

Abstract
Aerobic respiration is a major source of energy in eukaryotic cells. In this setting, ATP production by the mitochondrial respiratory chain relies on the availability of NADH and FADH2 to donate protons and electrons. The flux of electrons down the electron transport chain, based on a series of oxidation–reduction reactions, releases energy that allow for the transport of H+ ions across the inner mitochondrial membrane. The resulting proton-motive force is employed to drive ATP synthesis, while the final acceptor of the electrons flowing through the respiratory chain if molecular oxygen. A side effect of this process is the generation of reactive oxygen species (ROS). While mitochondrial ROS (mtROS) production has been linked to many pathological conditions (i.e., aging and tumorigenesis), recent evidence suggests that multiple cells, including malignant cells, employ these by-products of energy production as signals to control various cellular processes. Here, we describe protocols to use chemical probes for measuring mtROS production in intact cells by flow cytometry and spectrofluorometry.
1. INTRODUCTION
The image of the mitochondria (mt) as the powerhouse of the cell has undergone a dramatic transformation. Energy production is a major responsibility of the mt; however, these organelles perform a number of other cellular functions including calcium handling, production of reducing equivalents, cell death through intrinsic apoptosis, and participation in cell signaling. While once viewed in the negative light as damaging by-products of aerobic respiration, reactive oxygen species (ROS) production by mt are now viewed as essential signaling mediators that act in reduction–oxidation signaling pathways. The signaling pathways impacted by ROS generated by the mitochondrial ROS (mtROS) have been linked to both normal as well as aberrant conditions, including tumor growth. Advances in whole-cell detection of ROS will lead to an enhanced understanding of how these molecules influence cellular processes. In this chapter, we describe methods to detect mtROS that can be applied to a wide range of primary and transformed cell types from both human and animal models.
2. MITOCHONDRIAL ROS PRODUCTION
2.1. Physiology
There are four structural entities within the mitochondria: the inner and outer membranes, and the two compartments that they create, the intermembrane space and the matrix. These structural regions are the scaffolds upon which many important metabolic processes take place. Control of mitochondria comes from expression of genes from both the nuclear and mitochondrial genomes. Intergenomic interactions have been implicated in pathophysiology (Chen, Gusdon, Thayer, & Mathews, 2008; Chen, Lu, et al., 2008; Johnson, Zheng, Bykhovskaya, Spirina, & FischelGhodsian, 2001).
Mitochondrial respiration is the process in which water is formed and energy trapped in the bonds of the ATP molecules. The energy that drives respiration is obtained from the oxidation of fatty acids, amino acids, and carbohydrates. The product of macronutrient oxidation and glycolysis is acetyl-CoA which, when joined with oxaloacetate, forms citrate to be processed in the mitochondrial Kreb’s cycle. The Kreb’s cycle produces reducing equivalents as well as carbon dioxide, in a cycle of enzymatically catalyzed reactions. For every turn of the Kreb’s cycle, two carbon dioxide molecules and eight reducing equivalents are produced. The reducing equivalents (NADH and FADH2) are utilized by the respiratory chain to build an electrochemical potential across the mitochondrial inner membrane.
In the electron transport process, the free energy of electron transfer is coupled to ATP synthesis. The free energy necessary to generate ATP is extracted from the oxidation of NADH by NADH dehydrogenase (complex I) and FADH2 by the Kreb’s cycle component succinate dehydrogenase (complex II). The electron transport chain (ETC) passes electrons from lower to higher standard reduction potentials and ultimately to oxygen forming water. Oxidative reactions, catalyzed by inner mitochondrial membrane embedded enzyme complexes, generate an electrochemical or pH gradient, by proton translocation, across the mitochondrial inner membrane. ATP can then be made from ADP and Pi by the membrane-bound F1F0 ATP synthase using the proton gradient to drive synthesis. The oxidative phosphorylation (OXPHOS) system is composed of five respiratory complexes (complexes I–V) that are assembled from subunits derived from mtDNA and nDNA plus the adenine nucleotide translocator.
Complex I contains one molecule of flavin mononucleotide (FMN), a redox-active prosthetic group, and six to seven iron–sulfur [Fe–S] clusters that participate in the electron transfer (Efremov & Sazanov, 2012; Kim, Khalimonchuk, Smith, & Winge, 2012). Redox-active Fe–S clusters occur as prosthetic groups of nonheme Fe–S proteins. There are two common types, designated [2Fe–2S] and [4Fe–4S] clusters. Each cluster contains an equal number of iron and sulfide ions and is coordinated to four protein-cysteine-sulfhydryl groups, with the iron atoms coordinated between four sulfurs to form a tetrahedron. Electrons pass from the FMN through the multiple Fe–S centers to coenzyme Q [ubiquinone or CoQ]. The reduction of CoQ is coupled to proton translocation (Hatefi, 1985).
Complex II contains three small hydrophobic subunits, all of which are nuclear encoded (Shoffner & Wallace, 1994). The physiological function of complex II is to catalyze the transfer of electrons from succinate to CoQ, with the participation of a covalently bound flavin adenine dinucleotide (FAD), one [4Fe–4S] cluster, two [2Fe–2S] clusters, and one cytochrome b560. Electron transfer from succinate to CoQ has a standard redox potential that is insufficient to provide the free energy to drive ATP synthesis, but the complex is important in that it allows these relatively high-potential electrons to enter the electron transport system.
Complex III catalyzes the electron transfer from reduced CoQ to cytochrome c. This complex contains two b-cytochromes, one cytochrome c, and a [2Fe–2S] cluster. Cytochromes are redox-active heme-proteins that reversibly alternate between the Fe(II) and Fe(III) oxidation states. The electrons pass through cytochrome c to cytochrome c oxidase of complex IV (Wallace, 1992).
The reactions catalyzed by complex IV include the four-electron reductions of O2 by the single-electron donor, ferrocytochrome c. The redox-active centers are two a-type heme groups, which, unlike c-type heme groups contain no sulfur, and two Cu atoms that alternate between their +1 and +2 oxidation states. The identical hemes (a and a3) are each associated with a copper atom (Cua or CuA and Cua3 or CuB, respectively). Heme a and Cua are of low potential and are the recipients of electrons from cytochrome c. Heme a3 and CuB are of higher potential and are the sites of oxygen reduction (Hatefi, 1985).
ATP production by the mitochondria occurs through the coupling of the redox free energy to the phosphorylation of ADP (Hatefi, 1985). The free energy of the electrochemical proton gradient derived from the operation of respiratory complexes I, III, and IV, across the inner mitochondrial membrane, is harnessed in the synthesis of ATP from ADP and Pi, by the F1F0 ATP synthase (Hatefi, 1985). Also known as complex V of the respiratory chain, this enzyme is also capable of ATP hydrolysis linked to proton translocation from the matrix to the intermembrane space (Hatefi, 1985; Senior, 1988).
Previous reports over the past four decades have provided evidence for ROS production by mitochondria and have further implicated specific sites within the ETC as ROS generators. ETC inhibitors, such as rotenone and antimycin A, have been used extensively to localize mtROS production. Redox centers within Complex I and III have been noted as the major sites of mtROS production (St-Pierre, Buckingham, Roebuck, & Brand, 2002; Sugioka et al., 1988; Turrens & Boveris, 1980). Within complex I, both the FMN and a distal site presumably the ubiquinone-binding site have been shown to be capable of generating ROS with the direction of electron flow dictating the relative contribution from each site (Treberg, Quinlan, & Brand, 2011). ROS from Complex I are released into the mitochondrial matrix. Under ATP-generating conditions, the FMN site within complex I has been shown to be responsible for the majority of ROS production (Quinlan, Treberg, Perevoshchikova, Orr, & Brand, 2012). Complex III supported ROS production can occur at both the Qi site of the cytochrome bc1 complex promoted by a partially oxidized ubiquinone pool (Drose & Brandt, 2008) as well as the Qp site that faces the inner membrane space. For Complex III ROS production is directional, with Qi generating ROS in the mitochondrial matrix, while ROS from the Qp site of Complex III are released into the intermembrane space (Gusdon, Votyakova, & Mathews, 2008; Gusdon, Votyakova, Reynolds, & Mathews, 2007; Muller, Liu, & Van Remmen, 2004; St-Pierre et al., 2002; Tahara, Navarete, & Kowaltowski, 2009).
It is important to note that ROS production is dissimilar when comparing isolated mitochondria from different tissues (St-Pierre et al., 2002). Studies using rat models have demonstrated that during state 4 respiration, mitochondria from brain and heart produce significantly more ROS than kidney, skeletal muscle, or liver mitochondria. During state 3 respiration (after addition of ADP), a significant drop of peroxide production in brain as well as in heart and skeletal muscle mitochondria was observed; however, ROS production from liver was almost unchanged, while kidney mitochondria exhibited only a slight reduction in ROS. Further, mitochondria isolated from brain, cardiac muscle, and kidney produced high levels of ROS when respiring on succinate, while liver mitochondria exhibited elevated ROS production when complex I substrates were added. Skeletal muscle was uniformly low for all substrates tested. These distinctions could result from gene expression differences in mitochondrial proteins that control mitochondrial respiration or other mitochondrial functions (e.g., substrate transporters) among these tissues. These data also suggest that specific mtDNA mutations exhibit tissue-specific phenotypes due to tissue-specific ROS production that when combined can drive pathophysiology.
2.2. Pathophysiology/apoptosis
Mitochondrial dysfunction has been implicated in a growing number of disorders. The manifestations of these syndromes have in many cases been associated with an imbalance in mtROS generation. The ROS produced by the mitochondria principally consist of superoxide and hydrogen peroxide. ROS generated by the mitochondria have also been implicated in cancer (Sohn et al., 2013), aging (Shigenaga, Hagen, & Ames, 1994; Sohal & Weindruch, 1996; Terzioglu & Larsson, 2007; Tong, Schriner, McCleary, Day, & Wallace, 2007), as well as in cardiovascular disorders, including hypertension (Peterson, Sharma, & Davisson, 2006; Puddu, Puddu, Cravero, Rosati, & Muscari, 2008; Zimmerman et al., 2002), atherosclerosis (Bayir et al., 2006; Hsiai & Berliner, 2007; Pandya, 2001; Singh & Jialal, 2006; Steinberg & Witztum, 2002; Zhang et al., 2007), and myocardial infarction (Surekha et al., 2007; Venardos & Kaye, 2007). mtROS has been tightly linked to neurodegenerative conditions, including Alzheimer’s disease (Butterfield, Howard, & LaFontaine, 2001; Kennedy, Sandiford, Skerjanc, & Li, 2012; Lin & Beal, 2006), Parkinson’s disease (Gautier, Kitada, & Shen, 2008; Kennedy et al., 2012; Lin & Beal, 2006; Tretter, Takacs, Hegedus, & Adam-Vizi, 2007), amyotrophic lateral sclerosis (Lin & Beal, 2006; Panov et al., 2011), and Huntington’s disease (Butterfield et al., 2001; Lin & Beal, 2006; Siddiqui, Hanson, & Andersen, 2012). β-Cell dysfunction and death in both type 1 and type 2 diabetes have been linked to mtROS production (Chen, Gusdon, Piganelli, Leiter, & Mathews, 2011; Gusdon et al., 2007; Gusdon et al., 2008; Krauss et al., 2003), and increased superoxide production has been shown to cause DNA damage leading poly(ADP-ribose) polymerase activation, inhibition of glyceraldehyde 3-phosphate dehydrogenase, and induction of the main pathways of hyperglycemia induced pathology (Du et al., 2003).
In some of these pathophysiological cases, the mitochondria play a key role through participation in the intrinsic pathway of apoptosis by producing ROS, undergoing permeability transition, and releasing cytochrome c for apoptosome formation. Literature supports the activation of caspase 8 and downstream apoptotic events by H2O2 (Aggarwal, Misro, Maheshwari, Sehgal, & Nandan, 2010; Payton et al., 2007; Perez-Cruz, Carcamo, & Golde, 2007; Rigoulet, Yoboue, & Devin, 2011; Shenoy, Wu, & Pervaiz, 2009). Further, upon exposure to peroxide, TNFR-associated factor 2 (TRAF2), receptor-interacting protein (RIP), and FAS-associated death domain (FADD) dissociate from TNFR1 and this new complex is capable of recruiting and activating caspase 8 as well as ROS modulator (Romo1) to promote apoptosis (Kim, Lee, Park, & Yoo, 2010; Shen et al., 2004; Terry Powers et al., 2010). In sum, mtROS can directly damage cells leading to disease onset, the development of sequelae (e.g., Tumorigenesis or Diabetes complications), and impact cellular processes by acting as signaling intermediates to potentiate proliferative or apoptotic signals.
2.3. Redox signaling
ROS act as signaling mediators not only during cell death/apoptosis but also in processes that control cellular proliferation and differentiation. A clear role for cytoplasmic ROS generated by NADPH Oxidase 2 as well as Dual Oxidase 1 has been shown in T cell receptor signaling as well as downstream activation and differentiation of T cells (Jackson, Devadas, Kwon, Pinto, & Williams, 2004; Kwon et al., 2010; Thayer et al., 2011; Tse et al., 2010). Mitochondrial metabolic activity plays a central role in controlling T cell activation, proliferation, and programmed cell death (Chhabra, 2010). Specifically, during the process of T cell activation, mt are recruited to the immunological synapse where through balanced fusion/fission activity, they control transmembrane potential [∆Ψμ], ATP synthesis, and local calcium concentrations (Baixauli et al., 2011; Quintana et al., 2007; Schwindling, Quintana, Krause, & Hoth, 2010). Additionally, calcium-dependent mtROS production is induced. In T cells, ROS production by the Qp site of complex III is required for antigen-induced T cell activation. The loss of Qp generated ROS in T cells results in a failure of transcription factor activation and an inability to produce interleukin-2, a cytokine essential for T cell proliferation (Kaminski et al., 2010; Sena et al., 2013). However, the specific target of mtROS in this process has not yet been identified.
Mitochondria are now recognized as contributors during cell transformation and tumor growth. Production of mtROS in tumors is thought to result from oncometabolism, a term that describes the increased metabolic and energetic demands of transformed cells. Emerging evidence has associated mtROS productionbytumors to the dysregulated expressionof nuclear-encodedmitochondrial proteins, such as CISD1 (mitoNEET) (Sohn et al., 2013). Overexpression of the CISD1 in tumors results in iron accumulation, elevated mtROS, and heightened tumor growth, while conversely shRNA-mediated suppression of expression led to reduced cell proliferation and inhibited tumor growth. Overall, mtROS are an important characteristic of oncometabolism. By extensively characterizing the production and actions of mtROS in tumors, including the mechanisms of production and cellular targets, therapeutic strategies can be developed that target processes specific to cell transformation.
3. METHODS TO DETECT ROS PRODUCTION
3.1. Reagents
3.1.1. Culture medium
3.1.1.1. Culture medium for mouse beta cell lines (NIT-1 and NIT-4):
DMEM (Gibco, Catalog number 11885-084) supplemented with:
10% FBS (Hyclone, Catalog number SH30071.03, heat inactivated)
0.02% BSA (Sigma, Catalog number A8412)
0.15 mM HEPES (Cellgro, Catalog number 25-060-Cl)
MEM nonessential amino acid (Gibco, Catalog number 11140)
Add glucose to a final concentration of 16.5 mM
Penicillin and streptomycin (Gemini Bio-Products, Catalog number 400-109. Penicillin final concentration 100 U/mL and streptomycin final concentration 100 μg/mL)
3.1.1.2. Culture medium for human beta cell line (BetaLox 5):
DMEM (Cellgro, Catalog number 10-014-CV) supplemented with:
10% FBS (Hyclone, Catalog number SH30071.03, heat inactivated)
0.02% BSA (Sigma, Catalog number A8412)
15 mM HEPES (Cellgro, Catalog number 25-060-Cl)
MEM Nonessential amino acid (Gibco, 11140)
Penicillin and streptomycin (Gemini Bio-Products, Catalog number 400-109. Penicillin final concentration 100 U/mL and streptomycin final concentration 100 μg/mL)
3.1.1.3. Culture medium for mouse splenocytes and human PBMC, human T cells:
RPMI1640 (Cellgro, Catalog number 10-040-CV) supplemented with:
10% FBS (Heat Inactivated, HyClone, Catalog number SH30071.03)
10 mM HEPES (Cellgro, Catalog number 25-060-Cl)
Penicillin and streptomycin (Gemini Bio-Products, Catalog number 400-109. Penicillin final concentration 100 U/mL and streptomycin final concentration 100 μg/mL)
1 mM sodium pyruvate (Cellgro, Catalog number 25-000-cl)
50 μg/mL uridine (Sigma, Catalog number U3003)
25 mM glucose final concentration
3.1.1.4. Hemolytic Gey’s buffer
Stocks of A, B, and C can be made ahead and stored in the refrigerator; however, 1 × Gey’s buffer must be made fresh each day by mixing stocks A (4 parts), B (1 part), C (1 part), and water (14 parts).
| Solution A | |
| NH4Cl | 131.08 mM |
| KCl | 4.96 mM |
| Na2HPO4· 12H2O | 0.85 mM |
| KH2PO4 | 176.00 μM |
| Glucose | 5.55 mM |
| Phenol red | 28.22 μM |
| Dissolve in 1 L of distilled water and autoclave | |
| Solution B | |
| MgCl2·6H2O | 1.03 mM |
| MgSO4·6H2O | 0.29 mM |
| CaCl2 | 1.16 mM |
| Dissolve in 0.5 L of distilled water and autoclave | |
| Solution C | |
| NaHCO3 | 13.39 mM |
| Dissolve in 0.5 L of distilled water and autoclave | |
3.1.2. Fluorescent dyes:
MitoSox Red (Invitrogen, Catalog number M36008)
CYTOX Green (Invitrogen Catalog number S-7020)
Annexin V-APC (Invitrogen, Catalog number A35110)
DiOC6 (Invitrogen, Catalog number D273)
CM-H2DCFDA (Invitrogen, Catalog number C6827)
H2DCFDA (Sigma, Catalog number D6883)
Dihydroethidium (DHE) (Invitrogen, Catalog number D23806)
3.1.3. Buffers:
PBS (Cellgro, Catalog number 21-040-CV):
HBSS (Lonza, Catalog number10-527F)
FACS buffer: 1 × PBS+ 2% FBS, phenol red-free
3.1.4. Other reagents:
- Cytokines:
- Recombinant human tumor necrosis factor alpha (TNF-α) (R & D Systems, Catalog number 210-TA-010/CF)
- Recombinant human interferon gamma (IFN-γ) (BD Bio-sciences, Catalog number 554617)
- Recombinant mouse TNF-α (eBioscience, Catalog number 14-8321-63)
- Recombinant mouse IFN-γ (Sigma, Catalog number I4777)
Ficoll (GE Healthcare, Catalog number 17-1440-03)
T cell enrichment cocktail (Stemcell, Catalog number 15021/15061)
0.25% Trypsin–EDTA (Gemini Bio-product, Catalog number 400-151)
3.2. Cell preparation for flowcytometry analysis
3.2.1. Mouse beta cell lines (NIT-1 and NIT-4):
Seed cells in 12-well plates at 5 × 105 cells per well. Allow cells attach overnight.
On the second day, treat cells accordingly (see Section 3.3 as an example).
On the day of the assay, the dyes should be prepared fresh and the cells stained as described below.
3.2.2. Human beta cell line (BetaLox5):
BetaLox5 cells are seeded in 12-well Corning Costar culture plates (Fisher Scientific) at a density of 5 × 104 cells per well in a total of 500 μL and allowed to adhere for 24 h.
Treat cells accordingly (see Section 3.3 as an example).
On the day cells will be analyzed on a flow cytometer, stain cells with MitoSox Red and DiOC6 dye as described below.
3.2.3. Mouse splenocytes:
Harvest mouse spleens.
Put one spleen in 5 mL ice-cold HBSS and homogenize using a 7-mL glass Tenbroeck homogenizer (Kontes Glass Company).
Collect cells by centrifuging at 350 × g for 5 min at 4 °C.
Remove red blood cells by incubating cells in hemolytic Gey’s solution for 5 min on ice. Stop reaction by adding same volume of isotonic HBSS. Spin down at 350 × g for 5 min.
Resuspend cells in cold HBSS and pass through cell strainer [Falcon, Catalog number 352340] to remove dead cell clusters.
Count cells and proceed to staining.
3.2.4. Human PBMC:
Collect human blood samples using a BD Vacutainer with heparin sulfate [Green Top].
Leave samples on a rocker overnight.
Transfer blood to a 50-mL conical tube. Fill to 17.5 mL per tube.
Add DPBS without calcium or magnesium to a total volume of 35 mL. This achieves a 1:1 dilution v/v dilution.
Gently add 15 mL Ficoll beneath the sample. We use the following method: Place a 9″ glass Pasteur pipette into the conical so that the small end is at the bottom of the tube. Then use a 5-mL serological pipette to add Ficoll to the large end of Pasteur pipette.
Centrifuge at 800 × g for 20 min at room temperature with the centrifuge break set to off.
Aspirate plasma layer to within 1 cm of interphase layer.
Collect interphase layer (buffy coat) using a transfer pipette, slowly sweep top of the desired layer, constantly releasing the pipette bulb and transfer to a new 50-mL conical tube.
Wash with PBS up to 50 mL.
Centrifuge at 450 × g for 10 min at room temperature. Max brake and max acceleration.
Resuspend cells in 1 mL PBS. Remove red blood cells using NH4CL if necessary. Add 9 mL NH4Cl (0.8% NH4CL in H2O, 0.1 mM EDTA, buffered with NaHCO3 to pH 7.2–7.6) to 1 mL cell suspension, incubate on ice for 10 min. Wash twice with PBS as above.
Count cells and proceed to staining.
3.2.5. Human T cells:
Collect human blood samples using BD Vacutainers with heparin sulfate [Green Top]
Leave samples on the rocker for gentle shake overnight.
Enrich T cells using RosetteSep Human T cell enrichment cocktail (Stemcell, Catalog number 15021/15061) following the manufacturer’s instructions.
- Treat cells accordingly.
- We activate T cells in vitro by seeding 1 × 106 cells per well of enriched human T cells in 12-well plates precoated with stimulating antibodies or isotype controls. Activation generally follows a time course of 24, 48, and 72 h.
Proceed to staining and flow cytometry analysis.
3.3. Induction of apoptosis
Apoptosis is induced in mouse and human beta cell lines by combined inflammatory cytokine treatment: IFN-γ and TNF-α. Cell death including apoptosis is detected using CYTOX Green or Propidium Iodide and Annexin-V, or the mitochondrial membrane potential dye DiOC6. ROS production is detected simultaneously as described below.
3.3.1. For mouse beta cell lines NIT-1 and NIT-4, after allowing cells to attach to the plate for overnight, dilute recombinant mouse TNF-α 1000 U/mL and recombinant mouse IFN-γ 1000 U/mL in culture medium. Remove old medium from wells andfeed with fresh medium ormedium with cytokines. Cytokines areaddedatdifferent time points to achieve desired treatment durations. Control wells are fed with fresh medium on the second day. Since cytokines are dissolved in culture medium, this experimental design does not require vehicle control treatment of beta cells. If diluent of the treatment reagents is not culture medium, vehicle control wells should be added with correspondent dissolvent at the same concentration as treated wells.
3.3.2. For human beta cell line BetaLox 5: Allow cells to attach to the plate for 24 h. Add recombinant human TNF-α (2000 U/mL) and recombinant human IFN-γ (1000 U/mL). Incubate for 48 h. Proceed to flow cytometer analysis.
3.4. Instruments
Flow cytometer: We have used both the Accuri 6 as well as BD LSRFortessa cytometers for these protocols
Plate reader: SpectraMax M5 microplate reader
Cell culture facility and mouse work station
3.5. Steps using flow cytometer
3.5.1. Prepare MitoSox Red before use, by adding 13.2 μL DMSO to one 50 mg vial to yield a stock solution of 5 mM. Dilute the dye to working concentration in culture medium. Avoid light. Working concentrations vary for different cell types, titration prior to experiment is recommended. In our studies, we have used 2.5 μM for mouse and human beta cell lines and 5 μM for human PBMC and T cells.
3.5.2. For beta cell lines seeded in 12-well culture plates:
Remove old medium and add 1 mL diluted dye to each well except unstained controls.
Incubate at 37 °C for 15 min in the dark.
Remove dye, rinse once with HBSS.
Harvest cells by incubation with trypsin for 5 min at room temperature. Trypsin is neutralized with an equal volume of culture medium.
Wash cells once with phenol red-free FACS buffer (we use PBS + 2% FBS).
Add dyes to exclude dead cells. For mouse beta cells, we use CYTOX Green, diluted in FACS buffer to 10 μM, and Annexin-V-APC (Invitrogen, Catalog number A35110) with a 1/5 dilution in FACS buffer. Add 10 μL of each diluted dye to each tube. Stain for 1 min at room temperature and wash with FACS buffer. For human beta cell lines, we use DiOc6 20 nM, added together with MitoSox Red staining.
3.5.3. For mouse lymphocytes or human PBMCs:
Stain for cell surface markers as usual, if desired. Wash.
Add 500 μL culture medium containing dye and incubate at 37 °C for 15 min in dark.
Wash with 2-mL of phenol red-free FACS buffer.
Resuspend in 300 μL FACS buffer. Add 1:10 diluted Annexin-V 10 μL to stain dead cells.
3.5.4. Proceed to flow cytometer analysis. Depending on the instrument corresponding settings should be chosen. For example, using an Accuri C6 for mouse beta cells, the MitoSox Red signal is detected in both the FL-2 and FL-3 channels, while CYTOX green is collected by the FL-1 channel and Annexin-V-APC in the FL-4 channel. For human beta cells, mouse splenocytes, as well as human PBMCs or T cells, we have used a BD LSR Fortessa. With the Fortessa, Mitosox Red is detected in the PE channel and DiOC6 via the FITC channel.
3.5.5. Results are expressed as ratio to control. Figure 12.1 provides an example of the gating strategy for detection of human T cell mtROS using flow cytometry.
Figure 12.1. Gating strategy for the cytofluorometic detection of the mitochondrial ROS in human T cells.
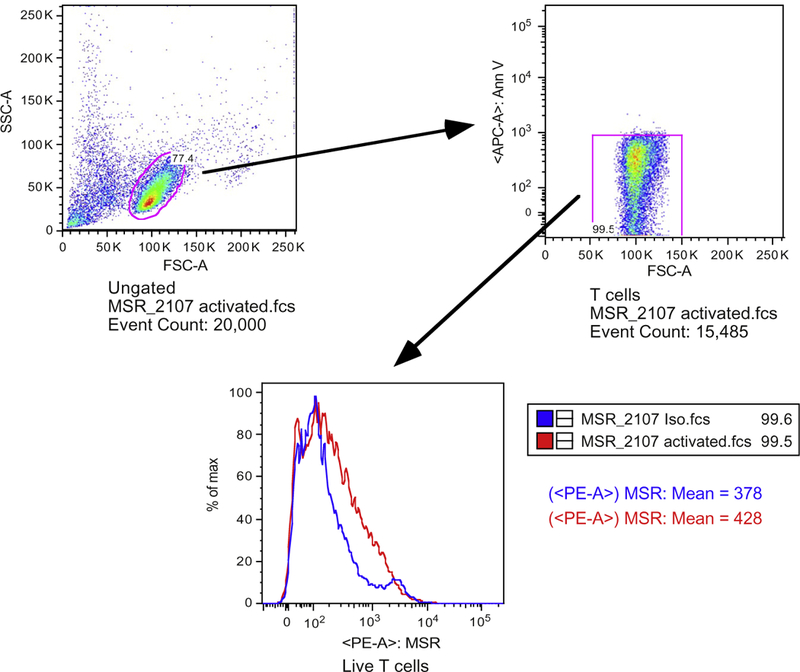
Samples are enriched human T cells as mentioned in the text (step 3.2.5). Lower T cell percentage is due to the fact that red blood cells are not lysed in this experiment. The top left panel is a dot plot for the T cell population that was gated on side-scatter and forward-scatter. Live T cells were gated by exclusion of Annexin-V positive cells (top right panel). Lower panel shows comparison of MitoSox Red histogram between activated T cells (red line) and isotype antibody treated controls (blue line).
In summary, the mitochondria-specific dye MitoSox Red can be used to detect basal as well as cytokine-induced mtROS production in mouse and human beta cell lines. Additionally, these methods can also be employed for the measurement of mtROS production in primary mouse and human lymphocytes. Selection of fluorescent dyes such as DHE or DCFDA will allow detection of total cell ROS production using this method. For mouse and human cells, the described method allows time course studies of ROS production during both short-(minutes) and long-term (days) experiments.
3.6. Steps using plate reader
3.6.1. Seed cells in 96-well flat bottom plate at 1 × 105 cells per well. Allow cells attach overnight.
3.6.2. On the second day, prewarm the plate reader at 37 °C.
3.6.3. Dilute ROS detection dye to the desired final concentration in culture medium. Remove medium from the wells of the plate and add 200 μL medium with dye. We use three dyes separately: CM-H2DCFDA at final concentration of 5 μM, DHE at final concentration of 100 M, H2DCFDA at final concentration of 10 μM. Dye concentrations are also cell type dependent and titration is recommended.
3.6.4. Read on a SpectraMax M5 or similar spectrofluorometer. Set at kinetic read mode with a temperature of 37 °C and read for 2 h with an interval of 1 min 20 s with 6 reads per well. Fluorescence reading Ex/Em/Cutoff: Lm1 510/590/570 (for DHE). Lm2 488/517/515 (for both H2DCFDA and CM-H2DCFDA). After a 15-min read to establish base line, open spectrofluorometer, add treatments to corresponding wells, reinsert plate into the spectrofluorometer, and continue reading.
In summary, the plate reader method is useful to detect acute ROS production (minutes to hours). While cytokine treatment of beta cells did not elicit ROS production over a short-term experiment, positive control wells where exogenous peroxide was added induced a sharp increase of fluorescence.
4. CONCLUDING REMARKS
ROS play essential roles for cell physiology and participate in many pathological processes. Mitochondria are an important source of cell ROS production. Here, we introduce techniques to detect mitochondrial-specific ROS production in several cell types using flow cytometry and spectrofluorometer plate reader. By selecting different ROS-detecting probes, it is possible to detect either total cellular ROS or mitochondrial-specific ROS. These techniques allow investigators to monitor basal ROS levels as well as dynamic ROS production in hours (using spectrofluorometer plate reader) or in days (using a flow cytometer). In addition to the detection of dynamic ROS production during long-term treatment, these flow cytometry methods also allow for the measurement of ROS in most specific types as well as in cells during different conditions or heterogeneous cell populations by costaining with cell surface markers.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institute of Health (AI056374, AI42288, and DK074656) and the Sebastian Family Endowment for Diabetes Research to C. E. M. as well as grants from the Juvenile Diabetes Research Foundation and the American Diabetes Association to J. C. and C. E. M.
REFERENCES
- Aggarwal A, Misro MM, Maheshwari A, Sehgal N, & Nandan D (2010). N-acetylcysteine counteracts oxidative stress and prevents hCG-induced apoptosis in rat Leydig cells through down regulation of caspase-8 and JNK. Molecular Reproduction and Development, 77, 900–909. [DOI] [PubMed] [Google Scholar]
- Baixauli F, Martin-Cofreces NB, Morlino G, Carrasco YR, Calabia-Linares C, Veiga E, et al. (2011). The mitochondrial fission factor dynamin-related protein 1 modulates T-cell receptor signalling at the immune synapse. The EMBO Journal, 30, 1238–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayir H, Fadeel B, Palladino MJ, Witasp E, Kurnikov IV, Tyurina YY, et al. (2006). Apoptotic interactions of cytochrome c: Redox flirting with anionic phospholipids within and outside the mitochondria. Biochimica et Biophysica Acta, 1757, 648–659. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Howard BJ, & LaFontaine MA (2001). Brain oxidative stress in animal models of accelerated aging and the age-related neurodegenerative disorders, Alzheimer’s disease and Huntington’s disease. Current Medicinal Chemistry, 8, 815–828. [DOI] [PubMed] [Google Scholar]
- Chen J, Gusdon AM, Piganelli J, Leiter EH, & Mathews CE (2011). mt-Nd2(a) Modifies resistance against autoimmune type 1 diabetes in NOD mice at the level of the pancreatic beta-cell. Diabetes, 60, 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Gusdon AM, Thayer TC, & Mathews CE (2008). Role of increased ROS dissipation in prevention of T1D. The Annals of the New York Academy of Sciences, 1150, 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Lu Y, Lee CH, Li R, Leiter EH, & Mathews CE (2008). Commonalities of genetic resistance to spontaneous autoimmune and free radical-mediated diabetes. Free Radical Biology & Medicine, 45, 1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra A (2010). Mitochondria-centric activation induced cell death of cytolytic T lymphocytes and its implications for cancer immunotherapy. Vaccine, 28, 4566–4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drose S, & Brandt U (2008). The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. The Journal of Biological Chemistry, 283, 21649–21654. [DOI] [PubMed] [Google Scholar]
- Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, et al. (2003). Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. The Journal of Clinical Investigation, 112, 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov RG, & Sazanov LA (2012). The coupling mechanism of respiratory complex I—A structural and evolutionary perspective. Biochimica et Biophysica Acta, 1817, 1785–1795. [DOI] [PubMed] [Google Scholar]
- Gautier CA, Kitada T, & Shen J (2008). Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proceedings of the National Academy of Sciences of the United States of America, 105, 11364–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusdon AM, Votyakova TV, & Mathews CE (2008). mt-Nd2a suppresses reactive oxygen species production by mitochondrial complexes I and III. The Journal of Biological Chemistry, 283, 10690–10697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusdon AM, Votyakova TV, Reynolds IJ, & Mathews CE (2007). Nuclear and mitochondrial interaction involving mt-Nd2 leads to increased mitochondrial reactive oxygen species production. The Journal of Biological Chemistry, 282, 5171–5179. [DOI] [PubMed] [Google Scholar]
- Hatefi Y (1985). The mitochondrial electron transport and oxidative phosphorylation system. Annual Review of Biochemistry, 54, 1015–1069. [DOI] [PubMed] [Google Scholar]
- Hsiai T, & Berliner JA (2007). Oxidative stress as a regulator of murine atherosclerosis. Current Drug Targets, 8, 1222–1229. [DOI] [PubMed] [Google Scholar]
- Jackson SH, Devadas S, Kwon J, Pinto LA, & Williams MS (2004). T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nature Immunology, 5, 818–827. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Bykhovskaya Y, Spirina O, & Fischel-Ghodsian N (2001). A nuclear-mitochondrial DNA interaction affecting hearing impairment in mice. Nature Genetics, 27, 191–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski MM, Sauer SW, Klemke CD, Suss D, Okun JG, Krammer PH, et al. (2010). Mitochondrial reactive oxygen species control T cell activation by regulating IL-2 and IL-4 expression: Mechanism of ciprofloxacin-mediated immunosuppression. Journal of Immunology, 184, 4827–4841. [DOI] [PubMed] [Google Scholar]
- Kennedy KA, Sandiford SD, Skerjanc IS, & Li SS (2012). Reactive oxygen species and the neuronal fate. Cellular and Molecular Life Sciences, 69, 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Khalimonchuk O, Smith PM, & Winge DR (2012). Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochimica et Bio-physica Acta, 1823, 1604–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Lee SB, Park JK, & Yoo YD (2010). TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L). Cell Death and Differentiation, 17, 1420–1434. [DOI] [PubMed] [Google Scholar]
- Krauss S, Zhang CY, Scorrano L, Dalgaard LT, St-Pierre J, Grey ST, et al. (2003). Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction. The Journal of Clinical Investigation, 112, 1831–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon J, Shatynski KE, Chen H, Morand S, de Deken X, Miot F, et al. (2010). The nonphagocytic NADPH oxidase Duox1 mediates a positive feedback loop during T cell receptor signaling. Science Signaling, 3, ra59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, & Beal MF (2006). Mitochondrial dysfunction and oxidative stress in neuro-degenerative diseases. Nature, 443, 787–795. [DOI] [PubMed] [Google Scholar]
- Muller FL, Liu Y, & Van Remmen H (2004). Complex III releases superoxide to both sides of the inner mitochondrial membrane. The Journal of Biological Chemistry, 279, 49064–49073. [DOI] [PubMed] [Google Scholar]
- Pandya DP (2001). Oxidant injury in coronary heart disease (Part-I). Comprehensive Therapy, 27, 284–292. [DOI] [PubMed] [Google Scholar]
- Panov A, Kubalik N, Zinchenko N, Hemendinger R, Dikalov S, & Bonkovsky HL (2011). Respiration and ROS production in brain and spinal cord mitochondria of transgenic rats with mutant G93a Cu/Zn-superoxide dismutase gene. Neurobiology of Disease, 44, 53–62. [DOI] [PubMed] [Google Scholar]
- Payton KS, Sheldon RA, Mack DW, Zhu C, Blomgren K, Ferriero DM, et al. (2007). Antioxidant status alters levels of Fas-associated death domain-like IL-1B-converting enzyme inhibitory protein following neonatal hypoxia-ischemia. Developmental Neuroscience, 29, 403–411. [DOI] [PubMed] [Google Scholar]
- Perez-Cruz I, Carcamo JM, & Golde DW (2007). Caspase-8 dependent TRAIL-induced apoptosis in cancer cell lines is inhibited by vitamin C and catalase. Apoptosis, 12, 225–234. [DOI] [PubMed] [Google Scholar]
- Peterson JR, Sharma RV, & Davisson RL (2006). Reactive oxygen species in the neuropathogenesis of hypertension. Current Hypertension Reports, 8, 232–241. [DOI] [PubMed] [Google Scholar]
- Puddu P, Puddu GM, Cravero E, Rosati M, & Muscari A (2008). The molecular sources of reactive oxygen species in hypertension. Blood Pressure, 17, 70–77. [DOI] [PubMed] [Google Scholar]
- Quinlan CL, Treberg JR, Perevoshchikova IV, Orr AL, & Brand MD (2012). Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radical Biology & Medicine, 53, 1807–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A, Schwindling C, Wenning AS, Becherer U, Rettig J, Schwarz EC, et al. (2007). T cell activation requires mitochondrial translocation to the immunological synapse. Proceedings of the National Academy of Sciences of the United States of America, 104, 14418–14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigoulet M, Yoboue ED, & Devin A (2011). Mitochondrial ROS generation and its regulation: Mechanisms involved in H(2)O(2) signaling. Antioxidants & Redox Signaling, 14, 459–468. [DOI] [PubMed] [Google Scholar]
- Schwindling C, Quintana A, Krause E, & Hoth M (2010). Mitochondria positioning controls local calcium influx in T cells. Journal of Immunology, 184, 184–190. [DOI] [PubMed] [Google Scholar]
- Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. (2013). Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity, 38, 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior AE (1988). ATP synthesis by oxidative phosphorylation. Physiological Reviews, 68, 177–231. [DOI] [PubMed] [Google Scholar]
- Shen HM, Lin Y, Choksi S, Tran J, Jin T, Chang L, et al. (2004). Essential roles of receptor-interacting protein and TRAF2 in oxidative stress-induced cell death. Molecular and Cellular Biology, 24, 5914–5922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy K, Wu Y, & Pervaiz S (2009). LY303511 enhances TRAIL sensitivity of SHEP-1 neuroblastoma cells via hydrogen peroxide-mediated mitogen-activated protein kinase activation and up-regulation of death receptors. Cancer Research, 69, 1941–1950. [DOI] [PubMed] [Google Scholar]
- Shigenaga MK, Hagen TM, & Ames BN (1994). Oxidative damage and mitochon-drial decay in aging. Proceedings of the National Academy of Sciences of the United States of America, 91, 10771–10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoffner JM, & Wallace DC (1994). Oxidative phosphorylation diseases and mitochondrial DNA mutations: Diagnosis and treatment. Annual Review of Nutrition, 14, 535–568. [DOI] [PubMed] [Google Scholar]
- Siddiqui A, Hanson I, & Andersen JK (2012). Mao-B elevation decreases parkin’s ability to efficiently clear damaged mitochondria: Protective effects of rapamycin. Free Radical Research, 46, 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh U, & Jialal I (2006). Oxidative stress and atheroschlerosis. Pathophysiology, 13, 129–142. [DOI] [PubMed] [Google Scholar]
- Sohal RS, & Weindruch R (1996). Oxidative stress, caloric restriction, and aging. Science, 273, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn YS, Tamir S, Song L, Michaeli D, Matouk I, Conlan AR, et al. (2013). NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth. Proceedings of the National Academy of Sciences of the United States of America, 110, 14676–14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg D, & Witztum JL (2002). Is the oxidative modification hypothesis relevant to human atherosclerosis? Do the antioxidant trials conducted to date refute the hypothesis? Circulation, 105, 2107–2111. [DOI] [PubMed] [Google Scholar]
- St-Pierre J, Buckingham JA, Roebuck SJ, & Brand MD (2002). Topology of super-oxide production from different sites in the mitochondrial electron transport chain. The Journal of Biological Chemistry, 277, 44784–44790. [DOI] [PubMed] [Google Scholar]
- Sugioka K, Nakano M, Totsune-Nakano H, Minakami H, Tero-Kubota S, & Ikegami Y (1988). Mechanism of O2-generation in reduction and oxidation cycle of ubiquinones in a model of mitochondrial electron transport systems. Biochimica et Bio-physica Acta, 936, 377–385. [DOI] [PubMed] [Google Scholar]
- Surekha RH, Srikanth BBMV, Jharna P, Ramachandra RV, Dayasagar RV, & Jyothy A (2007). Oxidative stress and total antioxidant status in myocardial infarction. Singapore Medical Journal, 48, 137–142. [PubMed] [Google Scholar]
- Tahara EB, Navarete FD, & Kowaltowski AJ (2009). Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radical Biology & Medicine, 46, 1283–1297. [DOI] [PubMed] [Google Scholar]
- Terry Powers JL, Mace KE, Parfrey H, Lee SJ, Zhang G, & Riches DW (2010). TNF receptor-1 (TNF-R1) ubiquitous scaffolding and signaling protein interacts with TNF-R1 and TRAF2 via an N-terminal docking interface. Biochemistry, 49, 7821–7829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzioglu M, & Larsson NG (2007). Mitochondrial dysfunction in mammalian ageing. Novartis Foundation Symposium, 287, 197–213. [DOI] [PubMed] [Google Scholar]
- Thayer TC, Delano M, Liu C, Chen J, Padgett LE, Tse HM, et al. (2011). Super-oxide production by macrophages and T cells is critical for the induction of autoreactivity and type 1 diabetes. Diabetes, 60, 2144–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong JJ, Schriner SE, McCleary D, Day BJ, & Wallace DC (2007). Life extension through neurofibromin mitochondrial regulation and antioxidant therapy for neurofibromatosis-1 in Drosophila melanogaster. Nature Genetics, 39, 476–485. [DOI] [PubMed] [Google Scholar]
- Treberg JR, Quinlan CL, & Brand MD (2011). Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). The Journal of Biological Chemistry, 286, 27103–27110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter L, Takacs K, Hegedus V, & Adam-Vizi V (2007). Characteristics of alpha-glycerophosphate-evoked H2O2 generation in brain mitochondria. Journal of Neurochemistry, 100, 650–663. [DOI] [PubMed] [Google Scholar]
- Tse HM, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, et al. (2010). NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. Journal of Immunology, 185, 5247–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF, & Boveris A (1980). Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. The Biochemical Journal, 191, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venardos KM, & Kaye D (2007). Myocardial ischemia-reperfusion injury, antioxidant enzyme systems, and selenium: A review. Current Medicinal Chemistry, 14, 1539–1549. [DOI] [PubMed] [Google Scholar]
- Wallace DC (1992). Diseases of the mitochondrial DNA. Annual Review of Biochemistry, 61, 1175–1212. [DOI] [PubMed] [Google Scholar]
- Zhang H, Luo Y, Zhang W, He Y, Dai S, Zhang R, et al. (2007). Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. The American Journal of Pathology, 170, 1108–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR, et al. (2002). Superoxide mediates the actions of angiotensin II in the central nervous system. Circulation Research, 91, 1038–1045. [DOI] [PubMed] [Google Scholar]