

Abstract
As a continuation of our project aimed at searching for new chemotherapeutic agents against American trypanosomiasis (Chagas disease), new selenocyanate derivatives were designed, synthesized and biologically evaluated against the clinically more relevant dividing form of Trypanosoma cruzi, the etiologic agent of this illness. In addition, in order to establish the role of each part of the selenocyanate moiety, different derivatives, in which the selenium atom or the cyano group were absent, were conceived, synthesized and biologically evaluated. In addition, in order to study the optimal position of the terminal phenoxy group, new regioisomers of WC-9 were synthesized and evaluated agaisnt T. cruzi. Finally, the resolution of a racemic mixture of a very potent conformationally rigid analogue of WC-9 was accomplished and further tested as growth inhibitors of T. cruzi proliferation. The results provide further insight into the role of the selenocyanate group in its antiparasitic activity.
Graphical Abstract
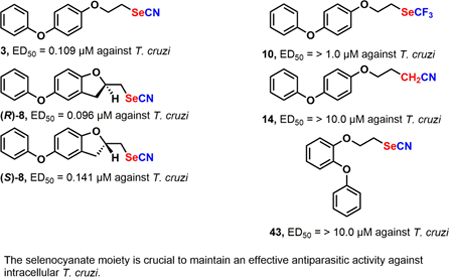
Introduction
Trypanosoma cruzi is the hemoflagellate protozoan parasite that causes American trypanosomiasis (Chagas disease), which is an endemic disease widespread from Southern United States to Southern Argentina.1 In addition, Chagas disease is a well-recognized opportunistic infection in AIDS patients. The number of infected people with T. cruzi diminished from 18 million in 1991 to 6 million in 2010, but it is still the most prevalent parasitic disease in the Americas.2 The two drugs available for Chagas disease treatment, nifurtimox and benznidazole require long-term treatment, which is associated to severe side effects. Besides, nifurtimox is not approved by the FDA and benznidazole has been recently approved but only for recent infections in children.3 In fact, until 2017, in the United States they were available only from CDC under investigational protocols. The isoprenoid pathway has been particularly useful for the identification of new targets against trypanosomatids. Enzymes studied so far that are involved in the synthesis of sterols and farnesyl diphosphate, and in protein prenylation, have been reported to be excellent drug targets against pathogenic parasites.4–6 Certainly, the isoprenoid pathway constitutes a major target for the treatment of parasitic diseases including Chagas disease, toxoplasmosis and others. In this sense, we were able to established a rigorous chemical structure / biological activity relationship on a vast number aryloxyethyl thiocyanate as growth inhibitors of T. cruzi growth targeting T. cruzi squalene synthase (TcSQS).7–15
We have recently described that the many isosteric analogues of our lead drug WC-9 (compound 1) (Figure 1) behaved as extremely potent growth inhibitors of the clinically relevant intracellular form (amastigotes) of T. cruzi acting in the low nanomolar concentrations.16 It is worth mentioning that WC-9 constitutes one of the few examples of a lead structure bearing a thiocyanate group covalently bonded to a main skeleton.17 WC-9 targets TcSQS, a membrane protein, being a non-competitive nanomolar inhibitor of the enzymatic activity of mitochondrial and glycosomal TcSQS (wild type) but inactive against truncated TcSQS, the soluble enzyme, which was cloned and expressed in Escherichia coli.16,18 Certainly, the replacement of the sulfur atom by a selenium one in WC-9 or its regioisomer 210 and in other closely related molecules brought about a dramatic enhancement of their effectiveness as inhibitors of T. cruzi proliferation making the selenocyanate derivatives16 almost two orders of magnitude more potent than the thiocyanate counterparts with excellent selectively index values.10–12,16 Compounds 3−8 emerge as representative members of this family of selenium-containing antiparasitic agents as shown in Figure 1. These selenium-containing analogues were extremely selective and were almost devoid of toxicity in in vitro assays.16 In fact, a covalently bonded selenocyanate moiety acting at the low nanomolar range against intracellular T. cruzi is definitely an innovation in Medicinal Chemistry.
Figure 1.
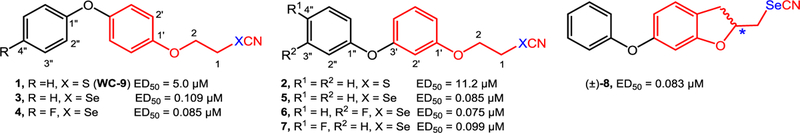
Chemical structure of WC-9 and other closely related inhibitors of T. cruzi proliferation.
As these isosteric analogues of WC-9 had shown improved effectiveness being an average of two order of magnitude more potent than the thiocyanate counterpart (same non-polar skeleton),16 it was realistic to consider their structural optimization taken into account that all of these reference molecules possessed drug-like characteristics.19
Ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one) is a selenium-containing inhibitor of Sporosarcina pasteurii and Helicobacter pylori ureases.20 It was postulated that the precise mode of action of ebselen could be attributed to the formation of a covalent bond with cysteine residue 322 of S. pasteurii urease acting at low nanomolar concentration, as shown in Scheme 1.21
Scheme 1.
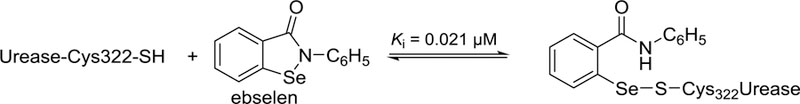
Precise mode of action of ebselen on S. pasteurii urease by forming a selenium-sulfur bond
Results and discussion
Bearing in mind that, positively, the selenium atom has a crucial role on biological activity and the target enzyme (TcSQS) possesses nine cysteine residues,22 it was reasonable to consider that the cellular activity would be associated to the formation of a selenium-sulfur bond at TcSQS. In this sense and based on our previous studies, it was very important to study the influence of each part of the selenocyanate moiety on biological activity. Therefore, compounds 9−12, where the cyano group was replaced by trifluoromethyl moiety, were synthesized to study its relevance on their biological action.23,24 The trifluoromethyl unit is an electron withdrawing group as it is the cyano unit. Then, the title compounds 9−12 were straightforwardly prepared from thiocyanates 1 and 2 and selenocyanates 3 and 4 via a nucleophilic displacement of the cyano group by the corresponding trimethyl(trifluoromethyl)silane via a Langlois-type reaction, as illustrated in Scheme 2.23–25
Scheme 2.
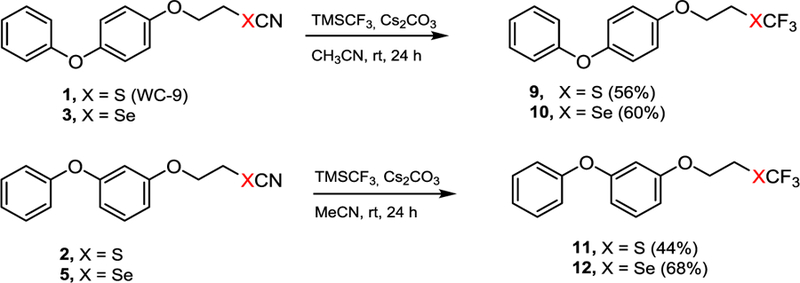
Preparation of trifluoromethyl derivatives of WC-9.
As the selenium-containing compounds turned out to be very potent growth inhibitors of intracellular T. cruzi, it seemed of interest to study the influence of compounds where the selenium atom was absent on the biological activity. Consequently, compound 13 and 14 were synthesized by deleting the selenium atom or by replacing it by a methylene group. These target molecules were straightforwardly synthesized employing 4-phenoxyphenol (15) as starting material via the already described alcohol 17,8 which was treated with N-bromosuccinimide and triphenyl phosphine to produce the bromide 18 that treated with potassium cyanide in N,N-dimethylformamide gave rise to the title compound 13. Similarly, 4-phenoxyphenol was reacted with 1,3-dibromopropane to produce 19, which on treatment with potassium cyanide gave rise to the target molecule 14 in excellent yields, as illustrated in Scheme 3.
Scheme 3.
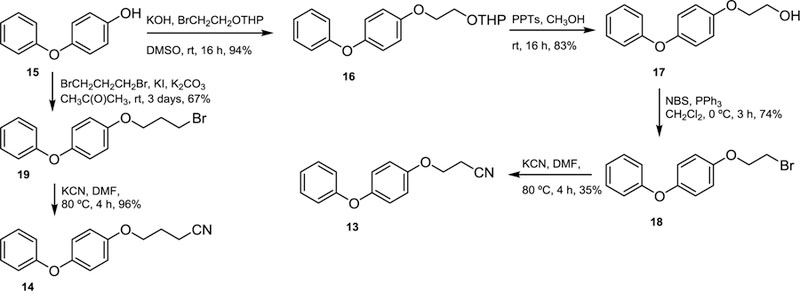
Synthesis of WC-9 derivatives where the sulfur or selenium atoms are missing.
Deleting of the terminal aromatic ring was another structural variations considered. We have demonstrated that selenocyanates structurally related to WC-9 were potent inhibitors of T. cruzi proliferation. All of them had the corresponding terminal aromatic ring B with different substitution patterns.16 Therefore, the preparation of simplified models of the reference structures 2−7 was done. In this case, the phenyl group and substituted phenyl ring with an electron withdrawing group and electron donor group was taken as molecular targets 20−22. Scheme 4 shows the synthetic approach to access these molecules.
Scheme 4.
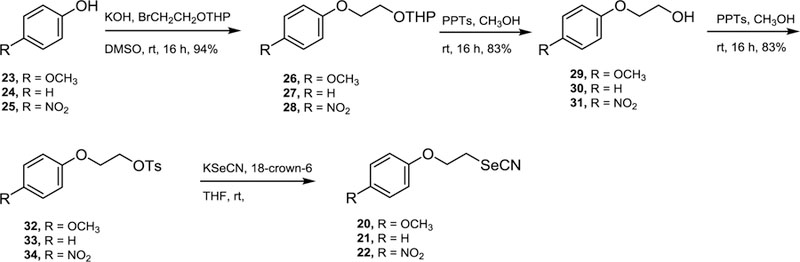
Synthetic strategy to access simplified analogues of selenium-containing analogues of WC-9.
Conformationally constrained derivatives turned out to be extremely potent inhibitors of intracellular T. cruzi proliferation, in particular, compound (±)−8 which had been evaluated as a racemic mixture (ED50 = 0.083 µM against intracellular T. cruzi),16 The biological evaluation of each enantiomer (+)−8 and (−)-8 would seem of interest in order to study if there is a molecular recognition preference by a particular enantiomer. Therefore, there was a strong evidence to believe that one particular enantiomer should have optimal space distribution and a better molecular recognition. It is well-established that a rigid optimum conformer is quite beneficial for a particular biological response.26 Therefore, the first approach that we took was a chiral resolution of alcohol (±)−35, a committed synthetic intermediate for the preparation of the title compound (±)−8. In this context, the O-acetyl mandelate derivatives of (±)−35 were prepared via a Steglich esterification.27 Thus, on treatment with (S)-(−)-O-acetilmandelic acid (36) and dicyclohexylcarbodiimide in the presence of 4-dimethylaminopyridine, (±)−35 was converted into the diastereomeric mixture 37 and 38 in almost theoretical yield, as illustrated in Scheme 5. All attempts to separate the diasteromers 37 and 38 were unsuccessful either under classic column chromatography or preparative HPLC, both molecules exhibiting similar chromatographic properties.
Scheme 5.
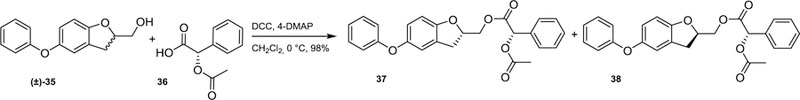
Formation of O-acetyl mandelates from racemic alcohol (±)−35 following a Steglich approach.
Enzymatic kinetic resolution arose as an interesting approach to separate both enantiomers of alcohol (±)−35. In this sense, Ramadas and Krupadanam had described a kinetic resolution of structurally related homochiral alcohols (2-hydroxymethyl-2,3-dihydrobenzofuran derivatives) by using a lipase from Pseudomonas cepacia and isopropenyl acetate as an acetyl donor.28 Therefore, (±)−35 treated with lipase from P. cepacia and isopropenyl acetate employing acetonitrile as a solvent at room temperature gave rise to (S)-alcohol 8 and the corresponding enriched acetate (R)-acetate 39 as illustrated in Scheme 6.
Scheme 6.
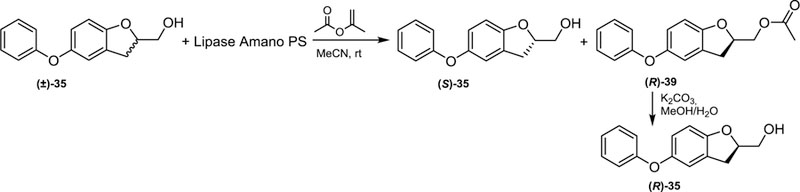
Kinetic resolution approach to resolve the racemic mixture (±)‒35.
The reaction was monitored by HPLC with the aid of a chiral column (analytical chiral column Lux 5µ Celullose-1 (4.60 mm × 25 cm) at a flow rate of 1.0 mL/min) employing a mixture of acetonitrile−water (7:3) as eluent. Figure 2 shows the elution profile of the racemic mixture (±)‒35. In fact, under these chromatographic conditions we were able to separate both enantiomers (±)‒35 into the (R)-(+)-35 and (S)-(‒)-35 enantiomers, which could be eluted at retention times of 5.92 min and 6.45 min, respectively.
Figure 2.
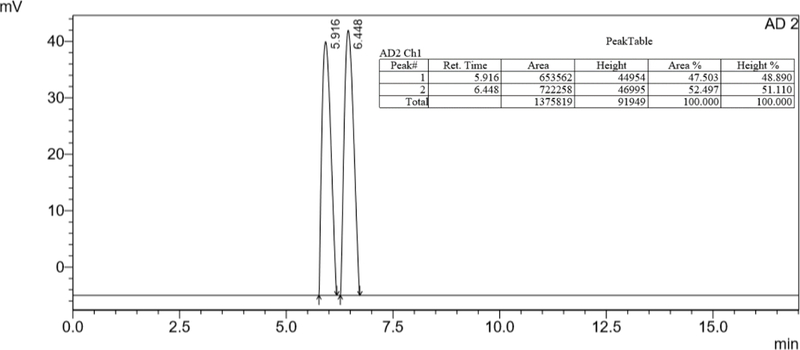
Elution profile for the resolution of the racemic mixture (±)‒35 into its enantiomers.
In addition, we were able to solve the racemic mixture of acetates (±)-39 into its enantiomers employing the same HPLC chiral column and the corresponding chromatographic conditions as illustrated in Figure 3. Once again, each enantiomer could be resolved into the (S)-(‒)-39 and (R)-(+)-39 isomers with retention times of 7.68 min and 8.34 min, respectively.
Figura 3.
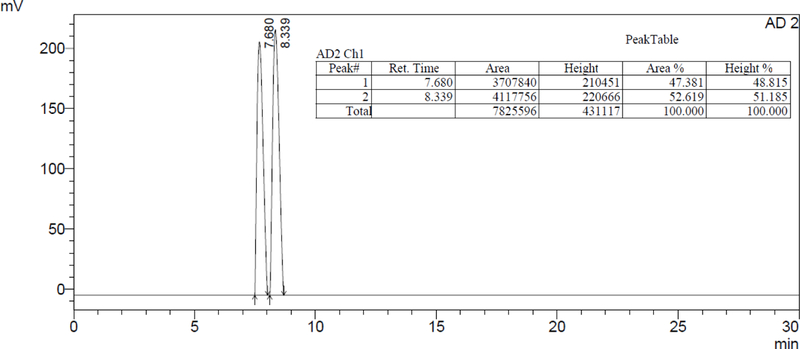
Elution profile of the racemic mixture of acetate (±)-39 employing a chiral Lux 5µ Celullose-1 column.
It is worth mentioning that each of the four compounds involved in the lipase-catalyzed kinetic resolution of alcohol (±)−35 could be separated by analytical HPLC resulting an excellent tool to monitor this enzymatic reaction. Thus, samples of the reaction mixture were taken every 30 minutes and analyzed by analytical HPLC. The (R)-alcohol would react faster than its (S)-enantiomer according to the chemical behavior of quite similar structurally related homochiral alcohols such as (±)−40a−j whose structure are drawn in Scheme 7.28,29 Therefore, from the analysis of the chromatograms versus time illustrated in Figure 4 it was possible to assign each enantiomer bearing in mind that the (R)-enantiomer would be acetylated faster than its corresponding (S)-enantiomer. In this sense, it was observed an area decrease of alcohol (R)-(+)-35 (tR = 5.93 min) with a concomitant area increase of the acetate assigned as the (R)-acetate (R)-(+)-39 (tR = 8.34 min). The (S)-(−)-35 alcohol reacted more slowly (tR = 6.45 min), which gave rise to the corresponding acetate (S)-(−)-39 (tR = 7.68 min).
Scheme 7.
Lipase-catalyzed resolution of homochiral alcohol structurally related to (±)-35.
Figure 4.
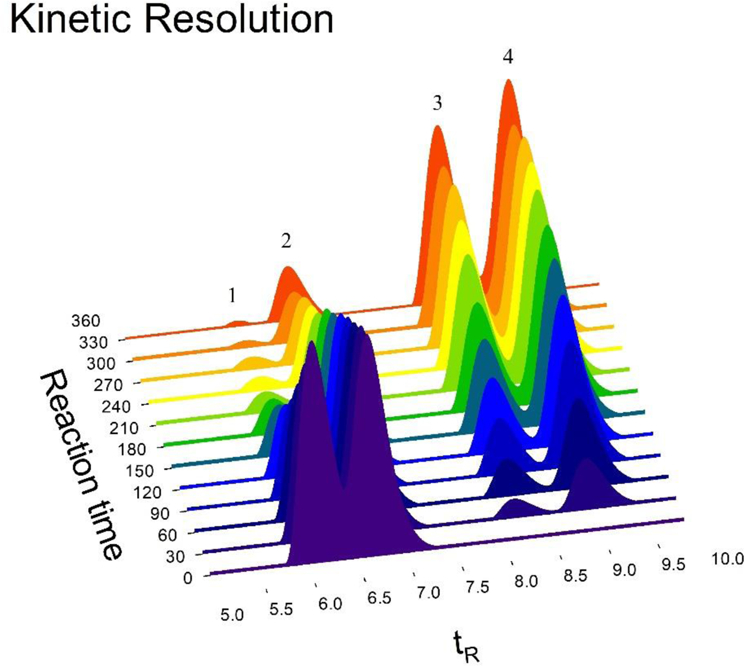
Analytical HPLC 2D elution profile of alcohol (±)-35 and acetate (±)-39 employing a chiral column to monitor lipase-catalyzed acetylation reaction of alcohol (±)-35.
Employing long reaction times (330 minutes), we were able to obtain, after purification by column chromatography, the (S)-alcohol with an enantiomeric excess of 84% and 22% yield and the (R)-enriched mixture of acetates 39.
The obtainment of the (R)-alcohol was conducted by hydrolysis of the (R)-enriched mixture of enantiomeric acetates 39 by treatment with potassium carbonate employing a mixture of methanol-water as a solvent to yield quantitatively the (R)-enriched alcohol 35, which was reacted under the same reaction conditions (lipase-catalyzed acetylation) to give a much more (R)-enriched acetate 39. The protocol was repeated four times producing the corresponding (R)-alcohol. In summary, after five turnovers of this lipase-mediated reaction it was possible the obtainment of (R)-(−)-35 in (ee 82% of enantiomeric excess and 38% yield. Table 1 reviews this achievement.
Table 1.
Retention times (tR), enantiomeric excess (ee), specific optical rotations and reaction yields for (S)-35 and (R)-35.
| Compound | tR (min) | ee (%) | (CHCL3) | Yield (%) |
|---|---|---|---|---|
| (S)-35 | 6.45 | 84 | +43.6 | 22 |
| (R)-35 | 5.92 | 82 | −45.1 | 39 |
It is important to mention that the racemic mixture (±)-35 is a solid (mp = 48 °C), initially depicted as an oil,16 but each isolated enantiomer was obtained as an oil. For that reason, further crystallographic studies to unambiguously establish the absolute configuration of each enantiomers resulted irrelevant. It is well-known that Mosher’s esters are not satisfactory to predict the absolute configuration in primary homochiral alcohols.
Once both homochiral alcohols were at hand, each compound was treated with tosyl chloride in pyridine at 0 °C to produce the corresponding chiral tosylates (S)-(+)-42 and (R)-(−)-42, respectively, which on reaction with potassium selenocyanate in the presence of 18-crown-6 were further converted into the title compounds (S)-(+)-8 and (R)-(−)-8 as shown in Scheme 8.
Scheme 8.
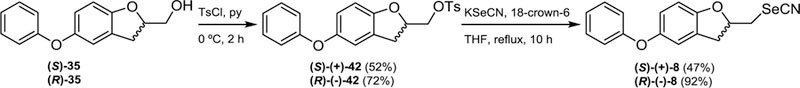
Synthetic approach for the preparation of chiral conformationally constrained selenocyanates (S)-(+)-8 and (R)-(−)-8.
We demonstrated that the position of the phenoxy group at the C-4′ position, as it was the case of compounds 1 and 3, was not an essentially required substitution pattern for biological activity bearing in mind that the regioisomers where the phenoxy moiety was bonded at the C-3′ position such as 2 and 5 exhibited similar or even better inhibitory action than 1 and 3.10,16 For the above reasons it seemed reasonable the study the antiparasitic activity of the C-2′ regiosiomers 43 and 44 whose preparation is described in Scheme 9. Then, the target molecules 43 and 44 were successfully synthesized starting from 2-iodophenol (45), which was reacted with benzyl bromide to yield the protected phenol 46. This compound was the substrate of the Buchwald coupling reaction,30–34 a key reaction step in this synthetic approach. Therefore, on reaction with phenol in the presence of copper(I) iodide, picolinic acid and potassium phosphate tribasic employing dimethyl sulfoxide as a solvent at 80 ºC for 5 days, 46 was converted into 47 in a low but reproducible yield. Cleavage of the benzyl group of 47 was performed by treatment with hydrogen in the presence of palladium on charcoal as catalyst to give rise to 48 in theoretical yield, which treated with bromoethyl tetrahydropyranyl ether yielded 49. The tetrahydropyranyl protecting group present in 49 was removed by treatment with pyridinium 4-toluenesulfonate producing the corresponding alcohol 50 in 86% yield, which was tosylated to give 51 in 61% yield. On treatment with potassium selenocyanate this compound was transformed into the target molecule 43, whereas on treatment with potassium thiocyanate, tosylate 51 was converted into the title compound 44.
Scheme 9.
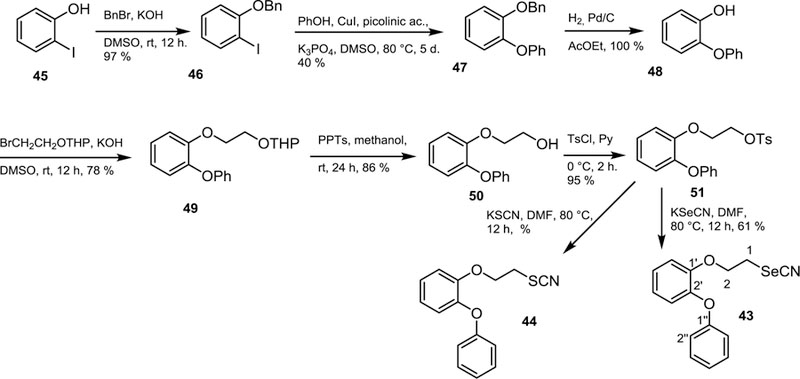
Synthetic approach for the preparation of regioisomers of WC-9 bearing the terminal aromatic ring at the C-2′ position.
Biological evaluation of these new isosteric analogues of WC-9 was very encouraging from the point of view of molecular reconition. Title compounds 9−12, where the cyano moiety in lead structures 1−3 and 5 was replaced by a trifluoromethyl group, were devoid of antiparasitic activity against intracellular T. cruzi indicating that the cyano part either of the selenocyanate or thiocyante play a crucial role on biological activity. The cyano group has an electrophilic center at the carbon atom and is a hydrogen bond acceptor due to the lone pair of electrons at the nitrogen atom.
Interestingly, cyanides 13 and 14 were inactive molecules as inhibitors of T. cruzi proliferation suggesting that the presence of the selenium or the sulfur atoms is vital for biological action and the existence of the cyano group is not sufficient to warrant antiparasitic activity.
The simple selenocyanate derivatives such as 20−22 are interesting molecules where the terminal aromatic ring B was absent but contained the pharmacophoric structure (aryloxyethyl selenocyanate) to warrant a pharmacological response. Surprisingly these compounds exhibited vanishing antiparasitic activity and also resulted to be cytotoxic against Vero cells.
The conformationally rigid (±)-8 is a potent growth inhibitor of the intracellular form of T. cruzi showing ED50 values at the low nanomolar concentrations (0.083 µM) and selectivity index value > 1,500.16 Consequently, the attempt to solve this racemic mixture into its enantiomers was quite sound considering that only one of them would be responsible for the antiparasitic activity. Unexpectedly, both enantiomers (S)-8 and (R)-8 exhibited practically the same inhibition potency against amastigotes of T. cruzi.
Finally, the regioisomers of the lead structures 1−3 and 5, that is, 43 and 44 where the phenoxy group was attached at the C-2′ position were free of anti-T. cruzi activity providing further insights into the chemical structure-biological activity relationship. Work aimed at exploiting the prospective antiparasitic activity of selenocyanate derivatives covalently bonded to a non-polar skeleton is currently being pursued in our laboratory.
Experimental
The glassware used in air-and/or moisture-sensitive reactions was flame dried, and the reactions were performed under a dry argon atmosphere. Unless otherwise noted, chemicals were commercially available and were used without further purification. Anhydrous N,N-dimethylformamide and anhydrous dimethyl sulfoxide were used as supplied from Aldrich. Nuclear magnetic resonance spectra were obtained using a Bruker Fourier 300 machine, or using a Bruker AM-500 MHz apparatus or using a Bruker Avance NEO 500 spectrometer. Chemical shifts are reported in parts per million δ relative to tetramethylsilane. 13C NMR spectra were fully decoupled. High-resolution mass spectra were carried out by using a Bruker micrOTOF-Q II spectrometer, which is a hybrid quadrupole time of flight mass spectrometer with MS–MS capability. Melting points were determined by using a Fisher–Johns apparatus. Column chromatography was performed with E. Merck silica gel plates (Kieselgel 60, 230–400 mesh). Analytical thin-layer chromatography was done by employing 0.2 mm coated commercial silica gel plates (E. Merck, DC-Aluminum sheets, Kieselgel 60 F254).
As judged from the homogeneity of the 1H, 13C, 19F and 77Se NMR spectra and HPLC analyses of the title compounds employing a Beckmann Ultrasphere ODS-2 column 5 μM, 250 × 10 mm eluting with acetonitrile–water (9:1) at 3.00 mL/min with a refractive index detector indicated a purity >97%.
(2-(4-Phenoxyphenoxy)ethyl)(trifluoromethyl)sulfane (9).
To a solution of WC-9 (50.0 mg, 0.18 mmol) in acetonitrile (2 mL) in the presence of cesium carbonate (59.9 mg, 0.18 mmol) was added trifluoromethyltrimethylsilane (39.2 mg, 40.8 µL, 0.28 mmol) under an argon atmosphere. The reaction mixture was stirred at room temperature for 24 h. Then, water (30 mL) was added and the mixture was extracted with methylene chloride (3 × 20 mL). The combined organic layers were washed with brine (3 × 20 mL), dried (MgSO4) and the solvent was evaporated. The product was purified by column chromatography (silica gel) eluting with a mixture of hexane−EtOAc (49:1) to give 32.3 mg (56% yield) of compound 9 as a white solid: mp = 29 °C; Rf = 0.73 (hexane−AcOEt, 4:1); IR (film, cm−1) 1589, 1496, 1215, 1091, 1070, 738, 691, 511; 1H NMR (500.13 MHz, CDCl3) δ 3.26 (t, J = 6.5 Hz, 2H, H-1), 4.23 (t, J = 6.5 Hz, 2H, H-2), 6.88 (d, J = 9.1 Hz, 2H, H-2′), 6.95 (dd, J = 8.7, 1.0 Hz, 2H, H-2′′), 6.98 (d, J = 9.1 Hz, 2H, H-3′), 7.05 (tt, J = 7.3, 1.1 Hz, 1H, H-4′′), 7.31 (dd, J = 8.6, 7.4, 2H, H-3′′); 13C NMR (125.77 MHz, CDCl3) δ 29.1 (q, J = 2.1 Hz, C-1), 67.1 (C-2), 115.8 (C-2′′), 117.8 (C-2′), 120.8 (C-3′), 122.6 (C-4′′), 129.7 (C-3′′), 130.9 (q, J = 306.2 Hz, SCF3), 150.91 (C-4′); 154.3 (C-1′), 158.2 (C-1′′); 19F NMR (470.59 MHz; CDCl3) δ −41,17 ppm. HRMS (ESI) calcd. for C15H14F3O2S [M+H]+ 315.0667; found 315.0662.
(2-(3-Phenoxyphenoxy)ethyl)(trifluoromethyl)sulfane (10).
To a solution of WC-9 (50.0 mg, 0.18 mmol) in acetonitrile (2.0 mL) in the presence of cesium carbonate (59.9 mg, 0.18 mmol) was treated with trifluoromethyltrimethylsilane (39.2 mg, 40.8 µL, 0.28 mmol) as described for the preparation of 9. The product was purified by column chromatography (silica gel) eluting with a mixture of hexane−EtOAc (99:1) to give 25.5 mg (44% yield) of 10 as a colorless oil: Rf = 0.67 (hexane−EtOAc, 4:1); IR (film, cm−1) 1585, 1484, 1214, 1105, 1028, 759, 687; 1H NMR (500.13 MHz, CDCl3) δ 3.27 (t, J = 6.5 Hz, 2H, H-1), 4.21 (t, J = 6.5 Hz, 2H, H-2), 6.59 (t, J = 2.3 Hz, 1H, H-2′), 6.66 (m, 2H, H-4′, H-6′), 7.06 (dd, J = 8.7, 1.1 Hz, 2H, H-2′′), 7.15 (tt, J = 7.4, 1.1 Hz, 1H, H-4′′), 7.26 (t, J = 8.2 Hz, 1H, H-5′), 7.38 (dd, J = 8.6, 7.4 Hz, 2H, H-3′′); 13C NMR (125.77 MHz; CDCl3) δ 28.9 (q, J = 2.1 Hz, C-1), 66.6 (C-2), 105.5 (C-2′), 109.2 (C-6′), 111.7 (C-4′), 119.2 (C-2′′), 123.6 (C-4′′), 129.8 (C-3′′), 130.3 (C-5′), 130.9 (q, J = 306.2 Hz, SCF3), 156.8 (C-1′′), 158.7 (C-1′), 159.4 (C-4′); 19F NMR (470.59 MHz, CDCl3) δ −41,19 ppm. HRMS (ESI) calcd. for C15H14F3O2S [M+H]+ 315.0667; found 315.0662.
(2-(4-Phenoxyphenoxy)ethyl)(trifluoromethyl)selane (11).
To a solution of WC-9-Se (50.0 mg, 0.16 mmol) in acetonitrile (2.0 mL) in the presence of cesium carbonate (61.2 mg, 0.19 mmol) was treated with trifluoromethyltrimethylsilane (33.5 mg, 35 µL, 0.24 mmol) as described for the preparation of 9. The reaction mixture was stirred for 4 h. The product was purified by column chromatography (silica gel) employing a mixture of hexane−EtOAc (99:1) as eluent to produce 34.2 mg (60% yield) of 11 as a white solid; mp = 29 °C; Rf = 0.59 (hexane−EtOAc, 4:1); 1H NMR (500.13 MHz, CDCl3) δ 3.33 (t, J = 6.4 Hz, 2H, H-1), 4.30 (t, J = 6.5 Hz, 2H, H-2), 6.88 (d, J = 9.1 Hz, 2H, H-2′), 6.95 (dd, J = 8.7, 1.1 Hz, 2H, H-2′′), 6.98 (d, J = 9.1 Hz, 2H, H-3′), 7.05 (tt, J = 7.6, 1,1 Hz, 1H, H-4′′), 7.30 (dd, J = 8.7, 7.4 Hz, 2H, H-3′′); 13C NMR (125.77 MHz, CDCl3) δ 24.2 (q, J = 1.6 Hz, C-1), 67.6 (C-2), 115.8 (C-2′′), 117.8 (C-2′), 120.8 (C-3′), 122.5 (q, J = 330.4 Hz, SeCF3), 122.6 (C-4′′), 129.6 (C-3′′), 150.9 (C-4′), 154.3 (C-1′), 158.2 (C-1′′); 19F NMR (470.59 MHz, CDCl3) δ −34.26 ppm; 77Se NMR (95.38 MHz; CDCl3) δ 434.30 ppm (q, J = 8.2 Hz). HRMS (ESI) calcd. for C15H14F3O2Se [M+H]+ 363.0111; found 363.0115.
(2-(3-Phenoxyphenoxy)ethyl)(trifluoromethyl)selane (12).
To a solution of WC-9-Se (50.0 mg, 0.16 mmol) in acetonitrile (2.0 mL) in the presence of cesium carbonate (61.2 mg, 0.19 mmol) was treated with trifluoromethyltrimethylsilane (33.5 mg, 35.0 µL, 0.24 mmol) as depicted for the preparation of 9. The product was purified by column chromatography (silica gel) eluting with a mixture of hexane−EtOAc (99:1) to give 38.5 mg (68% yield) of 12 as a colorless oil: Rf = 0.61 (hexane−EtOAc, 4:1); IR (film, cm−1) 1584, 1490, 1214, 1092, 738, 682; 1H NMR (500.13 MHz, CDCl3) δ 3.30 (t, J = 6.5 Hz, 2H, H-1), 4.28 (t, J = 6.5 Hz, 2H, H-2), 6.56 (t, J = 2.3 Hz, 1H, H-2′), 6.62 (m, 2H, H-4′, H-6′), 7.02 (dd, J = 8.7, 1.0 Hz, 2H, H-2′′), 7.12 (tt, J = 7.4, 1.0 Hz, 1H, H-4′′), 7.22 (t, J = 8.3 Hz, 1H, H-5′), 7.34 (dd, J = 8.7, 7.3 Hz, 2H, H-3′′); 13C NMR (125.77 MHz, CDCl3) δ 24.0 (q, J = 1.6 Hz, C-1), 67.1 (C-2), 105.5 (C-2′), 109.3 (C-6′), 111.6 (C-4′), 119.2 (C-2′′), 122.5 (q, J = 330.5 Hz, SeCF3), 123.5 (C-4′′), 129.8 (C-3′′), 130.3 (C-5′), 156.8 (C-1′′), 158.6 (C-1′), 159.4 (C-4′); 19F NMR (470.59 MHz, CDCl3) δ −34,28 ppm; 77Se NMR (95.38 MHz; CDCl3) δ 434.61 (q, J = 8.1 Hz). HRMS (ESI) calcd. for C15H14F3O2Se [M+H]+ 363.0111; found 363.0087.
4-Phenoxphenoxyethyl bromide (18).
A solution of 178 (369 mg, 1.60 mmol) in methylene chloride (5 mL) cooled at 0 °C was treated with triphenylphosphine (463 mg, 1.76 mmol) and N-bromosuccinimide (313 mg, 1.76 mmol). The reaction mixture was stirred at room temperature for 2 h. Then, water (25 mL) was added and the resulting mixture was extracted with methylene chloride (3 × 15 mL). The combined organic layers were washed with brine (3 × 50 mL), dried (MgSO4), and the solvent was evaporated. The residue was purified by column chromatography (silica gel) employing a mixture of CH2Cl2−methanol (24:1) to give 348 mg (74% yield) of 18 as a colorless oil: Rf = 0.60 (AcOEt-methanol, 4:1); 1H NMR (300.18 MHz, CDCl3) δ 3.64 (t, J = 6.1 Hz, 2H, H-1), 4.28 (t, J = 6.3 Hz, 2H, H-3), 6.90 (d, J = 9.2 Hz, 2H, H-2′), 6.98 (m, 2H, H-2′′), 6.98 (d, J = 9.2 Hz, 2H, H-3′), 7.05 (tt, J = 7.4, 0.9 Hz, 1H, H-4′′), 7.30 (dd, J = 8.6, 7.4, 2H, H- 3′′); 13C NMR (75.48 MHz, CDCl3) δ 29.1 (C-1), 68.5 (C-2), 116.0 (C-2′′), 117.8 (C-2′), 120.7 (C-3′), 122.6 (C-4′′), 129.6 (C-3′′), 150.9 (C-4′), 154.3 (C-1′), 158.2 (C-1′′).
4-Phenoxyphenoxyethyl cyanide (13).
To a solution of 18 (348 mg, 1.20 mmol) in anhydrous N,N-dimethylformamide (3 mL) was added potassium cyanide (234 mg, 3.60 mmol) and the reaction mixture was stirred at 80 °C for 1 h. Then, the reaction was allowed to cool to room temperature and water (20 mL) was added and the mixture was extracted with methylene chloride (3 × 20 mL). The combined organic phases were washed with brine (3 × 20 mL), dried (MgSO4), and the solvent was evaporated. The product was purified by column chromatography (silica gel) employing a mixture of hexane−EtOAc (87:13) as eluent to produce 101 mg (35% yield) of 13 as a white solid: mp = 67 °C; Rf = 0.21 (hexane−EtOAc, 4:1); 1H NMR (300.18 MHz, CDCl3) δ 2.83 (t, J = 6.4 Hz, 2H, H-1), 4.19 (t, J = 6.4 Hz, 2H, H-2), 6.89 (d, J = 9.2 Hz, 2H, H-2′), 6,97 (dd, J = 8,7; 1,1 Hz, 2H, H-2′′); 6.99 (d, J = 9.2 Hz, 2H, H-3′); 7.06 (tt, J = 7.4, 1.1 Hz, 1H, H-4′′), 7.31 (dd, J = 8.6, 7.6 Hz, 2H, H-3′′); 13C NMR (75.48 MHz, CDCl3) δ 18.7 (C-1); 63.3 (C-2), 116.0 (C-2′′), 117.2 (CN), 117.9 (C-2′), 120.8 (C-3′), 122.2 (C-4′′), 129.7 (C-3′′), 151.3 (C-4′), 153.9 (C-1′), 158.1 (C-1′′). HRMS (ESI) calcd. for C15H14NO2 [M+H]+ 240.1025; found 240.1013.
3-(4-Phenoxyphenoxy)propyl bromide (19).
A solution of 4-phenoxyphenol (15; 300 mg, 1.61 mmol), potassium carbonate (334 mg, 2.41 mmol) and potassium iodide (53 mg, 0.32 mmol) in acetone (8 mL) was treated with 1,3-dibromopropane (975 mg, 490 µL, 4.83 mmol). The reaction mixture was stirred at room temperature for 3 days. The solvent was evaporated and the product was purified by column chromatography (silica gel) eluting with a mixture of hexane−EtOAc (99:1) to give 331 mg (67% yield) of 19 as a colorless oil: Rf = 0.65 (hexane−EtOAc, 4:1); 1H NMR (300.18 MHz, CDCl3) δ 2.32 (p, J = 6.1 Hz, 2H, H-2), 3.62 (t, J = 6.4 Hz, 2H, H-1), 4.09 (t, J = 5.8 Hz, 2H, H-3), 6.88 (d, J = 9.1 Hz, 2H, H-2′), 6.94 (m, 4H, H-2′′, H-3′), 7,04 (tt, J = 7.4, 1.0 Hz, 1H, H-4′′), 7.30 (dd, J = 8.6, 7.4, 2H, H-3′′); 13C NMR (75.48 MHz, CDCl3) δ 30.0 (C-2), 32.4 (C-1), 65.8 (C-3), 115.6 (C-2′′), 117.7 (C-2′), 120.8 (C-3′), 122.5 (C-4′′), 129.6 (C-3′′), 150.4 (C-4′), 155.0 (C-1′), 158.4 (C-1′′).
3-(4-Phenoxyphenoxy)propyl cyanide (14).
A solution of 19 (42.4 mg, 0.14 mmol) in anhydrous N,N-dimethylformamide (3 mL) was treated with potassium cyanide (42.4 mg, 0.65 mmol). The reaction mixture was stirred at 80 ºC for 1 h. The reaction was quenched as depicted for the preparation of 13. The product was purified by column chromatography (silica gel) employing a mixture of hexane−EtOAc (19:1) to produce 33.5 mg (96% yield) of 14 as a colorless oil: Rf = 0.31 (hexane−EtOAc, 4:1); IR (film, cm−1) 2248, 1503, 1487, 1214, 841, 754, 691; 1H NMR (300.18 MHz, CDCl3) δ 2.17 (m, 2H, H-2), 2.62 (t, J = 7.1 Hz, 2H, H-1), 4.06 (t, J = 5.7 Hz, 2H, H-3), 6.89 (d, J = 9.1 Hz, 2H, H-2′), 6,98 (m, 4H, H-2′′, H-3′); 7.05 (tt, J = 7.4, 1.1 Hz, 1H, H-4′′), 7.30 (dd, J = 8.6, 7.4 Hz, 2H, H-3′′); 13C NMR (75.48 MHz, CDCl3) δ 14.2 (C-1), 25.5 (C-2), 65.8 (C-3), 115.5 (C-2′′), 117.7 (C-2′), 119.2 (CN), 120.8 (C-3′), 122.6 (C-4′′), 129.6 (C-3′′), 150.6 (C-4′), 154.6 (C-1′), 158.3 (C-1′′). HRMS (ESI) calcd. for C16H15NNaO2 [M+Na]+ 276.1000; found 276.0994.
4-Methoxyphenoxyethyl tetrahydro-2H-pyran-2-yl ether (26).
To a solution of 4- methoxyphenol (700 mg, 5.64 mmol) in methyl sulfoxide (5 mL) was added potassium hydroxide (634 mg, 11.3 mmol). The reaction mixture was stirred at room temperature for 10 min. Then, 2-(2-bromoethoxy)-tetrahydro-2H-pyrane (1.18 g, 5.64 mmol) was added and the reaction mixture was stirred at room temperature overnight. The reaction was worked up as described for the preparation of 18 yielding 1.34 g (94% yield) of 26 (colorless oil), which was used as such in the next step: Rf = 0.50 (hexane−EtOAc, 4:1); 1H NMR (300.18 MHz, CDCl3) δ 1.52−1.68 (m, 4H, H-4′′, H-5′′), 1.71−1.77 (m, 1H, H-3′′a), 1.79−1.89 (m, 1H, H-3′′b), 3.54 (m, 1H, H-6′′a), 3.77 (s, 3H, OCH3), 3.83 (m, 1H, H-6′′b), 3.89 (m, 1H, H-1a), 3.92 (m, 1H, H-1a), 4.05 (m, 1H, H-1b), 4.13 (m, 2H, H-2), 4.70 (dist t, J = 3.6 Hz, 1H, H-2′′), 6.84 (d, J = 9.4 Hz, 1H, H-2′), 6.90 (d, J = 9.4 Hz, 1H, H-3′); 13C NMR (75.48 MHz, CDCl3) δ 19.4 (C-4′′), 25.4 (C-5′′), 30.5 (C-3′′), 55.7 (OCH3), 62.2 (C-1), 65.9 (C-6′′), 68.1(C-2), 99.0 (C-2′′), 114.6 (C-3′), 115.7 (C-2′), 153.1 (C-1′), 153.9 (C-4′).
4-Metoxyphenoxyethanol (29).
A solution of 23 (1.33 g, 5.27 mmol) in methanol (15 mL) was treated with pyridinium 4-toluenesulfonate (30 mg). The reaction mixture was stirred at room temperature overnight. Then, water (50 mL) was added and the mixture was extracted with methylene chloride (3 × 50 mL). The combined organic layers were washed with brine (3 × 50 mL), dried (MgSO4), and the solvent was evaporated to produce 885 mg (100% yield) of pure alcohol 29 as a yellow pale solid: mp = 65 °C; Rf = 0.11 (hexane−EtOAc, 4:1); 1H NMR (300.18 MHz, CDCl3) δ 2.05 (br s, 1H, OH), 3.79 (s, 3H, OCH3), 3.96 (m, 2H, H-1), 4.06 (dd, J = 5.1, 3.4 Hz, 2H, H-2), 6.87 (m, 4H, H-2′, H-3′); 13C NMR (75.48 MHz, CDCl3) δ 55.8 (OCH3), 61.6 (C-1), 69.9 (C-2), 114.7 (C-3′), 115.6 (C-2′), 152.7 (C-1′), 154,1 (C-4′).
4-Methoxyphenoxyethyl 4-Toluenesulfonate (32).
A solution of alcohol 29 (884 mg, 5.25 mmol) in pyridine (3 mL) was treated with p-toluenesulfonyl chloride (546 mg, 2.9 mmol) at 0 °C and the mixture was stirred at 0 ºC for 4 h. Then, 5% HCl (50 mL) was added and the reaction mixture was stirred for an additional hour. The mixture was partitioned between methylene chloride (50 mL) and water (50 mL). The organic layer was washed with 5% HCl (3 × 50 mL) and water (3 × 50 mL). The organic phase was dried (MgSO4) and the solvent was evaporated. The product was purified by column chromatography (silica gel) employing a mixture of hexane−EtOAc (4:1) as eluent to produce 1.46 g of 32 (86% yield) as a white solid: mp = 89 °C; Rf = 0.28 (hexane−EtOAc, 4:1); 1H NMR (300.18 MHz; CDCl3) δ 2.45 (s, 3H, PhCH3), 3.76 (s, 3H, OCH3), 4,10 (m, 2H, H-1); 4,346 (m, 2H, H-2); 6.72 (d, J = 9.3 Hz, 2H, H-3′), 6.79 (d, J = 9.3 Hz, 2H, H-2′), 7.34 (d, J = 8.4 Hz, 2H, H-3′′), 7.82 (d, J = 8.3 Hz, 2H, H-2′′); 13C NMR (75.48 MHz, CDCl3) δ 21.6 (PhCH3), 55.7 (OCH3), 66.3 (C-1), 68.2 (C-2), 114.6 (C-3′), 115.8 (C-2′), 128.0 (C-2′′), 129.8 (C-3′′), 132.9 (C-4′′), 144.9 (C-1′′), 152.1 (C-1′), 154.3 (C-4′).
4-Methoxyphenoxyethyl selenocyanate (20).
A solution of tosylate 32 (1.45 g, 4.36 mmol) in anhydrous tetrahydrofuran (30 mL) was treated with potassium selenocyanate (681 mg, 4.8 mmol) in the presence of 18-crown-6 (0.1 mmol) and the reaction mixture was refluxed for 10 h. The solution was cooled to room temperature and the mixture was partitioned between brine (50 mL) and methylene chloride (30 mL). The aqueous phase was extracted with methylene chloride (3 × 25 mL). The combined organic layers were dried (MgSO4) and the solvent was evaporated. The product was purified by column chromatography (silica gel) employing a mixture of hexane−EtOAc (97:3) to give 826 mg (74% yield) of 20 as a white solid: mp = 40 °C; Rf = 0.30 (hexano−AcOEt, 4:1); 1H NMR (500.13 MHz, CDCl3) δ 3.41 (t, J = 6.0 Hz, 2H, H-1), 3.77 (s, 3H, OCH3), 4.33 (t, J = 6.0 Hz, 2H, H-2), 6.88 (mAB, 4H, H-2′ y H-3′); 13C NMR (125.77 MHz; CDCl3) δ 28.3 (C-1), 55.7 (OCH3), 67.3 (C-2), 101.3 (SeCN), 114.8 (C-3′), 115.9 (C-2′), 151.8 (C-1′), 154.6 (C-4′); 77Se NMR (95.38 MHz, CDCl3) δ 190.46 ppm. HRMS (ESI) calcd. for C10H11NNaO2Se [M+Na]+ 279.9853; found 279.9854.
Phenoxyethyl tetrahydro-2H-pyran-2-yl ether (27).
A solution of phenol (700 mg, 5.44 mmol) in methyl sulfoxide (5 mL) was treaded with potassium hydroxide (835 mg, 14.9 mmol) as depicted for the preparation of compound 26. After the usual work-up 1.50 g of 27 were obtained (90% yield) as a colorless oil, which was used as such in the next step: Rf = 0.53 (hexane−EtOAc, 4:1); 1H NMR (300.18 MHz, CDCl3) δ 1.52−1.68 (m, 4H, H-4′′, H-5′′), 1.70−1.77 (m, 1H, H-3′′a), 1.79−1.91 (m, 1H, H-3′′b), 3.55 (m, 1H, H- 6′′a), 3.85 (ddd, J = 11.0, 6.2, 4.7 Hz, 1H, H-6′′b), 3.93 (ddd, J = 11.4, 8.0. 3.5 Hz, 1H, H- 1a), 4.07 (dt, J = 11,1; 4,6 Hz, 1H, H-1b), 4.18 (m, 2H, H-2), 4.71 (dist t, J = 3.5 Hz 1H, H-2′′), 6.96 (m, 3H, H-2′, H-4′), 7.29 (m, 2H, H-3′); 13C NMR (75,48 MHz; CDCl3) δ 19.4 (C-4′′), 25.4 (C-5′′), 30.5 (C-3′′); 62,2 (C-1); 65,9 (C-6′′); 67,3(C-2); 99,0 (C-2′′); 114,7 (C-2′); 120,8 (C-4′); 129,4 (C-3′) 158,9 (C-1′).
Phenoxyethanol (30).
A solution of 27 (1.33 g, 5.27 mmol) in methanol (15 mL) was treated with pyridinium 4-toluenesulfonate (30 mg) according to the preparation of 29. Evaporation of the solvent gave 846 mg of 30 (92% yield) as a colorless oil: Rf = 0.23 (hexane−EtOAc, 4:1), 1H NMR (300.18 MHz, CDCl3) δ 2.10 (br s, 1H, OH), 3.98 (m, 2H, H-1), 4.11 (m, 2H, H-2), 6.95 (d, J = 8.8 Hz, 2H, H-2′), 6.99 (t, J = 7.4 Hz, 1H, H- 4′), 7.32 (dd, J = 8.6, 7.4 Hz, 2H, H-3′); 13C NMR (75.48 MHz, CDCl3) δ 61.5 (C-1), 69.0 (C-2), 114.5 (C-2′), 121.1 (C-4′), 129.5 (C-3′), 158.6 (C-1′).
Phenoxyetyl 4-toluenesulfonate (33).
To a solution of alcohol 30 (846 mg, 6.12 mmol) in pyridine (6 mL) cooled at 0 °C was added p-toluenesulfonyl chloride (4.65 g, 24.4 mmol). The reaction mixture was treated according the preparation of MNCC53 32. The residue was purified by column chromatography (silica gel) employing a mixture of hexane−EtOAc (17:3) to give 1.54 g (86% yield) of 33 as a white solid: mp = 73 ºC; Rf = 0.30 (hexane−EtOAc, 4:1); 1H NMR (300.18 MHz, CDCl3) δ 2.47 (s, 3H, PhCH3), 4.17 (m, 2H, H-1), 4.39 (m, 2H, H-2), 6.80 (dd, J = 9.7, 0.9 Hz, 2H, H-2′), 6.97 (tt, J = 7.4, 0.9 Hz, 1H, H-4′), 7.27 (dd, J = 8.7, 7.4 Hz, 2H, H-3′), 7.36 (d, J = 8.0 Hz, 2H, H-3′′), 7.82 (d, J = 8.3 Hz, 2H, H-2′′); 13C NMR (75.48 MHz, CDCl3) δ 21.7 (PhCH3), 65.3 (C-1), 68.1 (C-2), 114.5 (C-2′), 121.4 (C-4′), 128.0 (C-2′′), 129.5 (C-3′), 129.8 (C-3′′), 132.9 (C-4′′), 144.9 (C-1′′), 158.0 (C-1′).
Phenoxyethyl selenocyanate (21).
A solution of 33 (1.53 g, 5.22 mmol) in anhydrous tetrahydrofuran (10 mL) was treated with potassium selenocyanate (828 mg, 5.74 mmol) in the presence of 18-crown-6 (13.8 mg) according to the preparation of compound 20. The product was purified by column chromatography (silica gel) employing a mixture of hexane−EtOAc (9:1) as eluent to give 1.08 g (92% yield) of 21 as a white solid: mp = 49 °C; Rf = 0.44 (hexane−EtOAc, 4:1); 1H NMR (500.13 MHz, CDCl3) δ 3.44 (t, J = 6.0 Hz, 2H, H-1), 4.39 (t, J = 6.0 Hz, 2H, H-2), 6.92 (dd, J = 8.8, 1.0 Hz, 2H, H-2′), 7.01 (tt, J = 7.6, 1.0 Hz, 1H, H-4′), 7.31 (dd, J = 8.8, 7.4 Hz, 2H, H-3′); 13C NMR (125.77 MHz, CDCl3) δ 28.2 (C-1), 66.4 (C-2), 101.2 (SeCN), 114.7 (C-2′), 121.8 (C-4′), 129.7 (C-3′), 157.7 (C-1′); 77Se NMR (95.38 MHz, CDCl3) δ 191.67 ppm. HRMS (ESI) calcd. for C9H9NNaOSe [M+Na]+ 249.9747; found 249.9732.
4-Nitrophenoxyethyl tetrahydro-2H-pyran-2-yl ether (28).
To a solution de 4- nitrophenol (700 mg, 5.03 mmol) in dimethylsulfoxide (5 mL) was added potassium hydroxide (565 mg, 10.1 mmol). The mixture was stirred at room temperature for 10 minutes. Then, 2-bromoethyl tetrahydropyranyl ether (1.05 g, 5.03 mmol) was added as described for the preparation of 26. The product was purified by column chromatography (silica gel) eluting with hexane−EtOAc (21:4) to give 366 mg (27% yield) of compound 28 as a colorless oil: Rf = 0.28 (hexane−AcOEt, 4:1); 1H NMR (300.18 MHz, CDCl3) δ 1.53−1.66 (m, 4H, H-4′′, H-5′′), 1.71−1.76 (m, 1H, H-3′′a), 1,79−1,88 (m, 1H, H-3′′b), 3.55 (m, 1H, H-6′′a), 3.88 (m, 2H, H-6′′b, H-1a), 4.11 (dt, J = 11.6, 4.5 Hz, 1H, H-1b), 4.27 (m, 2H, H-2), 4.72 (dd, J = 3.9, 2.8 Hz, 1H, H-2′′), 7.00 (d, J = 9.3 Hz, 1H, H-2′), 8.22 (d, J = 9.3 Hz, 1H, H-3′); 13C NMR (75.48 MHz, CDCl3) δ 19.4 (C-4′′), 25.3 (C-5′′), 30.5 (C-3′′), 62.3 (C-1), 65.9 (C-6′′), 68.2(C-2), 99.2 (C-2′′), 114.6 (C-2′), 125.9 (C-3′), 141.6 (C-4′), 164.0 (C-1′).
4-Nitrophenoxyethanol (31).
To a solution of 28 (365 mg, 1.37 mmol) in methanol (10 ml) was added pyridinium p-toluenesulfonate (30 mg) and was treated according to the general procedure. It was obtained 251 mg (100% yield) of alcohol 31 as a white solid: mp = 87 ºC; Rf = 0.17 (hexane−AcOEt, 4:1); 1H NMR (300.18 MHz, CDCl3) δ 1.97 (br s, 1H, OH); 4.03 (dist t, 2H, H-1); 4.19 (m, 2H, H-2); 7,01 (d, J = 9,3 Hz, 2H, H-2′); 8,24 (d, J = 9,2 Hz, 2H, H-3′). 13C NMR (75,48 MHz; CDCl3) δ 61,1 (C-1); 70,0 (C-2); 114,5 (C-2′); 126,0 (C-3′).
4-Nitrophenoxyethyl 4-toluenesulfonate (34).
A solution of alcohol 31 (245 mg, 1.34 mmol) in pyridine (3 mL) cooled at 0 ºC was treated with p-toluenesulfonyl chloride (1.35 g, 4,01 mmol) according to the general procedure. The product was purified by column chromatography (silica gel) employing a mixture of hexane−EtOAc (4:1) to give 371 g (82% yield) of 34 as a white solid: mp = 123 ºC; Rf = 0.51 (hexane−EtOAc, 3:2); 1H NMR (300.18 MHz; CDCl3) δ 2.46 (s, 3H, PhCH3), 4.26 (m, 2H, H-1), 4.41 (m, 2H, H-2), 6.87 (d, J = 9.3 Hz, 2H, H-2′), 7.35 (d, J = 8.0 Hz, 2H, H-3′′), 7.81 (d, J = 8.3 Hz, 2H, H-2′′), 8.17 (d, J = 9.3 Hz, 2H, H-3′); 13C NMR (75.48 MHz; CDCl3) δ 21,7 (PhCH3); 66,1 (C-1); 67,4 (C-2); 114,5 (C-2′); 125,8 (C-3′); 128,1 (C-2′′); 129,9 (C-3′′); 132,7 (C-4′′); 142,0 (C-4′); 145,2 (C-1′′); 162,9 (C-1′).
4-Nitrophenoxyethyl selenocyanate (22).
A solution of 34 (363 mg, 1.08 mmol) in anhydrous tetrahydrofuran (10 mL) was treated with potassium selenocyanate (171 mg, 1.18 mmol) in the presence of 18-crown-6 (2.8 mg) according to the preparation of compound 20. The product was purified by column chromatography (silica gel) eluting a mixture of hexane−EtOAc (4:1) followed by HPLC purification eluting with acetonitrile−water (7:3) and employing a semi- preparative column Beckmann Ultrasphere-ODS-2 (5 µM) to give 135 mg (46% yield) of 22 as a colorless oil: Rf = 0.15 (hexane−EtOAc, 4:1); 1H NMR (500.13 MHz, CDCl3) δ 3.48 (t, J = 6.0 Hz, 2H, H-1), 4.52 (t, J = 6.0 Hz, 2H, H-2), 7.03 (d, J = 9.2 Hz, 2H, H-2′), 8.26 (d, J = 9.2 Hz, 1H, H-4′); 13C NMR (125.77 MHz, CDCl3) δ 27.2 (C-1); 67.1 (C-2), 100.5 (SeCN); 114.6 (C-2′), 126.0 (C-3′), 142.3 (C-4′), 162.6 (C-1′); 77Se NMR (95.38 MHz, CDCl3) δ 197.78 ppm. HRMS (ESI) calcd. for C9H8O3N2SeNa [M+Na]+ 294.9598, found 294.9578.
((S)-5-Phenoxy-2,3-dihydrobenzofuran-2-yl)methyl (S)-2-acetoxy-2-phenylacetate (37) and ((R)-5-phenoxy-2,3-dihydrobenzofuran-2-yl)methyl (S)-2-acetoxy-2- phenylacetate (38).
To a solution of alcohol (±)-3516 (10.0 mg, 4.13 × 10−3 mmol), S-(+)- O-acetylmandelic acid (36; 12.0 mg, 6.19 × 10−3 mmol) and 4-(dimethylamino)pyridine (0.8 mg, 6.61 × 10−4 mmol) in methylene chloride (2.0 mL) cooled at 0 °C was added dropwise a solution of dicyclohexylcarbodiimide (17.0 mg, 8.26 × 10−3 mmol) in methylene chloride (1.0 mL). The reaction mixture was stirred at 0 °C for 3 h. The mixture was filtered off to eliminate the resulting dicyclohexylurea. Then, methylene chloride (15 mL) was added and the organic layer was washed with an aqueous 1 M solution of hydrochloric acid (20 mL), an aqueous 1 M solution of sodium bicarbonate (20 ml), and brine (20 mL). The organic phase was dried (MgSO4), and the solvent was evaporated. The residue was purified by column chromatography (silica gel) employing a mixture of hexane‒EtOAc (97:3) as eluent to give 17.0 mg (98% yield) of a diastereomeric mixture 37/38 as a yellow pale oil: 1H NMR (CDCl3, 500.13 MHz) δ 2.16 2.18 (s, 3H, C(O)CH3), 2.78 (dd, J = 16.0, 7.0 Hz, 1H, H-3a), 2.92 (dd, J = 16.0, 7.1 Hz, 1H, H-3a), 3.18 (dd, J = 15.5, 9.6 Hz, 1H, H-3b), 3.21 (dd, J = 15.6, 9.6 Hz, 1H, H-3b), 4.31 (dd, J = 11.7, 5.5 Hz, 1H, CHaHOH), 4.37 (dt, J = 11.6, 3.8 Hz, 1H, CHbHOH), 4.95 (m, 1H, H-2), 5.92 (s, 1H, C(O)OCH), 6.67 6.71 (d, J = 8.4 Hz, 1H, H-7), 6.81–6.97 (m, 4H, aromatic protons), 7.03 (t, J = 7.4 Hz, 1H, aromatic proton), 7.29 (t, J = 7.6 Hz, 2H, aromatic protons); 13C NMR (125.76 MHz, CDCl3) δ 20.60 20.63 (CH3C(O)), 31.99 32.02 (C-3), 66.35 66.39 (CH2O), 74.40 74.42 (C-2′′), 79.89 79.90 (C-2), 109.75 109.77 (C-7), 116.89 116.92 (C-6), 117.5 (C-2′), 119.6 (C-4), 122.3 (C-4′), 127.10 127.15 (C-4a), 127.46 127.51 (C-3′′), 128.0 (C-3′′), 129.6 (C-3′), 129.23 129.27 (C-2′′), 133.4 133.5 (C-1′′), 150.4 (C-5), 155.5 (C-7a), 158.7 (C-1′), 168.69 168.76 (CH3C(O)), 170.3 (C-1′′).
(S)-(5-Phenoxy-2,3-dihydrobenzofuran-2-yl)methanol ((S)-(+)-35) and (R)-(5- Phenoxy-2,3-dihydrobenzofuran-2-yl)methyl acetate (39).
To a solution of (±)-(5- phenoxy-2,3-dihydrobenzofuran-2-yl)methanol ((±)-3516; 575 mg, 2.37 mmol) in acetonitrile (57 mL) was added Lipase Amano PS (395 mg). The resulting suspension was thermostatized at 23 ºC and stirred for 10 min. Then, isopropenyl acetate (3.97 g, 4.30 mL, 39.6 mmol) was added. The reaction was monitored by HPLC employing a chiral column Lux 5 µ Cellulose-1 (4.60 mm × 25 cm) eluting with a mixture of acetonitrile-water (7:3) at a flow rate of 1.0 mL/min. The mixture was stirred for 6 h. The reaction mixture was filtered off and the solvent was evaporated. The residue was purified by column chromatography (silica gel) employing a mixture of hexane‒EtOAc (4:1) as eluent to give the less reactive (S)-alcohol (+)-35 (64.2 mg) with 84% ee as a colorless oil and the enriched (R)-acetate 39 (598 mg) as a result from the acetylation of the more reactive (R)-alcohol (−)-35 (ee 13%).28 Then, to 598 mg (2.1 mmol) of 39 in methanol (30 mL) (1.16 g 8.41 mmol) of anhydrous powder of potassium carbonate were added while stirring, followed by water (9 mL) to obtain complete solution. The reaction was stirred at room temperature for 4 h. The mixture was neutralized with an aquous 10% solution of hydrochloric acid. The mixture was extracted with methylene chloride (3 × 20 mL), and the combined organic layers were dried (MgSO4), and the solvent was evaporated to produce 512 mg (100% yield) of more enriched (R)-alcohol 35. The resulting alcohol was acetylated under the presence of lipase as chiral catalyst to give the respective acetate that was hydrolyzed another time. This process was repeated four times to give 111 mg (39% yield) of the (R)-alcohol (R)-(−)-35 with 82% of enantiomeric excess. The 1H NMR and 13C NMR matched those previously described.16 Compound (S)-(+)-35: = +43.6 (c 1.0, CHCl3); (R)-(−)-35: = −45.1 (c 1.0, CHCl3); Compound (±)-39: Rf = 0.64 (hexane−EtOAc, 4:1); 1H NMR (CDCl3, 500.13 MHz) δ 2.11 (s, 3H, C(O)CH3), 2.96 (dd, J = 15.9, 7.5 Hz, 1H, H-3a), 3.30 (dd, J = 15.8, 9.5 Hz, 1H, H-3b), 4.23 (dd, J = 11.9, 7.0 Hz, 1H, CHaHOAc), 4.34 (dd, J = 11.9, 3.7 Hz, 1H, CHbHOAc), 5.02 (dddd, J = 9.4, 7.2, 7.2, 3.7 Hz, 1H, H-2), 6.76 (d, J = 8.7 Hz, 1H, H-7), 6.81 (ddt, J = 8.4, 2.6, 0.7 Hz, 1H, aromatic proton), 6.87 (m, 1H, aromatic proton), 6.94 (m, 2H, aromatic protons), 7.04 (tt, J = 7.4, 1.0 Hz, 1H, aromatic proton), 7.29 (dd, J = 8.7, 7.4 Hz, 2H, aromatic protons); 13C NMR (125.76 MHz, CDCl3) δ 20.8 (C(O)CH3), 32.3 (C-3), 65.8 (CH2OH), 80.5 (C-2), 109.9 (C-7), 116.9 (C-6), 117.5 (C- 2′), 119.7 (C-4), 122.4 (C-4′), 127.1 (C-4a), 129.6 (C-3′), 150.5 (C-5), 155.4 (C-7a), 158.6 (C-1′), 170.9 (C(O)CH3).
(+)-(S)-(5-Phenoxy-2,3-dihydrobenzofuran-2-yl)methyl 4-toluenenesulfonate ((S)- 42).
A solution of (S)-35 (45.6 mg, 0.19 mmol) in pyridine (3.0 mL) cooled at 0 ºC was treated with p-toluenesulfonyl chloride (108 mg, 0.56 mmol) according to the method described for the preparation of 32. The product was purified by column chromatography (silica gel) employing a mixture of hexane−EtOAc (4:1) as eluent to give 39.1 mg (52% yield) of tosylate (S)-38 as a colorless oil: NMR data matched with those previously depicted.16 = +58.3 (c 1.0, CHCl3)
(−)-(R)-(5-Phenoxy-2,3-dihydrobenzofuran-2-yl)methyl 4-toluenenesulfonate ((R)- 42).
To a solution of (R)-35 (50.3 mg, 0.21 mmol) in pyridine (3.0 mL) cooled at 0 ºC was added p-toluenesulfonyl chloride (119 mg, 0.62 mmol) following the method described for the preparation of 32. The product was purified by column chromatography (silica gel) employing a mixture of hexane−EtOAc (4:1) as eluent to give 59.3 mg (72% yield) of tosylate (R)-38 as a colorless oil: NMR data matched with those previously depicted.16 = −61.7 (c 1.0, CHCl3).
(+)-(S)-5-Phenoxy-2-(selenocyanatomethyl)-2,3-dihydrobenzofuran ((+)-(S)-8).
A solution of (S)-38 (28.8 mg, 7.26 × 10−2 mmol) in anhydrous tetrahydrofuran (3.0 ml) in the presence of 18-crown-6 (0.2 mg) was treated with potassium selenocyanate (11.5 mg, 7.99 × 10−2 mmol) according to the general procedure. The product was purified by column chromatography (silica gel) employing a mixture of hexane−EtOAc (93:7). The resulting partially purified compound was further purified by HPLC by using a semi- preparative Beckmann Ultrasphere ODS-2 (5µm, 10.0 mm × 25 cm) at a flow rate of 3 mL/min eluting with a mixture of acetonitrile-water (7:3) to yield 11.4 mg (47% yield) of (+)-(S)-8 as a yellow pale oil: = +36.0 (c 1.0, CHCl3). 77Se NMR (95.38 MHz, CDCl3) δ 171.29 ppm.
(−)-(R)-5-Phenoxy-2-(selenocyanatomethyl)-2,3-dihydrobenzofuran ((R)-8).
To a solution of (R)-38 (43.7 mg, 0.11 mmol) in anhydrous tetrahydrofuran (3.0 mL) in the presence of 18-crown-6 (0.3 mg) was added potassium selenocyanate (17.5 mg, 0.12 mmol) according to the general procedure. The product was purified by column chromatography (silica gel) employing a mixture of hexane−EtOAc (93:7) and was further purified by HPLC by using a semi-preparative Beckmann Ultrasphere ODS-2 (5µm, 10.0 mm × 25 cm) at a flow rate of 3 mL/min eluting with a mixture of acetonitrile-water (7:3) to give 33.6 mg (92% yield) of (R)-8 as a yellow pale oil: = −33.9 (CHCl3). 77Se NMR (95.38 MHz, CDCl3) δ 171.26 ppm.
1-(Benzyloxy)-2-iodobenzene (46).
A solution of 2-iodophenol (45; 1.32 g, 6.0 mmol) in N,N-dimethylformamide (15.0 mL) was added potassium carbonate (4.15 g, 30.0 mmol) and was stirred for 5 min. Then was added benzyl bromide (1.29 mg, 0.78 mL, 6.6 mmol) slowly. The reaction mixture was stirred at 50 ºC for 1 day. Then, the mixture was cooled to room temperature and was partitioned between methylene chloride (20 mL) and water (20 mL). The aqueous layer was extracted with methane chloride (2 × 20 mL) and the combined organic phases were washed with brine (5 × 50 mL), dried (MgSO4) and the solvent was evaporated affording 1.80 g (97% yield) of product 46 as colorless which was used as such in the next step: Rf = 0.75 (hexane–EtOAc, 4:1); 1H NMR (300.18 MHz, CDCl3) δ 5.12 (s, 2H, OCH2Ph), 6.75 (dt, J = 7.6, 1.3 Hz, 1H, H-5), 6.87 (dd, J = 8.2, 1.1 Hz, 1H, H-6), 7.29 (ddd, J = 8.2, 7.4, 1.5 Hz, 1H, H-4), 7.33–7.46 (m, 3H, H-3′, H-4′), 7.55 (d, J = 7.3 Hz, 2H, H-2′), 7.85 (dd, J = 7.8, 1.6 Hz, 1H, H-3); 13C-NMR (75.48 MHz, CDCl3) δ 70.8 (OCH2Ph), 87.0 (C-2), 112.9 (C-6), 123.0 (C-4), 127.1 (C-4′), 128.0 (C-2′), 128.7 (C-5), 129.6 (C-3′), 136.6 (C-1′), 139.6 (C-3), 157.2 (C-1).
1-(Benzyloxy)-2-phenoxybenzene (47).
A mixture of 46 (1.80 g, 7.76 mmol), phenol (1.30 g, 13.9 mmol), copper(I) iodide (132 mg, 6.93 mmol), 2-picolinic acid (170 mg, 1.38 mmol), and tripotassium phosphate (2.94 g, 13.9 mmol) under anhydrous conditions was evacuated and backfilled with argon twice. Then, dimethyl sulfoxide (12.0 mL) was added, and the mixture was stirred vigorously at 80 ºC for 5 days. The mixture was allowed to cool to room temperature and partitioned between ethyl acetate (20 mL) and water (20 mL). The aqueous layer was extracted with ethyl acetate (2 × 20 mL). The combined organic phases were washed with brine (5 × 50 mL), dried (MgSO4), and the solvent was evaporated. The product was purified by column chromatography (silica gel) eluting with hexane to produce 840 mg (39% yield) of 47 as a colorless oil: Rf = 0.64 toluene−hexane (9:1); 1H NMR (300.18 MHz, CDCl3) δ 5.11 (s, 2H, OCH2Ph), 6.95–7.00 (m, 3H, aromatic protons), 7.04–7.14 (m, 4H, aromatic protons), 7.19–7.21 (m, 2H, aromatic protons), 7.27–7.35 (m, 5H, aromatic protons); 13C-NMR (75.48 MHz, CDCl3) δ 70.8 (OCH2Ph), 115.3 (C-3′), 116.9 (C-2′′), 121.8 (C-6′), 122.0 (C-5′), 122.3 (C-4′′), 125.0 (C-4′), 127.1 (C-2′′′), 127.7 (C-4′′′), 128.4 (C-3′′), 129.5 (C-3′′′), 136.9 (C-1′′′), 145.4 (C-1′), 150.6 (C-2′), 158.4 (C-1′′). HRMS (ESI) calcd. for C19H16O2Na [M+Na]+ 299.1048; found 299.1033.
2-Phenoxyphenol (48).
A solution of 47 (744 mg, 2.69 mmol) in ethyl acetate (10 mL) in the presence of 5% palladium on charcoal (42 mg) was treated with hydrogen at 3 atm. The reaction was stirred at room temperature for 2 h. The mixture was filtered off and the solvent was evaporated to produce 503 mg (100% yield) of 48 as a white solid: mp = 105 ºC; Rf 0.74 (toluene−hexane, 4:1); 1H NMR (300.18 MHz, CDCl3) δ 5.58 (s, 1H, OH), 6.86 (m, 2H, aromatic protons), 7.04 (m, 4H, aromatic protons), 7.12 (m, 1H, aromatic proton), 7.34 (dd, J = 8.3, 7.7 Hz, 2H, H-3′); 13C NMR (75.48 MHz, CDCl3) δ 116.2 (C-6), 118.0 (C-2′), 118.8 (C-3), 120.6 (C-4), 123.6 (C-4′), 124.8 (C-5), 129.9 (C-3′), 143.5 (C- 1), 147.5 (C-2), 156.7 (C-1′). HRMS (ESI) calcd. C12H11O2 for [M + H]+ 187.0759; found 187.0744; calcd. C12H10NaO2 for [M+Na]+ 209.0578; found 209.0564.
2-Phenoxyphenoxyethyl tetrahydro-2H-pyran-2-yl ether (49).
To a solution of 2- phenoxyphenol (48; 383.3 mg, 2.06 mmol) in dimethyl sulfoxide (3.0 mL) was added potassium hydroxide (231 mg, 4.12 mmol) and the mixture was stirred at room temperature for 5 min. Then, bromoethyl tetrahydropyranyl ether (430 mg, 0.31 mL, 2.06 mmol) was added dropwise. The reaction mixture was stirred at room temperature for 20 h. The mixture was extracted with methylene chloride (2 × 25 mL) and the combined organic phases were washed with brine (5 × 50 mL), dried (MgSO4) and the solvent was evaporated. The product was purified by column chromatography (silica gel) using a mixture of hexane−EtOAc (24:1) as eluent to give 503.6 mg (78% yield) of 49 as a colorless oil: Rf = 0.50 (hexane–toluene, 4:1); 1H NMR (300.18 MHz, CDCl3) δ 1.41– 1.74 (m, 6H, H-2′′′, H-3′′′, H-4′′′), 3.44 (m, 1H, H-6′′′a), 3.67 (ddd, J = 11.2, 6.3, 4.8 Hz, 1H, H-6′′′b), 3.77 (ddd, J = 11.3, 8.4, 3.0 Hz, 1H, H-1a), 3.91 (dt, J = 11.3 Hz J = 4.7 Hz, 1H, H-1b), 4.18 (m, 2H, H-1 H-2), 4.55 (dist. t, J = 3.3 Hz, 1H, H-1′′′), 6.89–7.14 (m, 7H, aromatic protons), 7.26 (t, J = 8.0 Hz, 2H, aromatic protons); 13C NMR (75.48 MHz, CDCl3) δ 19.1 (C-3′′′), 25.4 (C-4′′′), 30.4 (C-2′′′), 61.8 (C-1), 65.6 (C-5′′′), 68.6 (C-1), 98.8 (C-1′′′), 115.0 (C-6), 116.7 (C-3′), 121.6 (C-2′′), 121.9 (C-4′), 122.1 (C-4′′), 125.0 (C-5′), 129.3 (C-3′′), 145.1 (C-2′), 151.0 (C-1′), 158.3 (C-1′′). HRMS (ESI) calcd. For C19H22O4Na [M+Na]+ 337.1416; found 337.1425.
2-Phenoxyphenoxyethanol (50).
A solution of 49 (490 mg, 1.57 mmol) in methanol (8 mL) was treated with pyridinium p-toluenesulfonate (20 mg). The mixture was stirred at room temperature overnight. Then, water (20 mL) was added, and the mixture was extracted with methylene chloride (3 × 20 mL). The combined organic layers were washed with brine (3 × 50 mL), dried (MgSO4), and the solvent was evaporated. The residue was purified by column chromatography (silica gel, hexane/EtOAc 95:5) to give pure alcohol 50 (230 mg, 86%) as a colorless oil: Rf = 0.26 (hexane/EtOAc 8:2); 1H NMR (300.18 MHz, CDCl3) δ 3.73 (dist. q, J = 5.0 Hz, H-2), 4.03 (dist. q, J = 4.4 Hz, H-1), 6.92–7.17 (m, 7H, aromatic H),; 7.28–7.33 (m, 2H, aromatic H); 13C NMR (75.48 MHz, CDCl3) δ 61.2 (C-2), 70.5 (C-1), 114.8 (C-6′), 116.5 (C-2′′), 122.0 (C-3′), 122.1 (C-4′), 122.4 (C-4′′), 125.2 (C-5′), 129.6 (C-3′′), 145.1 (C-2′), 150.4 (C-1′), 158.3 (C-1′′).
2-Phenoxyphenoxyethyl 4-toluenesulfonate (51).
A solution of 50 (310.0 mg, 1.35 mmol) in pyridine (3.0 mL) cooled at 0 °C was treated 4-toluenesulfonyl chloride (773.4 mg, 4.06 mmol) portion wise, and the mixture was stirred at 0 °C for 3 h. Then, a 5% aqueous solution of hydrochloric acid (10 mL) was added and the reaction mixture was stirred for an additional hour. The mixture was extracted with methylene chloride (30 mL) and the organic layer was washed with an aqueous 5% solution of hydrochloric acid (3 × 25 mL) and water (3 × 30 mL). The organic phase was dried (MgSO4) and the solvent was evaporated. The product was purified by column chromatography (silica gel) eluting with hexane to give 494.8 mg (95% yield) of 51 as a white solid: mp = 64−65 ºC; Rf = 0.19 (hexane–EtOAc; 4:1); 1H NMR (300.18 MHz, CDCl3) δ 2.43 (s, 3H, CH3PhSO3), 4.20–4.23 (m, 4H, H-1 H-2), 6.88–7.01 (m, 5H, aromatic protons), 7.03–7.12 (m, 2H, aromatic protons), 7.26–7.32 (m, 4H, aromatic protons), 7.73 (d, J = 8.3 Hz, 2H, aromatic protons); 13C NMR (75.48 MHz, CDCl3) δ 21.6 (CH3PhSO3), 67.0 (C-2), 67.9 (C-1), 116.0 (C-6′), 117.2 (C-2′′), 121.5 (C-3′), 122.5 (C-4′), 122.6 (C-4′′), 124.8 (C-5′), 127.9 (C-2′′′), 129.5 (C-3′′), 129.8 (C-3′′′), 132.7 (C-4′′′), 144.8 (C-1′′′), 145.9 (C-2′), 149.9 (C-1′), 157.8 (C-1′′).
2-Phenoxyphenoxyethyl selenocyanate (43).
A solution of 51 (100 mg, 0.26 mmol), potassium selenocyanate (41.2 mg, 0.29 mmol), and 18-crown-6 (7 mg) in anhydrous tetrahydrofuran (10 mL) was refluxed for 24 h. The solution was cooled to room temperature and the mixture was partitioned between brine (50 mL) and methylene chloride (30 mL). The aqueous phase was extracted with methylene chloride (3 × 25 mL). The combined organic layers were dried (MgSO4) and the solvent was evaporated. The product was purified by column chromatography (silica gel) using a mixture of hexane- EtOAc (96:4) as eluent to give 50.7 mg (61% yield) of 43 as a colorless oil: Rf = 0.58 (hexane–EtOAc, 7:3); 1H NMR (300.18 MHz, CDCl3) δ 3.27 (t, J = 6.0, 2H, H-1), 4.36 (t, J = 6.0 Hz, 2H, H-2), 6.90 (dd, J = 8.8, 1.1 Hz, 2H, H-2′′), 7.00–7.07 (m, 4H, H-3′, H- 4′, H-5′, H-6′), 7.14 (ddd, J = 8.1, 7.4 Hz, 1.9 Hz, 1H, H-4′′), 7.29 (dd, J = 8.7, 7.4 Hz, 2H, H-3′′); 13C NMR (75.48 MHz, CDCl3) δ 28.1 (C-1), 67.8 (C-2), 101.5 (SeCN), 115.6 (C-6′), 116.6 (C-2′′), 122.2 (C-3′), 122.5 (C-4′), 123.0 (C-4′′), 125.17 (C-5′), 129.6 (C-3′′), 145.3 (C-2′), 149.7 (C-1′), 158.1 (C-1′′); 77Se NMR (95.38 MHz, CDCl3) δ 196.06 ppm. HRMS (ESI) calcd. for C15H13O2NSeNa [M+Na]+ 342.0009; found 342.0012.
2-Phenoxyphenoxyethyl thiocyanate (44).
A solution of MV121 51 (100.0 mg, 0.26 mmol) in anhydrous N,N-dimethylformamide (5 mL) was treated with potassium thiocyanate (126 mg, 1.3 mmol). The reaction mixture was heated at 90 ºC for 5 h. The mixture was allowed to cool to room temperature and water (20 mL) was added. The aqueous phase was extracted with methylene chloride (2 × 30 mL) and the combined organic layers were washed with brine (5 × 30 mL) and water (2 × 30 mL). The organic phase was dried (MgSO4), and the solvent was evaporated. The residue was purified by column chromatography (silica gel) eluting with hexane–EtOAc (9:1) to give 55.0 mg (78 % yield) of 44 as a colorless oil: Rf 0.17 (hexane–EtOAc, 4:1), 1H NMR (500 MHz,CDCl3) δ 3.14 (t, J = 6.0 Hz, 2H, H-1), 4.28 (t, J = 6.0 Hz, 2H, H-2), 6.91 (dd, J = 8.6, 0.9 Hz, 2H, H-2′′), 7.00–7.06 (m, 4H, H-3′, H-4′, H-5′, H-6′), 7.14 (ddd, J = 8.2, 7.0, 1.9 Hz, 1H, H-4′′), 7.29 (dd, J = 8.6, 7.5 Hz, 2H, H-3′′); 13C NMR (125 MHz, CDCl3) δ 33.2 (C-1), 67.2 (C-2), 111.8 (SCN), 116.0 (C-6′), 116.8 (C-2′′), 122.0 (C-3′), 122.5 (C-4′), 123.0 (C-4′′), 125.1 (C-5′), 129.5 (C-3′′), 145.6 (C-2′), 149.7 (C-1′), 158.0 (C-1′′). HRMS (ESI) calcd. for C15H13O4NSNa [M+Na]+ 294.0565; found 294.0546.
T. cruzi amastigote assays
These experiments were performed as reported using tdTomato labeled trypomastigotes with the modifications described by Recher et al., 2013.35 Briefly, gamma-irradiated (2,000 Rads) Vero cells (3.4 × 104 cells/well) were seeded in 96 well plates (black, clear bottom plates from Greiner Bio-One) in 100 µL RPMI media (Sigma) with 10% FBS. Plates were incubated overnight at 35 oC and 7% CO2. Then, cells were tested with 3.4 × 105 trypomastigotes CL strain overexpressing a tdTomato red fluorescent protein /well in 50 µL volume and incubated for 5 h at 35 ºC and 7% CO2. After infection, cells were washed with Hanks solution (150 µL/well) and compounds were added in serial dilutions in RPMI media in 150 µL volumes. Each concentration was tested in quadruplicate. Each plate also contained controls with host cells where parasites are absent for background control, and evaluation with parasites without inhibitors to have a positive control. Inhibitors were tested against amastigotes at solutions of 1.56 µM, 3.125 µM, 6.25 µM, 12.5 µM, 25 µM. Benznidazole was employed as positive control at concentrations of 0.39 µM, 0.78 µM, 1.56 µM, 3.125 µM, and 6.25 µM, respectively for all set of testing. Once compound treatment, plates were incubated at 35 ºC and 7% CO2 and plates were assayed for fluorescence after 72 hours post-infection. ED50 values were determined by non-linear regression analysis using SigmaPlot.35
Cytotoxicity for Vero cells
The cytotoxicity was tested using the Alamar BlueTM assay as described by Recher et al., 2013. 35 Concisely, confluent monolayers of hTERT cells were seeded in 96 well plates in 150 µL DMEM high glucose no phenol red (Gibco Cat# 21063) with 10% Cosmic Calf Serum. Plates were incubated 16 hours at 35 ºC and 7% CO2. Then, wells were washed once with Hanks (150 µL/well), and inhibitors were added in sequential concentrations in DMEM media in 150 µL volumes. All dilutions was tested in quadruplicate. All plates contained controls with host cells without inhibitor. Plates containing drug dilutions were incubated at 35 ºC and 7% CO2 for 72 hours. Then, Alamar Blue indicator was aseptically added equivalent to 10% of the culture volume, which were incubated at 35 ºC for 6 hours. At that time, absorbance was measured at 570 and 600 nm.35 Different concentrations of DMSO were tested as positive control of toxicity.35
Supplementary Material
Table 2.
Growth inhibitory effect of selenocyanates derivatives and closely related molecules against intracellular T. cruzi.
| Compound | T. cruzi (amastigotes) ED50 (µM)a | Cytotoxicity ED50 (µM)b |
|---|---|---|
| 9 | > 10 µM 12% at 10 µM | > 20 |
| 10 | > 1 µM 6.7% at 1.0 µM | > 20 |
| 11 | > 10 µM | > 20 |
| 12 | > 1 µM 30.9% at 1.0 µM | > 20 |
| 13 | > 10.0 µM | > 20 |
| 14 | > 10.0 µM | > 20 |
| 20 | > 10.0 µM, no inhibition at 6 µM | **cytotoxic at 20 µM, 15 µM |
| 21 | > 10.0 | **cytotoxic at 20 µM |
| 22 | 3.87 ± 1.3 | **cytotoxic at 20 µM, 15 µM |
| (S)-8 | 0.141 ± 0.02 | > 20 |
| (R)-8 | 0.096 ± 0.014 | > 20 |
| (±)-8 | 0.0939 ± 0.025 | > 20 |
| 43 | > 10.0, 33% at 10.0µM | > 20 |
| 44 | > 10.0, 33% at 10.0 µM | > 20 |
| benznidazole | 2.48 ± 0.45 | > 10 |
The values are means ± S.D. of 3 independent biological replicates, each one done with 4 technical replicates.
The results are from three biological replicates, each one with 4 technical replicates. Vero cells were used and treatment, was for 72 h. Cytotoxicity was determined by visualization of cell detachment by optical microscopy or by using a BioTek plate reader as described by Recher et al., 2013.35
Acknowledgments:
We thank Melissa Storey for technical help with the drug screening. This work was supported by grants from the Consejo Nacional de Investigaciones Científicas y Técnicas (PIP 112–201501-00631 CO), Agencia Nacional de Promoción Científica y Tecnológica (PICT 2015 1349), and the Universidad de Buenos Aires (20020170100067BA) to J.B.R., and the U.S. National Institutes of Health to R.D. (AI- 107663).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
A. Supplementary data
Copies of the 1H NMR, 13C NMR, 19F NMR and 77Se NMR spectra are included as Supplementary Data.
References
- (1).Brener Z Biology of Trypanosoma cruzi. Annu. Rev. Microbiol 1973, 27, 347–382. [DOI] [PubMed] [Google Scholar]
- (2).Rodriguez JB; Falcone BN; Szajnman SH Detection and Treatment of Trypanosoma cruzi: A Patent Review (2011–2015). Expert Opin. Ther. Pat 2016, 26, 993–1015. [DOI] [PubMed] [Google Scholar]
- (3).Alpern JD; Lopez-Velez R; Stauffer WM Access to Benznidazole for Chagas Disease in the United States—Cautious Optimism? PLoS Negl. Trop. Dis 2017, 11, 10–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Urbina JA Specific Chemotherapy of Chagas Disease: Relevance, Current Limitations and New Approaches. Acta Trop 2010, 115, 55–68. [DOI] [PubMed] [Google Scholar]
- (5).Urbina JA New Insights in Chagas Disease Treatment. Drugs Future 2010, 35, 409–420. [Google Scholar]
- (6).Buckner FS; Urbina JA Recent Developments in Sterol 14-Demethylase Inhibitors for Chagas Disease. Int. J. Parasitol. Drugs Drug Resist 2012, 2, 236–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Szajnman SH; Yan W; Bailey BN; Docampo R; Elhalem E; Rodriguez JB Design and Synthesis of Aryloxyethyl Thiocyanate Derivatives as Potent Inhibitors of Trypanosoma cruzi Proliferation. J. Med. Chem 2000, 43, 1826–1840. [DOI] [PubMed] [Google Scholar]
- (8).Cinque GM; Szajnman SH; Zhong L; Docampo R; Schvartzapel AJ; Rodriguez JB; Gros EG Structure-Activity Relationship of New Growth Inhibitors of Trypanosoma cruzi. J. Med. Chem 1998, 41, 1540–1554. [DOI] [PubMed] [Google Scholar]
- (9).Elhalem E; Bailey BN; Docampo R; Ujváry I; Szajnman SH; Rodriguez JB Design, Synthesis, and Biological Evaluation of Aryloxyethyl Thiocyanate Derivatives against Trypanosoma cruzi. J. Med. Chem 2002, 45, 3984–3999. [DOI] [PubMed] [Google Scholar]
- (10).Elicio PD; Chao MN; Galizzi M; Li C; Szajnman SH; Docampo R; Moreno SNJ; Rodriguez JB Design, Synthesis and Biological Evaluation of WC-9 Analogs as Antiparasitic Agents. Eur. J. Med. Chem 2013, 69, 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chao MN; Li C; Storey M; Falcone BN; Szajnman SH; Bonesi SM; Docampo R; Moreno SNJ; Rodriguez JB Activity of Fluorine-Containing Analogues of WC-9 and Structurally Related Analogues against Two Intracellular Parasites: Trypanosoma cruzi and Toxoplasma gondii. ChemMedChem 2016, 11, 2690–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chao MN; Matiuzzi CE; Storey M; Li C; Szajnman SH; Docampo R; Moreno SNJ; Rodriguez JB Aryloxyethyl Thiocyanates Are Potent Growth Inhibitors of Trypanosoma cruzi and Toxoplasma gondii. ChemMedChem 2015, 10, 1094–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liñares GG; Gismondi S; Codesido NO; Moreno SNJ; Docampo R; Rodriguez JB Fluorine-Containing Aryloxyethyl Thiocyanate Derivatives Are Potent Inhibitors of Trypanosoma cruzi and Toxoplasma gondii Proliferation. Bioorganic Med. Chem. Lett 2007, 17, 5068–5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Schvartzapel AJ; Zhong L; Docampo R; Rodriguez JB; Gros EG Design, Synthesis, and Biological Evaluation of New Growth Inhibitors of Trypanosoma cruzi (Epimastigotes). J. Med. Chem 1997, 40, 2314–2322. [DOI] [PubMed] [Google Scholar]
- (15).Schvartzapel AJ; Fichera L; Esteva M; Rodriguez JB; Gros EG Design, Synthesis, and Anti-Trypanosoma cruzi Evaluation of a New Class of Cell Growth Inhibitors Structurally Related to Fenoxycarb. Helv. Chim. Acta 1995, 78, 1207–1214. [Google Scholar]
- (16).Chao MN; Storey M; Li C; Rodríguez MG; Di Salvo F; Szajnman SH; Moreno SNJ; Docampo R; Rodriguez JB Selenium-Containing Analogues of WC-9 Are Extremely Potent Inhibitors of Trypanosoma cruzi Proliferation. Bioorg. Med. Chem 2017, 25, 6435–6449. [DOI] [PubMed] [Google Scholar]
- (17).Bazzini Patrick; Wermuth CG Substituent Groups. In The Practice of Medicinal Chemistry; Wermuth CG; Aldous D; Raboisson P; Rognan D, Ed.; Academic Press, 2015; pp 348–349. [Google Scholar]
- (18).Urbina JA; Concepcion JL; Montalvetti A; Rodriguez JB; Docampo R Mechanism of Action of 4-Phenoxyphenoxyethyl Thiocyanate (WC-9) against Trypanosoma cruzi, the Causative Agent of Chagas’ Disease. Antimicrob. Agents Chemother 2003, 47, 2047–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- (20).Capper MJ; Wright GSA; Barbieri L; Luchinat E; Mercatelli E; McAlary L; Yerbury JJ; O’Neill PM; Antonyuk SV; Banci L; Hasnain SS The Cysteine-Reactive Small Molecule Ebselen Facilitates Effective SOD1 Maturation. Nat. Commun 2018, 9, 1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Macegoniuk K; Grela E; Palus J; Rudzińska E; Grabowiecka A; Biernat M; Berlicki Ł 1,2-Benzisoselenazol-3(2H)-One Derivatives As a New Class of Bacterial Urease Inhibitors. J. Med. Chem 2016, 59, 8125–8133. [DOI] [PubMed] [Google Scholar]
- (22).Liu C-I; Jeng W; Chang W-J; Shih M-F; Ko T-P; Wang AH-J Structural Insights into the Catalytic Mechanism of Human Squalene Synthase. Acta Crystallogr. Sect. D Biol. Crystallogr 2014, D70, 231–241. [DOI] [PubMed] [Google Scholar]
- (23).Billard T; Large S; Langlois BR Preparation of Trifluoromethyl Sulfides or Selenides from Trifluoromethyl Trimethylsilane and Thiocyanates or Selenocyanates. Tetrahedron Lett 1997, 38, 65–68. [Google Scholar]
- (24).Matheis C; Wang M; Krause T; Goossen LJ Metal-Free Trifluoromethylthiolation of Alkyl Electrophiles via a Cascade of Thiocyanation and Nucleophilic Cyanide–CF3 Substitution. Synlett 2015, 26, 1628–1632. [Google Scholar]
- (25).Jouvin K; Matheis C; Goossen LJ Synthesis of Aryl Tri- and Difluoromethyl Thioethers via a C H-Thiocyanation/Fluoroalkylation Cascade. Chem. - A Eur. J 2015, 21, 14324–14327. [DOI] [PubMed] [Google Scholar]
- (26).Rodriguez JB; Marquez VE; Nicklaus MC; Barchi JJ Jr. Synthesis of Cyclopropane-Fused Dideoxycarbocyclic Nucleosides Structurally Related to Neplanocin C. Tetrahedron Lett 1993, 34, 6233–6236. [Google Scholar]
- (27).Rodriguez JB; Markey SP; Ziffer H Preparation of 2(R) and 2(S) Methyl-2- Methylglycerates. Tetrahedron: Asymmetry 1993, 4, 101–108. [Google Scholar]
- (28).Ramadas S; Krupadanam GLD Enantioselective Acylation of 2- Hydroxymethyl-2,3-Dihydrobenzofurans Catalysed by Lipase from Pseudomonas Cepacia (Amano PS) and Total Stereoselective Synthesis of (−)- (R)-MEM-Protected Arthrographol. Tetrahedron Asymmetry 2000, 11, 3375–3393. [Google Scholar]
- (29).Ghanem A; Aboul-Enein HY Application of Lipases in Kinetic Resolution of Racemates. Chirality 2005, 17, 1–15. [DOI] [PubMed] [Google Scholar]
- (30).Ruiz-Castillo P; Buchwald SL Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Maiti D; Buchwald SL Orthogonal Cu- and Pd-Based Catalyst Systems for the O- and N-Arylation of Aminophenols. J. Am. Chem. Soc 2009, 131, 17423–17429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bruno NC; Buchwald SL Synthesis and Application of Palladium Precatalysts that Accommodate Extremely Bulky di-tert-Butylphosphino Biaryl Ligands. Org. Lett 2013, 15 (11), 2876–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bhayana B; Fors BP; Buchwald SL A Versatile Catalyst System for Suzuki-Miyaura Cross-Coupling Reactions of C(Sp2)-Tosylates and Mesylates. Org. Lett 2009, 11, 3954–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Fors BP; Watson DA; Biscoe MR; Buchwald SL A Highly Active Catalyst for Pd-Catalyzed Amination Reactions J. Am. Chem. Soc 2008, 130, 13552–13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Recher M; Barboza AP; Li Z-H; Galizzi M; Ferrer-Casal M; Szajnman SH; Docampo R; Moreno SNJ; Rodriguez JB Design, Synthesis and Biological Evaluation of Sulfur-Containing 1,1-Bisphosphonic Acids as Antiparasitic Agents. Eur. J. Med. Chem 2013, 60, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.