

Abstract
Identifying controlling features of responsiveness to checkpoint blockade therapies is an urgent goal in oncology research. Our group and others have previously shown melanoma tumors resistant to checkpoint blockade display features of mesenchymal transition including E-cadherin loss. Here, we present the first in vivo evidence tumor E-cadherin facilitates immune attack, using a B16F10 melanoma mouse model in which E-cadherin is exogenously expressed (B16.Ecad). We find, compared to vector control, B16.Ecad exhibits delayed tumor growth, reduced metastatic potential, and increased overall survival in vivo. Transplantation of B16.Ecad into Rag1−/− and CD103−/− mice abrogated the tumor growth delay. This indicates the anti-melanoma response against B16.Ecad is both immune and CD103+ mediated. Moreover, B16.Ecad showed increased responsiveness to combination immune checkpoint blockade (ICB) compared to vector control. This work establishes a rationale for ICB responses observed in high E-cadherin-expressing tumors and suggests therapeutic advancement through amplifying CD103+ immune cell subsets.
Keywords: Melanoma, cancer immunity, PD-1, CTLA-4, E-cadherin, CD103, immunotherapy
Introduction
The advent of immune checkpoint blockade (ICB) has transformed the clinical management of metastatic melanoma (1,2). Subsets of patients experience durable, lasting responses, while others face tumor resistance and progressive disease. Response rates are increased when immune checkpoints are blocked in combination regimens, as with the combination of ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1) (1,2). Still, approximately 40% of patients show no response to combination checkpoint blockade (2). There is an immediate need to determine factors which modulate or predict tumor response to ICB therapies and to further explore the underlying biology of anti-tumor immune activity.
Recent mechanisms proposed to influence responsiveness to ICB fall broadly into two categories: host-centric and tumor-centric factors. Responsive tumors have increased effector T-cell populations, higher mutational (and subsequently antigen) loads, and are contained within hosts who have a favorable microbiome composition (3–9). Mechanisms of tumor resistance to ICB include immunosuppressive oncogenic pathways, perturbed immune signaling, mesenchymal transition, and defects in antigen processing and presentation (10–13). Despite these advances, knowledge regarding patient specific responsiveness remains incomplete.
Previously, using a proteomic approach, we identified elevated E-cadherin in ICB responding (versus non-responding) pretreatment melanoma tumor biopsies (14). A recent transcriptomic study also found CDH1 (E-cadherin) to be more highly expressed in melanoma tumors responsive to PD-1 therapy (11). E-cadherin is an adherens junction protein that serves as the major mediator of melanocyte adhesion to neighboring keratinocytes, and loss of E-cadherin is a hallmark event in mesenchymal transition (15–17). E-cadherin has classical roles in embryonic development and tumor suppression, mainly via control of cellular adhesion and motility (16,18). Although the role of E-cadherin in tumor suppression is firmly established, the functional significance of E-cadherin in checkpoint blockade responses is unknown.
In normal tissues and organs, E-cadherin is abundant within epithelial tissues, and subsets of immune cells utilize a heterodimeric integrin termed αΕβ7 to bind to E-cadherin expressing cells and perform immunosurveillance functions (19,20). CD103 (αΕ) heterodimerizes with the integrin β7 to form αΕβ7 (21). It has been shown melanoma tumor CD103+ CD8+ T-cell counts more strongly predict patient survival than tumor CD8+ counts alone in immunotherapy naïve patients (22). However, the effect of E-cadherin expression in melanoma on ICB responses and anti-tumor activity of CD103+ immune cells has not been studied.
In this study, we examined E-cadherin expression in melanoma as a variable in the anti-tumor immune response. Using a B16F10 murine melanoma model, we demonstrate E-cadherin expression (B16.Ecad) results in B16F10 tumor growth delays in wild-type mice. Importantly, the tumor growth delays and survival advantage observed in wild-type mice bearing B16.Ecad is lost in Rag1−/− and CD103−/− hosts. Additionally, we show B16.Ecad has enhanced responses to combination immune checkpoint blockade. We propose these results could inform strategies aimed at potentiation of CD103 immune cell activity against E-cadherin expressing malignancies.
Materials and Methods
Western Blotting
For immunoblot analysis, harvested cells were lysed on ice for 30 minutes with radioimmunoprecipitation assay buffer (RIPA). Lysates were then centrifuged for clearing and protein concentrations were determined by bicinchoninic acid (BCA) protein assay (Pierce, ThermoFisher #23225). Samples were then heated at 95°C for 5 minutes. Electrophoresis was performed using bis-Tris gels 4-12% (Invitrogen) and then samples were transferred to polyvinylidene difluoride (PVDF; Millipore) membranes. For probing, the following antibodies were used: E-cadherin (1:1000; rabbit monoclonal, Cell signaling, Danvers, MA, #3195), and GAPDH (1:1000; rabbit monoclonal, Cell signaling, Danvers, MA, #5174). Detection was performed using Western Lightning Plus ECL enhanced chemiluminescent substrate (Perkin-Elmer Inc., #NEL103001EA) according to manufacturer’s instructions.
Immunohistochemistry
Immunohistochemistry on the Ventana Ultra platform was performed using the manufacturer’s recommended settings. 4 micron tissue sections were heated and then treated with Cell Conditioning 1 solution (Ventana) for heat-induced epitope retrieval. Slides were incubated with the ready-to-use anti-E-cadherin ((36) Mouse Monoclonal (Ventana, Catalog Number: 790-4497)) and visualized using the Ventana ultraView Detection Kit. Stained melanoma tumor slides were evaluated by a dermatopathologist to determine melanoma tumor margins and E-cadherin staining status.
Cell culture and protein isolation for proteomics
B16.Ecad and B16.Vector cell lines were cultured as five-biological replicates in DMEM supplemented with 10% FBS, penicillin and streptomycin at 37°C and 5% CO2. For protein isolation, harvested cells were lysed on ice for 30 minutes with radioimmunoprecipitation assay buffer (RIPA). Lysates were then centrifuged for clearing and protein concentrations were determined by bicinchoninic acid (BCA) protein assay (Pierce, ThermoFisher #23225).
Proteomics
Purified proteins were reduced, alkylated, and digested using filter-aided sample preparation (23). Tryptic peptides were labeled using a tandem mass tag 10-plex isobaric label reagent set (ThermoFisher #90309) following the manufacturer’s instructions. Labeled peptides were separated into 36 fractions on a 100 × 1.0 mm Acquity BEH C18 column (Waters) using an UltiMate 3000 UHPLC system (ThermoFisher) with a 40 min gradient from 99:1 to 60:40 buffer A:B ratio under basic pH conditions, and then consolidated into 12 non adjacent super-fractions (Buffer A = H2O with 10mM ammonium hydroxide; Buffer B = acetonitrile with 10mM ammonium hydroxide). Each super-fraction was then further separated by reverse phase XSelect CSH C18 2.5 um resin (Waters) on an in-line 120 × 0.075 mm column using an UltiMate 3000 RSLCnano system (ThermoFisher). Peptides were eluted using a 60 min gradient from 98:2 to 67:33 buffer A:B ratio. Eluted peptides were ionized by electrospray (2.15 kV) followed by mass spectrometric analysis on an Orbitrap Fusion Lumos mass spectrometer (ThermoFisher) using multi-notch MS3 parameters (24). MS data were acquired using the FTMS analyzer in top-speed (2.5 s cycle) mode at a resolution of 120,000 over a range of 375 to 1500 m/z. Following CID activation with normalized collision energy of 35.0, MS/MS data were acquired using the ion trap analyzer in centroid mode and normal mass range. Using synchronous precursor selection, up to 10 MS/MS precursors were selected for HCD activation with normalized collision energy of 65.0, followed by acquisition of MS3 reporter ion data using the FTMS analyzer in profile mode at a resolution of 50,000 over a range of 100-500 m/z.
Proteomics Data Analysis
A total of 8,052 proteins were identified and TMT-10plex reporter ions quantified by searching a custom UniprotKB database (Mus musculus plus E-cadherin (Homo sapiens) 83,100 entries) using MaxQuant (version 1.5.8.3, Max Planck Institute) with a parent ion tolerance of 3 ppm, a fragment ion tolerance of 0.5 Da, and a reporter ion tolerance of 0.001 Da. Modifications searched included fixed modifications of carbamidomethyl on cysteine and TMT-10plex on lysine and n-terminal; as well as variable modifications of oxidation on methionine and acetylation on the n-terminal peptide. We first searched a contaminants database (262 entries) to identify common contaminating proteins followed by the main search. Protein identifications were accepted if they could be established with less than 1.0% false discovery and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm (25).
The TMT sample consisted of five vector control (B16.Vector) samples mixed with five E-cadherin overexpressed samples (B16.Ecad). In order to identify the significantly differentiating proteins between the control and E-cadherin overexpression, the TMT reporter ion intensity values were normalized by adjusting the medians of each sample to the sample with the highest median value. The values were log2 transformed to correct for heteroscedasticity and to allow for parametric testing using a Student’s t-test. Fold changes were calculated by subtracting vector samples from E-cadherin expressing samples. Proteins with an adjusted p-value < 0.05 and a fold change > 1.5 were considered to be significant. P-values were adjusted using the Benjamini-Hochberg (FDR) procedure.
Immunofluorescence of human metastatic melanoma tumors
The protocol used by this study to obtain the clinical samples (study #204543) was approved by the UAMS Institutional Review Board. All aspects of this study were conducted in accordance with the ethical guidelines of the Belmont Report. This study was exempted from informed consent by the UAMS IRB as archival tissues were de-identified and no patients were enrolled per U.S. Department of Health and Human Services 45 CFR 46.102.
Formalin-fixed paraffin-embedded (FFPE) human melanoma tumor biopsies (sectioned at 4 μm) were shipped to Reveal Biosciences. Immunofluorescence was performed using a Leica Bond automated immunostainer. Heat induced antigen retrieval was performed using Leica Bond Epitope Retrieval Buffer 2 (EDTA solution, pH 9.0) for 20 minutes. Non-specific background was blocked with Novocastra Protein Block (Leica, #RE7102-CE, Lot #6056766) for 20 minutes. Rabbit anti-CD103 (Abcam, #ab129202, Lot #GR3185003-9) and rat anti-CD3 (Abcam, #ab11089, Lot #GR280475-11) antibodies were applied at 1:100 dilution for one hour. Secondary antibodies, Alexa Fluor® 647 Donkey anti-rabbit IgG (H+L) (lnvitrogen, #A31573, Lot #1964354) and Alexa Fluor® 546 goat anti-rat IgG (H+L) (lnvitrogen, #A11081, Lot #1744742) were applied at 1:200 for 30 minutes. Slides were mounted with DAPI in Fluorogel II for nuclear visualization. Whole slide images were generated in florescence using a Pannoramic SCAN (3D Histech) and quantified using the ImageDx platform.
Mice
The animal experiments described in this study were reviewed and approved by the University of Arkansas for Medical Sciences Institutional Animal Care and Use Committee (IACUC). C57BL/6, Rag1−/, and B6.129S2(C)-Itgaetm1Cmp/J (CD103−/−) mice were purchased from The Jackson Laboratory.
Cell lines
The cell lines B16.F10 and 293T were purchased and authenticated from American Type Culture Collection (ATCC). Cell lines were screened and found to be negative for all tested pathogens (Ectromelia, EDIM, LCMV, MAV1, MAV2, MHV, MPV, MVM, Mycoplasma pulmonis, Polyoma, PVM, REO3, Sendai, TMEV) by IDEXX. B16.F10 was cultured in DMEM supplemented with 10% FBS, penicillin and streptomycin at 37°C and 5% CO2. Standard B16 culture and passage methods were used as described previously (26). B16.SIY was a gift from Thomas Gajewski, University of Chicago (27) B16.SIY was screened for mycoplasma with the MycoAlert Detection Kit (LT07-318) by Lonza.
Retroviral vectors and virus production
The pMSCV-puro-E-Cadherin was generated by Gibson Assembly of human E-Cadherin (addgene #45769) and pMSCVpuro (Clonetech). Expression construct was verified by sequencing. Ecotropic retroviruses were produced by cotransfection of one of the following retroviral expression plasmids by using FuGene 6 (Roche Applied Bioscience, Indianapolis, IN) in 293T cells with packaging plasmids (pMD-old-Gag-Pol and pCAG4-Eco): pMSCV-puro, pMSCV-puro-E-Cadherin (human).
Antibodies & FACS
For checkpoint blockade: Anti-CTLA-4 (9D9), αPD-1 (RMP1-14), Rat Ig, and mIgG2b (controls) used in vivo were purchased from Bio X cell. For each 100 μl dose (suspended in PBS) 100 μg of 9D9, 250 μg of RMP114, 250 μg of Rat Ig, and 100 μg of mIgG2b was used (28).
Tumor dissociation was performed as described previously (29). For FACS, samples were acquired using an LSRFortessa instrument (Beckman Coulter) and analysis was performed using Flow Jo. The following fluorochrome-labeled anti-mouse Abs were purchased from Biolegend: CD3 FITC (#100204), CD4 BV711 (#001549), CD8 PerCP (#100731), CD45 PE-Cy7 (#552848), NK1.1 AF700 (#560515), and DAPI (#422801).
Tumor challenge
Mice were injected in the subcutaneous flank space with 1×105 tumor cells suspended in 100μl of PBS. Tumor growth was monitored daily with caliper measurements of tumor length and width. Tumor endpoints were 200-400 mm2. Survival plotted on Kaplan-meier curves was the number of days post-B16 engraftment where tumor was less than 150 mm2, with no ulceration, and no tumor measurement >20 mm in any direction. Functional studies were performed in all experiments at least twice, in an independent way. Unless otherwise noted in figure legends, data represents a summary of the repeated experiments.
For intravenous injection 2×105 tumor cells were injected into the tail vein of the animals. On day 18 mice were sacrificed and lungs dissected. Lungs were washed 3 times with PBS and lung tumors were counted by two independent observers. Counts were then averaged for each set of lungs.
Statistics
Statistical significance was determined by using unpaired Student’s T-test (2-tailed). A P value of less than 0.05 was considered significant and multiple test corrections were performed when appropriate. For Kaplan-Meier plots of survival log-rank test was used to determine P values. Data was analyzed using GraphPad Prism 7, R Studio, and Microsoft Excel.
Results
Metastatic melanoma tumors show variable E-cadherin expression.
Previously, our group and others have observed E-cadherin expression to be lost in patient tumors non-responsive to ICB therapies (11,14). E-cadherin provides clinical information in breast cancer and other carcinomas, but is not typically assayed in the setting of metastatic melanoma (30). To determine the variability of E-cadherin (CDH1) expression in human melanoma tumor cells, we accessed a single-cell RNA-Seq data set and profiled tumors based upon the CDH1 expression in the coterminous cells (GSE72056) (31). Fifteen of the nineteen tumors within the dataset contained melanoma cells, and E-cadherin expression varied widely across the sample group. The median tumor cell CDH1 expression was zero in 5 of the tumors (Fig. 1A). Next we examined 16 biopsies from patients with metastatic melanoma for E-cadherin protein by immunohistochemistry analysis (Fig. 1B). Twenty-five percent of tumors stained negative for E-cadherin (4/16; first row) while 12 showed weak to strong staining. The absence or presence of E-cadherin expression was found to be unrelated to lesion location as examples from soft tissue, lymph node, lung, colon, and skin show differences in expression levels between different patient tumors (Fig. 1B). In addition, tumor cell morphology (epithelioid versus spindled) did not predict E-cadherin expression.
Figure 1. Human melanoma E-cadherin expression and generation of B16.Ecad.
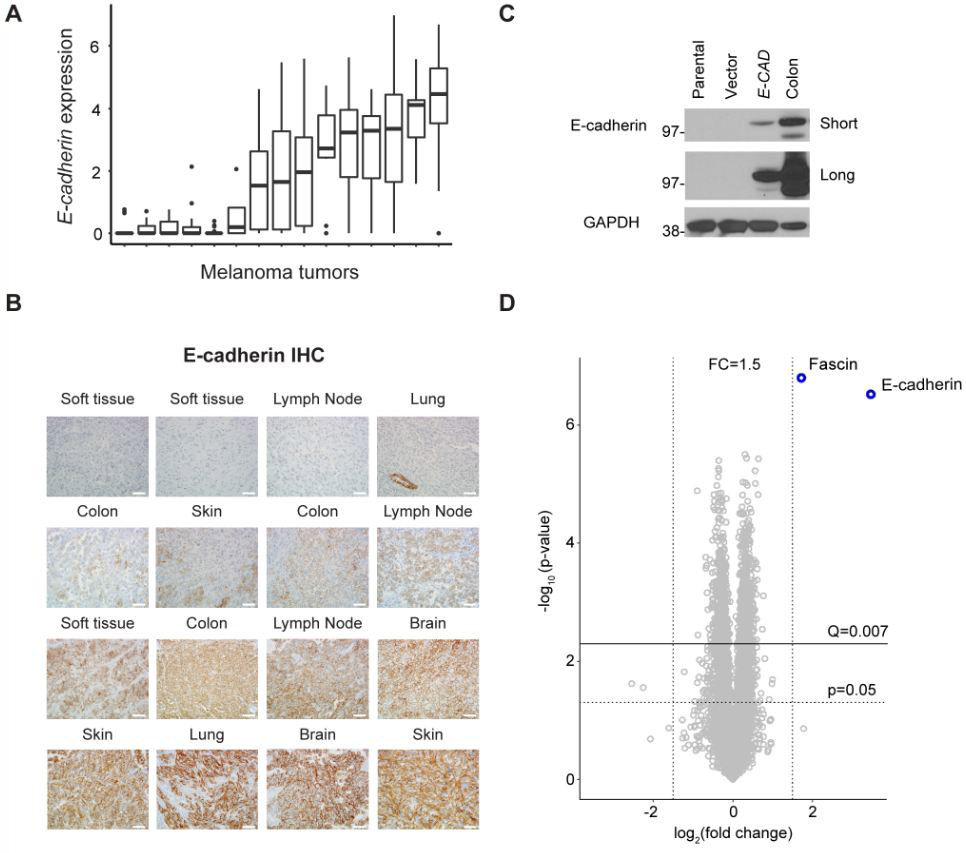
(A) E-cadherin (CDH1) expression in 15 human tumors measured by single-cell RNA-seq. Data extracted from NCBI GEO, accession GSE72056. (B) IHC analysis for E-cadherin was performed on 16 FFPE patient samples of melanoma metastatic to the site shown above each image. Image magnification, 400x. Scale bars, 50 μm. (C) Western blot analysis of E-cadherin in transduced murine melanoma. Parental line= B16F10. Vector= MSCV. GAPDH was used as a loading control. Mouse colon lysate was used as a positive control. Short and long exposure times are shown. (D) Volcano plot showing pairwise comparison between B16.Vector and B16.Ecad cells using tandem mass tag (TMT)-based quantitative proteomics. A total of 8,052 proteins were quantified. Q-value determined by Benjamini-Hochberg adjustment for multiple testing.
Exogenous expression of E-cadherin in B16F10 exerts minimal proteome changes
The varying clinical responses of melanoma tumors to ICB, paired with the knowledge that E-cadherin expression is variable and correlates with such responses, provides impetus to study the functional significance of E-cadherin within the context of immune regulation. To this end, we generated a B16F10 murine melanoma cell line, which stably expresses E-cadherin (B16.Ecad). B16F10 is poorly immunogenic, widely studied, and notoriously resistant to treatment (26). Immunoblotting revealed the parental B16F10 and vector control have undetectable levels of E-cadherin (Fig. 1C). In order to determine if E-cadherin overexpression resulted in proteome level changes that could perturb B16F10 biology, we performed quantitative mass spectrometry using tandem mass tags (TMT). Proteins from five biological replicates were isolated and labeled with isobaric mass tags, followed by mass spectrometric analysis on an Orbitrap Fusion Lumos Tribrid mass spectrometer which identified 8,052 quantifiable proteins. Only 2 proteins were differently regulated between B16.Ecad and vector by Q<0.007 and log2 fold change of >1.5 as illustrated by a volcano plot (Benjamini Hochberg correction for multiple tests (α input= 0.05) resulted in a corrected p value (Q)= 0.007) (Figure 1D). The two proteins with abundance differences meeting the significance criteria were the exogenously expressed E-cadherin (human) and fascin. Fascin is a globular actin cross-linking protein (F-actin), which is commonly observed to exhibit differential expression in melanomas, but has not been shown to impact disease progression or outcomes (32,33). Fascin has been observed to be up-regulated in B16F10 melanoma versus the less metastatic variant, B16F0 (34).
B16.Ecad displays delayed tumor growth and reduced lung metastases
In order to study the behavior of B16.Ecad within an in vivo setting, we utilized B16.SIY tumor cells as a positive control for immune-delayed tumor growth. B16.SIY cells express the model antigen (SIYRYYGL) fused to GFP which is recognized by T-cells (27). Immunoblotting for E-cadherin confirmed B16.SIY to not express E-cadherin (Fig. 2A). Importantly, exogenous expression of E-cadherin did not alter the in vitro proliferation rate of B16.Ecad when compared to vector or B16.SIY (Fig. 2B). Upon subcutaneous transplantation of 1×105 tumor cells into wild-type C57BL/6 mice, it was observed that mice bearing B16.Ecad tumors demonstrated significantly longer overall survival (tumors <150mm2, and no endpoint features) (Fig. 2C). Moreover, B16.Ecad tumors grew more slowly than vector tumor bearing mice (Fig. 2D, E). B16.Ecad demonstrated similar growth delays to B16.SIY (Fig. 2E, F). Tumor weights at endpoint between B16.Ecad and vector control showed no differences (Fig. 2G). In order to confirm B16.Ecad still expressed E-cadherin protein in vivo, immunoblotting was performed on an ex vivo tumor lysate, which revealed detectable E-cadherin protein (Fig. 2H). Tail vein injection of B16F10 cells resulted in many small primary tumors seeding in the lungs, and this was used as readout for a metastatic model (26). Upon tail vein injection of 2×105 tumor cells B16.Ecad resulted in fewer lung lesions at day 18 (Fig. 2I, J).
Figure 2. B16.Ecad displays delayed tumor growth and reduced lung metastases.
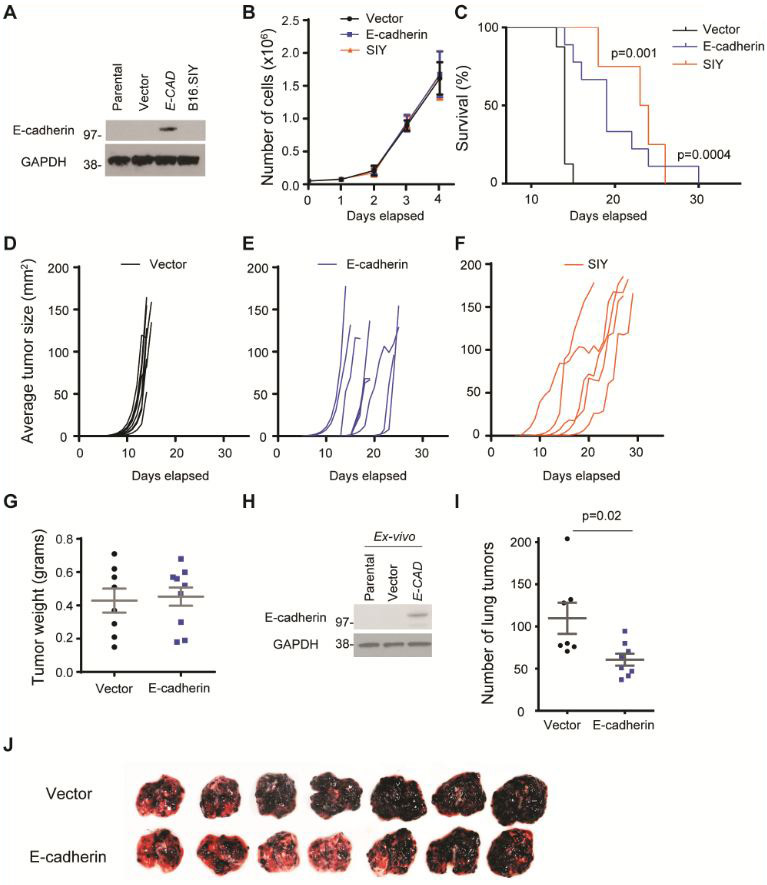
(A) Western blot analysis of E-cadherin in transduced murine melanoma cells B16.SIY. Parental line= B16F10. Vector= MSCV. GAPDH was used as a loading control. (B) In vitro proliferation of E-cadherin transfected B16F10 cells versus vector control. Cell counting was performed via trypan blue dye exclusion method. Experiment is representative of five independent experiments. (C) Kaplan-Meier plot of overall survival; p values, log-rank test. 1×105 tumor cells were injected into the subcutaneous space on the animal’s flank. (D-F) Tumor growth kinetics in individual mice (n=5-9 per group). (G) Tumor weights upon harvest. (H) Ex-vivo western blot of harvested tumors. (I) Average number of lung metastatic foci as counted by two independent observers. (J) Lung metastases as the result of tail vein injection of either vector or E-cadherin B16 melanoma on day 18 (2×105 tumor cells) (n=7 per group).
CD103 immune cells control B16.Ecad growth
To gain insight into mechanisms which confer the anti-tumor benefit to mice bearing E-cadherin expressing B16F10, we transplanted E-cadherin and vector expressing tumors into Rag1−/− mice, which are deficient in mature B and T lymphocytes. The tumor growth delay and survival advantage observed in WT mice bearing E-cadherin-expressing tumors was completely abrogated within the Rag1−/− hosts, confirming the immune system was generating the tumor growth delay (Fig. 3A-E). B16.Ecad tumors emerged rapidly and had indistinguishable growth patterns from vector control or B16.SIY (Fig. 3B-D). Tail vein injection of 2×105 B16.Ecad cells into Rag 1−/− mice resulted in fewer lung lesions (Fig. S1). E-cadherin has known roles in tumor suppression and prevention of metastasis via multiple pathways (35). We find this effect to be present without immune surveillance mechanisms intact in the Rag 1−/− mice.
Figure 3. CD103+ immune cells control B16.Ecad growth.
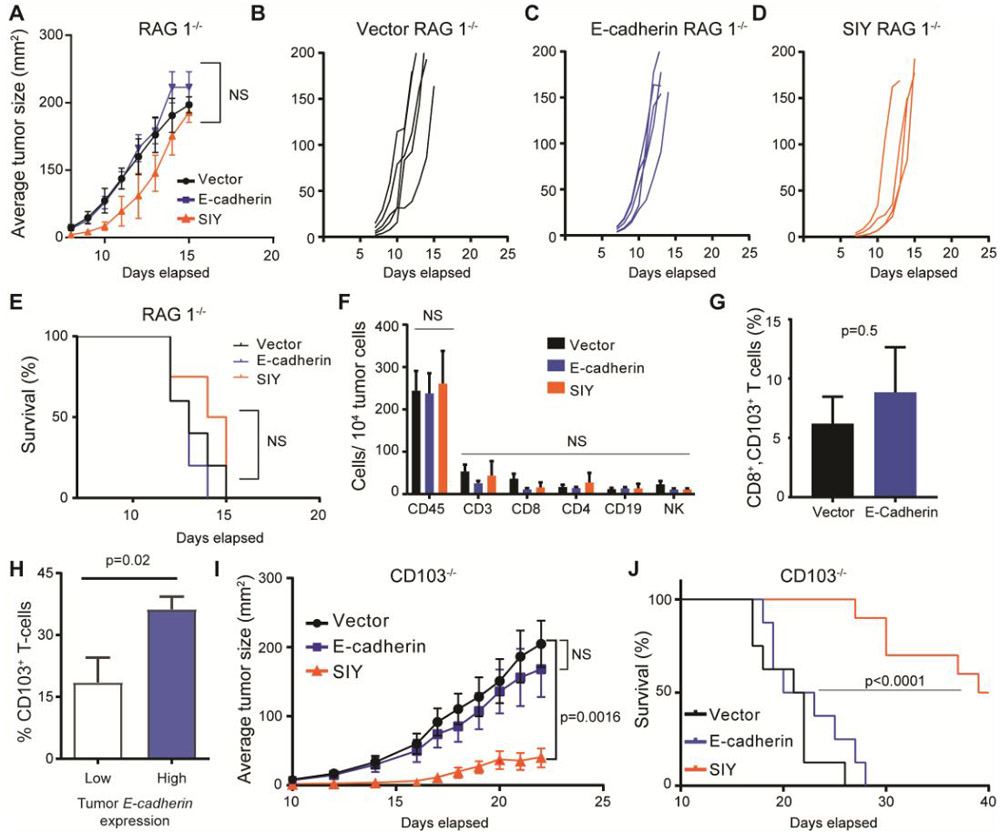
(A) Mean tumor size of each group. Data represent ±SEM. Statistical analysis by unpaired Student’s t test. (B-D) Tumor growth kinetics in Rag1−/− mice. (E) Kaplan-Meier plot of overall survival; p values, log-rank test. 1×105 tumor cells were injected into the subcutaneous space on the animal’s flank (n=4-5 per group. (F) Absolute numbers of immune cell populations per 10,000 total tumor cells as analyzed by flow cytometry. Graphs show mean ±SEM Immune cells were defined as the following: CD19 (CD45+, CD19+, CD3−), CD3 (CD45+, CD3+), CD4 (CD45+, CD3+, CD4+), CD8 (CD45+, CD3+, CD8+), NK cells (CD3−, NK1.1+). (G) Average percent of CD103+ cells per total CD8+ lymphocytes in each tumor. (H) CD103+ (ITGAE) T-cells as a percent of total T-cells present within human melanoma tumors with high and low E-cadherin (CDH1) expression. Data extracted from single-cell RNA-seq analysis: NCBI GEO, accession GSE72056. Inclusion criteria for analysis were: >5 melanoma cells and >5 T-cells per tumor. Average tumor analyzed for melanoma cell CDH1 and T-cell ITGAE contained single cell RNA-seq data on 97 tumor cells and 115 T-cells. (H-I) Tumor growth kinetics in CD103− mice, 1×105 tumor cells. (J) Mean tumor size of each group (n=7 per group). Data represent ±SEM Statistical analysis by unpaired Student’s t test.
Using wild-type mice, we isolated and analyzed tumors for immune infiltrates by flow cytometry. For fluorescence-activated cell sorting (FACS), tumors were harvested at 200mm2. Although E-cadherin and SIY expressing tumors demonstrated obvious growth delays, the percent of CD45+ cells remained unchanged from vector control (Fig. 3F). Moreover, analysis of CD45+ subpopulations by FACS revealed no differences in the composition of the immune infiltrates, including between CD8+ CD103+ tumor lymphocytes (Fig. 3F, G).
Loss of B16.Ecad tumor growth delay within the Rag1−/− hosts led us to consider more specific immune cell populations which may be mediating the control of the E-cadherin expressing melanoma. The single–cell RNA-seq dataset, used to show tumor cell E-cadherin expression in Figure 1A, was accessed and analyzed for CD103+ (Itgae) T-cells as they possess a unique ability to bind to E-cadherin expressing target cells. CD103+ T-cells were examined as a percent of total T-cells residing in melanoma tumors which were separated into high and low E-cadherin expression groups (NCBI GEO, accession GSE72056) (31,36,37). Tumors with high E-cadherin expression (>0 median E-cadherin expression level) had a significantly higher percent of CD103+ T-cells than low E-cadherin tumors (0 median E-cadherin expression level) (Fig. 3H).
To test whether CD103 was mediating an anti-tumor effect Itgae−/− mice were obtained. Itgae knockout mice (CD103−/−) are fully viable, fertile, normal in size, have no observable physical or behavioral abnormalities and demonstrate no endogenous expression of CD103 on intraepithelial lymphocytes or TGF-β treated splenocytes (38). Further, mice missing CD103 demonstrate reduced intraepithelial and lamina propria lymphocytes, confirming a loss in T-cell migration potential (38). CD103−/− mice were engrafted with B16.Ecad, vector, and SIY cells as before. The B16.Ecad tumor growth delay observed in wild-type mice was lost, in a similar fashion to the Rag1−/− hosts (Fig. 3I). Overall survival was not different between B16.Ecad and vector tumor bearing mice (Fig. 3J).
B16.Ecad shows enhanced responses to combination checkpoint blockade
Given the tumor growth delay observed in mice bearing B16.Ecad tumors, we next sought to characterize the role of tumor E-cadherin expression in the context of immune checkpoint blockade. Wild-type C57BL/6 mice were challenged with 1×105 vector or B16.E-cadherin cells and ICB treatment (anti-CTLA-4 and anti-PD-1) began at day four (Fig. 4A). E-cadherin-tumor bearing mice showed a significant survival benefit from combination ICB over vector treated mice (Fig. 4B). ICB treatment significantly reduced the rate of tumor growth in both B16.Ecad and vector compared to control immunoglobulin treated mice (Fig. 4C-F). All experimental mice, except the E-cadherin-expressing ICB group, had tumor volumes greater than 150 mm2 by day 20 (Fig. 4C-F, G). Three of seven mice injected with B16.E-cad and treated with ICB were tumor-free at the conclusion of the experiment on day 40.
Figure 4. B16.Ecad shows enhanced responses to combination checkpoint blockade.
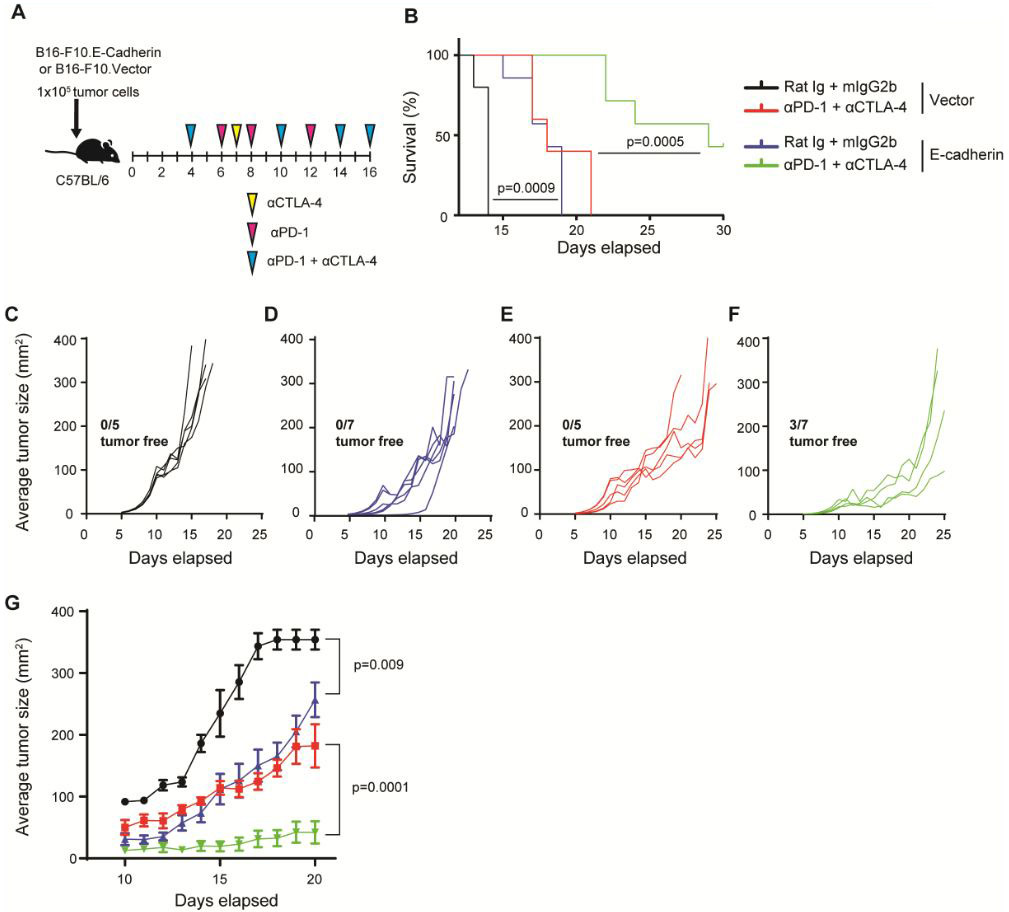
(A) Schema of experimental design showing time points of drug dosing. αCTLA4 or Rat Ig (100 μg) was administered on days 4, 7,10,14,16 i.p., αPD-1 or mIgG2b (250 μg) was administered on days 4,6,8,10,12,14,16 i.p. 1×105 tumor cells were injected into the subcutaneous space on the animal’s flank. (B) Kaplan-Meier plot of overall survival; p values, log-rank test. (C-F) Individual tumor outgrowth measured in mm2 once per day (n=5-7 per group). 3/7 B16.Ecad mice remained tumor free 40 days post injection. All other mice developed tumors. (G) Mean tumor size of each treatment group. Data represent mean ±SEM. Statistical analysis by unpaired Student’s t test.
CD103+ lymphocytes infiltrate E-cadherin expressing human metastatic melanoma tumors
A cohort of human metastatic melanoma tumor biopsies was interrogated by IHC analysis for the presence of CD103+ lymphocytes (CD3+) when segregated by tumor E-cadherin expression. IHC was performed for E-cadherin on 15 tumor samples (4 E-cadherin negative, 11 E-cadherin positive). Both E-cadherin negative and E-cadherin positive melanoma tumors showed the presence of CD103 lymphocytes by double staining (Fig. 5A, B). Overlay of CD3+ and CD103+ staining showed a strong trend of more infiltration of double positive (CD3+, CD103+) lymphocytes in E-cadherin positive patient melanoma tumors (Fig. 5B-D).
Figure 5. CD103+ lymphocytes infiltrate E-cadherin expressing human metastatic melanoma tumors.
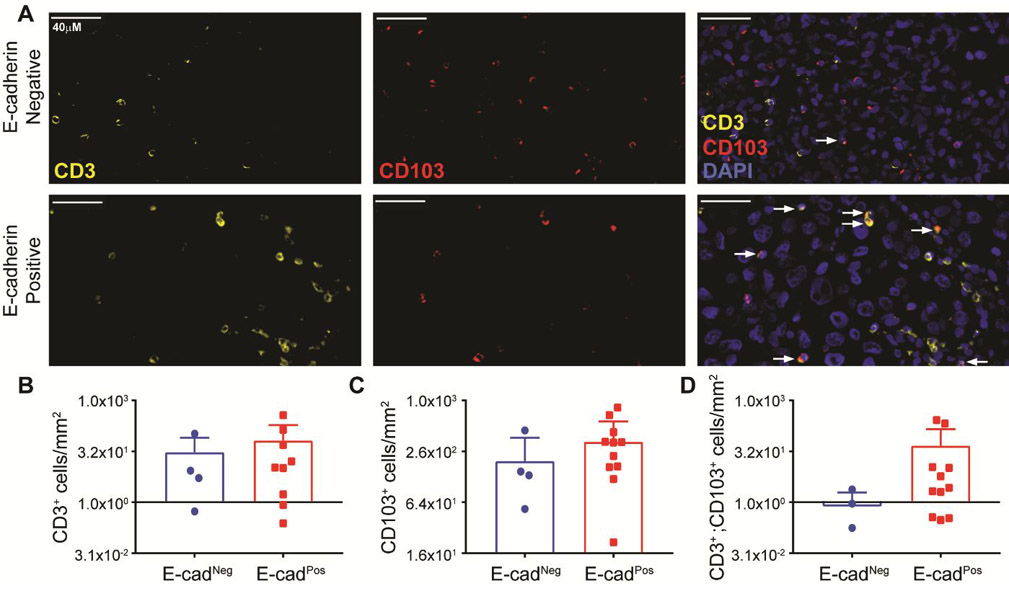
(A) Representative images of immunoflourescent staining against CD3 (yellow, left panel), CD103 (red, middle panel), and overlay with DAPI (blue, right panel). Fifteen tumors were assayed. Four E-cadherin negative (top row) and eleven positive tumors (bottom row) were examined. Tumor E-cadherin status was determined by a dermatopatholgist. E-cadherin negative tumors had no positive tumor cells, while any level of positive staining (low to high) was considered a positive E-cadherin tumor. (B) Number of CD3+ cells per mm2 tumor area. (C) Number of CD103+ cells per mm2 tumor area. (D) Number of CD3+ CD103+ cells per mm2 tumor area.
Discussion
Loss of E-cadherin expression is a common event in tumor progression, and provides cells a means of escape from the primary tumor for travel to distant body regions. However, non-canonical metastatic pathways or loss of mesenchymal signaling can result in melanoma metastases with E-cadherin expression. Analysis of single-cell melanoma transcription data and immunohistochemistry of patient samples, revealed a diversity of E-cadherin expression among human melanomas, which has been unexplored within the context of immune checkpoint blockade (31).
E-cadherin has known roles impacting the regulation of signaling pathways including: Wnt/β-catenin, PI3K/Akt, Rho-GTPase family proteins, and NF-κB (39–42). We hypothesized one potential effect of forced E-cadherin expression in the B16F10 line would be dampening of β-catenin signaling as active β-catenin signaling has been implicated in resistance to PD-1 checkpoint blockade (10). E-cadherin contains a cytoplasmic tail of 150 amino acids which sequesters β-catenin, preventing it from joining the nuclear pool of β-catenin that affects gene transaction (43). Due to the proteomic findings, along with the similar growth rates in vitro (Fig. 2B), we proceeded with data supporting that our E-cadherin overexpression had not drastically perturbed B16F10 biology within the measureable proteome.
One potential explanation for the apparent decrease in tumorigenicity in the B16.Ecad group, is via E-cadherin affecting tumor cell growth. Limited evidence suggests E-cadherin can have a growth inhibitory role in vitro through alteration of β-catenin activity (34). However, based upon the results of the in vitro proliferation assay and the proteomic analysis, we find no impact of exogenous E-cadherin on tumor cell growth. We therefore reasoned an underlying immune response was responsible for the tumor control observed after transplantation of B16.Ecad into wild-type mice.
Upon transplantation of tumor cell lines to wild-type mice, we observed no significant differences in the immune-cell infiltrate populations of B16.Ecad, vector control, and SIY tumors. It appears T-cell effector activity, Treg behavior, and APC function seem to be more relevant factors in anti-melanoma activity than the percent of immune cells residing within the tumor at endpoint (44). Our results support the previous finding that the main mechanism which increases anti-tumor T-cell function is via stimulation of T-cells already present within the tumor site (44).
Our data suggest the B16.Ecad tumors are more responsive to combination ICB therapy, due to contributions of CD103 immune cell populations, which have increased activity against E-cadherin expressing B16F10 melanoma (Figs. 3, 4). E-cadherin has been shown in vitro to have immunostimulatory effects, through binding to αEβ7 on T-cell subsets (45). Previously it has been reported tumor E-cadherin interaction with CD103 on tumor infiltrating lymphocytes plays a major role in effective tumor cell lysis in NSCLC (46). Additionally, besides facilitating retention within the tumor, CD103 is recruited to the immune synapse formed between the tumor cell and the cytotoxic lymphocyte, promoting cytotoxic functions (47,48). CD103+ immune cells of both myeloid and lymphoid lineages have been implicated in production of anti-tumor responses to melanoma, but CD103 function in melanoma has not previously been connected with tumor E-cadherin (22,49). Further characterization of CD103+ immune cell subsets is necessary to determine whether CD103 expressing dendritic cells, effector CD8 cells, or regulatory CD4 cells are functioning through E-cadherin binding to mediate anti-tumor activity. Three of seven E-cadherin ICB treated mice were able to completely reject the tumor challenge, indicating the immune delays observed in untreated wild-type mice were amplified upon checkpoint blockade therapy. The magnitude of increased survival of B16.Ecad ICB versus vector control ICB was greater than the increased survival of B16.E-cad relative to vector control minus blockade treatment. These data suggest a basis for the increased responsiveness observed in ICB treated patients with higher E-cadherin expressing melanomas (11,14). In pre-ICB treatment human metastatic melanoma tumors, double staining revealed a trend towards more CD103+ lymphocytes in E-cadherin positive tumors. Our work supports future studies to further characterize the role of these unique lymphocyte populations during immune checkpoint blockade therapy.
In human melanoma, CD103+ tumor-resident CD8+ T-cells are associated with improved survival in immunotherapy naïve patients and longitudinal biopsies taken from patients undergoing anti-PD-1 therapy showed significant expansion of this T-cell subpopulation (22). Here we show the first evidence melanoma tumor E-cadherin expression enhances CD103 anti-melanoma responses, and provide a rationale for a possible tumor resistance mechanism by downregulation of E-cadherin to avoid robust CD103-mediated immune attack. We find melanoma tumors with E-cadherin expression better recapitulate the natural tissue architecture in which T-cells perform immunosurveillance activities on E-cadherin expressing cells through CD103. Analysis of CD103 deficient CD8 lymphocytes is needed to elucidate this interaction and to support this observation. Animal models which employ tumor specific T-cells would allow for additional mechanistic studies, informing the specific mechanisms through which tumor E-cadherin/ host CD103 produce anti-tumor behavior. In vitro studies support the concept that E-cadherin/CD103 binding generates intracellular changes within T-cells, but this is yet to be determined in the context of melanoma immune checkpoint blockade therapies.
Immune-mediated destruction of tumor cells is the objective of immunotherapy. In order to generate a successful immune response, T-cells must traffic and adhere to the tumor cell targets and perform cytotoxic functions. This process is mediated by a wide array of receptor interactions which can have enhancing or dampening effects on the overall immune response. The immune system offers a diverse array of immune cell subsets, which are specialized to conduct immunosurveillance in normal tissues. Our results suggest, future therapeutic strategies may seek to amplify CD103 immune cell subsets to target E-cadherin expressing tumors from a variety of cancer types.
Supplementary Material
Significance:
Findings identify the mechanism behind checkpoint blockade resistance observed in melanoma that has undergone mesenchymal transition and suggest activation of CD103+ immune cells as a therapeutic strategy against other E-cadherin-expressing malignancies.
Acknowledgments:
The authors would like to thank the UAMS Division of Laboratory Animal Medicine; the UAMS Proteomics Core; the UAMS Flow Cytometry Core; the Arkansas Children’s Research Institute Bioinformatics Core.
Additional Information: We acknowledge support from NIH grant P20GM121293 (AJT) and the Scharlau Family Endowed Chair in Cancer Research to AJT. This study was additionally supported by the National Institutes of Health (UL1TR000039, P20GM103625, S10OD018445 and P20GM103429).
Footnotes
Conflict of interest statement: The authors have declared that no conflict of interest exists.
References:
- 1.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma N Engl J Med. Massachusetts Medical Society; 2015;373:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma N Engl J Med. Massachusetts Medical Society; 2017;377:1345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma N Engl J Med. Massachusetts Medical Society; 2014;371:2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (80- ). 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. NIH Public Access; 2015;350:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (80- ). 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. NIH Public Access; 2014;515:568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schumacher TN, Kesmir C, van Buuren MM. Biomarkers in cancer immunotherapy. Cancer Cell. 2015;27:12–4. [DOI] [PubMed] [Google Scholar]
- 10.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. Nature Research; 2015;523:231–5. [DOI] [PubMed] [Google Scholar]
- 11.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma Cell. Elsevier; 2016;165:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shields BD, Mahmoud F, Taylor EM, Byrum SD, Sengupta D, Koss B, et al. Indicators of responsiveness to immune checkpoint inhibitors Sci Rep. Springer; US; 2017;7:807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Roy F Beyond E-cadherin: roles of other cadherin superfamily members in cancer Nat Rev Cancer. Nature Publishing Group; 2014;14:121–34. [DOI] [PubMed] [Google Scholar]
- 16.van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008;65:3756–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene. 2008;27:6920–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pećina-Slaus N Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int. BioMed Central; 2003;3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schenkel JM, Masopust D. Tissue-Resident Memory T Cells. Immunity. 2014;41:886–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pauls K, Schön M, Kubitza RC, Homey B, Wiesenborn A, Lehmann P, et al. Role of Integrin αE(CD103)β7 for Tissue-Specific Epidermal Localization of CD8+ T Lymphocytes. J Invest Dermatol. 2001;117:569–75. [DOI] [PubMed] [Google Scholar]
- 21.Cepek KL, Shaw SK, Parker CM, Russell GJ, Morrow JS, Rimm DL, et al. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the αEβ7 integrin Nature. Nature Publishing Group; 1994;372:190–3. [DOI] [PubMed] [Google Scholar]
- 22.Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A, et al. CD103+ tumor-resident CD8+ T cells are associated with improved survival in immunotherapy naive melanoma patients and expand significantly during anti-PD1 treatment Clin Cancer Res. American Association for Cancer Research; 2018;clincanres.2257.2017. [DOI] [PubMed] [Google Scholar]
- 23.Wisniewski JR, Zougman A, Nagaraj N, Mann M, Wi JR. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:377–362. [DOI] [PubMed] [Google Scholar]
- 24.McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem. 2014;86:7150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–58. [DOI] [PubMed] [Google Scholar]
- 26.Ya Z, Hailemichael Y, Overwijk W, Restifo NP. Mouse model for pre-clinical study of human cancer immunotherapy. Curr Protoc Immunol. 2015;2015:20.1.1–20.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kline J, Brown IE, Zha Y-Y, Blank C, Strickler J, Wouters H, et al. Homeostatic Proliferation Plus Regulatory T-Cell Depletion Promotes Potent Rejection of B16 Melanoma. Clin Cancer Res. 2008;14:3156–67. [DOI] [PubMed] [Google Scholar]
- 28.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors Proc Natl Acad Sci U S A. National Academy of Sciences; 2010;107:4275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pachynski RK, Zabel BA, Kohrt HE, Tejeda NM, Monnier J, Swanson CD, et al. The chemoattractant chemerin suppresses melanoma by recruiting natural killer cell antitumor defenses. J Exp Med. 2012;209:1427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong SHM, Fang CM, Chuah L-H, Leong CO, Ngai SC. E-cadherin: Its dysregulation in carcinogenesis and clinical implications Crit Rev Oncol Hematol. Elsevier; 2018;121:11–22. [DOI] [PubMed] [Google Scholar]
- 31.Tirosh I, Izar B, Prakadan SM, Ii MHW, Treacy D, Trombetta JJ, et al. Dissecting the multicellular exosystem of metastatic melanoma by single-cell RNA-seq. Science (80- ). 2016;352:189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yildiz L, Kefeli M, Aydin O, Kandemir B. Fascin expression in melanocytic lesions of the skin. Eur J Dermatol. 19:445–50. [DOI] [PubMed] [Google Scholar]
- 33.Ma Y, Faller WJ, Sansom OJ, Brown ER, Doig TN, Melton DW, et al. Fascin expression is increased in metastatic lesions but does not correlate with progression nor outcome in melanoma. Melanoma Res. 2015;25:169–72. [DOI] [PubMed] [Google Scholar]
- 34.Kim SH, Kim Y, Kim M, Kim DS, Lee SC, Chi S-W, et al. Comparative proteomic analysis of mouse melanoma cell line B16, a metastatic descendant B16F10, and B16 overexpressing the metastasis-associated tyrosine phosphatase PRL-3. Oncol Res. 2009;17:601–12. [DOI] [PubMed] [Google Scholar]
- 35.Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008; [DOI] [PubMed] [Google Scholar]
- 36.Hadley W, Romain F, Lionel H, Kirill M. dplyr: A Grammar of Data Manipulation. 2017. [Google Scholar]
- 37.Wickham H Ggplot2. Elegant Graph. Data Anal. 2009. [Google Scholar]
- 38.Schön MP, Arya A, Murphy EA, Adams CM, Strauch UG, Agace WW, et al. Mucosal T lymphocyte numbers are selectively reduced in integrin alpha E (CD103)-deficient mice. J Immunol. 1999;162:6641–9. [PubMed] [Google Scholar]
- 39.Stairs DB, Bayne LJ, Rhoades B, Vega ME, Waldron TJ, Kalabis J, et al. Deletion of p120-Catenin Results in a Tumor Microenvironment with Inflammation and Cancer that Establishes It as a Tumor Suppressor Gene. Cancer Cell. 2011;19:470–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuphal S, Poser I, Jobin C, Hellerbrand C, Bosserhoff AK. Loss of E-cadherin leads to upregulation of NFκB activity in malignant melanoma. Oncogene. 2004;23:8509–19. [DOI] [PubMed] [Google Scholar]
- 41.Lau MT, Klausen C, Leung PCK. E-cadherin inhibits tumor cell growth by suppressing PI3K/Akt signaling via Β-catenin-Egr1-mediated PTEN expression. Oncogene. 2011;30:2753–66. [DOI] [PubMed] [Google Scholar]
- 42.Cowell CF, Yan IK, Eiseler T, Leightner AC, Döppler H, Storz P. Loss of cell-cell contacts induces NF-kappaB via RhoA-mediated activation of protein kinase D1. J Cell Biochem. 2009;106:714–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gottardi CJ, Wong E, Gumbiner BM. E-cadherin suppresses cellular transformation by inhibiting β-catenin signaling in an adhesion-independent manner. J Cell Biol. 2001;153:1049–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother cancer. 2014;2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van den Bossche J, Malissen B, Mantovani A, De Baetselier P, Van Ginderachter JA. Regulation and function of the E-cadherin/catenin complex in cells of the monocyte-macrophage lineage and DCs. Blood. 2012;119:1623–33. [DOI] [PubMed] [Google Scholar]
- 46.Le Floc’h A, Jalil A, Vergnon I, Le Maux Chansac B, Lazar V, Bismuth G, et al. Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med. 2007;204:559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Floc’h AL, Jalil A, Franciszkiewicz K, Validire P, Vergnon I, Mami-Chouaib F. Minimal Engagement of CD103 on Cytotoxic T Lymphocytes with an E-Cadherin-Fc Molecule Triggers Lytic Granule Polarization via a Phospholipase C -Dependent Pathway. Cancer Res. 2011;71:328–38. [DOI] [PubMed] [Google Scholar]
- 48.Franciszkiewicz K, Le Floc’h A, Boutet M, Vergnon I, Schmitt A, Mami-Chouaib F. CD103 or LFA-1 Engagement at the Immune Synapse between Cytotoxic T Cells and Tumor Cells Promotes Maturation and Regulates T-cell Effector Functions. Cancer Res. 2013;73:617–28. [DOI] [PubMed] [Google Scholar]
- 49.Lè Ne Salmon H, Idoyaga J, Rahman A, Brody J, Ginhoux F, Correspondence MM, et al. Expansion and Activation of CD103 + Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.