

Abstract
During stress, accumulation of misfolded proteins in the endoplasmic reticulum (ER) triggers activation of the adaptive mechanisms that restore protein homeostasis. One mechanism that eukaryotic cells use to respond to ER stress is through activation of the unfolded protein response (UPR) signaling pathway, which initiates degradation of misfolded proteins and leads to inhibition of translation and increased expression of chaperones and oxidative folding components that enhance ER protein folding capacity. However, the mechanisms of adaptation to ER stress are not limited to the UPR. Using yeast Saccharomyces cerevisiae, we recently discovered that the protein folding burden in the ER can be alleviated in a UPR-independent manner through duplication of whole chromosomes containing ER stress protective genes. Here we discuss our findings and their implication to our understanding of the mechanisms by which cells respond to protein misfolding in the ER.
Keywords: aneuploidy, genome instability, next-generation sequencing, ER stress resistance, unfolded protein response
Introduction
In eukaryotes, secretory and transmembrane proteins are folded in the endoplasmic reticulum (ER) before export to their target organelles (1). If the protein folding needs exceed the ER folding capacity, cells accumulate misfolded proteins and experience ER stress. In response to ER stress, cells activate the unfolded protein response (UPR) signaling pathway that restores protein homeostasis by improving ER protein folding and clearing out misfolded proteins (2). However, the mechanisms of adaptation to ER stress are not limited to the UPR. While the role of the UPR signaling in maintaining ER homeostasis has been extensively studied, there are still significant gaps in our understanding of the mechanisms by which cells adapt to the accumulation of misfolded proteins in the ER.
In S. cerevisiae, the activation of the UPR depends on the ER stress sensor protein Ire1 and the downstream activation of the Hac1 transcription factor that induces broad transcriptomic changes in order to restore the ER homeostasis (3, 4). The transcriptional output of the UPR includes increased expression of ER chaperones and genes involved in protein folding in the ER as well as components of the ER-associated degradation (ERAD) machinery leading to an increase in the ER folding capacity (5, 6). However, persistent activation of the UPR may lead to apoptosis and cell death when adaptive mechanisms fail (2). Because prolonged UPR signaling is detrimental for the cell survival, deactivating the UPR is as important as its activation (7, 8). Numerous human age-related diseases have been associated with the accumulation of misfolded proteins and protein aggregation, such as neurodegenerative diseases, atherosclerosis, cancer, cardiovascular disease, and type 2 diabetes (9, 10). Given that decline in cellular protein homeostasis contributes to the development and pathogenesis of aging-related disorders, improved understanding of the mechanisms by which cells maintain ER homeostasis may reveal new therapeutic targets for diseases associated with protein misfolding.
In order to better understand the mechanisms by which cells adapt to protein misfolding in the ER, we recently performed a genetic screen in yeast to identify genome adaptations that confer resistance to tunicamycin-induced ER stress (11). Intriguingly, these studies uncovered aneuploidy as an important mediator of ER stress resistance, which protected cells against protein misfolding in a UPR-independent manner (Figure 1A). This unanticipated mechanism revealed that protein homeostasis in the ER is maintained by a combined activity of multiple redundant pathways. Instead of activating UPR, aneuploidy restores ER protein homeostasis by increasing the copy number of ER stress protective genes. Although aneuploidy is perceived to induce proteotoxicity, our study highlights important mechanisms by which specific chromosome duplications may counteract accumulation of misfolded proteins in the ER and improve protein homeostasis. Here, we discuss our findings and their potential implications for understanding the metabolic pathways and mechanisms that contribute to the development of the ER stress resistance.
Fig. 1.
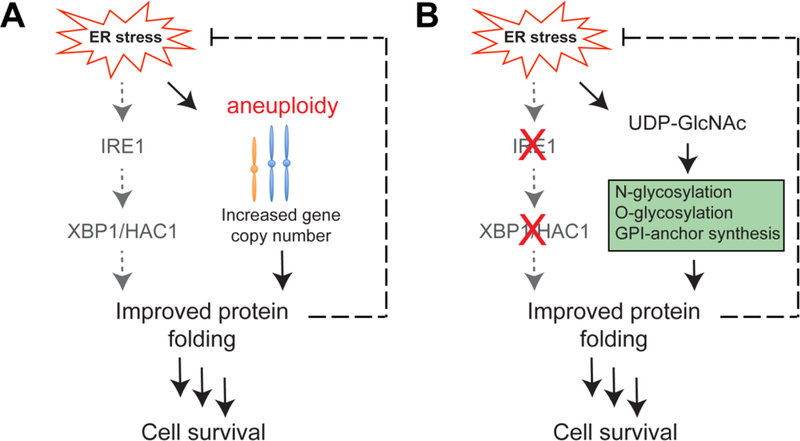
UPR-independent mechanisms of adaptation to ER stress. a Aneuploidy protects cells against protein misfolding in a UPR-independent manner by increasing the copy number of ER stress protective genes. b Increased levels of UDP-GlcNAc, a precursor for protein glycosylation and GPI-anchor synthesis, allow cells to compensate for the lack of the UPR.
Role of aneuploidy in adaptation to ER stress
To ensure survival, microorganisms such as yeast evolved quick adaptive mechanisms to withstand challenging environmental stresses (12). Recent studies have shown that gains of extra copies of chromosomes, or aneuploidy, can be used as a source of genetic variation that allows evolutionarily selection of adaptations in response to internal or environmental perturbations (13–17). Although aneuploidy, in general, is detrimental for the cell fitness and leads to proteotoxic, metabolic, replication, and mitotic stresses, gaining particular chromosomes in response to stress can confer growth advantages under specific conditions (18, 19).
In our recent study, we performed a genetic screen in S. cerevisiae to identify genome adaptations that confer resistance to tunicamycin-induced ER stress (11). Tunicamycin pharmacologically induces accumulation of misfolded proteins and ER stress by blocking N-linked glycosylation necessary for proper protein folding (20, 21). This screen led us to the discovery that aneuploidy plays an important role in adaptation of cells to ER stress. Our study uncovered that yeast cells can spontaneously evolve ER stress resistance by duplication of multiple chromosomes. We also found that the gain of an extra copy of chromosome (Chr) II alone was sufficient to induce protection from ER stress.
Although several other studies have previously demonstrated that aneuploidy allows adaptation to stress in general (including heat stress, oxidative stress, etc.), our study, for the first time, demonstrated that alterations in chromosome number may serve as a mechanism to protect cells against ER stress. What makes this finding surprising is that aneuploidy is known to cause proteotoxicity, because changing the dosage of multiple genes simultaneously imposes a significant burden on protein folding function of the ER (22). Moreover, it has been shown that aneuploidy impairs cell fitness leading to slow cell growth and shortened lifespan (13, 23). Therefore, it might seem counterintuitive that the duplication of chromosomes could help alleviate a drug-induced proteotoxic stress. However, we demonstrated that Chr II aneuploidy provides a growth advantage to the cell as a result of a delicate balance between beneficial effects of ER stress protective genes located on this chromosome and negative consequences of aneuploidy. While aneuploidy itself leads to proteotoxic stress, selective advantages provided by specific genes located on Chr II counteract the negative effects resulting in improved protein folding. Additionally, in contrast to spontaneous point mutations, increasing copy number of ER stress protective genes by gaining an extra chromosome provides a rapid solution to ensure cell survival, giving enough time for other less detrimental solutions to be selected (17).
Moreover, we went beyond the interesting and unexpected observation that Chr II aneuploidy can protect against ER stress to identify specific genes located on Chr II that mediate these effects. For this, we analyzed protein translation in the ER stress-resistant mutants using ribosome profiling. This analysis revealed that translation of genes on the duplicated chromosomes was in general doubled as a result of increased copy number. Therefore, we hypothesized that ER stress resistance can result from increased dosage of genes encoded on the duplicated chromosome. Our analyses revealed that the protective effect of aneuploidy against ER stress can be explained by a combined function of at least three genes located on Chr II, including ALG7, PRE7, and YBR085C-A. Overexpression of these genes was sufficient to induce tunicamycin resistance in wild-type cells, whereas deletion of all three genes completely reversed the tunicamycin-resistance phenotype (11). ALG7 encodes a UDP-N-acetylglucosamine-1-P transferase, a protein involved in the N-glycosylation (24, 25). N-glycosylation is required for the maturation and protein quality control in the ER, and deletion of ALG7 has been shown to increase the sensitivity of cells to pharmacologically induced ER stress (26). In turn, PRE7 encodes the β6 subunit of the 20S core proteasome that plays an important role in proteasome-mediated protein degradation (27, 28). Interestingly, other proteasome subunits were also upregulated in the ER stress-resistant mutants including PRE8 (α2 subunit), PRE5 (α6 subunit), along with UMP1, a chaperone involved in the 20S proteasome maturation and assembly of β subunits (29). However, overexpression of only PRE7, but not other subunits, induced tunicamycin resistance in our study. To our knowledge, this is the first time that the β6 subunit of the proteasome has been implicated in the drug-induced ER stress resistance. Despite its role in the assembly and maturation of the core 20S particle, β6 is not a catalytic subunit of the proteasome. One of the possible explanations could be that Pre7p is a limiting subunit and that its overexpression induces the formation of more core particles, or alternatively this protein may have additional roles independent of the proteasome function. The function of the third gene identified in our study, YBR085C-A, is currently unknown. Interestingly, none of the three genes identified in our screen have been previously reported as direct UPR targets. Moreover, overexpression of ALG7 and PRE7, but not YBR085C-A, in UPR-deficient cells protected against tunicamycin, suggesting that these genes induce ER stress resistance independently of the UPR. One striking observation in our study was that stress-resistant aneuploid cells were desensitized to the UPR activation in response to tunicamycin. Yet they were still capable of activating the UPR, demonstrating that the cells chose UPR-independent mechanisms to cope with ER stress.
UPR-independent mechanisms to restore the ER homeostasis
As an alternative strategy to identify UPR-independent mechanisms, we investigated the consequences of the inhibition of the UPR (11). Intriguingly, we found that deletion of IRE1 and HAC1 (a yeast homolog of XBP1) genes did not significantly affect cell growth and replicative lifespan, nor induced aneuploidy. These findings suggest that IRE1 and HAC1 deletion mutants activate UPR-independent mechanisms to compensate for the lack of the UPR. We used targeted metabolite analysis and ribosome profiling to characterize metabolic and protein synthesis profiles in UPR-deficient mutants to identify genes and metabolic pathways that regulate protein homeostasis in the UPR-deficient cells. We found that increased levels of UDP-GlcNAc, a precursor for protein glycosylation may be used as an adaptive mechanism to restore protein homeostasis (Figure 1B). It is possible that increased levels of UDP-GlcNAc may lead to increased glycosylation of proteins that improve their folding and export from the ER. Consistent with this possibility, activation of the UDP-GlcNAc synthesis pathway or the supplementation with GlcNAc have been recently shown to protect against ER stress in evolutionarily distant organisms, including C. elegans (30) and mice (31, 32). However, very little is currently known about the mechanisms that underlie molecular consequences of increased UDP-GlcNAc levels and how it affects protein folding. In future studies, it would be important to identify specific types of glycosylation that are responsible for the protective effect of UDP-GlcNAc against ER stress.
Perspectives
Over the past decades significant progress has been achieved in identifying and characterizing molecular components of the UPR and elucidating the mechanisms that regulate protein folding and quality control in the ER. While the role of the UPR signaling in ER stress adaptation has been well characterized, fewer studies have focused on UPR-independent pathways. Accumulating evidence indicates that protein homeostasis in the ER is maintained by a complex network containing multiple redundant pathways. This complexity allows cells to compensate for the lack of the UPR by activating alternative mechanisms. Our findings highlighted an important role of aneuploidy in different cellular processes, including stress resistance and protein folding/posttranslational modifications, and provided important insights into the mechanisms that are activated in eukaryotic cells under conditions of ER stress. Our study also demonstrated that stress-induced aneuploidy can be used as a tool to uncover new stress suppressor genes and pathways. Although others have performed traditional mutagenesis screens to study mechanisms by which cells adapt to ER stress, we have identified novel genes involved in ER stress resistance through the adaptive aneuploidy model. The advantage of the adaptive aneuploidy approach is that it allows gain-of-function screens and identification of complex multigenic mechanisms, e.g. when the protective effect requires a combined effect of multiple genes. This approach can be applied to other adaptive responses and might help identify specific aneuploidies that are selected in response to variety of stresses.
Among the open questions in understanding the mechanisms by which cells adapt to ER stress is how distinct protein quality control pathways in various cellular compartments (including the ER, mitochondria, lysosomes, Golgi, ribosome-bound quality control) are coordinately regulated (33–36) and what is the role of metabolism in these processes (37). To move forward, it will be necessary to develop tools to quantitatively measure the temporal activity of the UPR and UPR-independent mechanisms in live cells at the single-cell level and identify molecular components that are the most prone to failure. Moreover, challenging cells with physiological stress might help understand the contribution of the UPR and UPR-independent mechanisms to pathogenesis of diseases associated chronic protein misfolding. Improved understanding of these mechanisms may reveal new therapeutic targets for diseases associated with protein misfolding, such as cancer, diabetes and neurodegenerative diseases.
Acknowledgements
This work was supported by the National Institutes of Health grant AG054566 (to VML). This research was conducted while VML was an AFAR Research Grant recipient from the American Federation for Aging Research.
References
- 1.Gardner BM, Pincus D, Gotthardt K, Gallagher CM, & Walter P (2013) Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol 5(3):a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walter P & Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334(6059):1081–1086. [DOI] [PubMed] [Google Scholar]
- 3.Travers KJ, et al. (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101(3):249–258. [DOI] [PubMed] [Google Scholar]
- 4.Labunskyy VM, et al. (2014) Lifespan extension conferred by endoplasmic reticulum secretory pathway deficiency requires induction of the unfolded protein response. PLoS Genet 10(1):e1004019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pincus D, et al. (2010) BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol 8(7):e1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karagoz GE, et al. (2017) An unfolded protein-induced conformational switch activates mammalian IRE1. Elife 6:e30700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chawla A, Chakrabarti S, Ghosh G, & Niwa M (2011) Attenuation of yeast UPR is essential for survival and is mediated by IRE1 kinase. J Cell Biol 193(1):41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tabas I & Ron D (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13(3):184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshida H (2007) ER stress and diseases. FEBS J 274(3):630–658. [DOI] [PubMed] [Google Scholar]
- 10.Koga H, Kaushik S, & Cuervo AM (2011) Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res Rev 10(2):205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beaupere C, et al. (2018) Genetic screen identifies adaptive aneuploidy as a key mediator of ER stress resistance in yeast. Proc Natl Acad Sci U S A 115(38):9586–9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pereira T, et al. (2018) Quantitative Operating Principles of Yeast Metabolism during Adaptation to Heat Stress. Cell Rep 22(9):2421–2430. [DOI] [PubMed] [Google Scholar]
- 13.Torres EM, et al. (2007) Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317(5840):916–924. [DOI] [PubMed] [Google Scholar]
- 14.Kaya A, et al. (2015) Adaptive aneuploidy protects against thiol peroxidase deficiency by increasing respiration via key mitochondrial proteins. Proc Natl Acad Sci U S A 112(34):10685–10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen G, Bradford WD, Seidel CW, & Li R (2012) Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature 482(7384):246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selmecki A, Forche A, & Berman J (2006) Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313(5785):367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yona AH, et al. (2012) Chromosomal duplication is a transient evolutionary solution to stress. Proc Natl Acad Sci U S A 109(51):21010–21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu J, Tsai HJ, Gordon MR, & Li R (2018) Cellular Stress Associated with Aneuploidy. Dev Cell 44(4):420–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sunshine AB, et al. (2015) The fitness consequences of aneuploidy are driven by condition-dependent gene effects. PLoS Biol 13(5):e1002155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lennon K, Bird A, Chen YF, Pretel R, & Kukuruzinska MA (1997) The dual role of mRNA half-lives in the expression of the yeast ALG7 gene. Mol Cell Biochem 169(1–2):95–106. [DOI] [PubMed] [Google Scholar]
- 21.Barnes G, Hansen WJ, Holcomb CL, & Rine J (1984) Asparagine-linked glycosylation in Saccharomyces cerevisiae: genetic analysis of an early step. Mol Cell Biol 4(11):2381–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oromendia AB, Dodgson SE, & Amon A (2012) Aneuploidy causes proteotoxic stress in yeast. Genes Dev 26(24):2696–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pavelka N, et al. (2010) Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468(7321):321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bretthauer RK (2009) Structure, expression, and regulation of UDP-GlcNAc: dolichol phosphate GlcNAc-1-phosphate transferase (DPAGT1). Curr Drug Targets 10(6):477–482. [DOI] [PubMed] [Google Scholar]
- 25.Helenius A & Aebi M (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem 73:1019–1049. [DOI] [PubMed] [Google Scholar]
- 26.Giaever G, et al. (1999) Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat Genet 21(3):278–283. [DOI] [PubMed] [Google Scholar]
- 27.Groll M, et al. (1999) The catalytic sites of 20S proteasomes and their role in subunit maturation: a mutational and crystallographic study. Proc Natl Acad Sci U S A 96(20):10976–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finley D, Chen X, & Walters KJ (2016) Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem Sci 41(1):77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livneh I, Cohen-Kaplan V, Cohen-Rosenzweig C, Avni N, & Ciechanover A (2016) The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cell Res 26(8):869–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Denzel MS, et al. (2014) Hexosamine pathway metabolites enhance protein quality control and prolong life. Cell 156(6):1167–1178. [DOI] [PubMed] [Google Scholar]
- 31.Weimer S, et al. (2014) D-Glucosamine supplementation extends life span of nematodes and of ageing mice. Nat Commun 5:3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dephoure N, et al. (2014) Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. Elife 3:e03023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gottschling DE & Nystrom T (2017) The Upsides and Downsides of Organelle Interconnectivity. Cell 169(1):24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Defenouillere Q & Fromont-Racine M (2017) The ribosome-bound quality control complex: from aberrant peptide clearance to proteostasis maintenance. Curr Genet 63(6):997–1005. [DOI] [PubMed] [Google Scholar]
- 35.Koike N, Hatano Y, & Ushimaru T (2018) Heat shock transcriptional factor mediates mitochondrial unfolded protein response. Curr Genet 64(4):907–917. [DOI] [PubMed] [Google Scholar]
- 36.Stauffer B & Powers T (2017) Target of rapamycin signaling mediates vacuolar fragmentation. Curr Genet 63(1):35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Dalfsen KM, et al. (2018) Global Proteome Remodeling during ER Stress Involves Hac1-Driven Expression of Long Undecoded Transcript Isoforms. Dev Cell 46(2):219–235 e218. [DOI] [PMC free article] [PubMed] [Google Scholar]