

Abstract
Allosteric modulators of the metabotropic glutamate receptor subtype 5 (mGlu5) have been proposed as potential therapies for various CNS disorders. These ligands bind to sites distinct from the orthosteric (or endogenous) ligand, often with improved subtype selectivity and spatio-temporal control over receptor responses. We recently revealed that mGlu5 allosteric agonists and positive allosteric modulators exhibit biased agonism and/or modulation. To establish whether negative allosteric modulators (NAMs) engender similar bias, we rigorously characterized the pharmacology of eight diverse mGlu5 NAMs. Radioligand inhibition binding studies revealed novel modes of interaction with mGlu5 for select NAMs, with biphasic or incomplete inhibition of the radiolabeled NAM, [3H]methoxy-PEPy. We assessed mGlu5-mediated intracellular Ca2+ (iCa2+) mobilization and inositol phosphate (IP1) accumulation in HEK293A cells stably expressing low levels of mGlu5 (HEK293A-rat mGlu5-low) and mouse embryonic cortical neurons. The apparent affinity of acetylenic NAMs, MPEP, MTEP and dipraglurant, was dependent on the signaling pathway measured, agonist used, and cell type (HEK293A-rat mGlu5-low versus mouse cortical neurons). In contrast, the acetylenic partial NAM, M-5MPEP, and structurally distinct NAMs (VU0366248, VU0366058, fenobam), had similar affinity estimates irrespective of the assay or cellular background. Biased modulation was evident for VU0366248 in mouse cortical neurons where it was a NAM for DHPG-mediated iCa2+ mobilization, but neutral with DHPG in IP1 accumulation assays. Overall, this study highlights the inherent complexity in mGlu5 NAM pharmacology that we hypothesize may influence interpretation when translating into preclinical models and beyond in the design and development of novel therapeutics for neuropsychiatric and neurological disorders.
Keywords: biased modulation, kinetics, negative allosteric modulator, metabotropic glutamate receptor 5
1. Introduction
The metabotropic glutamate receptor subtype 5 (mGlu5) is a G protein-coupled receptor (GPCR) widely expressed throughout the brain and has been proposed as a therapeutic target in various central nervous system (CNS) disorders, ranging from anxiety and depression to Parkinson’s disease and autism (Gregory et al., 2013). mGlu5 is a well-established Gq-coupled receptor, with activation leading to production of inositol-1,2,3-trisphosphate (IP3) and mobilization of intracellular calcium (iCa2+) (Niswender and Conn, 2010). mGlu5 is one of eight mGlu subtypes, subdivided into group I (mGlu1/5), group II (mGlu2/3) and group III (mGlu4,6–8), which share a highly conserved glutamate binding site. Recent approaches in targeting mGlu5 have therefore focused on ligands that bind to less conserved, topographically distinct allosteric binding sites (Sengmany and Gregory, 2016). Allosteric ligands may modulate the activity of orthosteric ligands by influencing the affinity and/or efficacy (a property termed cooperativity), to either enhance (positive allosteric modulator; PAM), or diminish (negative allosteric modulator; NAM) endogenous receptor responses (Changeux and Christopoulos, 2016). Some PAMs also activate the receptor in the absence of orthosteric ligand and are categorized as PAM-agonists, while neutral allosteric ligands (NALs) bind to allosteric sites without influencing orthosteric ligand activity (Changeux and Christopoulos, 2016). In the absence of endogenous agonist, “pure” PAMs and NAMs offer the additional advantage of spatial and temporal fine-tuning of receptor responses – a desirable clinical outcome within the delicate CNS network.
Fine-tuning neurotransmitter receptor activity can also be achieved through biased agonism and modulation (Kenakin and Christopoulos, 2013), whereby individual ligands may differentially activate/modulate different receptor responses to the relative exclusion of others and, as such, have a unique “signaling fingerprint”. Biased agonism and modulation is operative across a wide range of GPCRs, from opioid and endocannabinoid systems to adrenergic and adenosine receptors, to name a few (Baltos et al., 2017; da Silva Junior et al., 2017; Khajehali et al., 2015; Priestley et al., 2017; Violin et al., 2014). Thus, determining ligand signaling fingerprints for different effectors offers the invaluable opportunity to design ligands that bias receptor signaling towards certain pathways over others. Such an approach may result in ligands that have improved efficacy and lower adverse effect liability.
While the notion of bias is gaining traction, the continued use of high-throughput single-assay drug screening approaches is not capable of capturing the full scope of such ligand activity. There remains a need to decipher which pathways, or bias profiles, are linked to preclinical or clinical efficacy, to stimulate a change in approach. Importantly, it is increasingly evident that mGlu5 is pleiotropically coupled to multiple G proteins and signaling partners (Francesconi and Duvoisin, 1998; Joly et al., 1995; Mao et al., 2005; Peavy et al., 2001; Rush et al., 2002; Thandi et al., 2002). For allosteric modulators, bias may be evident as pathway-dependent changes in three distinct parameters that dictate allosteric modulator activity; namely affinity, cooperativity and intrinsic efficacy. Indeed, we have clearly shown biased agonism and modulation to be operative amongst several chemotypes of mGlu5 ligands broadly classified as PAMs or PAM-agonists (Sengmany et al., 2017). Importantly, we revealed how previous classification of mGlu5 allosteric ligands based solely on their activity in iCa2+ mobilization assays failed to recognize the robust agonism characteristic of most mGlu5 allosteric ligands in IP1 accumulation and ERK1/2 phosphorylation assays (Sengmany et al., 2017). Thus, simply categorizing an mGlu5 allosteric ligand as a PAM or NAM based on a single functional assay precludes recognition of the rich complexity each ligand may offer.
While mGlu5 NAMs have been broadly classified as such based on their activity in iCa2+ mobilization assays, an insufficient understanding of mGlu5 modulator pharmacology is likely to have contributed to recent clinical failures. For instance, fenobam showed promise some 30 years ago in the treatment of generalized anxiety disorder (Pecknold et al., 1982), but it also impairs learning at therapeutic doses in preclinical models (Jacob et al., 2009). Cognition impairment, as well as abuse and psychoactive potential, remain common adverse effects amongst several mGlu5 NAMs including prototypical compounds such as MPEP and MTEP (Abou Farha et al., 2014; Dekundy et al., 2011; Hughes et al., 2013; Swedberg et al., 2014; Swedberg and Raboisson, 2014). It has been proposed that “partial NAMs”, i.e. NAMs with limited negative cooperativity, may offer the advantage of reduced adverse effect liability due to incomplete blockade of mGlu5 responses (Nickols et al., 2016). However, there remains a need to better quantify and assess the influence of chemically and pharmacologically diverse NAMs on mGlu5 activity to truly appreciate the underlying mechanisms of action that contribute to therapeutically beneficial versus adverse effects.
Here we aimed to determine the signaling profiles of select mGlu5 NAMs through rigorous assessment of interactions between mGlu5 orthosteric and negative allosteric ligands in iCa2+ mobilization and IP1 accumulation assays in both recombinant and neuronal systems. We show that some mGlu5 NAMs exhibit differential apparent affinities depending on cell background, pre-equilibration time, orthosteric agonist used, and, in mouse cortical neurons, co-application of CPCCOEt. Cooperativity of select NAMs was also influenced by co-application of CPCCOEt, and biased cooperativity was evident for VU0366248. In all, this study highlights the importance of robust evaluation of allosteric modulatory activity to appreciate the inherent complexity of ligand pharmacology. This study also highlights the potential pitfalls of applying standard high-throughput assays, with previously unappreciated activity at other signaling pathways potentially contributing to the efficacy and safety profiles of mGlu5 allosteric modulators in preclinical models.
2. Methods
2.1. Materials
Dulbecco’s modified Eagle’s medium (DMEM), Neurobasal medium, Fluo-4-AM and antibiotics were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was sourced from Thermo Electron Corporation (Melbourne, Australia). IP-ONE HTRF® assay kit was purchased from Cisbio, Genesearch (Arundel, Australia). Select mGlu5 ligands: 2-(1,3-benzoxazol-2-ylamino)-4-(4-fluorophenyl)pyrimidine-5-carbonitrile (VU0366058), 2-[2-(3-methoxyphenyl)ethynyl]-5-methylpyridine (M-5MPEP), 3-Fluoro-N-(4-methyl-2-thiazolyl)-5-(5-pyrimidinyloxy)benzamide (VU0409106), and N-(3-chloro-2-fluorophenyl)-3-cyano-5-fluorobenzamide (VU0366248) were synthesized as previously described (Felts et al., 2013; Mueller et al., 2012; Rodriguez et al., 2005; Sharma et al., 2008). (S)-3,5-dihydroxyphenylglycine (DHPG), 3-((2-methyl-1,3-thiazol-4-yl)ethynyl)pyridine hydrochloride (MTEP), and 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt) were purchased from Tocris Bioscience (Melbourne, Australia) and dipraglurant (ADX 48621) from ApexBio (Houston, TX). [3H]methoxy-PEPy was custom synthesized by Quotient Bioresearch (Rushden, Northamptonshire, UK) using the previously reported synthetic route (Cosford et al., 2003). Unless otherwise stated, all other reagents were purchased from Sigma-Aldrich (St. Louis, MO) and were of analytical grade.
2.2. Recombinant cell culture
HEK293A cells stably transfected with wild-type rat mGlu5 at low levels (HEK293A-rat mGlu5-low) comparable to those observed in primary cortical astrocytes (Noetzel et al., 2013) were maintained at 37°C and 5% CO2 in DMEM supplemented with 5% FBS, 16 mM HEPES and 500 µg/mL G418. A recombinant cell line with low mGlu5 expression was used as over-expression can result in pharmacological profiles that are not recapitulated in native cells, e.g. pure PAMs behaving as PAM-agonists (Noetzel et al., 2013). One day prior to experimentation, cells were plated onto poly-D-lysine coated, clear-bottom 96 well plates in assay buffer (glutamine-free DMEM supplemented with 5% dialyzed FBS, 16 mM HEPES and 500 µg/mL G418) at 40,000 cells/well.
2.3. Primary cell culture
All animal experiments were approved by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee (Protocol no. MIPS.2014.37). 8-week old female Asmu:Swiss wild-type mice were provided by the Monash Animal Research Platform (Clayton, VIC, Australia). Pregnant female mice were humanely sacrificed and E16 embryos collected for primary cell culture. Cortices were dissected from E16 Asmu:Swiss wild type mice and mechanically dissociated in Hank’s Balanced Salt Solution (HBSS: KCl 5.3 mM, KH2PO4 0.44 mM, NaHCO3 4.17 mM, NaCl 137.93 mM, Na2HPO4 0.34 mM, D-glucose 5.56 mM). Neurons were plated on poly-D-lysine, FBS-coated clear-bottom 96 well plates in Neurobasal media supplemented with 2 mM L-glutamine, 1 x B-27®, 50 U/mL penicillin, 50 U/mL streptomycin, 1.25 µg/mL Fungizone® antimycotic, at a density of 100,000 cells/well. Neurons were maintained at 37°C and 5% CO2 for 5–7 days prior to experimentation.
2.4. iCa2+ mobilization
iCa2+ mobilization was measured as previously described (Gregory et al., 2012). Briefly, changes in fluorescence of the Ca2+ indicator dye, Fluo-4-AM, were measured using a Flexstation I or III, with mGlu5 allosteric ligands added either 1 min or 30 min prior to orthosteric agonist. Experiments were conducted at room temperature (RT) for HEK293A-rat mGlu5-low cells, and at 37°C for mouse cortical neurons. A 5-point smoothing function was applied to raw fluorescence traces, with peak fluorescence normalized to the maximal responses of either glutamate (HEK293A-rat mGlu5-low) or the group I mGlu agonist DHPG (mouse cortical neurons).
2.5. Inositol monophosphate (IP1) accumulation assay
Recombinant cells or primary mouse cortical neurons were washed with phosphate buffered saline (PBS; 1.1 mM KH2PO4, 155 mM NaCl, 3 mM Na2HPO4, pH 7.4) and incubated with stimulation buffer (HBSS, with 20 mM HEPES, 30 mM LiCl2, 1.2 mM CaCl2, pH 7.4) supplemented with 1–10U/mL glutamic pyruvic transaminase and 6 mM sodium pyruvate for 1 h at 37°C and 5% CO2, followed by compound addition. After 1 h ligand incubation, cells were aspirated and lysed with lysis buffer (HTRF® IP-one assay kit) and IP1 levels detected as per kit instructions. Fluorescence was measured using the Envision plate reader (PerkinElmer), and expressed as fold over basal.
2.6. Whole cell radioligand binding
[3H]methoxy-PEPy whole-cell inhibition binding assays on mouse embryonic cortical neurons were performed in 24-well plates. For binding studies in HEK293A-rat mGlu5-low, cells were plated onto white-walled, clear bottom poly-D-lysine coated 96-well isoplates at a density of 40,000 cells/well in assay buffer (as described in section 2.2). Inhibition of ~2 nM [3H]methoxy-PEPy binding by increasing concentrations of the various allosteric ligands was assessed at RT for 1 h in HEPES-based binding buffer (145 mM NaCl, 10 mM D-glucose, 5 mM KCl, 1 mM MgSO4, 10 mM HEPES, 1.3 mM CaCl2, 15 mM NaHCO3, pH 7.4). The concentration of DMSO (0.3%) was kept constant throughout. Non-specific binding was determined using 10 µM MPEP. Assays were terminated by washing three times with ice-cold 0.9% NaCl. For mouse cortical neurons, cells were lysed with 250 µl/well of 0.2 M NaOH, lysates transferred to scintillation vials, 4 mL UltimaGold scintillation cocktail added and incubated for >2 h. For HEK293A-rat mGlu5-low cells, Microscint20 (40 µl/well) was added directly to isoplates, plates sealed and incubated for >2 h. Bound radioactivity was measured using either a MicroBeta2 plate counter or Tri-Carb 2900TR liquid scintillation counter (PerkinElmer, Waltham, MA).
2.7. Data analysis
Inhibition of [3H]methoxy-PEPy binding were fitted to either a one-site or two-site inhibition binding model as previously described (Gregory et al., 2012; Lazareno and Birdsall, 1995) and estimates of dissociation constants (KB) were derived using the Cheng-Prusoff equation (Cheng and Prusoff, 1973). For ligands that did not fully displace radioligand, inhibition binding curves were fitted with a modified allosteric ternary complex model to estimate KB (Lazareno and Birdsall, 1993):
| (1) |
where Y/Ymax is the fraction specific binding, the molar concentration of radioligand is [D] and KD is the radioligand equilibrium dissociation constant, [B] is the molar concentration of unlabeled allosteric modulator, and α is the affinity cooperativity factor between the radioligand and unlabeled allosteric modulator.
Allosteric modulation of glutamate or DHPG-mediated responses were fitted to the operational model of allosterism (Leach et al., 2007):
| (2) |
where [A] is the molar concentrations of orthosteric agonist (glutamate or DHPG). β is a scaling factor that denotes the magnitude of effect an allosteric modulator has on orthosteric agonist efficacy. In this respect, β is not a reciprocal efficacy cooperativity factor (Giraldo TIPS 2015). [B], KB, and α are as defined above. Here, affinity cooperativity (α) was assumed to be neutral between the orthosteric agonist and allosteric modulators as validated previously (Gregory et al., 2012) and thus constrained to a value of 1. KA is the equilibrium dissociation constant of the orthosteric agonist and was constrained to reported affinity estimates determined from inhibition binding assay as validated previously (Gregory et al., 2012; Mutel et al., 2000; Schoepp et al., 1994). Introducing this constraint to our analyses was necessary given that DHPG and glutamate behave as full agonists in the systems tested and therefore could not derive KA estimates directly from the functional data sets. τA and τB are operational measures of the respective ligand’s intrinsic efficacy, while Em and n represent the maximal system response and the transducer slope respectively.
Affinity and cooperativity estimates were also derived by globally fitting an orthosteric agonist concentration response curve (equation 3) and an allosteric modulator titration curve in the presence of a single concentration of agonist (equation 4):
| (3) |
| (4) |
where KA, τA, Em and basal values were shared across analyses. For equation 4, [A] was held constant to the molar agonist concentration added, that is, ~EC80 response in the particular assay.
All parameters were estimated as logarithms and expressed as mean ± SEM. Statistical analyses of binding data were performed as indicated using an extra sum-of-squares F test to determine the preferred model (one-site versus two-site binding) for each data set. Statistical analyses of functional assays were performed as indicated by using unpaired Student’s t-test or one-way analysis of variance (ANOVA) with Tukey’s or Sidak’s post hoc test, to compare affinity and cooperativity estimates between signaling assays and cell backgrounds.
3. Results
Eight mGlu5 allosteric ligands previously reported as NAMs of glutamate-mediated iCa2+ mobilization were chosen for this study (Fig. 1). All eight ligands have been proposed to interact with a “common allosteric mGlu5 site” located in the 7 transmembrane (7TM) spanning domain (Gregory et al., 2012; Porter et al., 2005). MPEP, a disubstituted alkyne, is a prototypical mGlu5 NAM (Gasparini et al., 1999) from which MTEP, reported to have greater mGlu5 selectivity (over mGlu1) and potency, was derived (Cosford et al., 2003b; Iso et al., 2006). M-5MPEP, also derived from MPEP, has limited negative cooperativity (also referred to as partial NAM activity) with glutamate (Nickols and Conn, 2014; Rodriguez et al., 2005). VU0409106, VU0366058 and VU0366248 represent chemotypes distinct from MPEP – namely the aryl ether series (Felts et al., 2013), 5-cyanopyrimides (Mueller et al., 2012) and 3-cyano-5-fluoro-N-arylbenzamides (Felts et al., 2010) respectively. Previous reports indicate VU0409106 and VU0366058 are full NAMs of glutamate-mediated iCa2+ mobilization, while VU0366248 is a partial NAM (Felts et al., 2013; Gregory et al., 2012). Fenobam and dipraglurant were included to represent mGlu5 NAMs that have progressed to clinical trials (Pecknold et al., 1982; Tison et al., 2016).
Figure 1. Inhibition of [3H]methoxy-PEPy binding to HEK293A-rat mGlu5-low cells and mouse cortical neurons.
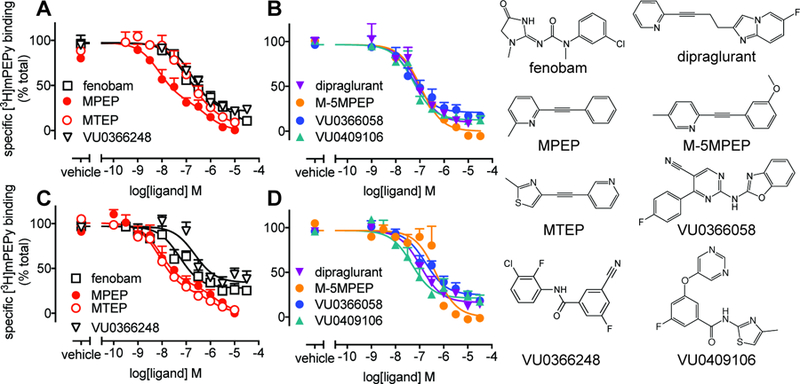
Using intact adherent cells, inhibition of [3H]methoxy-PEPy binding was determined in HEK293A-rat mGlu5-low cells (A & B) and mouse cortical neurons (C & D). Data were fitted to either a one-site or two-site inhibition-binding model as determined by an F-test on each individual experiment. Data are mean + SEM from 3–6 independent experiments performed in duplicate.
3.1. Affinity estimation for diverse mGlu5 NAMs using radioligand binding studies in native and recombinant cells.
Radioligand inhibition binding studies were conducted on intact adherent HEK293A-rat mGlu5-low cells and primary mouse cortical neurons (Fig. 1). A recombinant cell line with comparable expression to native cells was selected as over-expression of mGlu5 can influence allosteric modulator pharmacology (Noetzel et al., 2013). Interestingly, MPEP and MTEP inhibition binding curves were best fitted to a two-site model in both recombinant and native cells. Fenobam inhibition curves were also consistent with two-site binding in recombinant cells, but best fitted to a one-site allosteric ternary complex model in neurons. M-5MPEP, VU0366248, VU0409106 and dipraglurant inhibition curves were consistent with one-site binding in both cell types. VU0366058, VU0366248, VU0409106 and dipraglurant did not completely displace [3H]methoxy-PEPy binding in either cell type and curves were fitted with an allosteric ternary complex model to derive affinity (pKB) and cooperativity (logα) estimates (Table 1). With the exception of MTEP, allosteric ligands had similar pKB estimates between HEK293A-rat mGlu5-low and mouse cortical neurons (Table 1).
Table 1. Binding affinities of mGlu5 allosteric ligands derived from inhibition of [3H]methoxy-PEPy binding in intact and adherent HEK293A-rat mGlu5-low cells and mouse cortical neurons.
Data represent mean ± SEM of 3–6 independent experiments performed in duplicate.
| HEK293A-rat mGlu5-low pKB | Mouse cortical neurons pKB | |||||||
|---|---|---|---|---|---|---|---|---|
| High | Low | Fraction High | Logα | High | Low | Fraction High | Logα | |
| MPEP | 8.26 ±0.23 | 6.25 ±0.11 | 0.62 ±0.11 | n.a. | 8.38 ±0.30 | 5.92 ±0.20 | 0.73 ±0.11 | n.a. |
| fenobam | 7.15 ±0.28 | 4.66 ±0.22 | 0.71 ±0.09 | n.a. | 7.53 ±0.37a | n.a. | 1a | −0.66 ±0.10 |
| VU0409106 | 7.47 ±0.07 | n.a. | 1 | −1.12 ±0.12 | 7.48 ±0.15 | n.a. | 1 | −0.78 ±0.13 |
| VU0366248 | 6.94 ±0.13 | n.a. | 1 | −0.77 ±0.06 | 6.72 ±0.06 | n.a. | 1 | −0.54 ±0.06 |
| VU0366058 | 7.26 ±0.10 | n.a. | 1b | −0.82 ±0.07 | 6.76 ±0.23 | n.a. | 1b | −0.71 ±0.07 |
| M-5MPEP | 7.13 ±0.07 | n.a. | 1 | n.a. | 6.43 ±0.25 | n.a. | 1 | n.a. |
| dipraglurant | 7.31 ±0.11 | n.a. | 1 | −1.24 ±0.17 | 6.98 ±0.14 | n.a. | 1 | −0.89 ±0.08 |
| MTEP | 7.20 ±0.28 | 5.45 ±0.30 | 0.83 ±0.04 | n.a. | 8.16 ±0.14* | 6.09 ±0.45 | 0.74 ±0.04 | n.a. |
One individual experiment (from n=6) was best fitted to a two-site binding curve (F test p>0.05).
Two individual experiments (from n=5–6) were best fitted to a two-site binding curve.
Denotes p<0.05, One-way ANOVA, Sidak’s multiple comparisons test, compared to HEK293A-rat mGlu5-low
n.a. not applicable due to one-site or competitive binding curve fit
3.2. mGlu5 allosteric ligands are NAMs of glutamate-mediated iCa2+ mobilization in HEK293A-rat mGlu5-low cells.
In agreement with previous studies (Felts et al., 2010; Felts et al., 2013; Gregory et al., 2012; Mueller et al., 2012; Porter et al., 2005), all eight allosteric ligands were NAMs of glutamate-mediated iCa2+ mobilization in HEK293A-rat mGlu5-low cells after 1 min pre-incubation (Fig. 2). In particular, MPEP, MTEP, fenobam, dipraglurant and VU0409106 were full NAMs, while M-5MPEP and VU0366248 displayed limited negative cooperativity. VU0366058 is fluorescent, which limited the testing of concentrations above 1 µM, however, inhibition of glutamate-mediated iCa2+ mobilization was consistent with high negative cooperativity. In order to quantify functional affinity and cooperativity estimates, NAM interactions with glutamate were fitted to an operational model of allosterism (Leach et al., 2007). With the exception of dipraglurant, functional affinity estimates (pKB values) were in good agreement with binding estimates (for the high affinity site where applicable) determined from inhibition binding assays in HEK293A-rat mGlu5-low cells (Tables 1 & 2). The affinity of dipraglurant was significantly higher in iCa2+ mobilization assays (7 fold) relative to the inhibition binding estimate. All NAMs had similar magnitudes of negative cooperativity with glutamate as previously reported (Table 2, Gregory et al., 2010).
Figure 2. Negative allosteric modulation of glutamate-mediated iCa2+ mobilization in HEK293A-rat mGlu5-low cells.
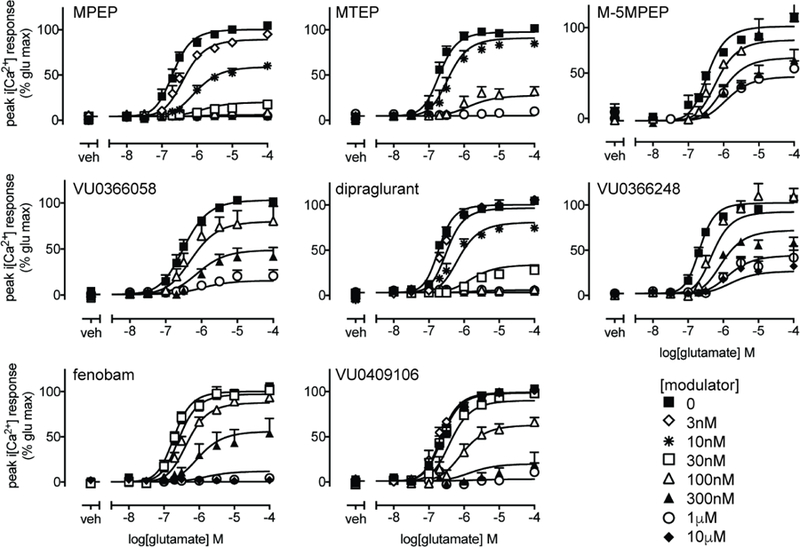
Concentration response curves for glutamate mediated iCa2+ mobilization in the absence or presence of indicated allosteric ligands following 1 min pre-incubation. Data are expressed as mean + SEM of 3–9 experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbols.
Table 2. Comparison of affinity and cooperativity estimates for allosteric modulation of DHPG and glutamate-mediated iCa2+ mobilization following 1 min vs 30 min pre-incubation of mGlu5 allosteric ligands in HEK293A-rat mGlu5-low cells.
Data represent mean ± SEM of indicated number (n) of independent experiments performed in duplicate.
| Glutamate | DHPG | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 min | 30 min | 1 min | 30 min | |||||||||
| pKBa | log βb | n | pKB | log β | n | pKB | log β | n | pKB | log β | n | |
| MPEP | 8.39±0.13 | Full NAMc | 7 | 7.90±0.08 | Full NAM | 4 | 9.08±0.14d,f | full NAM | 7 | 8.29 ±0.16e | full NAM | 5 |
| fenobam | 7.04±0.09 | Full NAM | 9 | 7.31±0.08 | Full NAM | 7 | 6.91±0.13 | full NAM | 5 | 6.98 ±0.14 | full NAM | 5 |
| VU0409106 | 7.27±0.12 | Full NAM | 7 | 7.38±0.12 | Full NAM | 6 | 7.03±0.14 | full NAM | 5 | 7.67 ±0.21e | full NAM | 3 |
| VU0366248 | 7.22±0.09 | −0.90±0.12 | 7 | 6.57±0.18e | −0.58±0.08 | 7 | 7.43±0.19 | −1.23±0.48 | 5 | 6.45 ±0.24e | −0.40 ±0.08 | 4 |
| VU0366058 | 6.98±0.11 | Full NAM | 5 | 7.11±0.28 | Full NAM | 5 | 6.71±0.13 | full NAM | 5 | 7.02 ±0.24 | full NAM | 4 |
| M-5MPEP | 7.00±0.07 | −0.59±0.03 | 3 | 6.72±0.26 | −0.52±0.06 | 3 | 6.70±0.15 | −0.97±0.24 | 5 | 6.49 ±0.24 | −0.47 ±0.09 | 3 |
| dipraglurant | 8.16±0.06d | Full NAM | 6 | 7.47±0.07e | Full NAM | 6 | 8.06±0.14d | full NAM | 6 | 7.31 ±0.16e | full NAM | 4 |
| MTEP | 7.83±0.09 | Full NAM | 3 | 6.97±0.15e | Full NAM | 6 | 8.37±0.14d | full NAM | 6 | 7.81 ±0.18f | full NAM | 5 |
pKB, negative logarithm of the equilibrium dissociation constant determined using an operational model of allosterism
log β, logarithm of the efficacy cooperativity scaling factor determined using an operational model of allosterism where α was assumed to be equal to 1
“full NAM” denotes complete inhibition of DHPG response, such that β approaches zero.
Denotes p<0.05, One-way ANOVA, Tukey’s multiple comparisons test, compared to binding pKi estimates
Denotes p<0.05, One-way ANOVA, Tukey’s multiple comparisons test, compared to pKB estimate derived from iCa2+ mobilization assays (1 min versus 30 min paradigm)
Denotes p<0.05, One-way ANOVA, Tukey’s multiple comparisons test, compared to pKB estimate derived from glutamate iCa2+ mobilization assays (equivalent incubation paradigm)
3.3. Ligand-receptor equilibrium influences mGlu5 NAM apparent affinity in HEK293A-rat mGlu5-low based on inhibition of glutamate stimulated iCa2+ mobilization
Differing ligand incubation times between different assays may result in potential bias within a kinetic context (Klein Herenbrink et al., 2016; Lane et al., 2017), especially for non-equilibrium assays. Non-equilibrium conditions may also contribute to differences in affinity estimates. Therefore, we extended the NAM pre-incubation period to 30 min prior to conducting glutamate-mediated iCa2+ mobilization assays in HEK293A-rat mGlu5-low cells (Fig 3). With an increased pre-incubation time for dipraglurant prior to iCa2+ mobilization assays, the pKB value was in much closer agreement with the binding estimate (Table 2). Extended pre-incubation with MTEP and VU0366248 resulted in lower pKB estimates (7 and 4-fold respectively) when compared with the 1 min assay. VU0366248 also had 2-fold lower negative cooperativity with glutamate although this did not reach significance (Table 2). Cooperativity with glutamate for all other NAMs was similar between the two temporal conditions.
Figure 3. Negative allosteric modulation of glutamate-mediated iCa2+ mobilization in HEK293A-rat mGlu5-low cells with extended pre-incubation.
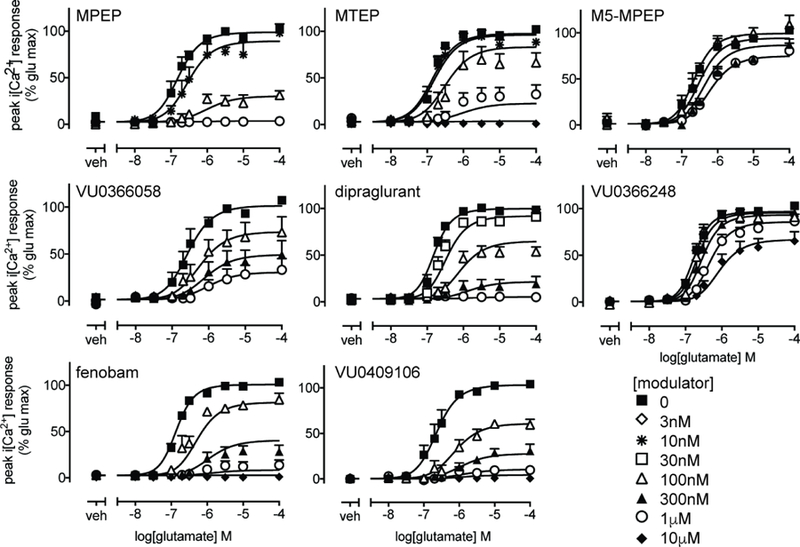
Concentration response curves for glutamate mediated iCa2+ mobilization in the absence or presence of indicated allosteric ligands following 30 min pre-incubation. Data are expressed as mean + SEM of 3–7 experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbols.
3.4. mGlu5 NAMs are inverse agonists for IP1 accumulation in HEK293A-rat mGlu5-low cells
IP1 accumulation was assessed as an alternative signaling endpoint, given previous observations of biased agonism/modulation for mGlu5 PAMs between iCa2+ mobilization and IP1 accumulation (Sengmany et al., 2017). Measurement of IP1 accumulation provides insight into receptor activity at ligand equilibrium relative to transient, non-equilibrium, iCa2+ mobilization responses. In the presence of glutamic pyruvic transaminase (GPT), which eliminates ambient glutamate, all eight mGlu5 NAMs decreased baseline IP1 accumulation in a concentration dependent manner in the absence of agonist (Fig. 4). In the absence of GPT, most NAMs exhibited a greater maximal inhibition of baseline IP1 accumulation, attributable to the presence of ambient glutamate (Supplementary Figure 1). However, in the presence of both 1U/mL and 10U/mL GPT, baseline inhibition is reduced to a similar extent. This indicates that the assay conditions have eliminated ambient glutamate and that these data are consistent with constitutive mGlu5 activity and inverse agonism. The potencies of MTEP and dipraglurant as inverse agonists were similar to binding affinity estimates, whereas for the remaining six NAMs, potencies were 2–4 fold lower than binding pKB values (Table 3). Due to appreciable inverse agonism for all eight NAMs for IP1 accumulation in HEK293A-rat mGlu5-low cells, it was not feasible to assess interactions between NAMs and glutamate using the operational model of allosterism applied here. Alternative operational models that incorporate constitutive activity are available, however, these models also require the ability to measure ligand responses in systems with different degrees of constitutive activity (Slack and Hall, 2012; Hall 2013).
Figure 4. Inverse agonism of constitutive IP1 accumulation in HEK293A-rat mGlu5-low cells but not in primary mouse cortical neuron cultures.
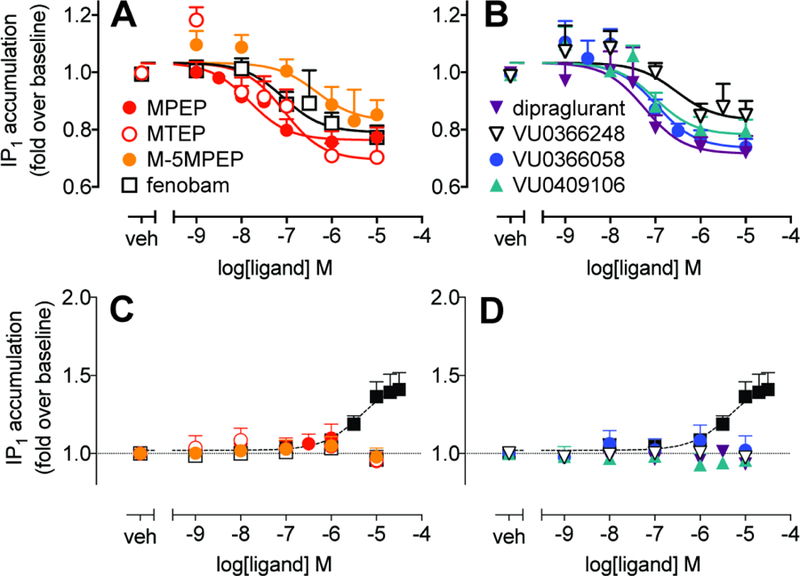
A & B) Concentration response curves for inhibition of constitutive mGlu5-mediated IP1 accumulation by indicated allosteric ligands in HEK293A-rat mGlu5-low cells. Data are expressed as mean + SEM of 5–9 experiments performed in duplicate. C &D) No change in IP1 accumulation basal levels were observed following 60 min exposure of primary mouse cortical neuron cultures to NAMs. Glutamic pyruvic transaminase (1U/mL) was included to eliminate ambient glutamate. A DHPG control curve from parallel experiments is shown for reference (closed black squares). Data are mean + SEM from 3–4 independent experiments performed in parallel. For all panels, error bars not shown lie within the dimensions of the symbols.
Table 3. Potency and efficacy of mGlu5 NAMs as inverse agonists of IP1 accumulation in HEK293A-rat mGlu5-low cells.
Data represent mean ± SEM of indicated number (n) of independent experiments performed in duplicate.
| pIC50a | Imaxb | n | |
|---|---|---|---|
| MPEP | 7.86 ±0.27 | 0.74 ±0.03 | 8 |
| fenobam | 6.83 ±0.26 | 0.76 ±0.03 | 9 |
| VU0409106 | 6.89 ±0.26 | 0.74 ±0.05 | 7 |
| VU0366248 | 6.48 ±0.16 | 0.81 ±0.05 | 7 |
| VU0366058 | 6.98 ±0.17 | 0.73 ±0.03 | 9 |
| M-5MPEP | 6.53 ±0.17 | 0.81 ±0.05 | 7 |
| dipraglurant | 7.36 ±0.15 | 0.73 ±0.04 | 6 |
| MTEP | 7.36 ±0.16 | 0.70 ±0.05 | 5 |
negative logarithm of the molar concentration required to give a half maximal inhibitory response
maximal inhibitory response, expressed as fold over basal IP1 accumulation levels.
3.5. mGlu5 allosteric ligands are NAMs of DHPG-mediated iCa2+ mobilization in HEK293A-rat mGlu5-low cells and cultured mouse cortical neurons.
We next sought to confirm the pharmacology of all eight ligands in primary mouse embryonic cortical neuron cultures. The complex cell background in neuronal cells limits the use of glutamate as an orthosteric agonist due to the presence of other glutamate receptors and transporters. Thus, we adopted a commonly used approach to measure mGlu signaling in response to the mGlu1/5 orthosteric agonist, DHPG, co-added with the mGlu1 NAM, CPCCOEt (to minimize mGlu1 signaling) (Jong et al., 2009; Kettunen et al., 2002; Luccini et al., 2007; Sengmany et al., 2017; Viwatpinyo and Chongthammakun, 2009). The nature of allosteric interactions are dependent on the two chemical entities present, a phenomenon known as probe dependence, therefore we first assessed affinity and cooperativity profiles of the NAMs with DHPG in HEK293A-rat mGlu5-low cells. To do so, we analyzed NAM inhibition of an EC80 DHPG iCa2+ mobilization response, in parallel with a control DHPG concentration-response curve, using both 1 min and 30 min pre-incubation paradigms (Supplementary Figure 2 and Table 2). NAM affinity and cooperativity estimates derived from DHPG iCa2+ mobilization inhibition curves were generally similar to those derived from glutamate inhibition curves, although there were a few exceptions (Table 2). For instance, MPEP and MTEP had higher affinity estimates when DHPG was used as the agonist compared to glutamate, with significant differences observed for MPEP using the 1 min paradigm (3-fold) and for MTEP using the 30 min paradigm (10 fold). Differential apparent affinities for these two NAMs under the same assay conditions but derived from interaction studies with different agonists is suggestive of probe dependence.
In mouse cortical neurons, inclusion of 30 µM CPCCOEt had no effect on DHPG potency or Emax for iCa2+ mobilization (Supplementary Figure 3). None of the eight NAMs influenced basal iCa2+ mobilization in the absence of agonist in mouse cortical neurons (Supplementary Figure 4). All compounds inhibited the response to an EC80 DHPG concentration in a concentration dependent manner following 1 min pre-incubation (Fig. 5A–B). In line with pharmacological profiles in recombinant cells, maximal concentrations of MPEP, fenobam, VU0409106, dipraglurant or MTEP resulted in complete inhibition of the response to 1µM DHPG (an approximately EC80 concentration for iCa2+ mobilization), whereas VU0366248 or M-5MPEP had limited negative cooperativity, as evidenced by incomplete inhibition of DHPG-mediated iCa2+ mobilization (Fig 5A–B). VU0366058 also showed incomplete inhibition of DHPG but could not be definitively characterized as a limited or full NAM due to the restricted concentration range. Similar to observations in recombinant cells, quantification of these data with the operational model of allosterism revealed there were anomalies with respect to binding versus functional pKB estimates for some ligands. For dipraglurant the pKB estimate derived from iCa2+ mobilization assays was significantly greater than the binding value (5-fold, Table 4). Extending the pre-incubation time in mouse cortical neurons had no significant effect on NAM pKB estimates, although there was a trend for reduced affinity for MTEP and dipraglurant (Fig 5C, D, Table 4). Increasing the pre-incubation time had no effect on negative cooperativity with DHPG for any of the NAMs (Table 5). Interestingly, extending the pre-incubation time resulted in VU0366058 becoming a full NAM at the concentration range tested.
Figure 5. Negative allosteric modulator activity at iCa2+ mobilization in embryonic mouse cortical neurons in the presence or absence of CPCCOEt 30 µM.
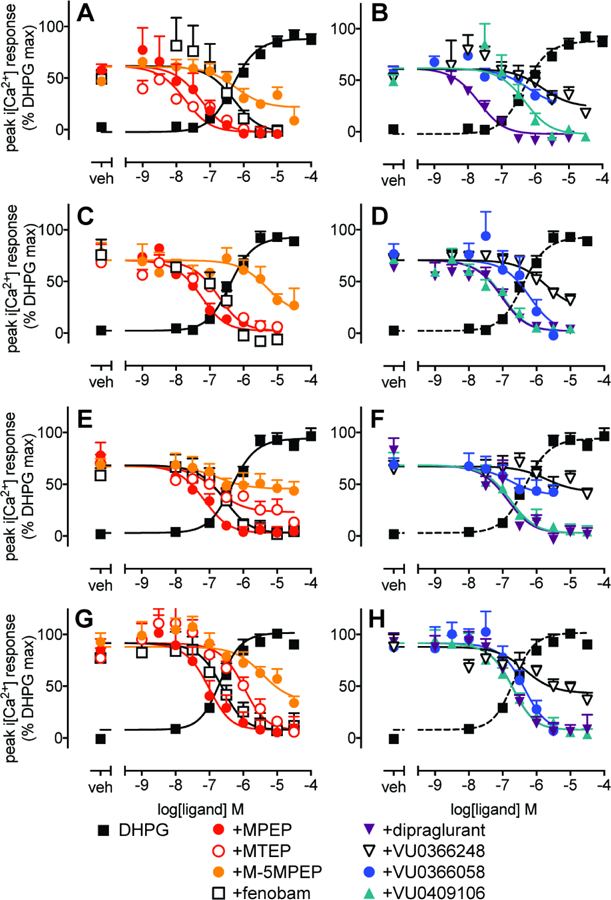
Concentration response curves for modulation of DHPG EC80 (1 µM)-mediated iCa2+ mobilization with 1min (A & B) or 30 min (C & D) pre-incubation with mGlu5 NAMs were performed in parallel with DHPG concentration-response curves. Modulation of DHPG-stimulated iCa2+ mobilization was also assessed in the absence of CPCCOEt with 1 min (E & F) or 30 min (G & H) pre-incubation with mGlu5 NAMs. Data are expressed as mean + SEM of 4–8 experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbols.
Table 4: Comparison of mGlu5 NAM functional affinity estimates (pKB) across different measures of receptor activity in primary mouse cortical neurons in the presence and absence of 30 µM CPCCOEt.
Data represent mean ± SEM of indicated number (n) of independent experiments performed in duplicate.
| With CPCCOEt | no CPCCOEt | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| iCa2+ (1min) | n | iCa2+ (30min) | n | IP1 accumulation | n | iCa2+ (1min) | n | iCa2+ (30min) | n | IP1 accumulation | n | |
| MPEP | 7.61 ±0.20a | 6 | 7.61 ±0.15 | 5 | 7.69 ±0.19 | 11 | 7.46 ±0.16a | 5 | 7.59 ±0.17 | 7 | 7.42 ±0.22 | 10 |
| fenobam | 6.62 ±0.23 | 4 | 7.10 ±0.17 | 4 | 6.63±0.28 | 9 | 6.85 ±0.18 | 4 | 7.20 ±0.17 | 6 | 6.51 ±0.24 | 7 |
| VU0409106 | 6.63 ±0.20 | 6 | 7.35 ±0.15 | 5 | 6.32 ±0.26b | 10 | 7.08 ±0.17 | 4 | 7.28 ±0.18 | 6 | 6.68 ±0.24 | 8 |
| VU0366248 | 5.99 ±0.35a | 7 | 6.04 ±0.33 | 5 | n.r. | 5 | 5.91 ±0.62a | 5 | 6.86 ±0.29 | 5 | n.r. | 5 |
| VU0366058 | 6.43 ±0.45 | 6 | 6.55 ±0.14 | 6 | 6.36 ±0.24 | 10 | 7.00 ±0.50 | 6 | 6.94 ±0.17 | 6 | 6.24 ±0.24 | 8 |
| M-5MPEP | 6.47 ±0.30 | 6 | 5.64 ±0.25 | 8 | 6.24 ±0.39 | 10 | 6.93 ±0.48 | 8 | 5.98 ±0.29 | 5 | 5.94 ±0.31 | 11 |
| dipraglurant | 8.03 ±0.23b | 5 | 7.39 ±0.15 | 5 | 6.94 ±0.20c | 9 | 7.16±0.16a | 5 | 7.28 ±0.17 | 6 | 6.88 ±0.12 | 7 |
| MTEP | 8.06 ±0.22 | 5 | 7.09 ±0.16 | 4 | 6.89 ±0.24c | 8 | 7.11±0.28a | 5 | 6.55 ±0.18a,b | 6 | 6.90 ±0.28 | 8 |
Denotes p<0.05, One-way ANOVA, Sidak’s multiple comparisons test, compared to pKB estimate derived from modulation of DHPG mediated iCa2+ mobilization in HEK293A-rat mGlu5-low cells under comparable assay paradigm (see Table 2).
Denotes p<0.05, One-way ANOVA, Sidak’s multiple comparisons test, compared to binding estimate
Denotes p<0.05, One-way ANOVA, Sidak’s multiple comparisons test, compared with pKB estimate derived from iCa2+ mobilization assays using a 1min paradigm
n.r. no modulatory response was evident.
Table 5: Comparison of mGlu5 NAM cooperativity (logβ) values with DHPG across different measures of receptor activity in primary mouse cortical neurons in the presence and absence of 30 µM CPCCOEt.
Data represent mean ± SEM of indicated number (n) of independent experiments performed in duplicate.
| With CPCCOEt | no CPCCOEt | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| iCa2+ (1min) | n | iCa2+ (30min) | n | IP1 accumulation | n | iCa2+ (1min) | n | iCa2+ (30min) | n | IP1 accumulation | n | |
| MPEP | full NAMa | 6 | full NAM | 5 | −0.56 ±0.11 | 11 | full NAM | 5 | full NAM | 7 | −0.55 ±0.09 | 10 |
| fenobam | full NAM | 4 | full NAM | 4 | −0.59 ±0.17 | 9 | full NAM | 4 | full NAM | 6 | −0.67 ±0.17 | 7 |
| VU0409106 | full NAM | 6 | full NAM | 5 | −0.39 ±0.09 | 10 | full NAM | 4 | full NAM | 6 | −0.52 ±0.09 | 8 |
| VU0366248 | −0.60 ±0.21 | 7 | −0.75±0.31 | 5 | n.r. | 5 | −0.36±0.15 | 5 | −0.69 ±0.09 | 5 | n.r. | 5 |
| VU0366058 | −0.59 ±0.32b | 6 | full NAM | 6 | −0.46 ±0.11 | 10 | −0.41 ±0.15 | 6 | full NAM | 6 | −0.69 ±0.20 | 8 |
| M-5MPEP | −0.65 ±0.19 | 6 | −0.88 ±0.35 | 8 | −0.24 ±0.06 | 10 | −0.31 ±0.08 | 8 | −0.80 ±0.18 | 5 | −0.33 ±0.08 | 10 |
| dipraglurant | full NAM | 5 | full NAM | 5 | full NAM | 9 | full NAM | 5 | full NAM | 6 | full NAM | 7 |
| MTEP | full NAM | 5 | full NAM | 4 | −0.52 ±0.13 | 8 | −0.68±0.16 | 5 | full NAM | 6 | −0.38 ±0.07 | 8 |
“full NAM” denotes complete inhibition of DHPG response, such that β values approach zero.
limited concentration range was tested due to compound fluorescence and therefore cannot definitively conclude cooperativity is limited.
n.r. no modulatory response was evident.
3.6. Inhibition of mGlu1 affects mGlu5 NAM affinity and cooperativity with DHPG in mouse cortical neuron iCa2+ mobilization assays
Given the observed differences between NAM pKB estimates determined in neuronal radioligand binding assays versus functional assay estimates derived in the presence of CPCCOEt, we sought to ensure that CPCCOEt did not influence mGlu5 NAM affinity and/or cooperativity. This was of particular concern for two reasons: (i) we recently showed that CPCCOEt binds to mGlu5 (with comparable affinity for mGlu1) albeit it exhibits neutral cooperativity with mGlu5 agonists (Hellyer et al., 2018), (ii) mGlu1 and mGlu5 can heterodimerize to form higher order oligomers (Correa et al., 2017), thus CPCCOEt could influence mGlu5 across an mGlu1/mGlu5 dimer. Hence, we re-evaluated the pharmacology of the eight studied mGlu5 allosteric ligands in the absence of CPCCOEt in mouse cortical neurons (Fig 5E–H). In general, in the absence of CPCCOEt, iCa2+ mobilization assays with either 1 or 30 min NAM pre-incubation yielded NAM pKB estimates that were in good agreement with binding estimates (Table 4). The single exception was MTEP, which had a 41-fold lower pKB estimate in the 30 min paradigm in the absence of CPCCOEt (Table 4). There were no significant differences between NAM pKB estimates in the absence versus presence of CPCCOEt when equivalent paradigms were assessed. Cooperativity of NAMs with DHPG were similar in the absence and presence of CPCCOEt, with the exception of VU0366058 and MTEP which transformed from full to partial NAMs in the 1 min pre-incubation paradigm (Table 5). Thus, inclusion of CPCCOEt in the cortical neuron functional assays influenced mGlu5 NAM pharmacology in a ligand-dependent manner. It is presently not clear whether this is due to unappreciated CPCCOEt activity at mGlu5 alone, or mediated across an mGlu1/mGlu5 heteromer.
3.7. mGlu5 allosteric ligands have differing degrees of cooperativity with DHPG when assessed in IP1 accumulation in cultured mouse cortical neurons.
In contrast to recombinant cells, there was no evidence for inverse agonist activity for all eight mGlu5 NAMs for IP1 accumulation in mouse cortical neurons (Figure 4), indicating that the receptor has low or no constitutive activity in these native cells. Inclusion of CPCCOEt, had no significant effect on DHPG potency or Emax for IP1 accumulation (Supplementary Figure 3) compared to untreated neurons. In the absence and presence of CPCCOEt, with the exception of VU0366248, all of the allosteric modulators were NAMs of DHPG-mediated IP1 accumulation in mouse cortical neurons (Fig 6). VU0366248 showed negligible modulatory activity of DHPG-mediated IP1 accumulation (Fig 6B, D), suggesting a loss of negative cooperativity with DHPG. Interestingly, only dipraglurant was able to completely abolish the response to an EC80 DHPG concentration in both conditions, whereas all other NAMs were partial (Fig 6, Table 5). Analysis of interactions with DHPG in the presence of CPCCOEt revealed that dipraglurant and MTEP had significantly lower (~10-fold) affinity estimates in IP1 accumulation relative to 1 min pre-incubation iCa2+ mobilization assays, and VU0409106 pKB in IP1 accumulation was 14-fold lower than the binding estimate (Table 4). In the absence of CPCCOEt, affinity estimates from IP1 accumulation assays were not significantly different to inhibition binding for all ligands (Table 4). There were no significant differences in functional affinity estimates for any of the eight NAMs between the presence and absence of CPCCOEt, based on modulation of DHPG-stimulated IP1 accumulation (Fig 6C–D; Table 4).
Figure 6. Inhibition of DHPG EC80-mediated IP1 accumulation by mGlu5 NAMs in mouse cortical neurons in the presence or absence of CPCCOEt 30 µM.
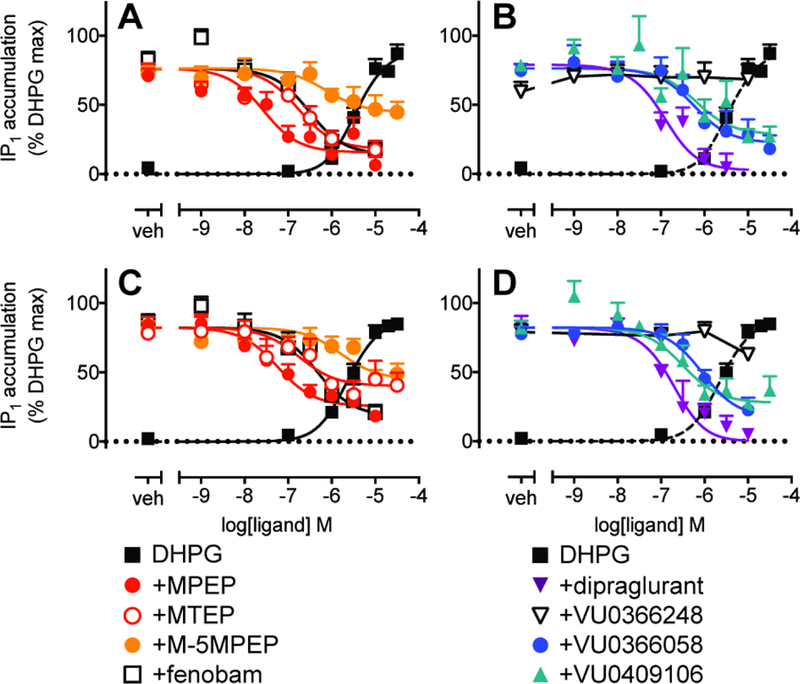
Concentration response curves for modulation of DHPG EC80 (20 µM)-mediated IP1 accumulation in the presence (A & B) or absence (C & D) of CPCCOEt. Data are expressed as mean + SEM of 5–10 experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbols.
3.8. Comparison of signaling fingerprints between recombinant and native cells based on affinity and cooperativity estimates.
To better visualize the differences and similarities in mGlu5 NAM pharmacology in recombinant and native systems, we plotted the functional affinity estimates (based on modulation of DHPG) relative to binding estimates side by side (Fig. 7). Of note, when comparing affinity estimates between the different cell types in equivalent iCa2+ mobilization assays with the same orthosteric agonist, MPEP, MTEP, dipraglurant and VU0366248 had lower apparent affinities in mouse cortical neurons compared to recombinant cells although this was assay dependent (Table 4). The switch of VU0366248 to neutral cooperativity in cortical neuron IP1 accumulation assays precluded estimation of affinity. Acetylenic full NAMs (MPEP, MTEP and dipraglurant) had similar pKB fingerprints, with affinity estimates influenced by assay conditions and cell type (Fig 7). In contrast, functional affinity estimates of the acetylenic partial NAM M-5MPEP and the structurally distinct NAMs, VU0366058, fenobam and VU0409106, were generally consistent across all measures in both cell systems. Importantly, these data suggest that the differences in functional affinity estimates observed between IP1 accumulation and iCa2+ mobilization assays are ligand dependent and not simply attributable to differences in assay conditions. For the most part, magnitudes of negative cooperativity between NAMs and DHPG were similar in neurons to HEK293A-rat mGlu5-low cells (Table 5, Table 2).
Figure 7. Comparison of affinity estimates in HEK293A-rat mGlu5-low cells and mouse cortical neurons.

The difference in affinity estimates from functional assays in HEK293A-mGlu5 low cells or mouse cortical neurons versus inhibition binding. Black bars: 1 min iCa2+ mobilization, red bars: 30 min iCa2+ mobilization, blue bars: IP1 accumulation. Data are mean + SEM from 3–10 experiments. Comparisons were performed using one-way ANOVA with Sidak’s post-test, where significance (*) was considered p<0.05. # denotes not determined due to lack of appreciable modulation.
4. Discussion
Inhibition of mGlu5 is proposed to offer therapeutic potential for CNS disorders, ranging from anxiety and depression to autism and addiction (Sengmany and Gregory, 2016). However, adverse effect liability in the form of cognitive impairments (Simonyi et al., 2010), abuse and psychotomimetic potential (Abou Farha et al., 2014; Swedberg et al., 2014; Swedberg and Raboisson, 2014), highlights the need to better understand the underlying pharmacology of mGlu5 inhibitors. A deeper understanding of mGlu5 NAM pharmacology may provide insights into the differential profiles of these compounds in preclinical animal models and allow for the rational design of ligands that display improved efficacy and safety. Our quantitative pharmacological profiling of eight ligands, previously classified as mGlu5 NAMs of glutamate in iCa2+ mobilization assays, revealed differences in apparent affinities between binding and functional assays (iCa2+ mobilization and IP1 accumulation) for acetylenic full NAMs (MPEP, MTEP and dipraglurant) in recombinant and/or neuronal systems, whereas, non-acetylenic NAMs or the prototypical acetylenic “partial NAM” M-5MPEP were similar across all measures. Biased NAM pharmacology was evident at the level of cooperativity with DHPG; most strikingly in mouse cortical neurons where VU0366248 behaved as a neutral allosteric ligand in IP1 accumulation assays but was a NAM of DHPG-mediated iCa2+ mobilization. Importantly, M-5MPEP and dipraglurant had similar levels of cooperativity with DHPG and glutamate, independent of cell type or assay paradigm, demonstrating an absence of bias at the level of cooperativity. Collectively, our study highlights the importance of rigorous assessment of allosteric ligands to quantify affinity and cooperativity, thereby enriching our appreciation of how aspects of mGlu5 NAM pharmacology may contribute to efficacy and safety profiles in preclinical models.
Several orthosteric agonists of mGlu receptors are well-established biased agonists (Emery et al., 2012; Hathaway et al., 2015), while positive allosteric modulators of Class C GPCRs also engender bias with respect to agonism and/or cooperativity (Cook et al., 2015; Sengmany et al., 2017; Zhang et al., 2005). At the class C calcium sensing receptor, the NAM NPS2143 demonstrated biased modulation toward iCa2+ mobilization relative to phosphorylation of ERK1/2 (Davey et al., 2012; Leach et al., 2016; Leach et al., 2013). However, for many of the mGlu5 NAMs studied herein, a single in vitro functional assay (iCa2+ mobilization) has been used to classify allosteric pharmacology. There are some exceptions, namely MPEP, M-5MPEP, VU0366248 and VU0366058, for which two different functional assays (iCa2+ mobilization and ERK1/2 phosphorylation) using glutamate as the orthosteric agonist in recombinant cells have been assessed (Gregory et al., 2012). No significant bias in functional affinity or cooperativity estimates between iCa2+ mobilization and ERK1/2 phosphorylation were observed (Gregory et al., 2012). In contrast, herein multiple NAMs showed differential magnitudes of cooperativity with DHPG in native cells depending on the measure (iCa2+ mobilization versus IP1 accumulation). Importantly, these differences were not observed for all NAMs, with M-5MPEP and dipraglurant retaining similar cooperativity with DHPG across all measures and cell types tested in the present study. Thus, our results highlight the need to assess multiple receptor endpoints to probe the full scope of allosteric ligand pharmacology.
Our results are also distinct from previous work in demonstrating the presence of two-site inhibition binding. Inhibition of [3H]methoxy-PEPy binding by MPEP, MTEP and fenobam were best fitted to a two-site inhibition binding model, contrary to the one-site binding previously reported (Gregory et al., 2012; Lea and Faden, 2006; Porter et al., 2005). Furthermore, dipraglurant, VU0366058, VU0409106 and VU0366248 did not fully displace [3H]methoxy-PEPy binding under our assay conditions. There are multiple possible underlying explanations for these observations. An important difference in the current study was the use of intact and adherent cells as opposed to membrane preparations, which have typically been employed (Cosford et al., 2003a; Gregory et al., 2012; Lindemann et al., 2011; Porter et al., 2005; Raboisson et al., 2012; Rodriguez et al., 2010a; Rodriguez et al., 2005). Incomplete radioligand displacement is generally considered evidence for non-competitive binding interactions (Flanagan, 2016; Pagano et al., 2000). However, in light of the two-site binding observed for select ligands, incomplete displacement may be due to very low (pKB <4.5) dipraglurant, VU0366058, VU0409106 and VU0366248 affinity for this apparent second site. The complex binding isotherms could also be attributable to allosteric ligands stabilizing distinct receptor conformations that are only evident within intact cells. Dynamic cellular processes such as G protein coupling, interactions with transducers or scaffolding partners, receptor dimerization and subcellular localization could contribute to an apparent “second site”. Indeed, each of the eight NAMs tested were also inverse agonists for IP1 accumulation in recombinant cells, but not cortical neurons, and could conceivably be influenced by receptor-G protein coupling. Moreover, mGlu5 is well-known to be expressed at the plasma membrane as well as on intracellular membranes (Jong et al., 2014). Differential membrane permeability or access to subcellular compartments may contribute to the complex binding isotherms. In keeping with this idea, all ligands that fully displaced [3H]methoxy-PEPy belong to a similar chemotype and have low molecular weights <250. Irrespective of mechanism, it is clear that mGlu5 NAMs, both within and across chemotypes, can stabilize different receptor conformational states.
Assessment of ligand pharmacology within physiologically relevant systems aims to most closely predict a drug response within the body. However, complex native cell backgrounds and the need to use surrogate ligands raises additional issues: 1) probe dependence; and 2) system bias, where different cell backgrounds result in different ligand pharmacology. Probe dependence between glutamate and DHPG was evident in cooperativity of select mGlu5 PAMs (Sengmany et al., 2017). Under certain conditions both MPEP and MTEP showed higher apparent affinity when DHPG was used as the agonist over glutamate, indicative of probe dependence. DHPG is a membrane impermeable agonist, and unlike glutamate is not actively transported into cells (Jong et al., 2005); therefore mGlu5 modulator probe dependence may arise from differential access of ligands to subcellular compartments. Context-dependent pharmacology, or system bias, has also previously been described for mGlu7 NAMs (Niswender et al., 2010). Here the influence of the system was evident for select mGlu5 NAMs, with differential functional affinities observed between HEK293A cells and neurons. However, despite these caveats, distinct NAM fingerprints were evident which may be linked to the known adverse effect and/or preclinical efficacy of these agents. Acetylenic full NAMs exhibit differential affinities in both cell types, whereas different NAM chemotypes and the partial acetylenic NAM do not. MPEP and MTEP have undergone extensive preclinical testing, and have shown anxiolytic and antidepressant efficacy along with adverse effect liability (cognitive and psychotomimetic) (Balschun and Wetzel, 2002; Belozertseva et al., 2007; Campbell et al., 2004; Chojnacka-Wojcik et al., 2001; Kumar et al., 2013; Lea and Faden, 2006; Li et al., 2006; Palucha-Poniewiera et al., 2014; Schulz et al., 2001; Spooren et al., 2000; Swedberg et al., 2014; Tatarczynska et al., 2001). In a rat model of cocaine addiction, the unbiased partial NAM, M-5MPEP, does not induce adverse psychotomimetic effects at doses that show efficacy in attenuating addiction-associated behaviors (Gould et al., 2016). For the structurally diverse NAMs (VU0409106, VU0366058, VU0366248) fewer preclinical studies have been performed, therefore it remains to be seen if the NAM pKB fingerprints established here can be used to inform future discovery efforts for mGlu5, which may seek to identify biased NAMs that more selectively target therapeutic signaling over adverse effects. Assessment of the biochemical effects of mGlu5 NAMs in vivo, such as inhibition of mGlu5 mediated phosphoinositide hydrolysis or MAP kinase activation, would also provide important information regarding the translatability of these compounds into preclinical animal models and beyond.
Another contributing factor to system bias in the more complex neuronal system is the co-expression of mGlu1, which is known to influence mGlu5 activity via heteromerization and/or signaling cross-talk (Bonsi et al., 2005; Lujan et al., 1996; Neyman and Manahan-Vaughan, 2008). In order to minimize the mGlu1 contribution we initially included CPCCOEt in all experiments (Jong et al., 2009; Sengmany et al., 2017). However, we recently showed that CPCCOEt has similar affinity for mGlu1 and mGlu5, and displays a negative allosteric interaction with the ‘MPEP site’ radioligand (Hellyer et al., 2018). Exclusion of CPCCOEt influenced estimation of MTEP, VU0409106 and dipraglurant affinity. Given that the majority of NAMs were unaffected, this suggests that CPCCOEt may allosterically modulate select mGlu5 NAMs via heteromers, distinct sites, and/or changes in co-localization (Jensen and Brauner-Osborne, 2007; Noetzel et al., 2013; Pandya et al., 2016; Pin and Prézeau, 2007; Rodriguez et al., 2010b; Sevastyanova and Kammermeier, 2014). Collectively, the data highlight that while assessment in recombinant cells may be convenient and have greater reproducibility, it is important to recognize the potential for disconnect between differing cell backgrounds.
Another key consideration is the influence of the kinetic context on biased pharmacology (Klein Herenbrink et al., 2016; Lane et al., 2017), where ligand-receptor and receptor-effector interactions require time to achieve equilibrium. Previously, we showed that the majority of mGlu5 PAMs had strong biased agonism toward IP1 accumulation over iCa2+ mobilization, which may have been associated with differences in ligand-receptor equilibrium between the two measures (Sengmany, et al., 2017). Experiments performed at equilibrium (radioligand binding and IP1 accumulation) for multiple, but not all, mGlu5 NAMs yielded lower affinity estimates relative to the transient non-equilibrium iCa2+ mobilization assay. Higher affinity upon shorter preincubation was somewhat unexpected, however, if we appreciate that the impact of kinetics involves multiple arms – ligand binding, transducer coupling and downstream cell signaling processes (Lane et al., 2017), the paradoxical results observed for mGlu5 NAM affinity may well be explained. While iCa2+ mobilization and IP1 accumulation are endpoints traditionally linked through the Gq activation pathway, it is well established that mGlu5 couples to ion channels (Kammermeier et al., 2000; Latif-Hernandez et al., 2016; Lu et al., 1999; McCool et al., 1998; Tu et al., 1999), resulting in a rapid influx of extracellular Ca2+. Therefore, the iCa2+ mobilization measured is a composite of both intracellular and extracellular calcium influx (Sengmany et al., 2017). Due to the rapid opening of ion channels relative to GPCR signaling cascades, it is possible that initial responses may be via extracellular calcium influx through ion channels. Therefore, select mGlu5 NAMs (MTEP, dipraglurant, MPEP) may have higher affinity for mGlu5 states that couple to plasma membrane channels, as opposed to the canonical Gq/11-PLC-IP3-pathway. Thus, this study highlights the importance of recognizing different determinants of observed allosteric modulator pharmacology (allosteric ligand, orthosteric agonist, receptor, system) and how these can be dissected to reveal new insights into negative allosteric modulator activity.
Overall, this study provides a rigorous characterization of mGlu5 NAMs, highlighting the stabilization of unique receptor conformations in both recombinant and native cells. In particular, ligand-dependent disparities in affinity and cooperativity between iCa2+ mobilization and IP1 accumulation highlight differential modulation of DHPG responses by select mGlu5 NAMs. Differing kinetic and cellular contexts also underscore the pharmacological fingerprints observed, underlining the necessity to carefully consider signaling pathways and cellular background when interpreting pharmacology. Acetylenic full mGlu5 NAMs showed the most divergent pharmacological fingerprints. It is tempting to speculate that a biased signaling fingerprint may be linked to their propensity to engender psychotomimetic and cognitive impairments in preclinical animal models, especially because a partial and unbiased NAM from the same scaffold does not demonstrate such adverse effects. Discovery efforts targeting GPCRs for CNS disorders continue to suffer high attrition rates, attributable in part to the difficulties in translating agents with promising in vitro pharmacology to efficacy in preclinical models and extending this to the clinic. Our findings highlight the inherent complexity in quantifying allosteric modulator pharmacology, with previously unappreciated bias at the level of affinity and/or cooperativity potentially contributing to efficacy and safety profiles of mGlu5 allosteric modulators in preclinical animal models. .
Supplementary Material
Highlights.
-
-
mGlu5 negative allosteric modulators can stabilise distinct affinity states
-
-
mGlu5 NAM affinity is both context and orthosteric probe dependent
-
-
biased mGlu5 modulation is evident at the level of cooperativity and affinity
Acknowledgements
This work was supported by the National Health & Medical Research Council of Australia (NHMRC): CJ Martin Overseas Biomedical postdoctoral training fellowship (KJG: APP1013709), Project Grants APP1084775 (KJG) and APP1127322 (KJG, LTM), Program Grant APP1055134 (AC) and Senior Principal Research Fellowship APP1102950 (AC). KJG and KL are supported by Australian Research Council Future Fellowships: FT160100075 and FT170100392. LTM is supported by a Heart Foundation Future Leader Fellowship (Award ID 101857). Work within the Vanderbilt Center for Neuroscience Drug Discovery on mGlu5 allosteric modulators was supported by the NIH/NIMH R01MH062646. PJC is an inventor on patents that protect multiple classes of mGlu5 allosteric modulators and receives research support from AstraZeneca.
Abbreviations
- CNS
central nervous system
- CPCCOEt
7-(hydroxyimino)cyclopropa[b] chromen-1a-carboxylate ethyl ester
- DHPG
(S)-3,5-dihydroxyphenylglycine
- dipraglurant
6-fluoro-2-[4-(2-pyridinyl)-3-butyn-1-yl]-Imidazo[1,2-a]pyridine
- DMEM
Dulbecco’s modified Eagle’s medium
- ERK1/2
extracellular signal-regulated kinases 1 and 2
- FBS
fetal bovine serum
- fenobam
1-(3-chlorophenyl)-3-[(2e)-1-methyl-4-oxoimidazolidin-2-ylidene]urea
- GPCR
G protein-coupled receptor
- GPT
glutamic pyruvic transaminase
- HBSS
Hank’s Balanced Salt Solution
- HEK293A
human embryonic kidney 293
- iCa2+
intracellular calcium
- IP1
inositol 1-phosphate
- M-5MPEP
2-[2-(3-methoxyphenyl)ethynyl]-5-methylpyridine
- mGlu1
metabotropic glutamate receptor subtype 1
- mGlu5
metabotropic glutamate receptor subtype 5
- MPEP
2-Methyl-6-(phenylethynyl)pyridine
- MTEP
2-methyl-4-(pyridin-3-ylethynyl)thiazole; 3-((2-Methyl-1,3-thiazol-4-yl)ethynyl)pyridine
- NAL
neutral allosteric ligand
- NAM
negative allosteric modulator
- PAM
positive allosteric modulator
- VU0366058
2-(1,3-benzoxazol-2-ylamino)-4-(4-fluorophenyl)pyrimidine-5-carbonitrile
- VU0366248
N-(3-Chloro-2-fluorophenyl)-3-cyano-5-fluoro-benzamide
- VU0409106
3-Fluoro-N-(4-methyl-2-thiazolyl)-5-(5-pyrimidinyloxy)benzamide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Chemical compounds studied in this article:
Glutamate (L-glutamic acid; PubChem CID: 33032); DHPG (PubChem CID: 108001); MPEP (PubChem CID: 3025961), fenobam (PubChem CID: 162834), MTEP (PubChem CID: 9794218), M-5MPEP (PubChem CID: 16036762), dipraglurant (PubChem CID: 44557636), VU0366058 (PubChem CID: 57328392).
Conflict of interest
For each author all relevant financial relationships have been acknowledged and declared within the manuscript.
References
- Abou Farha K, Bruggeman R, Balje-Volkers C, 2014. Metabotropic glutamate receptor 5 negative modulation in phase I clinical trial: potential impact of circadian rhythm on the neuropsychiatric adverse reactions-do hallucinations matter? ISRN Psychiatry 2014, 652750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balschun D, Wetzel W, 2002. Inhibition of mGluR5 blocks hippocampal LTP in vivo and spatial learning in rats. Pharmacol. Biochem. Behav 73, 375–380. [DOI] [PubMed] [Google Scholar]
- Baltos JA, Vecchio EA, Harris MA, Qin CX, Ritchie RH, Christopoulos A, White PJ, May LT, 2017. Capadenoson, a clinically trialed partial adenosine A1 receptor agonist, can stimulate adenosine A2B receptor biased agonism. Biochem. Pharmacol 135, 79–89. [DOI] [PubMed] [Google Scholar]
- Belozertseva IV, Kos T, Popik P, Danysz W, Bespalov AY, 2007. Antidepressant-like effects of mGluR1 and mGluR5 antagonists in the rat forced swim and the mouse tail suspension tests. Eur. Neuropsychopharmacol 17, 172–179. [DOI] [PubMed] [Google Scholar]
- Bonsi P, Cuomo D, De Persis C, Centonze D, Bernardi G, Calabresi P, Pisani A, 2005. Modulatory action of metabotropic glutamate receptor (mGluR) 5 on mGluR1 function in striatal cholinergic interneurons. Neuropharmacology 49 Suppl 1, 104–113. [DOI] [PubMed] [Google Scholar]
- Campbell UC, Lalwani K, Hernandez L, Kinney GG, Conn PJ, Bristow LJ, 2004. The mGluR5 antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP) potentiates PCP-induced cognitive deficits in rats. Psychopharmacol. (Berl) 175, 310–318. [DOI] [PubMed] [Google Scholar]
- Changeux JP, Christopoulos A, 2016. Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell 166, 1084–1102. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH, 1973. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Chojnacka-Wojcik E, Klodzinska A, Pilc A, 2001. Glutamate receptor ligands as anxiolytics. Curr. Opin. Investig. Drugs 2, 1112–1119. [PubMed] [Google Scholar]
- Cook AE, Mistry SN, Gregory KJ, Furness SG, Sexton PM, Scammells PJ, Conigrave AD, Christopoulos A, Leach K, 2015. Biased allosteric modulation at the CaS receptor engendered by structurally diverse calcimimetics. Br. J. Pharmacol 172, 185–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa AMB, Guimaraes JDS, Dos Santos EAE, Kushmerick C, 2017. Control of neuronal excitability by Group I metabotropic glutamate receptors. Biophys. Rev 9, 835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosford ND, Roppe J, Tehrani L, Schweiger EJ, Seiders TJ, Chaudary A, Rao S, Varney MA, 2003a. [3H]-methoxymethyl-MTEP and [3H]-methoxy-PEPy: potent and selective radioligands for the metabotropic glutamate subtype 5 (mGlu5) receptor. Bioorg. Med. Chem. Lett 13, 351–354. [DOI] [PubMed] [Google Scholar]
- Cosford ND, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, Bristow L, Brodkin J, Jiang X, McDonald I, Rao S, Washburn M, Varney MA, 2003b. 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J. Med. Chem 46, 204–206. [DOI] [PubMed] [Google Scholar]
- da Silva Junior ED, Sato M, Merlin J, Broxton N, Hutchinson DS, Ventura S, Evans BA, Summers RJ, 2017. Factors influencing biased agonism in recombinant cells expressing the human alpha1A -adrenoceptor. Br. J. Pharmacol 174, 2318–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey AE, Leach K, Valant C, Conigrave AD, Sexton PM, Christopoulos A, 2012. Positive and negative allosteric modulators promote biased signaling at the calcium-sensing receptor. Endocrinology 153, 1232–1241. [DOI] [PubMed] [Google Scholar]
- Dekundy A, Gravius A, Hechenberger M, Pietraszek M, Nagel J, Tober C, van der Elst M, Mela F, Parsons CG, Danysz W, 2011. Pharmacological characterization of MRZ-8676, a novel negative allosteric modulator of subtype 5 metabotropic glutamate receptors (mGluR5): focus on L: -DOPA-induced dyskinesia. J. Neural Transm. (Vienna) 118, 1703–1716. [DOI] [PubMed] [Google Scholar]
- Emery AC, DiRaddo JO, Miller E, Hathaway HA, Pshenichkin S, Takoudjou GR, Grajkowska E, Yasuda RP, Wolfe BB, Wroblewski JT, 2012. Ligand bias at metabotropic glutamate 1a receptors: molecular determinants that distinguish beta-arrestin-mediated from G protein-mediated signaling. Mol. Pharmacol 82, 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts AS, Lindsley SR, Lamb JP, Rodriguez AL, Menon UN, Jadhav S, Jones CK, Conn PJ, Lindsley CW, Emmitte KA, 2010. 3-Cyano-5-fluoro-N-arylbenzamides as negative allosteric modulators of mGlu(5): Identification of easily prepared tool compounds with CNS exposure in rats. Bioorg. Med. Chem. Lett 20, 4390–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts AS, Rodriguez AL, Morrison RD, Venable DF, Manka JT, Bates BS, Blobaum AL, Byers FW, Daniels JS, Niswender CM, Jones CK, Conn PJ, Lindsley CW, Emmitte KA, 2013. Discovery of VU0409106: A negative allosteric modulator of mGlu5 with activity in a mouse model of anxiety. Bioorg. Med. Chem. Lett 23, 5779–5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan CA, 2016. GPCR-radioligand binding assays. Methods Cell Biol 132, 191–215. [DOI] [PubMed] [Google Scholar]
- Francesconi A, Duvoisin RM, 1998. Role of the second and third intracellular loops of metabotropic glutamate receptors in mediating dual signal transduction activation. J. Biol. Chem 273, 5615–5624. [DOI] [PubMed] [Google Scholar]
- Gasparini F, Lingenhohl K, Stoehr N, Flor P, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Verney M, Johnson E, Hess S, Rao S, Sacaan A, Santori E, Velicelebi G, Kuhn R, 1999. 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology 38, 1493–1503. [DOI] [PubMed] [Google Scholar]
- Giraldo J, 2015. Operational Models of Allosteric Modulation: Caution is Needed. Trends Pharmacol Sci 36, 1–2. [DOI] [PubMed] [Google Scholar]
- Gould RW, Amato RJ, Bubser M, Joffe ME, Nedelcovych MT, Thompson AD, Nickols HH, Yuh JP, Zhan X, Felts AS, Rodriguez AL, Morrison RD, Byers FW, Rook JM, Daniels JS, Niswender CM, Conn PJ, Emmitte KA, Lindsley CW, Jones CK, 2016. Partial mGlu(5) Negative Allosteric Modulators Attenuate Cocaine-Mediated Behaviors and Lack Psychotomimetic-Like Effects. Neuropsychopharmacol 41, 1166–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Noetzel MJ, Niswender CM, 2013. Pharmacology of metabotropic glutamate receptor allosteric modulators: structural basis and therapeutic potential for CNS disorders. Prog. Mol. Biol. Transl 115, 61–121. [DOI] [PubMed] [Google Scholar]
- Gregory KJ, Noetzel MJ, Rook JM, Vinson PN, Stauffer SR, Rodriguez AL, Emmitte KA, Zhou Y, Chun AC, Felts AS, Chauder BA, Lindsley CW, Niswender CM, Conn PJ, 2012. Investigating metabotropic glutamate receptor 5 allosteric modulator cooperativity, affinity, and agonism: enriching structure-function studies and structure-activity relationships. Mol. Pharmacol 82, 860–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DA, 2013. Application of Receptor Theory to Allosteric Modulation of Receptors. Prog. Mol. Biol. Transl. Sci 115, 217–290. [DOI] [PubMed] [Google Scholar]
- Hathaway HA, Pshenichkin S, Grajkowska E, Gelb T, Emery AC, Wolfe BB, Wroblewski JT, 2015. Pharmacological characterization of mGlu1 receptors in cerebellar granule cells reveals biased agonism. Neuropharmacology 93, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellyer SD, Albold S, Wang T, Chen AN, May LT, Leach K, Gregory KJ, 2018. “Selective” Class C G protein-coupled receptor modulators are neutral or biased mGlu5 allosteric ligands. Mol. Pharmacol [DOI] [PubMed]
- Hughes ZA, Neal SJ, Smith DL, Sukoff Rizzo SJ, Pulicicchio CM, Lotarski S, Lu S, Dwyer JM, Brennan J, Olsen M, Bender CN, Kouranova E, Andree TH, Harrison JE, Whiteside GT, Springer D, O’Neil SV, Leonard SK, Schechter LE, Dunlop J, Rosenzweig-Lipson S, Ring RH, 2013. Negative allosteric modulation of metabotropic glutamate receptor 5 results in broad spectrum activity relevant to treatment resistant depression. Neuropharmacology 66, 202–214. [DOI] [PubMed] [Google Scholar]
- Iso Y, Grajkowska E, Wroblewski JT, Davis J, Goeders NE, Johnson KM, Sanker S, Roth BL, Tueckmantel W, Kozikowski AP, 2006. Synthesis and structure-activity relationships of 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine analogues as potent, noncompetitive metabotropic glutamate receptor subtype 5 antagonists; search for cocaine medications. J. Med. Chem 49, 1080–1100. [DOI] [PubMed] [Google Scholar]
- Jacob W, Gravius A, Pietraszek M, Nagel J, Belozertseva I, Shekunova E, Malyshkin A, Greco S, Barberi C, Danysz W, 2009. The anxiolytic and analgesic properties of fenobam, a potent mGlu5 receptor antagonist, in relation to the impairment of learning. Neuropharmacology 57, 97–108. [DOI] [PubMed] [Google Scholar]
- Jensen AA, Brauner-Osborne H, 2007. Allosteric modulation of the calcium-sensing receptor. Curr. Neuropharmacol 5, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly C, Gomeza J, Brabet I, Curry K, Bockaert J, Pin JP, 1995. Molecular, functional, and pharmacological characterization of the metabotropic glutamate receptor type 5 splice variants: comparison with mGluR1. J. Neurosci 15, 3970–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong YJ, Kumar V, Kingston AE, Romano C, O’Malley KL, 2005. Functional metabotropic glutamate receptors on nuclei from brain and primary cultured striatal neurons. Role of transporters in delivering ligand. J. Biol. Chem 280, 30469–30480. [DOI] [PubMed] [Google Scholar]
- Jong YJ, Kumar V, O’Malley KL, 2009. Intracellular metabotropic glutamate receptor 5 (mGluR5) activates signaling cascades distinct from cell surface counterparts. J. Biol. Chem 284, 35827–35838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammermeier PJ, Xiao B, Tu JC, Worley PF, Ikeda SR, 2000. Homer proteins regulate coupling of group I metabotropic glutamate receptors to N-type calcium and M-type potassium channels. J. Neurosci 20, 7238–7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A, 2013. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discov 12, 205–216. [DOI] [PubMed] [Google Scholar]
- Kettunen P, Krieger P, Hess D, El Manira A, 2002. Signaling mechanisms of metabotropic glutamate receptor 5 subtype and its endogenous role in a locomotor network. J. Neurosci 22, 1868–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajehali E, Malone DT, Glass M, Sexton PM, Christopoulos A, Leach K, 2015. Biased Agonism and Biased Allosteric Modulation at the CB1 Cannabinoid Receptor. Mol. Pharmacol 88, 368–379. [DOI] [PubMed] [Google Scholar]
- Klein Herenbrink C, Sykes DA, Donthamsetti P, Canals M, Coudrat T, Shonberg J, Scammells PJ, Capuano B, Sexton PM, Charlton SJ, Javitch JA, Christopoulos A, Lane JR, 2016. The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun 7, 10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J, Hapidin H, Bee Y-T, Ismail Z, 2013. Effects of the mGluR5 antagonist MPEP on ethanol withdrawal induced anxiety-like syndrome in rats. Behav. Brain Funct 9, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane JR, May LT, Parton RG, Sexton PM, Christopoulos A, 2017. A kinetic view of GPCR allostery and biased agonism. Nat. Chem. Biol 13, 929–937. [DOI] [PubMed] [Google Scholar]
- Latif-Hernandez A, Faldini E, Ahmed T, Balschun D, 2016. Separate Ionotropic and Metabotropic Glutamate Receptor Functions in Depotentiation vs. LTP: A Distinct Role for Group1 mGluR Subtypes and NMDARs. Front. Cell. Neurosci 10, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazareno S, Birdsall NJ, 1993. Estimation of competitive antagonist affinity from functional inhibition curves using the Gaddum, Schild and Cheng-Prusoff equations. Br. J. Pharmacol 109, 1110–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazareno S, Birdsall NJ, 1995. Detection, quantitation, and verification of allosteric interactions of agents with labeled and unlabeled ligands at G protein-coupled receptors: interactions of strychnine and acetylcholine at muscarinic receptors. Mol. Pharmacol 48, 362–378. [PubMed] [Google Scholar]
- Lea P. M. t., Faden AI, 2006. Metabotropic glutamate receptor subtype 5 antagonists MPEP and MTEP. CNS Drug Rev 12, 149–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K, Gregory KJ, Kufareva I, Khajehali E, Cook AE, Abagyan R, Conigrave AD, Sexton PM, Christopoulos A, 2016. Towards a structural understanding of allosteric drugs at the human calcium-sensing receptor. Cell Research 26, 574–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A, 2007. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol. Sci 28, 382–389. [DOI] [PubMed] [Google Scholar]
- Leach K, Wen A, Cook AE, Sexton PM, Conigrave AD, Christopoulos A, 2013. Impact of clinically relevant mutations on the pharmacoregulation and signaling bias of the calcium-sensing receptor by positive and negative allosteric modulators. Endocrinology 154, 1105–1116. [DOI] [PubMed] [Google Scholar]
- Li X, Need AB, Baez M, Witkin JM, 2006. Metabotropic glutamate 5 receptor antagonism is associated with antidepressant-like effects in mice. J. Pharmacol. Exp. Ther 319, 254–259. [DOI] [PubMed] [Google Scholar]
- Lindemann L, Jaeschke G, Michalon A, Vieira E, Honer M, Spooren W, Porter R, Hartung T, Kolczewski S, Büttelmann B, Flament C, Diener C, Fischer C, Gatti S, Prinssen EP, Parrott N, Hoffmann G, Wettstein JG, 2011. CTEP: A Novel, Potent, Long-Acting, and Orally Bioavailable Metabotropic Glutamate Receptor 5 Inhibitor. J. Pharmacol. Exp.Ther 339, 474–486. [DOI] [PubMed] [Google Scholar]
- Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF, 1999. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat. Neurosci 2, 331–338. [DOI] [PubMed] [Google Scholar]
- Luccini E, Musante V, Neri E, Brambilla Bas M, Severi P, Raiteri M, Pittaluga A, 2007. Functional interactions between presynaptic NMDA receptors and metabotropic glutamate receptors co-expressed on rat and human noradrenergic terminals. Br. J. Pharmacol 151, 1087–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P, 1996. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur. J. Neurosci 8, 1488–1500. [DOI] [PubMed] [Google Scholar]
- Mao L, Yang L, Tang Q, Samdani S, Zhang G, Wang JQ, 2005. The scaffold protein Homer1b/c links metabotropic glutamate receptor 5 to extracellular signal-regulated protein kinase cascades in neurons. J. Neurosci 25, 2741–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool BA, Pin JP, Harpold MM, Brust PF, Stauderman KA, Lovinger DM, 1998. Rat group I metabotropic glutamate receptors inhibit neuronal Ca2+ channels via multiple signal transduction pathways in HEK 293 cells. J. Neurophysiol 79, 379–391. [DOI] [PubMed] [Google Scholar]
- Mueller R, Dawson ES, Meiler J, Rodriguez AL, Chauder BA, Bates BS, Felts AS, Lamb JP, Menon UN, Jadhav SB, Kane AS, Jones CK, Gregory KJ, Niswender CM, Conn PJ, Olsen CM, Winder DG, Emmitte KA, Lindsley CW, 2012. Discovery of 2-(2-benzoxazoyl amino)-4-aryl-5-cyanopyrimidine as negative allosteric modulators (NAMs) of metabotropic glutamate receptor 5 (mGlu(5)): from an artificial neural network virtual screen to an in vivo tool compound. ChemMedChem 7, 406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutel V, Ellis GJ, Adam G, Chaboz S, Nilly A, Messer J, Bleuel Z, Metzler V, Malherbe P, Schlaeger EJ, Roughley BS, Faull RL, Richards JG, 2000. Characterization of [(3)H]Quisqualate binding to recombinant rat metabotropic glutamate 1a and 5a receptors and to rat and human brain sections. J. Neurochem 75, 2590–2601. [DOI] [PubMed] [Google Scholar]
- Neyman S, Manahan-Vaughan D, 2008. Metabotropic glutamate receptor 1 (mGluR1) and 5 (mGluR5) regulate late phases of LTP and LTD in the hippocampal CA1 region in vitro. Eur. J. Neurosci 27, 1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickols HH, Conn PJ, 2014. Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol. Dis 61, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickols HH, Yuh JP, Gregory KJ, Morrison RD, Bates BS, Stauffer SR, Emmitte KA, Bubser M, Peng W, Nedelcovych MT, Thompson A, Lv X, Xiang Z, Daniels JS, Niswender CM, Lindsley CW, Jones CK, Conn PJ, 2016. VU0477573: Partial Negative Allosteric Modulator of the Subtype 5 Metabotropic Glutamate Receptor with In Vivo Efficacy. J. Pharmacol. Exp. Ther 356, 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ, 2010. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Johnson KA, Miller NR, Ayala JE, Luo Q, Williams R, Saleh S, Orton D, Weaver CD, Conn PJ, 2010. Context-dependent pharmacology exhibited by negative allosteric modulators of metabotropic glutamate receptor 7. Mol. Pharmacol 77, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noetzel MJ, Gregory KJ, Vinson PN, Manka JT, Stauffer SR, Lindsley CW, Niswender CM, Xiang Z, Conn PJ, 2013. A novel metabotropic glutamate receptor 5 positive allosteric modulator acts at a unique site and confers stimulus bias to mGlu5 signaling.[Erratum appears in Mol Pharmacol. 2013 Oct;84(4):654]. Mol. Pharmacol 83, 835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano A, Ruegg D, Litschig S, Stoehr N, Stierlin C, Heinrich M, Floersheim P, Prezeau L, Carroll F, Pin JP, Cambria A, Vranesic I, Flor PJ, Gasparini F, Kuhn R, 2000. The non-competitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J. Biol. Chem 275, 33750–33758. [DOI] [PubMed] [Google Scholar]
- Palucha-Poniewiera A, Szewczyk B, Pilc A, 2014. Activation of the mTOR signaling pathway in the antidepressant-like activity of the mGlu5 antagonist MTEP and the mGlu7 agonist AMN082 in the FST in rats. Neuropharmacology 82, 59–68. [DOI] [PubMed] [Google Scholar]
- Pandya NJ, Klaassen RV, van der Schors RC, Slotman JA, Houtsmuller A, Smit AB, Li KW, 2016. Group 1 metabotropic glutamate receptors 1 and 5 form a protein complex in mouse hippocampus and cortex. Proteomics 16, 2698–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peavy RD, Chang MS, Sanders-Bush E, Conn PJ, 2001. Metabotropic glutamate receptor 5-induced phosphorylation of extracellular signal-regulated kinase in astrocytes depends on transactivation of the epidermal growth factor receptor. J. Neurosci 21, 9619–9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecknold JC, McClure DJ, Appeltauer L, Wrzesinski L, Allan T, 1982. Treatment of anxiety using fenobam (a nonbenzodiazepine) in a double-blind standard (diazepam) placebo-controlled study. J. Clin. Psychopharmacol 2, 129–133. [PubMed] [Google Scholar]
- Pin J-P, Prézeau L, 2007. Allosteric Modulators of GABA(B) Receptors: Mechanism of Action and Therapeutic Perspective. Curr. Neuropharmacol 5, 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter RH, Jaeschke G, Spooren W, Ballard TM, Buttelmann B, Kolczewski S, Peters JU, Prinssen E, Wichmann J, Vieira E, Muhlemann A, Gatti S, Mutel V, Malherbe P, 2005. Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J. Pharmacol. Exp.Ther 315, 711–721. [DOI] [PubMed] [Google Scholar]
- Priestley R, Glass M, Kendall D, 2017. Functional Selectivity at Cannabinoid Receptors. Adv. Pharmacol 80, 207–221. [DOI] [PubMed] [Google Scholar]
- Raboisson P, Breitholtz-Emanuelsson A, Dahllof H, Edwards L, Heaton WL, Isaac M, Jarvie K, Kers A, Minidis AB, Nordmark A, Sheehan SM, Slassi A, Strom P, Terelius Y, Wensbo D, Wilson JM, Xin T, McLeod DA, 2012. Discovery and characterization of AZD9272 and AZD6538-Two novel mGluR5 negative allosteric modulators selected for clinical development. Bioorg. Med. Chem. Lett 22, 6974–6979. [DOI] [PubMed] [Google Scholar]
- Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, Blatt TN, Jadhav S, Menon UN, Vinson PN, Rook JM, Stauffer SR, Niswender CM, Lindsley CW, Weaver CD, Conn PJ, 2010a. Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol. Pharmacol 78, 1105–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AL, Nong Y, Sekaran NK, Alagille D, Tamagnan GD, Conn PJ, 2005. A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol. Pharmacol 68, 1793–1802. [DOI] [PubMed] [Google Scholar]
- Rodriguez AL, Tarr JC, Zhou Y, Williams R, Gregory KJ, Bridges TM, Daniels JS, Niswender CM, Conn PJ, Lindsley CW, Stauffer SR, 2010b. Identification of a glycine sulfonamide based non-MPEP site positive allosteric potentiator (PAM) of mGlu5. Probe Reports from the NIH Molecular Libraries Program National Center for Biotechnology Information (US), Bethesda (MD). [PubMed] [Google Scholar]
- Rush AM, Wu J, Rowan MJ, Anwyl R, 2002. Group I metabotropic glutamate receptor (mGluR)-dependent long-term depression mediated via p38 mitogen-activated protein kinase is inhibited by previous high-frequency stimulation and activation of mGluRs and protein kinase C in the rat dentate gyrus in vitro. J. Neurosci 22, 6121–6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepp DD, Goldsworthy J, Johnson BG, Salhoff CR, Baker SR, 1994. 3,5-dihydroxyphenylglycine is a highly selective agonist for phosphoinositide-linked metabotropic glutamate receptors in the rat hippocampus. J. Neurochem 63, 769–772. [DOI] [PubMed] [Google Scholar]
- Schulz B, Fendt M, Gasparini F, Lingenhohl K, Kuhn R, Koch M, 2001. The metabotropic glutamate receptor antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP) blocks fear conditioning in rats. Neuropharmacology 41, 1–7. [DOI] [PubMed] [Google Scholar]
- Sengmany K, Gregory KJ, 2016. Metabotropic glutamate receptor subtype 5: molecular pharmacology, allosteric modulation and stimulus bias. Br. J. Pharmacol 173, 3001–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengmany K, Singh J, Stewart GD, Conn PJ, Christopoulos A, Gregory KJ, 2017. Biased allosteric agonism and modulation of metabotropic glutamate receptor 5: Implications for optimizing preclinical neuroscience drug discovery. Neuropharmacology 115, 60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevastyanova TN, Kammermeier PJ, 2014. Cooperative signaling between homodimers of metabotropic glutamate receptors 1 and 5. Mol. Pharmacol 86, 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Rodriguez AL, Conn PJ, Lindsley CW, 2008. Synthesis and SAR of a mGluR5 allosteric partial antagonist lead: unexpected modulation of pharmacology with slight structural modifications to a 5-(phenylethynyl)pyrimidine scaffold. Bioorg. Med. Chem. Lett 18, 4098–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack RJ, Hall DA, 2012. Development of Operational Models of Receptor Activation Including Constitutive Receptor Activity and Their Use to Determine the Efficacy of the Chemokine CCL17 at the CC Chemokine Receptor CCR4. Br. J. Pharmacol 166, 1774–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spooren WP, Vassout A, Neijt HC, Kuhn R, Gasparini F, Roux S, Porsolt RD, Gentsch C, 2000. Anxiolytic-like effects of the prototypical metabotropic glutamate receptor 5 antagonist 2-methyl-6-(phenylethynyl)pyridine in rodents. J. Pharmacol. Exp. Ther 295, 1267–1275. [PubMed] [Google Scholar]
- Swedberg MD, Ellgren M, Raboisson P, 2014. mGluR5 antagonist-induced psychoactive properties: MTEP drug discrimination, a pharmacologically selective non-NMDA effect with apparent lack of reinforcing properties. J. Pharmacol. Exp. Ther 349, 155–164. [DOI] [PubMed] [Google Scholar]
- Swedberg MD, Raboisson P, 2014. AZD9272 and AZD2066: selective and highly central nervous system penetrant mGluR5 antagonists characterized by their discriminative effects. J. Pharmacol. Exp. Ther 350, 212–222. [DOI] [PubMed] [Google Scholar]
- Tatarczynska E, Klodzinska A, Chojnacka-Wojcik E, Palucha A, Gasparini F, Kuhn R, Pilc A, 2001. Potential anxiolytic- and antidepressant-like effects of MPEP, a potent, selective and systemically active mGlu5 receptor antagonist. Br. J. Pharmacol 132, 1423–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thandi S, Blank JL, Challiss RA, 2002. Group-I metabotropic glutamate receptors, mGlu1a and mGlu5a, couple to extracellular signal-regulated kinase (ERK) activation via distinct, but overlapping, signalling pathways. J. Neurochem 83, 1139–1153. [DOI] [PubMed] [Google Scholar]
- Tison F, Keywood C, Wakefield M, Durif F, Corvol JC, Eggert K, Lew M, Isaacson S, Bezard E, Poli SM, Goetz CG, Trenkwalder C, Rascol O, 2016. A Phase 2A Trial of the Novel mGluR5-Negative Allosteric Modulator Dipraglurant for Levodopa-Induced Dyskinesia in Parkinson’s Disease. Mov. Disord 31, 1373–1380. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF, 1999. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23, 583–592. [DOI] [PubMed] [Google Scholar]
- Violin JD, Crombie AL, Soergel DG, Lark MW, 2014. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol. Sci 35, 308–316. [DOI] [PubMed] [Google Scholar]
- Viwatpinyo K, Chongthammakun S, 2009. Activation of group I metabotropic glutamate receptors leads to brain-derived neurotrophic factor expression in rat C6 cells. Neurosci. Lett 467, 127–130. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Rodriguez AL, Conn PJ, 2005. Allosteric potentiators of metabotropic glutamate receptor subtype 5 have differential effects on different signaling pathways in cortical astrocytes. J. Pharmacol. Exp. Ther 315, 1212–1219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.