients rarely had up to two markers of exhaustion and T cells from healthy controls almost uniformly had no markers of exhaustion except for a small percentage with only one marker. Additionally, we examined the proportion of CD3+ cells within each combination of exhaustion markers. There was a greater percentage of CD3+ TILs expressing two or more exhaustion markers across all the different combinations of exhaustion markers than PBMCs from the patients or controls (Fig. 1E). Our results suggest that malignant glioma patients possess elevated frequencies of PD-1 and other exhaustion markers on T cells from the tumor and in the peripheral blood compared with normal healthy volunteers.
To explore further the findings from our standard fluorescent antibody-based flow cytometry, we conducted CyTOF mass cytometry (21,25,26) on an additional 28 matching high-grade glioma patient TILs and PBMCs. A panel of immune lineage, memory, trafficking, activation, and exhaustion markers were used to stain these paired immune cell populations simultaneously (Suppl Table. 1). To analyze the CD3+ PD-1+ population from this set of patients, we employed FlowSom (22), an R package that uses machine learning algorithms to analyze mass cytometry data in an unbiased manner. Using this package, we ran a principal component analysis using the median expression levels of all the T cell markers from each individual patient as variables. An unsupervised clustering algorithm was used to generate a tSNE map of phenotypically defined population clusters. The principal component analysis showed a clear separation between the CD3+ PD-1+ PBMCs and the CD3+ PD-1+ TILs (Fig 2A). The unsupervised clustering algorithm grouped a majority of the CD3+ PD-1+ TILs in distinct clusters from the CD3+ PD-1+ PBMCs (Fig. 2B, Suppl. Fig. 2a).
Figure 2: High dimensional analysis of mass cytometry data shows elevated expression of markers of activation and exhaustion on CD3+ PD-1+ TILs and markers of activation and antigen experience on CD3+ PD-1+ PBMCs from malignant glioma patients.

A) A PCA plot showing CD3+ PD-1+ TILs (green dots) and PBMCs (red dots). Each dot represents an individual patient.
B) tSNE plots showing which clusters the CD3+ PD-1+ TILs were grouped in (green box) and which clusters the CD3+ PD-1+ PBMCs were grouped into (red box) by the clustering algorithm.
C) The median expression levels of various activation markers in TILs (green dots) and PBMCs (red dots). The same tSNE plot as (B) was colored to show which cells expressed the particular marker (*P≤0.05, **P≤0.005, ***P≤0.0005).
D) Antigen Experience/Memory markers.
E) Exhaustion markers.
F) A heat map of the median expression pattern for each of the exhaustion, activation, and memory/antigen experience markers for each individual patient.
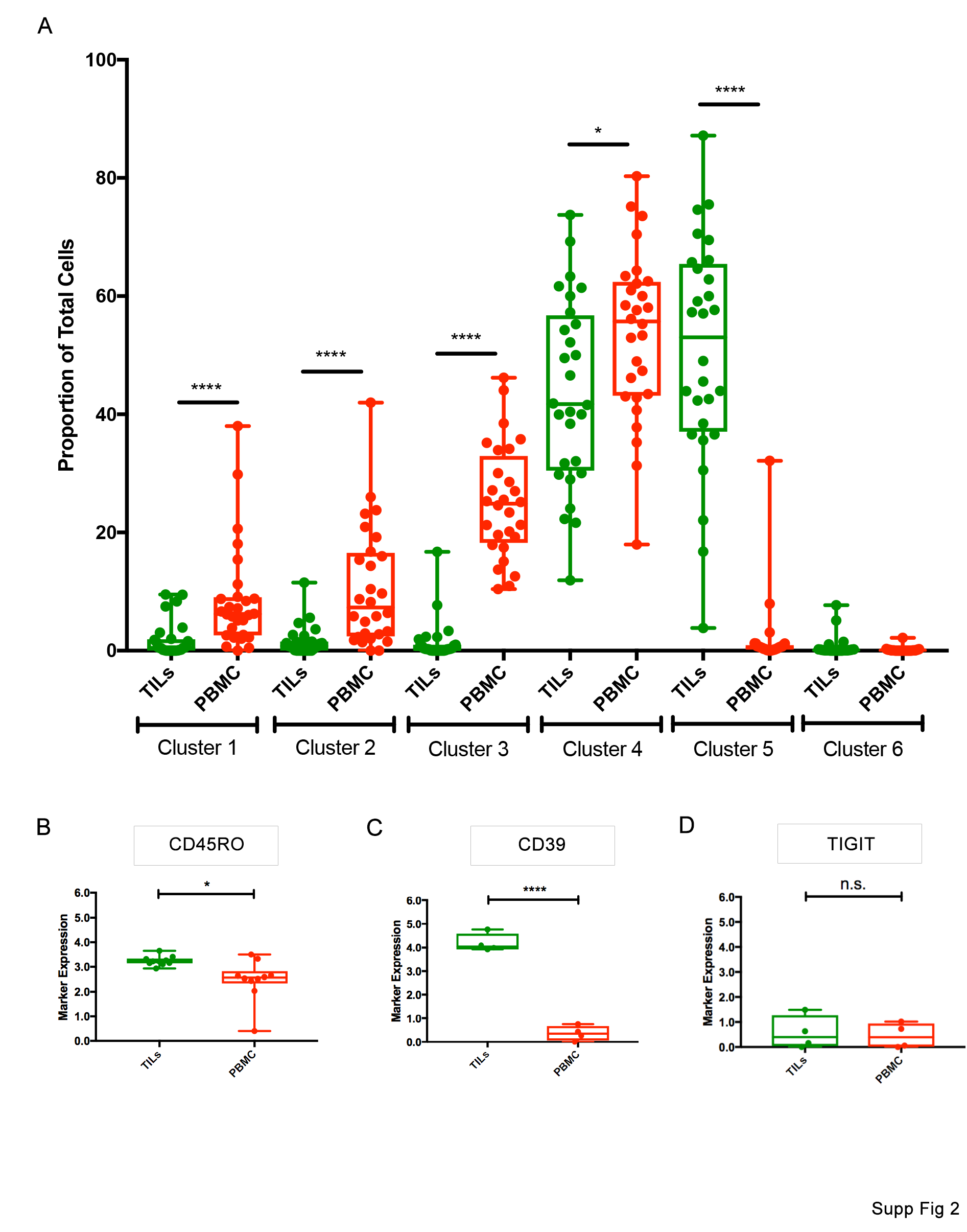
We next looked at each individual marker and its median expression levels in all the PBMCs and TILs. For each individual marker, we highlighted the regions on the generated tSNE map where the marker was most highly expressed. For the T cell exhaustion markers, there was significant increase in TIM-3, PD-L1, and CTLA-4 in the TIL population compared to the PBMC population (Fig. 2C). Among T cell activation markers, there was a significant increase in expression of CD38 and HLA-DR in the TIL population compared to the PBMC population (Fig. 2D). Meanwhile, among the antigen experience and T cell memory markers: CD45RA, CD27, and CD127, there was a significant decrease in the TIL population compared to the PBMC population (Fig. 2E). In 4 recent patients, two additional T cell exhaustion markers CD39 and TIGIT were incorporated into our panel and the expression levels were measured between the PBMC and TIL groups. We found a significant increase in CD39 in the TIL population (Suppl. Fig. 2c) but a non-significant difference in TIGIT expression between the two groups (Suppl. Fig. 2d). A heat map of each analyzed patient’s marker expression level was also generated. There was increased expression of various exhaustion and activation markers in the TIL population and increased expression of antigen experience markers in the PBMC population (Fig. 2F). The results generated from the CyTOF data strongly suggest phenotypic differences exist between CD3+ PD-1+ cells from malignant glioma patient TILs and PBMCs.
In order to further investigate the possible relationship of PD-1 expression with T cell exhaustion, activation, and memory phenotypes we sought to characterize the gene expression of the PD-1− and PD-1+ cells by performing a tailored comparison panel on the PBMCs and TILs from three randomly selected patients and PBMCs from one healthy individual using RNA hybridization probe technology (23). When the normalized RNA counts of PD-1+ T cells and PD-1− T cells were divided to create a ratio, we found that the ratio of PD-1+/PD-1− gene expression in malignant glioma TILs had an elevated fold change for markers of exhaustion (TIM-3, CTLA-4, TIGIT) when compared to the peripheral blood T cells (Suppl. Fig 3A). Gene expression analysis of the same samples uncovered an increase in several activation markers (OX-40, 4–1BB, CD25, BIM, ICOS) of TILs when compared to patient peripheral blood T cells (Suppl. Fig. 3B). We saw a reversal of this PD1+/PD1− fold change for markers of memory (Suppl. Fig. 3C). These data suggest that PD-1 expression reflects activation and exhaustion in TILs and may be a marker of antigen experience in peripheral blood T cells of malignant glioma patients, similar to our findings from our phenotypic analyses.
PD-1+ T cells demonstrate decreased T cell receptor diversity
We used high throughput DNA sequencing of the T cell receptor (TCR) V-β region to determine the TCR diversity of CD3+ PD-1+ and CD3+ PD-1− T cells FACS-sorted from one matched patient tumor and PBMC samples, two additional distinct patient PBMC samples, and normal donor PBMCs. Since our data suggested that PD-1 reflects antigen experience, activation, and exhaustion in our malignant glioma patient samples, it was unclear what the TCR repertoire would reflect. In fact, we saw an increase in the diversity of PD-1– T cells from all populations of sorted T cells (Fig. 3A). As expected, we saw a trend toward the least amount of TCR diversity in the TIL population, but our limited number of sorted TIL precludes a clear assessment. Among the predicted productive TCR rearrangements, we also saw an expansion of clonal T cell populations in the PBMC and TIL populations in malignant glioma patients, possibly signifying expansion of tumor antigen-specific T cells not seen in healthy donors (Fig. 3B). These findings suggest that PD-1+ T cells are enriched for a restricted TCR population, regardless of where the cells were obtained.
Figure 3: PD-1+ T cells have decreased TCR diversity compared to PD-1− cells.
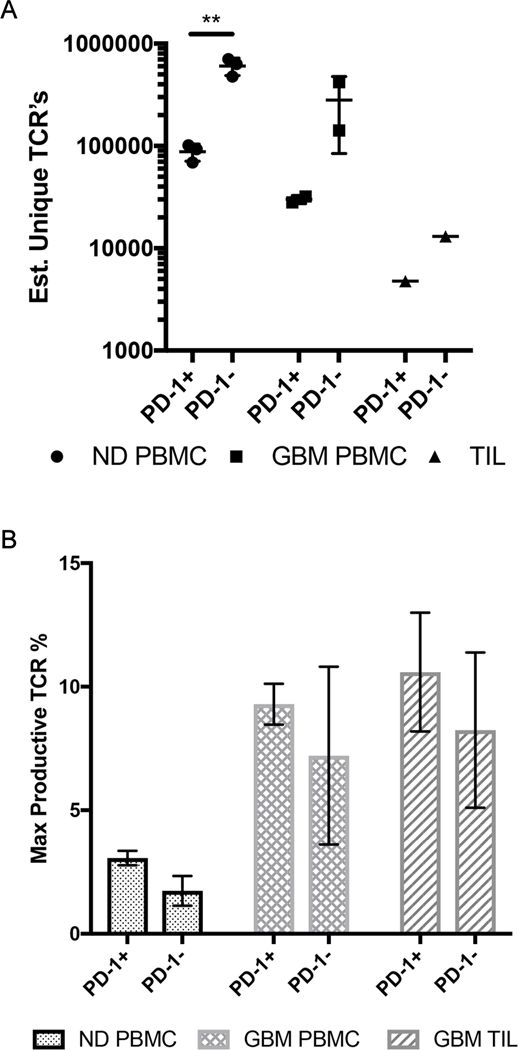
A) The number of estimated unique TCR sequences in PD-1− and PD-1+ populations in healthy donor PBMC, glioma patient PBMC, and glioma patient TILs. Each point represents a single patient. Circle points represent healthy donor PBMCs. Square points represent glioma patient PBMCs. Triangle points represent glioma patient TILs. (**P≤0.005).
B) The frequency of a specific TCR rearrangement in the populations. The populations shown are the CD3+ PD-1− or CD3+ PD-1+ populations from healthy donor PBMCs (dotted bar), glioma patient PBMCs (the checkerboard bar), and glioma patient TILs (slanted stripes bar)
PD-1+ peripheral T cells of GBM patients exhibit an increased rate of activation compared with PD-1− T cells.
To examine the functional differences between these T cell populations, we sorted PD-1+ and PD-1− CD3+ peripheral blood T cells from malignant glioma patients. Due to limited numbers of PD-1+ T cells in non-expanded PBMC, we pooled together PBMCs from multiple patients. The sorted T cells were placed in culture and activated using agonistic CD3/28 antibodies. At 24 and 48 hours post activation, supernatants were collected and the interferon gamma (IFN-γ) levels measured. At 24 hours, the amount of IFN-γ produced by the CD3+ PD-1+ cells was 2.4x higher than the IFN-γ produced by the CD3+ PD-1− cells (Fig. 4a). By 48 hours post stimulation, the amount of IFN-γ produced by the CD3+ PD-1+ cells was 8.8x higher than the IFN-γ produced by the CD3+ PD-1− cells (Fig. 4b).
Figure 4: CD3+ PD-1+ T cells from pooled glioma patient PBMCs produce higher levels of IFN-γ compared to the CD3+ PD-1− T cell population after T cell activation.
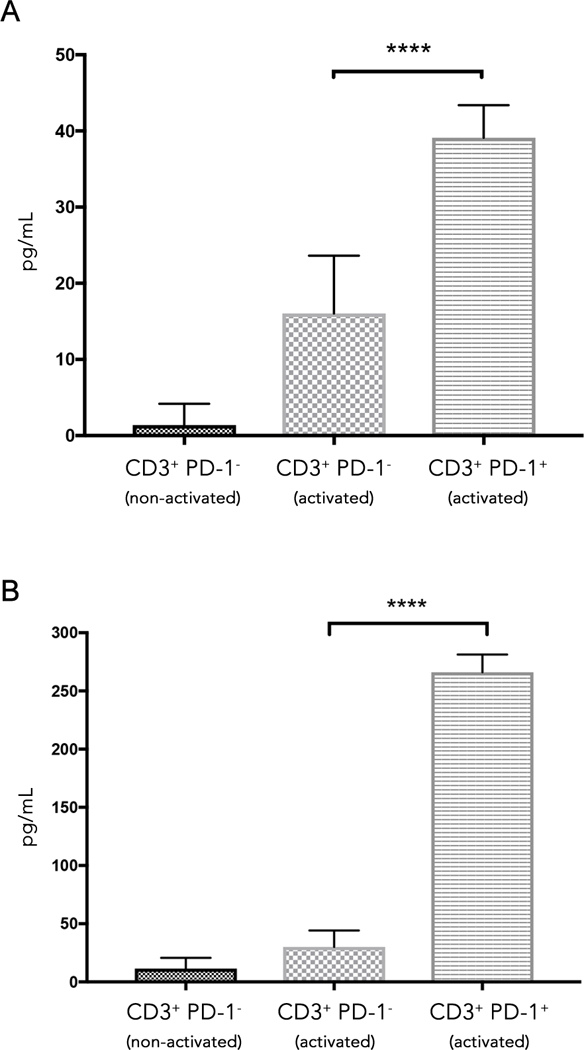
A) The amount of IFN-γ produced in CD3+ PD-1−, and CD3+ PD-1+ T cells from four glioma patient PBMCs 24 hours after CD3/CD28 activation. The dotted bar represents CD3+ PD-1− T cells that were not activated. The checkerboard bar represents CD3+ PD-1− T cells that were activated. The horizontal stripes bar represents CD3+ PD-1+ T cells that were activated. The results presented are a representative experiment that has been repeated multiple times with similar findings.
B) IFN-γ produced at 48hrs from the same set of cells as (A).
DISCUSSION
We previously demonstrated that the PD-1/PD-L1 axis plays an important immunoregulatory role in anti-tumor T cell function induced by DC vaccination (10,12). Several other studies have described T cell exhaustion and dysfunction within the tumor microenvironment and in relation to PD-1 expression. Thommen et al. showed in non-small cell lung cancer patients that PD-1 expressing T cells from the tumor had higher expression of various exhaustion markers, had higher upregulation of genes related to proliferation and activation, and were more clonal (27). Analyzing TILS from both human GBM and murine tumor models, Woroniecka et al. showed that TILs had a more prominent exhaustion signature compared to the peripheral blood from GBM patients and from healthy donor PBMCs (28). However, a comprehensive, high-dimensional analysis of PD-1 expression by human T lymphocytes in malignant glioma patients has not been described. In our study, we found several differences in both phenotypic characteristics and function between PD-1+ TILs and matched PD-1+ peripheral blood T lymphocytes extracted from malignant glioma patients. Notably, we found that PD-1 expression in the glioma TIL context may reflect T cell activation in addition to T cell exhaustion, but in the periphery, PD-1 expression on T cells may indicate activation and antigen experience (Fig. 5).
Figure 5:
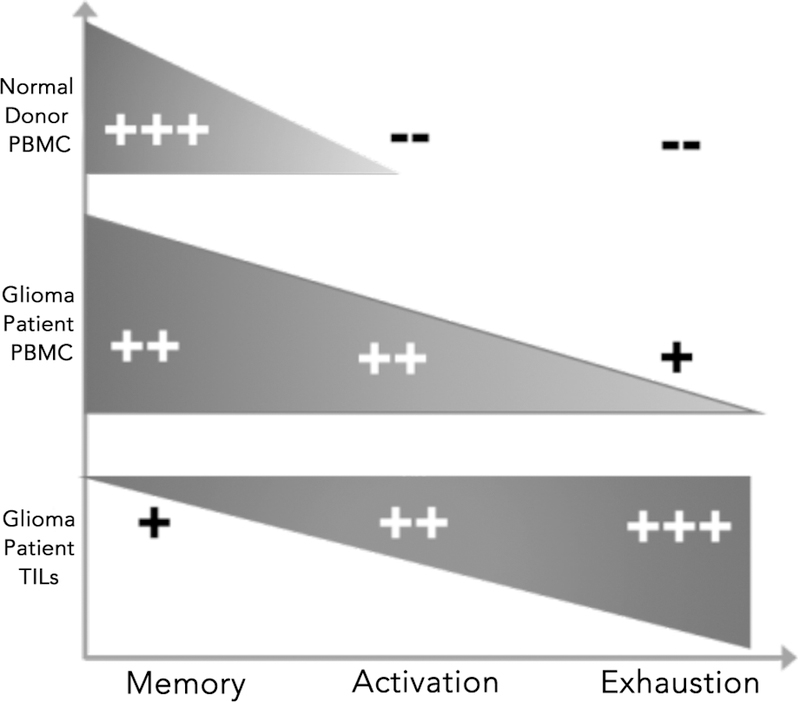
A schematic indicating what PD-1 expression means in the context of malignant glioma.
Expression of PD-1 on peripheral blood T cells of patients with malignant tumors, including malignant glioma, has previously been reported (29–34). In most of these previous studies, PD-1 was described as a marker of exhaustion by terminally differentiated effector T cells. But these reports failed to describe the phenotypic and functional differences between PD-1+ T cells in the peripheral blood and PD-1+ TILs. In our study, we performed a comprehensive analysis of PD-1 expression on T lymphocytes isolated from the tumors and peripheral blood of malignant glioma patients. We found that PD-1 was significantly elevated in glioma patient TILs when compared to T cells in glioma patient peripheral blood and T cells from healthy controls. PD-1+ TILs are enriched for markers of activation and exhaustion (CTLA-4, TIM-3, HLA-DR, and CD38) when compared to PD-1+ peripheral blood T cells. In contrast, peripheral blood PD-1+ T cells express a complete array of central memory, effector memory, and even the CD45RA+ effector memory T cell subsets (CD45RO, CD45RA, CD27, and CD127), suggesting diverging functionality between PD-1+ T cells obtained from distinct anatomical locale.
In the setting of chronic viral infections, similar trends have been observed. PD-1 expression has been associated with T cell exhaustion in both mouse LCMV models and humans infected with HIV (35,36). In studies examining the transcriptional profile of the exhausted T cell population from a chronic LCMV mouse model, the exhausted population remained distinct from the memory T cell population and had increased transcription of various exhaustion markers such as PD-1, TIM-3 and CTLA-4 and activation markers such as ICOS and OX40 (37,38). In contrast, previous reports have shown that in healthy controls, PD-1+ peripheral CD8+ T cells do not show exhausted characteristics, but instead exhibit hallmarks of effector memory T cells (39). In a subset of the patients, CD45RO was included in the CyTOF analysis, which allowed a more thorough exploration of the T cell memory subsets. We found that the expression CD45RO is uniformly expressed on all CD3+ PD1+ T cells from the tumor but variably expressed by the peripheral blood population. In the peripheral blood, there are multiple populations of PD-1+ T cells that do not express CD45RO and are likely long-lived stem memory TSCM or effector memory TEMRA cells. Thus, our data support the conclusion that the phenotype of PD-1+ T cells obtained from tumor tissue represents a chronically activated, exhausted effector T cell population (CD45RO+ CD27− IL-7R− HLA-DR+ CD38+ TIM-3+ LAG-3+ CTLA-4+), while the PD-1+ T cell population obtained from peripheral blood represents the full complement of memory T cell populations (TCM, CD45RO+ CD27+ CD45RA− IL-7R+; TEM, CD45RO+ CD45RA− CD27− IL-7R−; and TEMRA, CD45RO− CD27− CD45RA+ IL-7R−). Therefore, our data would suggest that PD-1 expression by peripheral blood T cells may not be a good surrogate or indicator for exhaustion or tumor immune evasion. Such information must be considered when applied as a biomarker in clinical immunotherapy trials.
Interestingly, we found that PD-1 expression was associated with increased T cell receptor clonality and decreased diversity in both the TIL population and peripheral blood T cells of malignant glioma patients. As such, increased clonal expansion of the PD-1+ T cell population may reflect the activation and expansion of specific anti-tumor antigen T cells when compared to PD-1- T cells, though future studies are needed to confirm this. Additionally, our CyTOF data revealed a significant increase in expression of CD39 in the CD3+ PD-1+ TIL population compared to the CD3+ PD-1+ PBMC population. CD39, an extracellular ATPase commonly associated with regulatory T cells, has been shown to be co-expressed with PD-1 on antigen-specific but exhausted CD8+ T cells in a chronic viral setting (40). More recently, CD39 has been proposed to be expressed by tumor specific T cells (41,42). The increase of clonality and CD39 expression in the PD-1+ TILs further supports their dual function phenotype of anti-tumor specific activation and exhaustion. Increased TCR clonality has been correlated with better outcomes after anti-PD-1 therapy in melanoma and has been associated with higher PD-1 expression in soft tissue sarcomas (11,43). Further investigation is needed to study the association between TCR clonality and PD-1 expression in the context of tumor antigen-specific T lymphocytes.
Finally, to characterize PD-1+ peripheral blood T cells in malignant glioma patients, we investigated the functional activation differences between PD-1+ and PD-1− peripheral blood T cells from GBM patients. Traditionally, it has been held that effector, memory, and naïve T cells have different response rates, as measured by amount of IFN-γ produced, after activation. We found that PD-1+ GBM patient PBMC T cells exhibited an increased rate of IFN-γ production when compared to PD-1− PBMC T cells from the same patients. Increased IFN-γ is consistent with an activated memory T cell phenotype further highlighting that PD-1 expression in peripheral T cells is not a marker of exhaustion as we would expect in the PD-1+ population in the tumor microenvironment.
In conclusion, we demonstrate that PD-1 expression in malignant glioma patients reflects chronically-activated effector T cells that display both hallmarks of memory and exhaustion depending on its location. The use of anti-PD-1/PD-L1 antibody blockade is currently being studied in malignant glioma clinical trials. Further exploration of the effect of these check point inhibitors on PD-1 expressing T cells in different anatomical environments is warranted both to understand the use of PD-1 expression as a therapeutic biomarker as well as its importance for clinical efficacy.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Statement of Significance:
PD-1 expression on T cells from malignant glioma patients reflects chronic activation and exhaustion in the tumor but exhibits the hallmarks of memory and activation in the peripheral blood.
Translational Relevance:
Inhibition of the PD-1/PD-L1 axis is currently being explored as one potential immunotherapy for patients with malignant gliomas. Typically, PD-1 expression on T cells in the context of cancer has been exclusively thought of as a marker of exhaustion and current PD-1 checkpoint therapies are thought to be effective due to their ability to reactivate exhausted T cell populations. In our study, we sought out to define and characterize the PD-1 expression on T cells in the context of malignant gliomas. We found that PD-1 expression on T cells in the tumor reflects both exhaustion and chronic activation. However, in peripheral blood T cells, PD-1 expression reflects antigen experience and chronic activation more than exhaustion. The results from this study may help inform our use of PD-1 as a biomarker and also be used to assess the clinical efficacy of using PD-1 checkpoint blockade in this malignancy.
ACKNOWLEDGEMENTS
This work was supported in part by NIH/NCI grants R21-CA186004 and R01CA154256 (RMP), RO1 CA125244 (LML), KL2 ULITR001881 (TBD), R25 NS079198 and the UCLA SPORE in Brain Cancer P50 CA211015 (RMP and LML). We also acknowledge support from the Parker Institute for Cancer Immunotherapy (PICI) (grant number 20163828), the Musella Foundation for Brain Tumor Research (RMP), the Joseph Drown Foundation (MH and TBD), and the UCLA Medical Scientist Training Program (MSTP) (MG). Alexander Hao Lee is a pre-doctoral fellow supported by the UCLA Tumor Immunology Training Grant (USHHS Ruth L. Kirschstein Institutional National Research Service Award # T32 CA009056). We would like to thank the Janis V. Giorgi Flow Cytometry Core Laboratory for their assistance in constructing our CyTOF panel and running the Helios CyTOF system. We would like to thank the Center for Systems Biomedicine (Integrated Molecular Technologies Core) for their technical assistance with the Nanostring studies, which is supported by CURE/P30DK41301. We also thank the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility that is supported by National Institutes of Health awards P30 CA016042 and 5P30 AI028697. We would also like to thank the UCLA Brain Tumor Translational Resource (BTTR) and the Translational Pathology Core Laboratory (TPCL) for their assistance with paraffin-embedding and histology. Finally, we would like to express our deepest gratitude to the patients and their families for donating the samples that allowed this project to be successful.
Abbreviations:
- TIL
tumor-infiltrating lymphocyte
- PD-1
programmed death-1
- PD-L1
programmed death ligand-1
- TCR T
cell receptor
- GBM
Glioblastoma
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest
References
- 1.Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro Oncol 2016;18(suppl_5):v1–v75 doi 10.1093/neuonc/now207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Preusser M, de Ribaupierre S, Wohrer A, Erridge SC, Hegi M, Weller M, et al. Current concepts and management of glioblastoma. Ann Neurol 2011;70(1):9–21 doi 10.1002/ana.22425. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352(10):987–96 doi 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Miranda A, Blanco-Prieto M, Sousa J, Pais A, Vitorino C. Breaching barriers in glioblastoma. Part II: Targeted drug delivery and lipid nanoparticles. Int J Pharm 2017. doi 10.1016/j.ijpharm.2017.07.049. [DOI] [PubMed] [Google Scholar]
- 5.Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, Giovannone AJ, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res 2005;11(15):5515–25 doi 10.1158/1078-0432.CCR-05-0464. [DOI] [PubMed] [Google Scholar]
- 6.Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res 2011;17(6):1603–15 doi 10.1158/1078-0432.CCR-10-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prins RM, Wang X, Soto H, Young E, Lisiero DN, Fong B, et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J Immunother 2013;36(2):152–7 doi 10.1097/CJI.0b013e3182811ae4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heimberger AB, Crotty LE, Archer GE, Hess KR, Wikstrand CJ, Friedman AH, et al. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin Cancer Res 2003;9(11):4247–54. [PubMed] [Google Scholar]
- 9.Kanaly CW, Ding D, Heimberger AB, Sampson JH. Clinical applications of a peptide-based vaccine for glioblastoma. Neurosurg Clin N Am 2010;21(1):95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antonios JP, Soto H, Everson RG, Orpilla J, Moughon D, Shin N, et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight 2016;1(10) doi 10.1172/jci.insight.87059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515(7528):568–71 doi 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antonios JP, Soto H, Everson RG, Moughon D, Orpilla JR, Shin NP, et al. Immunosuppressive tumor-infiltrating myeloid cells mediate adaptive immune resistance via a PD-1/PD-L1 mechanism in glioblastoma. Neuro-oncology 2017;19(6):796–807 doi 10.1093/neuonc/now287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-oncology 2015;17(8):1064–75 doi 10.1093/neuonc/nou307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDermott DF, Atkins MB. PD-1 as a potential target in cancer therapy. Cancer Med 2013;2(5):662–73 doi 10.1002/cam4.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maxwell R, Jackson CM, Lim M. Clinical Trials Investigating Immune Checkpoint Blockade in Glioblastoma. Curr Treat Options Oncol 2017;18(8):51 doi 10.1007/s11864-017-0492-y. [DOI] [PubMed] [Google Scholar]
- 16.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nature reviews Cancer 2016;16(5):275–87 doi 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Speiser DE, Utzschneider DT, Oberle SG, Munz C, Romero P, Zehn D. T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nature reviews Immunology 2014;14(11):768–74 doi 10.1038/nri3740. [DOI] [PubMed] [Google Scholar]
- 18.Yuzefpolskiy YBF, Penny L, Kalia V, Sarkar S. PD-1 signals instruct a cfritical metabolic switch for maintenance of T cell memory. the Journal of Immunology 2017;198(1 supplement). [Google Scholar]
- 19.Charlton JJ, Chatzidakis I, Tsoukatou D, Boumpas DT, Garinis GA, Mamalaki C. Programmed death-1 shapes memory phenotype CD8 T cell subsets in a cell-intrinsic manner. J Immunol 2013;190(12):6104–14 doi 10.4049/jimmunol.1201617. [DOI] [PubMed] [Google Scholar]
- 20.Hong JJ, Amancha PK, Rogers K, Ansari AA, Villinger F. Re-evaluation of PD-1 expression by T cells as a marker for immune exhaustion during SIV infection. PLoS One 2013;8(3):e60186 doi 10.1371/journal.pone.0060186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nowicka M, Krieg C, Weber LM, Hartmann FJ, Guglietta S, Becher B, et al. CyTOF workflow: differential discovery in high-throughput high-dimensional cytometry datasets. F1000Res 2017;6:748 doi 10.12688/f1000research.11622.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, et al. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 2015;87(7):636–45 doi 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- 23.Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nature biotechnology 2008;26(3):317–25 doi 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 24.Hsu MS, Sedighim S, Wang T, Antonios JP, Everson RG, Tucker AM, et al. TCR Sequencing Can Identify and Track Glioma-Infiltrating T Cells after DC Vaccination. Cancer Immunol Res 2016;4(5):412–8 doi 10.1158/2326-6066.CIR-15-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011;332(6030):687–96 doi 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leipold MD, Maecker HT. Mass cytometry: protocol for daily tuning and running cell samples on a CyTOF mass cytometer. J Vis Exp 2012(69):e4398 doi 10.3791/4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med 2018;24(7):994–1004 doi 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin Cancer Res 2018. doi 10.1158/1078-0432.CCR-17-1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waki K, Yamada T, Yoshiyama K, Terazaki Y, Sakamoto S, Matsueda S, et al. PD-1 expression on peripheral blood T-cell subsets correlates with prognosis in non-small cell lung cancer. Cancer Sci 2014;105(10):1229–35 doi 10.1111/cas.12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng H, Liu X, Zhang J, Rice SJ, Wagman M, Kong Y, et al. Expression of PD-1 on CD4+ T cells in peripheral blood associates with poor clinical outcome in non-small cell lung cancer. Oncotarget 2016;7(35):56233–40 doi 10.18632/oncotarget.9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rutkowski J, Cyman M, Slebioda T, Bemben K, Rutkowska A, Gruchala M, et al. Evaluation of peripheral blood T lymphocyte surface activation markers and transcription factors in patients with early stage non-small cell lung cancer. Cell Immunol 2017. doi 10.1016/j.cellimm.2017.09.007. [DOI] [PubMed] [Google Scholar]
- 32.Wang W, Shen G, Wu S, Song S, Ni Y, Suo Z, et al. PD-1 mRNA expression in peripheral blood cells and its modulation characteristics in cancer patients. Oncotarget 2017;8(31):50782–91 doi 10.18632/oncotarget.15006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacFarlane AWt, Jillab M, Plimack ER, Hudes GR, Uzzo RG, Litwin S, et al. PD-1 expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor resection. Cancer Immunol Res 2014;2(4):320–31 doi 10.1158/2326-6066.CIR-13-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei B, Wang L, Zhao X, Du C, Guo Y, Sun Z. The upregulation of programmed death 1 on peripheral blood T cells of glioma is correlated with disease progression. Tumour Biol 2014;35(4):2923–9 doi 10.1007/s13277-013-1376-9. [DOI] [PubMed] [Google Scholar]
- 35.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006;439(7077):682–7 doi 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 36.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006;443(7109):350–4 doi 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 37.Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, et al. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity 2014;40(2):289–302 doi 10.1016/j.immuni.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007;27(4):670–84 doi 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 39.Duraiswamy J, Ibegbu CC, Masopust D, Miller JD, Araki K, Doho GH, et al. Phenotype, function, and gene expression profiles of programmed death-1(hi) CD8 T cells in healthy human adults. J Immunol 2011;186(7):4200–12 doi 10.4049/jimmunol.1001783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gupta PK, Godec J, Wolski D, Adland E, Yates K, Pauken KE, et al. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PLoS Pathog 2015;11(10):e1005177 doi 10.1371/journal.ppat.1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun 2018;9(1):2724 doi 10.1038/s41467-018-05072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018;557(7706):575–9 doi 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 43.Pollack SM, He Q, Yearley JH, Emerson R, Vignali M, Zhang Y, et al. T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer 2017;123(17):3291–304 doi 10.1002/cncr.30726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.