

Abstract
Ketamine is a fast acting experimental antidepressant with significant therapeutic potential for emotional disorders such as major depressive disorder and alcohol use disorders. Of particular interest is binge alcohol use, which during intermittent withdrawal from drinking involves depressive-like symptoms reminiscent of major depressive disorder. Binge drinking has been successfully modeled in mice with the Drinking in the Dark (DID) paradigm, which involves daily access to 20% ethanol, for a limited duration and selectively during the dark phase of the circadian light cycle. Here we demonstrate that DID exposure reduces the cell surface expression of NMDA- and AMPA-type glutamate receptors in the prelimbic cortex (PLC) of female but not male mice, along with reduced activity of the mammalian target of rapamycin (mTOR) signaling pathway. Pretreatment with an acute subanesthetic dose of ketamine suppresses binge-like ethanol consumption in female but not male mice. Lastly, DID-exposure reduces spontaneous glutamatergic synaptic transmission in the PLC of both sexes, but synaptic transmission is rescued by ketamine selectively in female mice. Thus, ketamine may have therapeutic potential as an ethanol binge suppressing agent selectively in female subjects.
Keywords: alcohol, prelimbic cortex, drinking in the dark, ketamine
1. INTRODUCTION
Alcohol use disorder (AUD) is a chronic relapsing disorder, in which social alcohol consumption escalates to compulsive intake and physiological dependence, and is one of the costliest public health disorders in the US (Koob and Le Moal, 2008; Prevention, 2006; Rehm et al., 2014). Binge drinking is defined as consuming a large amount of alcohol such as a minimum of four drinks for women and five drinks for men in a period of 2 h or less, resulting in a blood alcohol concentration of at least 0.08 mg/dl (NIAAA, 2017). Finding treatments for AUDs has been hampered by the complex mechanism of action of ethanol and the large and diverse number of receptors, ion channels, neurotransmitters, and neuromodulators implicated in its actions (Abrahao et al., 2017). In particular, acute ethanol has been shown to inhibit the function of N-methyl-D-aspartate (NMDA)- (Lovinger et al., 1989; Ren et al., 2013; Woodward and Pava, 2009) and a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors (NMDARs and AMPARs) (Frye and Fincher, 2000) and to reduce corresponding synaptic currents (Roberto et al., 2006), suggesting that these receptors might serve as targets for treatment of AUDs (Lovinger, 1996).
AUDs are highly comorbid with major depressive disorder (MDD) (Brière et al., 2014), and both these disorders exhibit marked sex differences (Erol and Karpyak, 2015; Kessler et al., 2005; Kornstein et al., 2000). Males are more likely to self-medicate for mood disorders with alcohol (Turner et al., 2018). However, early life stress, which is an established vulnerability factor for both AUDs and MDD, results in more frequent diagnosis of AUDs in women than men (Enoch, 2011). Moreover, the “gender gap” in AUDs is rapidly closing, highlighting the need for novel therapeutic interventions especially for women (reviewed by Keyes et al., 2008).
The prefrontal cortex has emerged as a primary substrate of pathology for both AUDs and MDD. Chronic intermittent ethanol (CIE) leads to increased neural excitability, alterations in apical dendritic morphology, and decreased NMDA receptor currents in layer 2/3 pyramidal cells of the prelimbic cortex (PLC) (Holmes et al., 2012; Pleil et al., 2015b). These phenotypes together are reminiscent of corresponding changes in human and rodent studies of MDD and chronic stress models of depressive disorder (Banasr et al., 2007; Drevets et al., 2008; McEwen and Morrison, 2013; Rajkowska et al., 1999). By contrast, layer 5 neurons in the medial prefrontal cortex (mPFC) of a similar CIE model of mice were shown to exhibit increased NMDA receptor currents (Kroener et al., 2012), suggesting that exposure to alcohol induces distinct layer specific forms of neural plasticity.
Ketamine is a rapid acting antidepressant with efficacy even in otherwise treatment resistant MDD (Lener et al., 2017; Wilkinson et al., 2017). In preclinical models, ketamine has been shown to be effective also at attenuating fear-related behavior (McGowan et al., 2017), stress-induced depression-related behavior (Brachman et al., 2016), alcohol withdrawal-induced emotional behavior (Holleran and Winder, 2017; Vranjkovic et al., 2018) and in reducing alcohol preference in a strain of alcohol-preferring rats (Rezvani et al., 2017). Thus, ketamine may be a promising therapeutic for AUDs. Evidence from MDD patients (Abdallah et al., 2017; Chowdhury et al., 2017) and stress-induced rodent models of MDD indicate that structural alterations of neurons and functional defects in glutamatergic transmission in the prefrontal cortex can be reversed for a prolonged period by a single dose of ketamine, along with antidepressant-like behavioral effects (Li et al., 2011; Ren et al., 2016). The above mentioned alterations in glutamatergic transmission in the mPFC of models of CIE exposure suggest that ketamine may act on the same substrate also for AUDs.
The mechanism of acute ketamine-induced neuroplasticity that is associated with it’s therapeutic effects is currently of intense interest. Consistent with ketamine’s established function as a non-competitive NMDAR antagonist, its mechanism of action upon administration at subanesthetic doses is thought to involve selective and transient antagonism of NMDARs on tonically active interneurons that leads to transient disinhibition of neural networks, followed by a glutamate surge and activation of the mammalian target of rapamycin (mTOR) signaling pathway and changes in dendritic mRNA translation (reviewed in Monteggia et al., 2013; Wohleb et al., 2017). However, some of ketamine’s active metabolites are known to act as agonists of AMPARs rather than antagonists of NMDARs, and they appear to more directly lead to activation of glutamatergic neurons by a mechanism independent of mTOR (Zanos et al., 2018). Nevertheless, withdrawal from chronic ethanol consumption and chronic stress-induced depressive-like behavior seem to converge on similarly impaired prefrontal glutamatergic transmission (Holmes et al., 2012; Kristen E. Pleil et al., 2015b; Yuen et al., 2012) and altered mTOR signaling (Chandran et al., 2013; Neasta et al., 2014). Moreover, ketamine- induced activation of the mTOR pathway contributes to normalization of both alcohol- and chronic stress-induced impairments of NMDA-receptor function, as well as to normalization of ethanol drinking behavior and emotional reactivity (Sabino et al., 2013; Tang et al., 2015; Zhou et al., 2014).
Similar to the CIE exposure protocol mentioned above, the Drinking in the Dark (DID) model of binge-like ethanol consumption of rodents is designed to mimic repeated cycles of intoxication and withdrawal observed among human social drinkers with AUD, with the important distinction that the animals self-administer ethanol (Lowery-Gionta et al., 2012; Pleil et al., 2015b; Rhodes et al., 2007, 2005; Sparta et al., 2008). Using this protocol, we here found that female but not male mice subjected to DID display reductions in the cell surface expression of AMPARs and NMDARs, along with reduced activity of the mTOR pathway. Accordingly, we hypothesized that ketamine might suppress binge-like ethanol consumption, perhaps in a sex-specific manner. Indeed, pretreatment with a single subanesthetic dose of ketamine effectively suppressed binge drinking in female but not male mice. Functionally, the DID protocol resulted in a selective reduction of the frequency but not amplitude of spontaneous glutamatergic currents recorded from pyramidal cells, in a sex-specific manner. Moreover, ketamine restored glutamatergic synapse function selectively in female mice. These data suggest that ketamine and similarly acting rapid experimental antidepressants hold promise for the treatment of binge drinking behavior.
2. MATERIALS AND METHODS
2.1. Subjects
All animal experiments were approved by the Pennsylvania State University Institutional Animal Care and Use Committee. C57BL/6J mice (males and females, strain 000664, The Jackson Laboratory) were bred in house and maintained on a 12:12 h light cycle in a temperature-controlled and humidity-controlled vivarium. All mice were 6 weeks of age when transferred to a reverse light cycle room (lights off at 7 a.m.) and single housed, and they were allowed to acclimate for one week prior to the start of the DID protocol. They were maintained on a reversed 12:12 h light cycle throughout the experiment. Mice had ad libitum access to food and water, except as described during the DID protocol. The behavioral assessments were all conducted during the dark phase of the light cycle, starting at approximately 10 a.m. and 3 h after the lights went off. Mice used for cell surface biotinylation, western blot, and electrophysiology were euthanized at 10 a.m. on the binge day, a time that coincided with behavioral assessment.
2.2. Drugs
Ketamine (Ketaset HCI, Zoetis, Parsippanv-Trov Hills, NJ) was diluted freshly with sterile 0.9% saline immediately prior to injection.
2.3. Cell Surface Biotinylation
Cell surface biotinylation of coronal mouse brain slices was performed as described (Kilpatrick et al, 2016), Briefly, the mice were euthanized by cervical dislocation and the freshly isolated brains sliced into 1 mm coronal sections using a brain matrix, incubated in 1mg/ml sulfo-NHS-SS-biotin (ThermoFisher, Waltham, MA, USA) in pre-chilled oxygenated aCSF on ice for 30 min and then washed twice with 50 mM glycine and three times with 1mg/ml BSA in pre-chilled oxygenated aCSF. The mPFC was dissected from slices, extracted, and split into aliquots used as total extracts or purification of biotinylated cell surface fractions using NeutrAvidin beads (ThermoFisher, Waltham, MA, USA) followed by analyses of protein concentrations and then analyzed by western blot. Based on routine analyses of β-actin as a cytosolic protein reference, the affinity purified cell surface fractions were enriched for cell surface proteins at least 120fold (Kilpatrick et al, 2016).
2.4. Western Blotting
Western blotting was completed as described (Fuchs et al 2017, Kilpatrick et al 2016). For analyses of mTor and eukaryotic elongation factor 2 (eEF2) phosphorylation, whole tissue lysates from mPFC were prepared in a solution containing 50 mM Tris-HCI (pH 8.0), 150mMNaCI, 2 mM EDTA, 0.1% sodium dodecyl sulfate, 1% Triton X-100, 1 mM NaV03, 5 mM NaF and a protease inhibitor cocktail (Roche, Basel, Switzerland) and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins separated on gels were transferred to polyvinylidene difluoride membranes and probed with rabbit anti-phosphor-mTOR (Ser2448) (1:500, no. 5536), mouse anti-mTOR (1:500, no. 4517), rabbit anti-phosphor-eEF2 (T56) (no. 2331), rabbit anti-eEF2 (no. 2332) (all from Cell Signaling Technology, Danvers, MA, USA) and mouse anti^-tubulin antibodies (no. T8328, Sigma Aldrich, St. Louis, MO, USA). For quantitation, the mTOR and eEF2 protein bands were normalized to β-tubulin analyzed on the same or parallel blots, as described (Fuchs et al., 2017). Ionotropic glutamate receptors of total extracts and affinity-purified cell surface biotinylated protein fractions from mPFC brain slices were probed with mouse anti-GluN1 (1:500,556308, BD Biosciences, Franklin Lakes, NJ, USA), and rabbit anti-GluA2/3 (1:500, 07–598, Millipore, Burlington, MA, USA). For quantitation, the corresponding protein bands were normalized to β-actin quantitated in blots of total mPFC lysates of these same samples, using mouse anti-β- actin (1:5000, A00702, Genscript. Piscataway, NE, USA)(Kilpatrick et al 2016). Blots were developed with goat anti-mouse IRDye 680RD and goat anti-rabbit IRDye 800CW (1:5000, LI-COR, Lincoln, NE, USA) and quantitated using an Odyssey CLx imager (LI-COR, Lincoln, NE, USA) and Image Studio software (LI-COR).
2.5. Behavioral analyses
Drinking in the Dark.
DID experiments were conducted as described previously (Pleil et al, 2015b) with male and female mice subjected to DID in sequential, separate experiments. The mice received 20% ethanol (Koptec, Decon Labs, King of Prussia, PA) w/v for 2 h in drinking water, 3 h into the dark cycle (i.e. 10 a.m.) on three sequential days. On the fourth day, they received 20% ethanol for 4 h. Following the binge day, mice had three days of abstinence before repeating the one-week cycle in the subsequent week. The mice underwent four cycles of DID in total. The mice that were subjected to behavioral and electrophysiological analyses (Figures 2 and 3) received saline habituation injections (i.p.) 12 h before the binge of DID cycle 2 and 3; during cycle 4 the mice received saline or various doses of ketamine, i.p., (Ketaset HCI, Zoetis, diluted in 0.9% saline, i.p.), either three days or 12 h before the binge, as indicated in Figure 1. As a behavioral measure, the amount of ethanol consumed during the binge cycle 4 (see Figure 2A) was compared to that consumed during cycle 3. Control mice were housed under the same condition (singly housed) in the same room. All mice were weighed and cages changed weekly. The mice underwent no additional handling.
Figure 2. Ketamine prophylactically reduces binge like drinking in female, but not male mice.

(A) Saline treated male and female mice consumed similar amounts of ethanol during the week 4/day 5 binge. (B) Female mice treated with ketamine (3 mg/kg) three days before the binge drank less ethanol compared to mice administered saline. (C) Female mice administered ketamine (3 mg/kg) 12 h before the binge reduced their drinking compared to saline-injected controls. (D) Female mice were assessed for differences in ethanol-induced loss of righting reflex (LORR) following ketamine or saline. 3 mg/kg ketamine or saline was injected i.p. 12 h prior to 3.2 g/kg ethanol, i.p. No significant differences in recovery from LORR were seen. (E) Female mice were subjected to two weeks of 10% sucrose drinking-in-the-dark. Ketamine (3 mg/kg, 12 h before sucrose DID) did not alter sucrose consumption. (F) Male mice did not show ketamine-induced changes in DID, across a variety of timepoints and doses.
Figure 3. Ketamine sex-specifically normalizes defects in glutamatergic synaptic transmission of DID exposed mice.
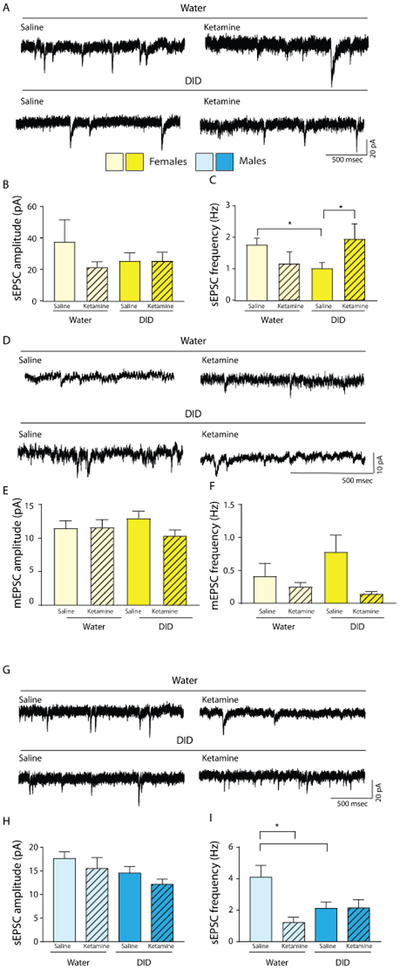
(A-C). Ketamine rescues DID- induced deficits in glutamatergic transmission in female mice. (A) Representative traces of sEPSCs in the PLC of water or DID exposed female mice, 12 h after either saline or 3 mg/kg ketamine treatment. (B) There were no significant changes in the sEPSC amplitude across groups. (C) There was an interaction between ethanol history and ketamine treatment on sEPSC frequency. DID mice showed a reduced sEPSC frequency vs. water controls. When DID mice treated with ketamine were compared to DID mice treated with water, there was a significant increase in the sEPSC frequency back to levels of water controls. (D-F) Ketamine fails to rescue DID induced deficits in glutamatergic transmission in male mice. (D) Representative traces of mEPSCs in the PLC of water or DID exposed female mice, 12 h after either saline or 3mg/kg ketamine. (E) The mEPSC amplitude was unaltered following DID. (F) mEPSC frequency was altered following DID and ketamine treatment. (F) Ketamine reduced mEPSC frequency in DID- exposed mice only. (G) Representative traces of sEPSCs in the PLC of water or DID exposed male mice, 12 h after either saline or 6 mg/kg ketamine treatment. (H) There were no significant changes in sEPSC amplitude between groups. (I) While DID-exposure reduced the sEPSC frequency comparably to that in female mice, and ketamine reduced sEPSC frequency in water exposed mice, ketamine did not rescue the DID induced sEPSC frequency deficit.
Figure 1: Binge drinking reduces expression of glutamate receptors in the PLC of female, but not male, mice.

(A) Mice were subjected to four cycles of DID or water control conditions followed by analyses of glutamate receptor and mTOR pathway proteins in cell surface or total extracts of the PLC (glutamate receptors) or total extracts of the mPFC (mTOR, eEF2)(Figure 1B-O), behavior (Figure 2), or electrophysiology (Figure 3). In addition to DID exposure, this schematic indicates that the animals analyzed in Figures 2 and 3 (but not the animals used to collect the data in Figure 1B-O) received ketamine or saline either 3 days or 12 h before the ethanol binge. (B-F) DID induced downregulation of cell surface NMDA and AMPARs in female mice. Representative western blots for GluN1, GluA2/3 (surface and total), and actin (total) for water exposed and DID exposed female mice (B). Female mice subjected to four cycles of DID showed significant reductions in cell surface GluN1-containing NMDA receptors (C), with no change in total receptor expression (D). Female mice subjected to four cycles of DID showed significant reductions in cell surface GluA2/3-containing AMPA receptors (E), with no change in total receptor (F). (G-K) Unaltered expression of NMDA and AMPARs of DID exposed male mice. Representative western blots for GluN1, GluA2/3 (surface and total), and actin (total) for water and DID exposed male mice (G). Male mice showed unaltered cell surface (H) and total (I) GluN1-containing NMDARs, or surface (J) and total (K) GluA2/3-containing AMPARs. (L-M) Downregulation of mTOR signaling in mPFC of DID exposed female mice. Representative western blots for p-eEF2, total eEF2, p-mTOR, total mTOR, and β-tubulin (L). Female DID exposed mice showed decreased overall mTOR pathway activity in the mPFC as compared to water controls (M) with near significant reductions in total eEF2 /p-eEF2. (N-O) DID resulted in a similar but insignificant trend in mTOR signaling in the mPFC of male mice. Representative western blots for p-eEF2, total eEF2, p-mTOR, total mTOR, and β-tubulin, male mice (N). Male DID exposed mice showed no significant reductions in mTOR pathway activity in the mPFC as compared to water controls. Data represent means ± SE. *p < 0.05, ** p < 0.01, t-tests.
Sucrose DID.
Sucrose DID, a measurement of alcohol-independent reward, was conducted as follows. The mice received 10% sucrose in water for two weeks (Rinker et al., 2017). The mice received 10% sucrose in water three h into the dark cycle on three sequential days. On the fourth day, they received 10% sucrose in water for 4 h. The mice underwent two cycles total. During cycle 2, the mice received ketamine (3 mg/kg, i.p.) 12 h before the sucrose binge. The amount of sucrose consumed during week 2 was compared to that during week 1.
Loss of Righting Reflex (LORR).
The latency to recover from LORR after alcohol administration was assessed as described (Lee et al., 2011). Briefly, 12 h following 3.0 mg/kg ketamine or vehicle injection (i.p.), the mice were injected with 3.2 g/kg ethanol (i.p.) and immediately placed on their backs. The animals that righted themselves were immediately returned to their backs. The latency to recover the righting reflex was defined as the total amount of time post ethanol it took the animals to right themselves to all four paws, three consecutive times.
2.6. Slice Electrophysiology
Whole-cell patch clamp electrophysiology recordings were done as previously described (Crowley et al., 2016; Pleil et al., 2015a). The mice were deeply anesthetized by exposure to 5% isoflurane and rapidly decapitated. The brains were removed and immediately placed in ice cold high-sucrose artificial cerebrospinal fluid (high sucrose aCSF, in mM: 194 sucrose, 20 NaCI, 4.4 KCI, 2 CaCI2, 1 MgCI2, 1.2 NaH2P04, 10.0 glucose, and 26.0 NaHC03 (pH 7.4), and saturated with 95% 02/5% C02) and 300 pm coronal sections containing the PFC were prepared on a vibratome (Leica VT 1200s). Slices were transferred to oxygenated and heated aCSF (31°C, in mM: 124 NaCI, 4.4 KCI, 2 CaCI2, 1.2 MgS04, 1 NaH2P04, 10.0 glucose, and 26.0 NaHC03), where they remained until use. Recording electrodes (3–7 mO) were pulled on a Flaming- Brown MicroPipette Puller and filled with a cesium-methanesulfonate based intracellular solution (in mM: 135 Cs- methanesulfonate, 10 KCI, 10 HEPES, 1 MgCI2, 0.2 EGTA, 4 Mg-ATP, 0.3 GTP, 20 phosphocreatine, pH 7.4) allowing for the recording of both IPSCs (spontaneous inhibitory post-synaptic currents, not shown) and EPSCs (spontaneous excitatory post-synaptic currents) (Crowley et al., 2016). Recordings were performed in PLC layer 2/3 pyramidal neurons, identified in coronal sections by anatomical landmarks such as the emergence of the corpus callosum and anterior commissure and corresponding to sections between +2.19 mm and −1.50 mm relative to bregma and dorso-ventrally as the region proximal to the midline between 0.3 and 1.3 mm from the pia (Crowley et al., 2016; Pleil et al., 2015a).
Pyramidal cells were identified by location from the midline and morphology, consistent with previously published electrophysiology in PLC layer 2/3 pyramidal neurons (Pleil et al., 2015b; Lowery-Gionta et al., 2018). Pyramidal cells were held at-55 mV to isolate sEPSCs and 10 mV to isolate sIPSCs. mEPSCs and mIPSCs were recorded under identical conditions, but in the presence of 500 nM tetrodotoxin. Input resistance and access resistance were continuously monitored throughout all experiments, and cells in which changes in access resistance exceeded 20% were excluded from data analyses. Signals were digitized at 10 kHz and filtered at 3 kHz using a Multiclamp 700B amplifier and analyzed using Clampfit 10.3 software (Molecular Devices, Sunnyvale, CA). Recordings were performed in a maximum of two neurons per mouse, with a minimum of four mice per condition, and n’s reported reflect the number of neurons for each measure.
2.7. Statistics
All datasets were confirmed to be normally distributed, and no statistical outliers were identified (Grubb’s test). All groups were compared by unpaired two sided f-tests or two-way ANOVA (alcohol history x drug treatment) as appropriate. Significant main effects were followed by Fisher’s LSD tests. For all experiments, p < 0.05 was used as the threshold for significance. For comparisons between drug and baseline effects, week 3 of the DID protocol was used as baseline. Data were analyzed and graphed in GraphPad Prism version 7.0 and figures were assembled in Adobe Illustrator. All data represent means ± standard errors of the mean of biological replicates.
3. RESULTS
3.1. Binge drinking reduces expression of glutamate receptors in the PFC of female, but not male, mice
Glutamatergic transmission and mTOR signaling are recognized as key effectors of ethanol consumption (Lovinger, 1996; Morisot and Ron, 2017). To begin to assess whether altered expression or function of these same substrates play a role in escalated ethanol consumption in a repeated binge consumption and withdrawal protocol, we made use of the DID paradigm (Material and Methods, Figure 1A). To begin to assess possible changes in glutamatergic transmission, we assessed DID-induced changes in the cell surface and total expression of NMDARs and AMPARs, using antibodies directed against GluN1 and GluA2/3, respectively as essential subunits required for assembly and representative of these receptors. Female and male mice were exposed to water or DID for four weeks, followed by quantitation of GluN1 and GluA2/3 subunits in cell surface-biotinylated fractions and total extracts from mPFC (Figure 1B). DID-exposed female mice showed reductions in cell surface GluN1 (t8 = 4.118, p = 0.003, n = 5 water and DID, Figure 1C) with no measurable change in total expression (t8 = 1.855, p = 0.10, n = 5 water and DID, Figure 1D). Cell surface GluA2/3 subunits were similarly decreased (t8 = 2.742, p = 0.025, n =5 water and DID, Figure 1E) with no alteration in total expression (t8 = 1.132, p = 0.29, n = 5 water and DID, Figure 1F). Interestingly, and in contrast to female mice, the DID protocol did not affect expression of these same proteins in male mice (GluN1 surface: t8 = 0.29, p = 0.78, n = 5 water and DID, total, t8 = 0.83, p = 0.43, n = 5 water and DID; GluA2/3 surface: t8 = 1.87, p = 0.098, n = 5 water and DID, total, t8 = 1.79, p = 0.11, n = 5 water and DID, Figure 1 G-K), Thus, the DID protocol result in female-specific downregulation of cell surface AMPARs and NMDARs.
In order to further assess whether ethanol binge drinking involved altered signaling by the mTOR pathway, we next assessed changes in the phosphorylation state of mTOR and eEF2, a key downstream target of mTOR that is known to be regulated by stress and antidepressant drug treatments (Chandran et al., 2013; Li et al., 2010; Opal et al., 2014). While phosphorylation of mTOR at Ser2448 results in increased mTOR pathway activity, this results in reduced inhibitory phosphorylation of its downstream effector, eEF2 at T56 and overall increased mRNA translation (Heise et al., 2014). Therefore, reductions in the p-mTOR/mTOR and eEF2/p-eEF2 ratios are both indicative of reduced mTOR activity. Analyses of these two proteins in medial prefrontal cortex (mPFC) extracts of female mice subjected to the DID protocol showed a significant main effect of alcohol on phosphorylation of these two proteins (two-way ANOVA, F(1,15) = 6.55, p = 0.0218, n = 5 water mTOR, n = 5 DID mTOR, n = 5 water eEF2, n = 4 DID eEf2, Figure 1L,M), indicative of reduced mTOR pathway activity. In addition, post hoc tests revealed a near-significant effect of the DID protocol also on eEF2/p-eEF2 (p = 0.067, Figure 1M). Corresponding analyses of male mice, however, showed only a trend of DID induced downregulation of mTOR pathway activity (two-way ANOVA, F1,10 = 4.499, p = 0.060, n = 6 all groups) (Figure 1N-O) and insignificant changes in p-mTOR and p-eEF2 (Fisher’s LSD, p = 0.07 mTOR, p = 0.1895 eEF2). Thus, the DID protocol results in female specific downregulation of glutamate receptors, and this effect was associated with reduced mTOR pathway activity that was more readily evident in female than male mice.
3.2. Ketamine attenuates binge drinking in female but not male mice
The DID exposure induced defects of mPFC glutamate receptor expression and mTOR pathway activity were reminiscent of similar alterations described in preclinical models of MDD. Moreover, antidepressant behavioral effects of ketamine have been shown to involve normalization of these defects (Li et al., 2011b; Ren et al., 2016; Zhou et alv 2014). Therefore, to assess whether ketamine suppresses binge-like ethanol consumption, male and female mice were subject to the DID protocol, followed by assessment of ethanol drinking behavior and analyses of synaptic transmission in brain slices. The mice were treated with ketamine or vehicle either three days before the fourth week ethanol binge (i.e. before the start of the fourth week DID cycle) or 12 h before the binge (i.e. 10 p.m. the night prior to ethanol binge in the fourth week, Figure 1A). There was no sex difference in ethanol consumption in saline-treated mice, indicating that males and females binged at roughly the same levels (t12 = 1.789, p = 0.097, n = 8 females, n = 6 males, Figure 2A). When a subanesthetic dose of ketamine (3 mg/kg i.p.) was administered to female mice, either three days (Figure 2B) or 12 h before the final binge (Figure 2C), there was a significant decrease in g/kg ethanol consumed as compared to week 3 baseline levels (3 days: t21 = 2.037, p = 0.05, n = 11 saline, n = 12 ketamine; 12 h: t21 = 3.088, p = 0.0056, n = 11 saline, n = 12 ketamine). In order to assess whether ketamine altered the sedative properties of ethanol, a separate cohort of female mice was subject to loss of righting reflex (LORR) tests. Analogous to the 12 h ketamine treatment protocol above, the mice were treated with 3 mg/kg ketamine or saline 12 h before injection with a sedative dose of ethanol (3.2 g/kg). There was no difference in the time to recovery from LORR in mice treated with ketamine vs. saline (t20 = 0.6172, p = 0.52, n = 11 saline, n = 12 ketamine), indicating that the ketamine-induced reduction in ethanol consumption was not due to a ketamine-induced alteration of ethanol’s sedative effects (Figure 2D). In addition, we assessed whether ketamine alters rewarding properties of other appetitive liquids. Mice were subjected to two weeks of sucrose DID (Rinker et al., 2017), with daily access to 10% sucrose during a limited period in the dark phase of the light cycle, analogous to the ethanol DID protocol. Pretreatment with 3 mg/kg ketamine 12 h before the sucrose binge did not alter sucrose consumption (t5 = 0.3682, p = 0.7278, n = 6 both groups), suggesting that ketamine-induced suppression of the ethanol binge was not due to an unspecific reduction of hedonic drive (Figure 2E).
We next tested for predicted effects of ketamine on binge ethanol drinking in male mice (Figure 2F). At a dose of 3 mg/kg, ketamine failed to alter binge ethanol consumption at both the 12 hour and 3 day pretreatment timepoints (12 hour: t13 = 1.583, p = 0.1375, n = 7 saline, n = 9 ketamine; 3 days: t14 = 1.065, p = 0.3047, n = 7 saline, n = 9 ketamine). To address whether the absence of ketamine effects in male mice were due to sex-specific differences in ketamine’s efficacy, we further tested whether higher doses of ketamine would be effective. However, neither pretreatment with 6 mg/kg nor 10 mg/kg ketamine measurably affected the ethanol binge at 12 h post ketamine (6 mg/kg, t18 = 1.014, p = 0.3239, n = 6 saline, n = 14 ketamine; 10 mg/kg, t18 = 1.668, p = 0.1125, n = 6 saline, n = 14 ketamine) (Figure 2F). Thus, ketamine sex-specifically suppresses ethanol binge drinking in female but not male mice, in accordance with ethanol binge drinking induced reductions of glutamate receptor expression in female but not male mice.
3.3. Ketamine rescues alterations in binge-drinking induced spontaneous neurotransmission.
In search of a sex-specific cellular functional correlate of binge drinking and its suppression by ketamine we next assessed possible changes in glutamatergic synapse function. We recorded spontaneous excitatory postsynaptic currents (sEPSCs) from layer 2/3 pyramidal neurons of the PLC (representative traces, Figure 3A,D). Mice were exposed to four cycles of DID or standard drinking water. Female mice were administered 3 mg/kg ketamine 12 h before sacrifice, while males were administered 6 mg/kg ketamine 12 h before sacrifice. In female DID-exposed mice, the sEPSC amplitude remained unaffected (two-way ANOVA, F1,25 = 0.065, p = 0.56 (ethanol), F1,25 = 0.027, p = 0.24 (ketamine), F1,25 = 0.053, p = 0.25 (interaction), cell numbers for female sEPSCs, n = 5 water saline, n = 7 water ketamine, n = 10 DID saline, n = 10 DID ketamine) (Figure 3B). However, the sEPSC frequency of female DID-exposed mice showed a significant interaction between ethanol history and ketamine treatment (two-way ANOVA, F1,28 = 5.261, p = 0.030), without main effects of ethanol (F1,25 = 0.040, p = 0.84) or ketamine (F1,25 = 0.3147, p = 0.58) (Figure 3C). Most remarkably, the DID-induced reduction of the sEPSC frequency was completely restored to control levels 12 h after ketamine treatment (p = 0.026). By contrast, ketamine had no effect on the same parameter of ethanol-naive mice (p = 0.285).
We found no effect of ethanol exposure or ketamine treatment on mEPSC amplitude, matching the results seen with sEPSCs [two-way ANOVA, F1,32 = 1.44, p = 0.239 (ketamine), F1,32 = 0.0183, p = 0.8932 (ethanol), F1,32 = 1.777, p = 0.192 (interaction), cell number for all mEPSC experiments, n = 7 water saline, n = 11 water ketamine, n = 9 DID saline, n = 10 DID ketamine] (Figure 3E). Interestingly and in contrast to the sEPSC frequency measures reported above, the mEPSC frequency measurements showed no interaction between DID exposure and ketamine treatment (two-way ANOVA, F1,32 = 2.722, p = 0.108), suggesting that the ketamine-induced increase in sEPSC frequency was driven entirely by increased presynaptic activity.
Male DID-exposed mice showed a small but significant reduction in sEPSC amplitude (two-way ANOVA, main effect of ethanol F1,32 = 4.2 99, p = 0.050), cell number for all male sEPSC experiments, n = 8 water saline, n = 8 water ketamine, n = 8 DID saline, n = 11 DID ketamine). There was no main effect of ketamine F1,32 = 2.122, p = 0.173) nor an interaction of ethanol and ketamine effects (F1,32 = 0.006187, p = 0.961) and post hoc tests were not significant (representative traces, Figure 3G, H). Male mice also showed a main effect of ketamine on the sEPSC frequency (two-way ANOVA, F1,32 = 6.882, p = 0.0044) and an interaction of ketamine and ethanol (F1,32 = 7.132, p = 0.0039) but no main effect of ethanol (F1,32 = 0.9115, p = 0.170). Post hoc tests revealed that DID reduced the sEPSC frequency of saline treated mice as compared to water–exposed saline treated mice (Figure 3I), thereby reproducing the effects seen in females (Figure 3C). In addition, ketamine reduced the sEPSC of water-exposed mice. However, in contrast to female mice, ketamine failed to rescue the reduced sESPC frequency of DID-exposed male mice (Figure 3F). Thus, ketamine induced rescue of DID induced reductions in sEPSCs may contribute to sex-specific ketamine-induced suppression of binge drinking in female mice.
4. DISCUSSION
Alcohol exposure is known to produce a variety of functional deficits throughout the brain, including within the medial prefrontal cortex (Pleil et al., 2015b; Rinker et al., 2017). Here, we found that DID exposure results in reduced cell surface expression of AMPARs and NMDARs selectively in female mice. Moreover, a subanesthetic dose of ketamine prophylactically reduced binge-like ethanol consumption selectively in female but not male mice. Functionally, glutamatergic synaptic inputs to L2/3 pyramidal cells of the PLC were downregulated by DID independent of sex, yet ketamine was effective in normalizing this defect again selectively in female but not male mice. An imperfect correspondence of sex differences observed between biochemical and functional electrophysiological measurement should not be surprising as the first measure is representative of all neurons throughout the entire medial prefrontal cortex while the other is representative of a narrowly defined cell population specifically in layer 2/3 of the PLC. Future experiments will have to address whether DID induced impairments of glutamatergic synapses are layer specific and limited to only glutamatergic neurons or also evident in GABAergic neurons and layer 5 of the PLC and beyond, and whether sex specificity of DID effects can be attributed to specific synapses.
The antidepressant potency of ketamine is known to be greater in female than male mice (Carrier and Kabbaj, 2013; Franceschelli et al., 2015), perhaps due to more rapid drug conversion into its active metabolite (2S,6S;2R,6R)- hydroxynorketamine in females than males (Zanos et al., 2016). Nevertheless, we showed that ketamine remained ineffective in males even when administered at twice the dose that was effective in females, suggesting that sex specificity did not simply reflect such differences in pharmacokinetics of ketamine metabolites. Notably, others have consistently demonstrated sexual dimorphism in ketamine’s behavioral effects also in rats (Carrier and Kabbaj, 2013; Franceschelli et al., 2015). Male but not female rats display conditioned place preference to ketamine at 10 mg/kg, while female rats are more responsive to locomotor stimulating effects of ketamine when assessed immediately after ketamine administration (Schoepfer et al., 2017). Importantly, we showed that ketamine’s binge suppressing effects were not due to unspecific reductions in reward seeking, as sucrose consumption after DID exposure was unaltered by ketamine. Moreover, the behavioral effects of ketamine were specific to ethanol drinking and did not involve increased sedation as the ethanol induced LORR remained unaffected. Lastly, blood ethanol concentrations (BECs) measured in a separate cohort of DID exposed mice showed no sex difference (males 56 ± 5.0 mg/dl, females 64.12 ± 8.7 mg/dl, p = 0.46, n = 5, t-test). However, it is possible that even insignificantly increased BECs in females vs. males, over the course of 4 weeks of alcohol exposure, led to greater or qualitatively different forms neural plasticity in female compared to male mice that then were responsible for the observed female specific effects of ketamine.
Glutamatergic synaptic transmission has long been proposed as a potential target for treatment of AUDs (Lovinger, 1996; Morisot and Ron, 2017), and ketamine has been suggested as a treatment for drug addiction (Ivan Ezquerra-Romano et al., 2018). Acamprosate, a promising and effective treatment for alcohol use disorders, is thought to act through NMDARs, and to some extent, via secondary GABA targets (Kalk and Lingford-Hughes, 2014), suggesting pharmacological mechanisms similar to those of ketamine. Indeed, ketamine treatment of animals with defects in prefrontal glutamatergic synaptic transmission leads to prominent potentiation of not only glutamatergic synaptic transmission but also GABAergic synaptic transmission (Ren et al., 2016). Topiramate, an antiepileptic drug thought to target both glutamatergic and GABAergic signaling (though its precise mechanism is unclear), has also been used successfully, albeit off-label, to treat alcoholism (Soyka and Müller, 2017). Notably, ketamine has recently been shown to attenuate alcohol preference in alcohol-preferring rats (Rezvani et al., 2017). Our work builds upon these findings by extending ketamine’s ability to reduce drinking in a clinically relevant model of a wildtype strain of mice.
The PLC and infralimbic cortex are implicated as key brain regions involved in alcohol addiction (Koob and Volkow, 2009), stress (Caruso et al., 2018; Lowery-Gionta et al., 2018) and major depressive disorder (Banasr and Duman, 2008; Drevets, 2000; Rajkowska et al., 1999), making it a likely candidate for therapeutic intervention via ketamine. Previous work using the CIE model has shown mixed changes in PLC glutamatergic and GABAergic transmission. Male C57BL/6J mice did not reveal differences in glutamatergic transmission in the PLC following four weeks of CIE, although differences were seen in the infralimbic cortex (Pleil et al., 2015b). However, seven weeks of CIE plus one week of withdrawal led to increased spine density in the PLC of mice (Varodayan et al., 2018). Adolescent alcohol exposure can also increase spine density of long/thin spines in layer 5 pyramidal neurons of the PLC when assessed later in adulthood in rats (Trantham-Davidson et al., 2017). We add to this literature by demonstrating DID induced reductions in sEPSC frequency in PLC layer 2/3 pyramidal neurons of both male and female C57BL/6J mice as well as differences of surface expression of NMDARs and AMPARs.
Alcohol has previously been shown to have profound effects on NMDAR and AMPARs in the PLC. Holmes et al. (2012) have proposed downregulation of NMDARs in layer 2/3 of the mPFC following CIE, similar to the work shown here. Others have shown that CIE increases GluN1, and subsequently alters the AMPA/NMDA ratio, in PLC layer 5 (Kroener et al., 2012). This increase in NMDAR expression is opposite to the one we found in PLC layer2/3, suggesting that ethanol withdrawal induces layer-specific forms of neural plasticity. Alternatively, the relatively short term ethanol exposure that is integral to the DID protocol used here may capture an initial downregulation of NMDARs that precedes the homeostatic-like increase in NMDAR function seen following chronic ethanol exposure observed by Kroener et al. Consistent with this idea, layer 2/3 neurons of the prefrontal cortex project to layer 5 (Song et al., 2012), suggesting that neural plasticity in layer 2/3 may lead to secondary changes in layer 5. Taken together, the literature suggests that glutamatergic transmission in the PLC may undergo dynamic shifts in synaptic transmission dependent on animal age, species, route of ethanol administration, and duration of exposure and withdrawal.
While our studies focused on DID and ketamine-induced alterations in the PLC, our findings by no means imply that plasticity is limited to this brain region. Indeed, the increase in sEPSC, but not mEPSC recorded from Layer 2/3 glutamatergic neurons points to altered activity of presynaptic neurons that project to the PLC from elsewhere. A similar ketamine-induced increase in presynaptic activity has also been reported in a GABAA receptor mutant mouse model of MDD (Ren et al 2016). Consistent with these findings, Carreno et al (2016) have shown that optogenetic or pharmacogenetic activation of neural projections from the ventral hippocampus to the mPFC (but not to the nucleus accumbens) mimicked the delayed effect of ketamine in alleviating depressive-like behavior. Moreover, pharmacologic blockade of neural activity in the ventral hippocampus blocked the behavioral effects of ketamine. Future experiments should address whether activation of these same projections from the hippocampus regulate binge drinking of mice.
DID exposure was associated with reduced activity of the mTOR pathway in a sex-independent manner, although the effect was significant only in females. Reduced mTOR activity has previously been described in MDD (Jernigan et al., 2011), and stress based models of MDD (Chandran et al., 2013), while increased mTOR activity is implicated in the antidepressant mechanism of ketamine and other rapid acting experimental antidepressants (Li et al., 2010; Tang et al., 2015; Voleti et al., 2013). Future experiments will need to address whether ketamine also normalizes prefrontal mTOR pathway activity in DID exposed mice and whether blocking mTOR prevents ketamine-mediated suppression of ethanol consumption. Experimentally addressing the latter is not trivial because blocking mTOR in the nucleus accumbens or systemically prevents alcohol seeking and drinking also independently of ketamine treatment (Neasta et al., 2010).
In conclusion, we have shown that binge-like ethanol exposure leads to gross perturbations of glutamatergic transmission in the PLC and that ketamine may act as an effective therapeutic for binge drinking in female mice. Future work should investigate the layer cell-type specific, and circuitry specific effects of ketamine modulation of glutamate transmission in the PFC, elucidate the mechanisms underlying sex differences of treatment efficacy.
Supplementary Material
HIGHLIGHTS.
Drinking in the Dark (DID) reduces surface expression of glutamate receptors and mTOR pathway signaling in the medial prefrontal cortex and glutamatergic synaptic inputs to pyramidal cells in layer2/3 of the prelimbic cortex
Ketamine prophylactically reduces binge-like ethanol consumption in female, but not male mice.
Ketamine normalizes glutamatergic synaptic inputs to pyramidal cells selectively in female mice, suggesting that normalization of glutamatergic synaptic transmission in the prelimbic cortex reverses excessive alcohol drinking.
5. ACKNOWLEDGEMENTS
NAC and BL conceived of the experiments, analyzed the data, and prepared the manuscript. NAC, SNM, MF, SNJ, NCD, DFB, and GM conducted experiments.
6. FUNDING AND DISCLOSURES
This publication was made possible by grants MFI099851 to B.L. from the National Institutes of Mental Health (NIMFI) and the Social Science Research Institute (L2 to N.A.C and B.L.). Its contents are solely the responsibility of the authors and do not necessarily represent the views of the funding agencies. The authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this: early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7. REFERENCES
- Abdallah CG, Averill CL, Salas R, Averill LA, Baldwin PR, Krystal JH, Mathew SJ, Mathalon DH, 2017. Prefrontal Connectivity and Glutamate Transmission: Relevance to Depression Pathophysiology and Ketamine Treatment. Biol, psychiatry. Cogn. Neurosci. neuroimaging 2, 566–574. 10.1016/j.bpsc.2017.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahao KP, Salinas AG, Lovinger DM, 2017. Alcohol and the Brain: Neuronal Molecular Targets, Synapses, and Circuits. Neuron 96, 1223–1238. 10.1016/J.NEURON.2017.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banasr M, Duman RS, 2008. Glial Loss in the Prefrontal Cortex Is Sufficient to Induce Depressive-like Behaviors. Biol. Psychiatry 64, 863–870. 10.1016/j.biopsych.2008.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banasr M, Valentine GW, Li X-Y, Gourley SL, Taylor JR, Duman RS, 2007. Chronic unpredictable stress decreases cell proliferation in the cerebral cortex of the adult rat. Biol. Psychiatry 62, 496–504. 10.1016/j.biopsych.2007.02.006 [DOI] [PubMed] [Google Scholar]
- Brachman RA, McGowan JC, Perusini JN, Urn SC, Pham TH, Faye C, Gardier AM, Mendez-David L, David DJ, Hen R, Denny CA, 2016. Ketamine as a Prophylactic Against Stress-Induced Depressive-like Behavior. Biol. Psychiatry 79, 776–86. 10.1016/j.biopsych.2015.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brière FN, Rohde P, Seeley JR, Klein D, Lewinsohn PM, 2014. Comorbidity between major depression and alcohol use disorder from adolescence to adulthood. Compr. Psychiatry 55, 526–33. 10.1016/j.comppsych.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier N, Kabbaj M, 2013. Sex differences in the antidepressant-like effects of ketamine. Neuropharmacology 70, 27–34. 10.1016/j.neuropharm.2012.12.009 [DOI] [PubMed] [Google Scholar]
- Caruso MJ, Seemiller LR, Fetherston TB, Miller CN, Reiss DE, Cavigelli SA, Kamens HM, 2018. Adolescent social stress increases anxiety-like behavior and ethanol consumption in adult male and female C57BL/6J mice. Sci. Rep 8, 10040 10.1038/s41598-018-28381-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran A, lyo AH, Jernigan CS, Legutko B, Austin MC, Karolewicz B, 2013. Reduced phosphorylation of the mTOR signaling pathway components in the amygdala of rats exposed to chronic stress. Prog. Neuro-Psychopharmacology Biol. Psychiatry 40, 240–245. https://doi.Org/10.1016/j.pnpbp.2012.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury GMI, Zhang J, Thomas M, Banasr M, Ma X, Pittman B, Bristow L, Schaeffer E, Duman RS, Rothman DL, Behar KL, Sanacora G, 2017. Transiently increased glutamate cycling in rat PFC is associated with rapid onset of antidepressant-like effects. Mol. Psychiatry 22, 120–126. 10.1038/mp.2016.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley NA, Bloodgood DW, Hardaway JA, Kendra AM, McCall JG, Al-Hasani R, McCall NM, Yu W, Schools ZL, Krashes MJ, Lowell BB, Whistler JL, Bruchas MR, Kash TL, 2016. Dynorphin Controls the Gain of an Amygdalar Anxiety Circuit. Cell Rep 14, 2774–2783. https://doi.Org/10.1016/j.celrep.2016.02.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC, 2000. Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression, in: Progress in Brain Research, pp. 413–431. 10.1016/S0079-6123(00)26027-5 [DOI] [PubMed] [Google Scholar]
- Drevets WC, Price JL, Furey ML, 2008. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct. Funct 213, 93–118. 10.1007/s00429-008-0189-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoch M-A, 2011. The role of early life stress as a predictor for alcohol and drug dependence. Psychopharmacology (Berl). 214, 17–31. 10.1007/s00213-010-1916-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erol A, Karpyak VM, 2015. Sex and gender-related differences in alcohol use and its consequences: Contemporary knowledge and future research considerations. Drug Alcohol Depend 156, 1–13. 10.1016/j.drugalcdep.2015.08.023 [DOI] [PubMed] [Google Scholar]
- Franceschelli A, Sens J, Herchick S, Thelen C, Pitychoutis PM, 2015. Sex differences in the rapid and the sustained antidepressant-like effects of ketamine in stress-naive and “depressed” mice exposed to chronic mild stress. Neuroscience 290, 49–60. 10.1016/j.neuroscience.2015.01.008 [DOI] [PubMed] [Google Scholar]
- Frye GD, Fincher A, 2000. Sustained ethanol inhibition of native AMPA receptors on medial septum/diagonal band (MS/DB) neurons. Br. J. Pharmacol 129, 87–94. 10.1038/sj.bjp.0703039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs T, Jefferson SJ, Flooper A, Yee P-H, Maguire J, Luscher B, 2017. Disinhibition of somatostatin-positive GABAergic interneurons results in an anxiolytic and antidepressant-like brain state. Mol. Psychiatry 22, 920–930. 10.1038/mp.2016.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise C, Gardoni F, Culotta L, di Luca M, Verpelli C, Sala C, 2014. Elongation factor-2 phosphorylation in dendrites and the regulation of dendritic mRNA translation in neurons. Front. Cell. Neurosci 8, 35 10.3389/fncel.2014.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holleran KM, Winder DG, 2017. Preclinical voluntary drinking models for alcohol abstinence-induced affective disturbances in mice. Genes, Brain Behav 16, 8–14. 10.1111/gbb.12338 [DOI] [PubMed] [Google Scholar]
- Holmes A, Fitzgerald PJ, MacPherson KP, DeBrouse L, Colacicco G, Flynn SM, Masneuf S, Pleil KE, Li C, Marcinkiewcz CA, Kash TL, Gunduz-Cinar O, Camp M, 2012. Chronic alcohol remodels prefrontal neurons and disrupts NMDAR-mediated fear extinction encoding. Nat. Neurosci 15, 1359–61. 10.1038/nn.3204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan Ezquerra-Romano I, Lawn W, Krupitsky E, Morgan CJA, 2018. Ketamine for the treatment of addiction: Evidence and potential mechanisms. Neuropharmacology. https://doi.Org/10.1016/j.neuropharm.2018.01.017 [DOI] [PubMed] [Google Scholar]
- Jernigan CS, Goswami DB, Austin MC, lyo AH, Chandran A, Stockmeier CA, Karolewicz B, 2011. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 1774–9. https://doi.Org/10.1016/j.pnpbp.2011.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalk NJ, Lingford-Hughes AR, 2014. The clinical pharmacology of acamprosate. Br. J. Clin. Pharmacol. 77, 315–323. 10.1111/bcp.12070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Dernier O, Jin R, Merikangas KR, Walters EE, 2005. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 62, 593–602. https://doi.Org/10.1001/archpsyc.62.6.593 [DOI] [PubMed] [Google Scholar]
- Keyes KM, Grant BF, Hasin DS, 2008. Evidence for a closing gender gap in alcohol use, abuse, and dependence in the United States population. Drug Alcohol Depend 93, 21–9. https://doi.Org/10.1016/j.drugalcdep.2007.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND, 2009. Neurocircuitry of Addiction. Neuropsychopharmacology 35, 217–238. 10.1038/npp.2009.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornstein SG, Schatzberg AF, Thase ME, Yonkers KA, McCullough JP, Keitner GI, Gelenberg AJ, Ryan CE, Hess AL, Harrison W, Davis SM, Keller MB, 2000. Gender differences in chronic major and double depression. J. Affect. Disord 60, 1–11. 10.1016/S0165-0327(99)00158-5 [DOI] [PubMed] [Google Scholar]
- Kroener S, Mulholland PJ, New NN, Gass JT, Becker HC, Chandler LJ, 2012. Chronic alcohol exposure alters behavioral and synaptic plasticity of the rodent prefrontal cortex. PLoS One 7, e37541 10.1371/journal.pone.0037541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lener MS, Kadriu B, Zarate CA, 2017. Ketamine and Beyond: Investigations into the Potential of Glutamatergic Agents to Treat Depression. Drugs 77, 381–401. 10.1007/s40265-017-0702-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Lee B, Liu R-J, Banasr M, Dwyer JM, Iwata M, Li X-Y, Aghajanian G, Duman RS, 2010. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–64. 10.1126/science.1190287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Liu R-J, Dwyer JM, Banasr M, Lee B, Son H, Li X-Y, Aghajanian G, Duman RS, 2011a. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol. Psychiatry 69, 754–61. https://doi.Org/10.1016/j.biopsych.2010.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Liu R-J, Dwyer JM, Banasr M, Lee B, Son H, Li X-Y, Aghajanian G, Duman RS, 2011b. Glutamate N-methyl-D-aspartate Receptor Antagonists Rapidly Reverse Behavioral and Synaptic Deficits Caused by Chronic Stress Exposure. Biol. Psychiatry 69, 754–761. https://doi.Org/10.1016/j.biopsych.2010.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, 1996. Interactions between ethanol and agents that act on the NMDA-type glutamate receptor. Alcohol. Clin. Exp. Res 20, 187A–191A. https://doi.Org/10.1111/j.1530-0277.1996.tb01773.x [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF, 1989. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 243, 1721–4. DOI: 10.1126/science.2467382 [DOI] [PubMed] [Google Scholar]
- Lowery-Gionta EG, Crowley NA, Bukalo O, Silverstein S, Holmes A, Kash TL, 2018. Chronic stress dysregulates amygdalar output to the prefrontal cortex. Neuropharmacology 139, 68–75. https://doi.Org/10.1016/j.neuropharm.2018.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery-Gionta EG, Navarro M, Li C, Pleil KE, Rinker JA, Cox BR, Sprow GM, Kash TL, Thiele TE, 2012. Corticotropin Releasing Factor Signaling in the Central Amygdala is Recruited during Binge-Like Ethanol Consumption in C57BL/6J Mice. J. Neurosci 32, 3405–3413. 10.1523/JNEUROSCI.6256-ll.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, Morrison JH, 2013. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron 79, 16–29. https://doi.Org/10.1016/j.neuron.2013.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan JC, LaGamma CT, Lim SC, Tsitsiklis M, Neria Y, Brachman RA, Denny CA, 2017. Prophylactic Ketamine Attenuates Learned Fear. Neuropsychopharmacology 42, 1577–1589. 10.1038/npp.2017.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, Gideons E, Kavalali ET, 2013. The role of eukaryotic elongation factor 2 kinase in rapid antidepressant action of ketamine. Biol. Psychiatry 73, 1199–203. https://doi.Org/10.1016/j.biopsych.2012.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisot N, Ron D, 2017. Alcohol-dependent molecular adaptations of the NMDA receptor system. Genes. Brain. Behav 16, 139–148. 10.1111/gbb.12363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neasta J, Barak S, Hamida S. Ben, Ron D, 2014. mTOR complex 1: a key player in neuroadaptations induced by drugs of abuse. J. Neurochem 130, 172–184. 10.1111/jnc.12725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neasta J, Ben Hamida S, Yowell Q, Carnicella S, Ron D, 2010. Role for mammalian target of rapamycin complex 1 signaling in neuroadaptations underlying alcohol-related disorders. Proc. Natl. Acad. Sci. U. S. A 107, 20093–8. 10.1073/pnas.1005554107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIAAA, 2017. Drinking Levels Defined | National Institute on Alcohol Abuse and Alcoholism (NIAAA) [WWW Document]. NIAAA Website. URL https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/moderate-binge-drinking (accessed 4.2.18). [Google Scholar]
- Opal MD, Klenotich SC, Morais M, Bessa J, Winkle J, Doukas D, Kay LJ, Sousa N, Dulawa SM, 2014. Serotonin 2C receptor antagonists induce fast-onset antidepressant effects. Mol. Psychiatry 19, 1106–14. 10.1038/mp.2013.144 [DOI] [PubMed] [Google Scholar]
- Pleil KE, Lowery-Gionta EG, Crowley NA, Li C, Marcinkiewcz CA, Rose JH, McCall NM, Maldonado-Devincci AM, Morrow AL, Jones SR, Kash TL, 2015b. Effects of chronic ethanol exposure on neuronal function in the prefrontal cortex and extended amygdala. Neuropharmacology 99, 735–749. https://doi.Org/10.1016/j.neuropharm.2015.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleil KE, Rinker JA, Lowery-Gionta EG, Mazzone CM, McCall NM, Kendra AM, Olson DP, Lowell BB, Grant KA, Thiele TE, Kash TL, 2015a. NPY signaling inhibits extended amygdala CRF neurons to suppress binge alcohol drinking. Nat. Neurosci 18, 545–552. 10.1038/nn.3972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, Overholser JC, Roth BL, Stockmeier CA, 1999. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiatry 45,1085–98. DOI: 10.1016/S0006-3223(99)00041-4 [DOI] [PubMed] [Google Scholar]
- Ren H, Zhao Y, Wu M, Peoples RW, 2013. A Novel Alcohol-Sensitive Position in the N-Methyl-D-Aspartate Receptor GluN2A Subunit M3 Domain Regulates Agonist Affinity and Ion Channel Gating. Mol. Pharmacol 84, 501–510. 10.1124/mol.113.085993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Pribiag H, Jefferson SJ, Shorey M, Fuchs T, Stellwagen D, Luscher B, 2016. Bidirectional Homeostatic Regulation of a Depression-Related Brain State by Gamma-Aminobutyric Acidergic Deficits and Ketamine Treatment. Biol. Psychiatry 80, 457–468. https://doi.Org/10.1016/j.biopsych.2016.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani AH, Levin ED, Cauley M, Getachew B, Tizabi Y, 2017. Ketamine Differentially Attenuates Alcohol Intake in Male Versus Female Alcohol Preferring (P) Rats. J. Drug Alcohol Res 6, 1–6. 10.4303/jdar/236030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC, 2005. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol. Behav https://doi.Org/10.1016/j.physbeh.2004.10.007 [DOI] [PubMed] [Google Scholar]
- Rinker JA, Fulmer DB, Trantham-Davidson H, Smith ML, Williams RW, Lopez MF, Randall PK, Chandler LJ, Miles MF, Becker HC, Mulholland PJ, 2017. Differential potassium channel gene regulation in BXD mice reveals novel targets for pharmacogenetic therapies to reduce heavy alcohol drinking. Alcohol 58, 33–45. https://doi.Org/10.1016/j.alcohol.2016.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinker JA, Marshall SA, Mazzone CM, Lowery-Gionta EG, Gulati V, Pleil KE, Kash TL, Navarro M, Thiele TE, 2017. Extended Amygdala to Ventral Tegmental Area Corticotropin-Releasing Factor Circuit Controls Binge Ethanol Intake. Biol. Psychiatry 81, 930–940. 10.1016/J.BIOPSYCH.2016.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Bajo M, Crawford E, Madamba SG, Siggins GR, 2006. Chronic ethanol exposure and protracted abstinence alter NMDA receptors in central amygdala. Neuropsychopharmacology 31, 988–96. 10.1038/sj.npp.1300840 [DOI] [PubMed] [Google Scholar]
- Sabino V, Narayan AR, Zeric T, Steardo L, Cottone P, 2013. mTOR activation is required for the anti-alcohol effect of ketamine, but not memantine, in alcohol-preferring rats. Behav. Brain Res 247, 9–16. 10.1016/j.bbr.2013.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepfer KJ, Strong CE, Saland SK, Wright KN, Kabbaj M, 2017. Sex- and dose-dependent abuse liability of repeated subanesthetic ketamine in rats. Physiol. Behav 10.1016/J.PHYSBEH.2017.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song D, Opris I, Chan RHM, Marmarelis VZ, Hampson RE, Deadwyler SA, Berger TW, 2012. Functional connectivity between Layer 2/3 and Layer 5 neurons in prefrontal cortex of nonhuman primates during a delayed match-to-sample task, in: 2012 Annual International Conference of the IEEE Engineering in Medicine and Biology Society IEEE, pp. 2555–2558. 10.1109/EMBC.2012.6346485 [DOI] [PubMed] [Google Scholar]
- Soyka M, Müller CA, 2017. Pharmacotherapy of alcoholism – an update on approved and off-label medications. Expert Opin. Pharmacother 18, 1187–1199. 10.1080/14656566.2017.1349098 [DOI] [PubMed] [Google Scholar]
- Sparta DR, Sparrow AM, Lowery EG, Fee JR, Knapp DJ, Thiele TE, 2008. Blockade of the corticotropin releasing factor type 1 receptor attenuates elevated ethanol drinking associated with drinking in the dark procedures. Alcohol. Clin. Exp. Res 32, 259–65. 10.1111/j.1530-0277.2007.00575.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Xue W, Xia B, Ren L, Tao W, Chen C, Zhang H, Wu R, Wang Q, Wu H, Duan J, Chen G, 2015. Involvement of normalized NMDA receptor and mTOR-related signaling in rapid antidepressant effects of Yueju and ketamine on chronically stressed mice. Sci. Rep 5, 13573 10.1038/srepl3573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trantham-Davidson H, Centanni SW, Garr SC, New NN, Mulholland PJ, Gass JT, Glover EJ, Floresco SB, Crews FT, Krishnan HR, Pandey SC, Chandler LJ, 2017. Binge-Like Alcohol Exposure During Adolescence Disrupts Dopaminergic Neurotransmission in the Adult Prelimbic Cortex. Neuropsychopharmacology 42, 1024–1036. 10.1038/npp.2016.190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner S, Mota N, Bolton J, Sareen J, 2018. Self-medication with alcohol or drugs for mood and anxiety disorders: A narrative review of the epidemiological literature. Depress. Anxiety, 10.1002/da.22771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varodayan FP, Sidhu H, Kreifeldt M, Roberto M, Contet C, 2018. Morphological and functional evidence of increased excitatory signaling in the prelimbic cortex during ethanol withdrawal. Neuropharmacology 133, 470–480. 10.1016/J.NEUROPHARM.2018.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voleti B, Navarria A, Liu R-J, Banasr M, Li N, Terwilliger R, Sanacora G, Eid T, Aghajanian G, Duman RS, 2013. Scopolamine Rapidly Increases Mammalian Target of Rapamycin Complex 1 Signaling, Synaptogenesis, and Antidepressant Behavioral Responses. Biol. Psychiatry 74, 742–749. 10.1016/j.biopsych.2013.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranjkovic O, Winkler G, Winder DG, 2018. Ketamine administration during a critical period after forced ethanol abstinence inhibits the development of time-dependent affective disturbances. Neuropsychopharmacology 1 10.1038/s41386-018-0102-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson ST, Wright D, Fasula MK, Fenton L, Griepp M, Ostroff RB, Sanacora G, 2017. Cognitive Behavior Therapy May Sustain Antidepressant Effects of Intravenous Ketamine in Treatment-Resistant Depression. Psychother. Psychosom 86, 162–167. 10.1159/000457960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, Gerhard D, Thomas A, Duman RS, 2017. Molecular and Cellular Mechanisms of Rapid-Acting Antidepressants Ketamine and Scopolamine. Curr. Neuropharmacol 15, 11–20. doi: 10.2174/1570159X14666160309114549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward JJ, Pava MJ, 2009. Effects of ethanol on persistent activity and up-States in excitatory and inhibitory neurons in prefrontal cortex. Alcohol. Clin. Exp. Res 33, 2134–40. 10.1111/j.1530-0277.2009.01059.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z, 2012. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 73, 962–77. 10.1016/j.neuron.2011.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, Alkondon M, Yuan P, Pribut HJ, Singh NS, Dossou KSS, Fang Y, Huang X-P, Mayo CL, Wainer IW, Albuquerque EX, Thompson SM, Thomas CJ, Zarate CA Jr, Gould TD, 2016. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533, 481–486. 10.1038/naturel7998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P, Thompson SM, Duman RS, Zarate CA, Gould TD, 2018. Convergent Mechanisms Underlying Rapid Antidepressant Action. CNS Drugs 32, 197–227. 10.1007/s40263-018-0492-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Wang N, Yang C, Li X-M, Zhou Z-Q, Yang J-J, 2014. Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur. Psychiatry 29, 419–23. 10.1016/j.eurpsy.2013.10.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.