

Abstract
The C57BL/6 mouse strain is represented by distinct substrains, increasingly recognized to differ genetically and phenotypically. The current study compared stroke vulnerability among C57BL/6 J (J), C57BL/6JEiJ (JEiJ), C57BL/6ByJ (ByJ), C57BL/6NCrl (NCrl), C57BL/6NJ (NJ) and C57BL/6NTac (NTac) substrains, using a model of permanent distal middle cerebral artery and common carotid artery occlusion. Mean infarct volume was nearly two-fold smaller in J, JEiJ and ByJ substrains relative to NCrl, NJ and NTac (N-lineage) mice. This identifies a previously unrecognized confound in stroke studies involving genetically modified strain comparisons if control substrain background were not rigorously matched. Mean infarct size was smaller in females of J and ByJ substrains than in the corresponding males, but there was no sex difference for NCrl and NJ mice. A higher proportion of small infarcts in J and ByJ substrains was largely responsible for both substrain- and sex-dependent differences. These could not be straightforwardly explained by variations in posterior communicating artery patency, MCA anatomy or acute penumbral blood flow deficits. Their larger and more homogeneously distributed infarcts, together with their established use as the common background for many genetically modified strains, may make N-lineage C57BL/6 substrains the preferred choice for future studies in experimental stroke.
Keywords: C57BL/6, focal ischemia, mouse substrain, posterior communicating artery, stroke vulnerability
Introduction
Differences in stroke vulnerability among mouse strains have been explored for some time1–4 with an early emphasis on defining the potential impact of strain background on studies involving targeted genetic manipulations. Strain differences also have been exploited to define the genetic loci underlying such naturally occurring variation.5,6 However, the potential importance of substrain divergence among physically separated populations of the commonly used C57BL/6 (B6) mouse has not yet been considered in the context of experimental stroke.
An initial transfer of B6 mice from The Jackson Laboratory (JAX) to NIH in 1951 gave rise to separate J and N lineages, and it is increasingly recognized that there is considerable genetic and phenotypic variation between these major substrain groups.7–14 This is a critical practical issue. The default wild type mouse for many investigations is often C57BL/6 J (J). However, embryonic stem cells used to produce mice under the International Knockout Mouse Consortium (IKMC) are of the N lineage,15 and subsequent breeding has the potential to generate mixed substrain backgrounds.16 Previously unappreciated background heterogeneity has been noted for a subset of commercially available knockout mouse strains.17 Indeed, gene variants that distinguish B6 substrains from specific vendors have been identified within both J and N lineages, including mutations in alpha-synuclein,18 nicotinamide nucleotide transhydrogenase (Nnt), impacting glucose tolerance,19 the guanine nucleotide exchange factor, Dock2, that affects immune function,20 the Crb1 gene involved in retinal degeneration,21 and cytoplasmic familial mental retardation 1-interacting protein 2 (Cyfip2).10,14 Recent sequencing efforts by the Wellcome Trust Sanger institute revealed that the J and NJ substrains differ at over 10,000 genomic loci, including single nucleotide polymorphisms and large structural variants,22 and over 50 of these are validated coding variants.11 Thus, B6 substrains represent a unique system in which to investigate the impact of a relatively small number of highly penetrant genetic variants on phenotypic variation.
We initially noted systematic differences in infarct size between mice obtained from JAX (J) and Charles River Laboratories (C57BL/6NCrl, NCrl), and there has been a recent report of corresponding substrain differences in vulnerability to neonatal hypoxia-ischemia.23 Additional substrain and sex comparisons were therefore undertaken to better define the variation in stroke vulnerability among C57BL/6 mouse lineages. Some of these results have been presented in abstract.24
Materials and methods
Experimental animals and study design
Both male and female mice were used in the study. C57BL/6 substrains included J, C57BL/6JEiJ (JEiJ), C57BL/6ByJ (ByJ), NCrl, C57BL/6NJ (NJ) and C57BL/6NTac (NTac). J and NJ mice were obtained both from JAX (Bar Harbor, ME, USA) and from an in-house breeding colony maintained less than five generations from JAX stock. All JEiJ and ByJ were from the same in-house breeding colony founded by JAX stock. NCrl and NTac mice were obtained directly from Charles River Laboratories (Wilmington, MA, USA) and Taconic Biosciences (Hudson, NY, USA), respectively. All procedures were approved by the Institutional Animal Care and Use Committee, University of Tennessee Health Science Center, and were conducted according to United States Public Health Service Policy on Humane Care and Use of Laboratory Animals. Results are reported in compliance with ARRIVE guidelines. Mice were subjected to experimental stroke at two to six months of age. Infarct volumes were evaluated after 24 h. Estrous status was evaluated in female mice at the time of occlusion. In total, 210 mice were subjected to experimental stroke, using 30, 22, 24, 15, 4 and 6 males, and 50, 14, 14, 20, 5 and 6 females, of J, ByJ, NCrl, NJ, JEiJ and NTac substrains, respectively. Circle of Willis anatomy was evaluated in another 110 mice of J, ByJ, NCrl, NJ and JEiJ substrains, allocated as indicated below in the study description. Since this was an exploratory study with no a priori expectations regarding outcomes, there was no attempt to blind the surgeon with respect to mouse identity at the time of occlusion. However, final assessments of experimental endpoints were done without awareness of substrain. Data for half of the J and NCrl mice were obtained several years prior to the completion of the study, providing initial evidence for the substrain difference. All remaining animals were then used over a six-month period, during which mice of each substrain and sex were subjected to ischemia at three to four different time points.
Stroke modeling
Experimental stroke was produced by permanent tandem occlusion of the right middle cerebral artery (MCA) and ipsilateral common carotid artery (CCA) using methods originally described for rats,25 and generally similar approaches have been used previously in mice.1 Spontaneously breathing animals were maintained under anesthesia with 1–2% isoflurane in 70% N2, 30% O2, delivered to a nose restraint that permitted axial rotation. Body temperature was maintained using a rectal probe and feedback-controlled heating pad. The right CCA was exposed and cauterized between two ligations. The right MCA was exposed at the level of the rhinal fissure by dissecting the temporalis and masseter muscles and drilling a 1 mm burr hole through the temporal-squamous bone. Brain surface temperature was maintained using a 37℃ saline drip (TC-324B, Warner Instruments, Inc., Hamden, CT, USA). The MCA was snared with a micromanipulator-controlled wire hook, raised approximately 0.5 mm, and cauterized to produce permanent focal brain ischemia. Incisions were closed with suture and swabbed with povidone/iodine. Subcutaneous buprenorphine (0.1 mg/kg) was administered, anesthesia was discontinued, and animals were allowed to recover consciousness and thermal equilibrium before being returned to their cages. The procedure required 15–20 min.
The distribution of ischemic territory was examined by laser speckle contrast perfusion imaging in a subset of animals. Following occlusion, the dorsal surface of the skull was exposed and blood flow images were collected for approximately 10 min using a commercially available system (FLPI, Moor Instruments, Wilmington, DE, USA) as previously described.26 Flow values were obtained from the stored images at circular 1 mm diameter regions of interest in the margin of the ipsilateral MCA territory (centered at 1.5 mm lateral, approximately 3 mm caudal to bregma) and contralateral positions, avoiding major blood vessels. We have previously calibrated this method with absolute autoradiographic blood flow measurements in the rat,26 demonstrating its linearity over the range of values observed in mice.27 Variations in signal attenuation between the thinned rat and intact mouse skull preclude inference of absolute CBF in the present study, but this establishes the validity of relative comparisons of contralateral and ipsilateral perfusion.
Infarct measurement
Preliminary data in mice of both N and J substrain lineages documented no differences in infarct volumes measured at survival intervals of 6, 24 and 72 h in the model under study, so 24 h was selected as the time point for analysis. Mice were killed by cervical dislocation under isoflurane anesthesia. Brains were placed in phosphate-buffered saline (PBS) on ice, and 1 mm coronal slices were cut using a brain matrix. Slices were stained with 2% triphenyltetrazolium chloride in PBS and fixed in formalin overnight at 4℃. Calibrated images were collected (ImageJ), and the area of pallor in each slice was summed to obtain infarct volume. Edema swelling was modest and consistent (averaging 15% of total infarct volume), and attempts to correct for this would introduce additional error, so direct measures of lesion size were used in these analyses.
Vascular filling
Mice were killed by thoracotomy under isoflurane anesthesia and heparin (50 units) was injected into the left cardiac ventricle, through which a length of PE-50 tubing heat-widened near the tip was inserted into the aorta. After clamping the descending aorta and opening the right atrium, the vasculature was manually flushed by syringe with several mL of saline, followed by approximately 0.3 mL of Microfil silicone MV-120 (blue) or MV-122 (yellow) (Flow Tech Inc., Carver, MA), prepared essentially according to the rapid cure procedure described by the supplier. Carcasses were refrigerated until dissection, after which brains were stored in buffered formalin. Posterior communicating artery (PcomA) anatomy was scored essentially as previously described:28,29 0, absent; 1, capillary; 2, clear vascular trunk; 3, robust arterial communication. Scores were then grouped as grade 0/1 (hypoplastic) or grade 2/3 (well-developed),28 with independent assessments on right and left sides. As a technical note, the yellow silicone achieved somewhat better filling of small vessels, consistent with its widespread use in microvascular imaging studies,6 but the two products produced identical results with respect to PcomA patency scoring (not shown). However, its use permitted evaluation of MCA distribution and anastomoses in a subset of animals. For purposes of illustration, images were captured in color, converted in Photoshop to black and white with enhancement of blue or yellow tones, respectively, followed by brightness and contrast adjustment.
Estrous cycle evaluation
While under anesthesia for occlusion surgery, the vaginal canal was flushed several times with 5 µl of physiological saline using a micropipette. The sample was spread on a glass slide, air dried, and subsequently formalin fixed and stained with hematoxylin/eosin for microscopic evaluation using established criteria.30
Exclusions and data analysis
Of 210 operated mice, there were 14 exclusions (7%), based either on anomalies in MCA branching recognized postmortem (n = 9), or the presence of a striatal lesion presumed secondary to local trauma impacting lenticulostriate artery perfusion (n = 5). Anatomical exclusions involved 6 of 44 J females, 2 of 27 J males and 1 of 14 NCrl females, whereas surgical trauma involved individual NCrl, EiJ and ByJ males, and NCrl and NTac females. Two outliers in the NCrl group were excluded from the statistical analyses based on the robust regression and outlier exclusion (ROUT) method. These happened to be from a single shipment obtained at two months of age used three days after arrival, suggesting acclimation as a potential contributing variable. Vascular filling failed in 7 of 110 mice. Normality of infarct volume data was assessed by the D’Agostino-Pearson omnibus K2 test. Substrain differences in infarct volume were compared by Kruskal–Wallis and Dunn’s Multiple Comparison tests. Sex differences within a substrain were evaluated by two-tailed Mann–Whitney test. In all cases, P < 0.05 was considered statistically significant. Analyses were implemented in GraphPad Prism 5.0 c (GraphPad Software, San Diego, CA, USA). Results are variably illustrated using either Tukey box plots (showing median, first and third quartiles, ±1.5 quartile range and outliers), scatter plots that include all data points, histograms of infarct volume distributions and PcomA scores, or group means ± SEM. The latter is appropriate for purposes of these comparisons since it provides an index of the variability in mean infarct volumes expected to be obtained for multiple sample populations of a given substrain.
Results
Substrain differences in infarct size
J and ByJ mice exhibited smaller infarcts than NCrl and NJ substrains, and this was evident for both males and females (Figure 1(a)). There was no sex difference in infarct volume for NCrl and NJ mice, but females of both J and ByJ substrains had significantly smaller infarcts than the corresponding males (Figure 1(b)). The basis for this sex difference became evident in an analysis of infarct size distributions for these groups (Figure 2), showing a skew toward very small infarcts in females of J and ByJ substrains. The range of infarct volumes was comparable for their male counterparts but was weighted more strongly toward the larger end. This was particularly evident in ByJ mice, resulting in essentially mirror distributions for males and females of this substrain. Non-Gaussian distributions were suggested by failure of the normality test for the relatively larger J female and ByJ male groups, whereas those of J males and ByJ females were probably underpowered for such analysis. In contrast, for NCrl and NJ, both males and females had symmetrically distributed infarcts, with slightly greater dispersion for females of both substrains. Although NCrl and NJ exhibited a number of infarcts larger than any seen in J and ByJ, group differences were largely driven by the proportion of small infarcts in the latter substrains.
Figure 1.
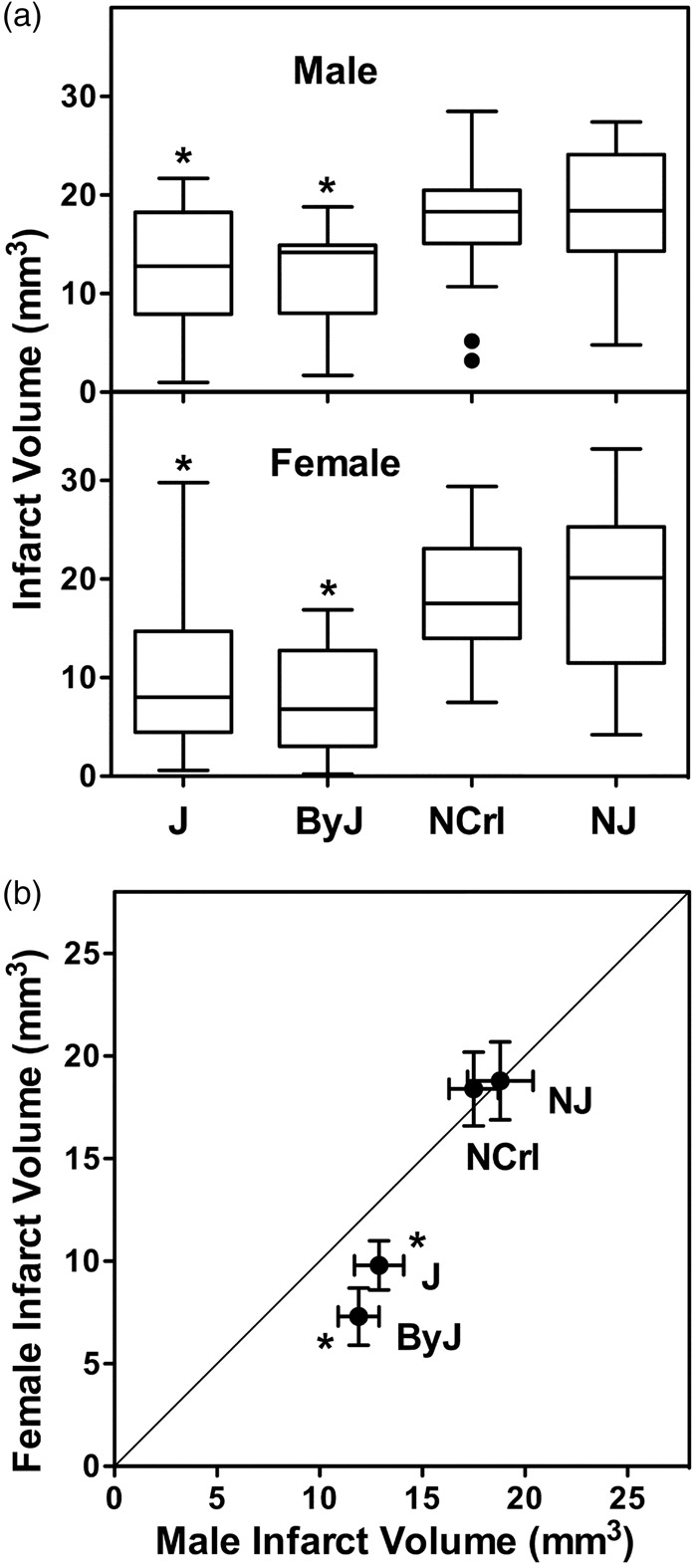
Substrain and sex differences in infarct size after tandem MCA/CCA occlusion. (a) Substrain differences. For both males and females, J and ByJ substrains exhibited smaller infarcts than NCrl and NJ mice (*, P < 0.05 vs. both N substrains). (b) Sex differences. Mean infarct volumes were identical for males and females of NJ and NCrl substrains, but J and ByJ females had smaller infarcts than their male counterparts (*, P < 0.05, male vs. female). Line indicates identity. Group sizes for J, ByJ, NCrl and NJ substrains were 25, 21, 23 and 15 for males and 38, 14, 12 and 20 for females, respectively.
Figure 2.

Infarct size distributions vary among B6 substrains. Bimodal patterns were evident for J and ByJ substrains, with smaller infarcts observed more frequently in females, and this sex difference was particularly prominent in ByJ. The larger infarcts in NCrl and NJ mice exhibited more symmetrical distributions in both males and females, with only occasional small infarcts.
A number of potential sources of variability were excluded as significant factors that could influence the above results. The same experienced individual was responsible for all surgery. There was no detectable impact of occlusion sequence or timing on outcome for a given substrain, and no effect of age over the range used in these studies. Infarct size variation was independent of estrous status in females (Figure S1).
Genetic divergence and stroke vulnerability
Additional results for limited numbers of JEiJ and NTac mice yielded stroke vulnerabilities corresponding to the J/ByJ and NCrl/NJ phenotypes, respectively, confirming the expected relationship to substrain lineage shown in Figure 3. The divergence between small and large infarct phenotypes appears to have occurred at NIH following separation of the ByJ substrain in 1961, but prior to establishing NCrl in 1974, after which the difference in vulnerability appears to have been stably maintained in the respective lineages.
Figure 3.
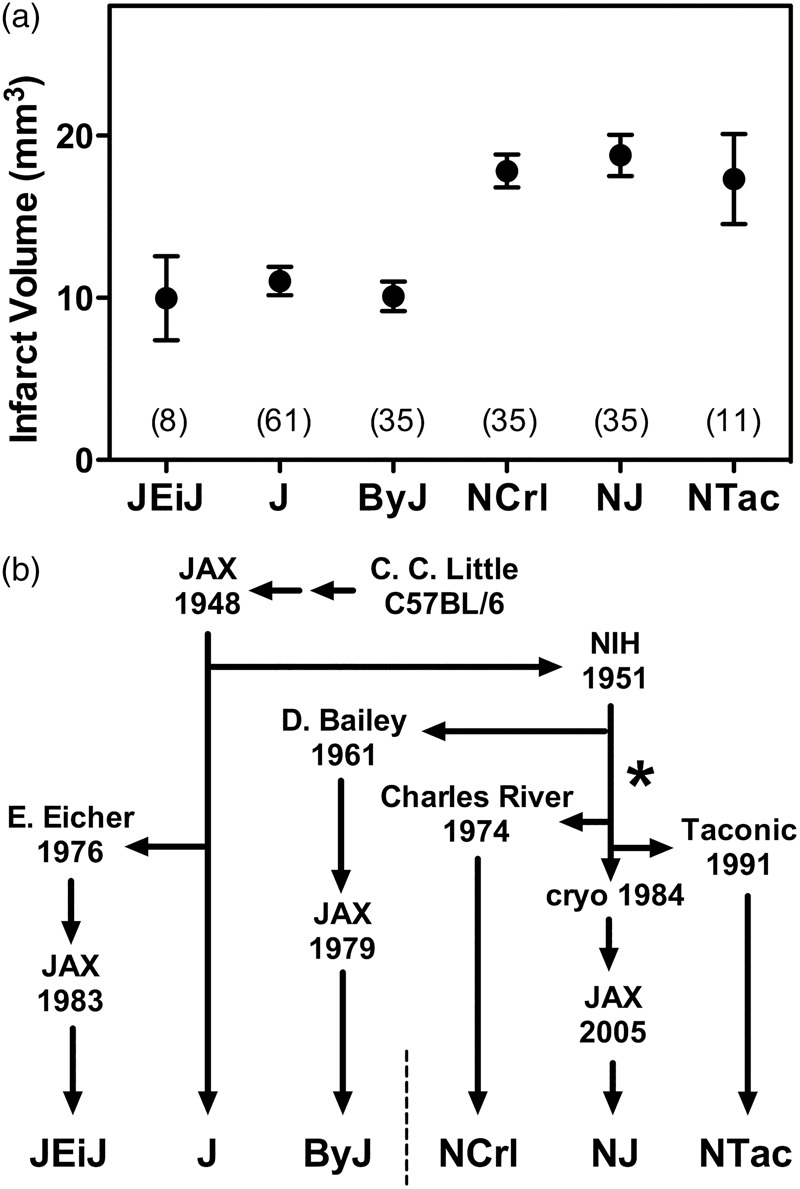
C57BL/6 lineages and the divergence of stroke vulnerability. (a) Infarct size variation across an expanded substrain set. Results are shown for pooled male and female mice of J, ByJ, NCrl and NJ substrains from Figure 1, together with more limited data for JEiJ and NTac substrains. Symbols indicate mean ± SEM with group sizes in parentheses. (b) C57BL/6 substrain lineages. Origins of the six evaluated substrains illustrate their relationship to the pattern of stroke vulnerability. The responsible mutation appears to have occurred at NIH (*), subsequent to the separation of the ByJ lineage in 1961 and prior to the transfer of mice to Charles River in 1974.
Anatomical and perfusion correlates of substrain vulnerability
Circle of Willis anatomy was highly variable, but a pattern of sex and substrain differences emerged (Figure 4). The PcomA on the right side was absent or poorly developed (grade 0/1) in the majority of males (60–70%) of all substrains. This was also the case for females, except that essentially all ByJ females showed robust communication between the basilar and posterior cerebral arteries. This would appear to correlate with the striking sex-dependence of infarct volume distributions in ByJ mice (Figure 2). On the other hand, right Pcom A scores were identical in male and female J/JEiJ mice that nevertheless showed significant differences in infarct volumes. Although perhaps less relevant to the current study that involved right MCA/CCA occlusion, there was a surprising discrepancy between right and left PcomA development in male ByJ mice. In contrast to their generally poor communication on the right, ByJ males had a consistently robust PcomA on the left. Scores for other strain pools also tended to be somewhat better on the left, except for J/JEiJ males. Choice of occlusion side therefore emerges as a potential substrain- and sex-dependent variable.
Figure 4.
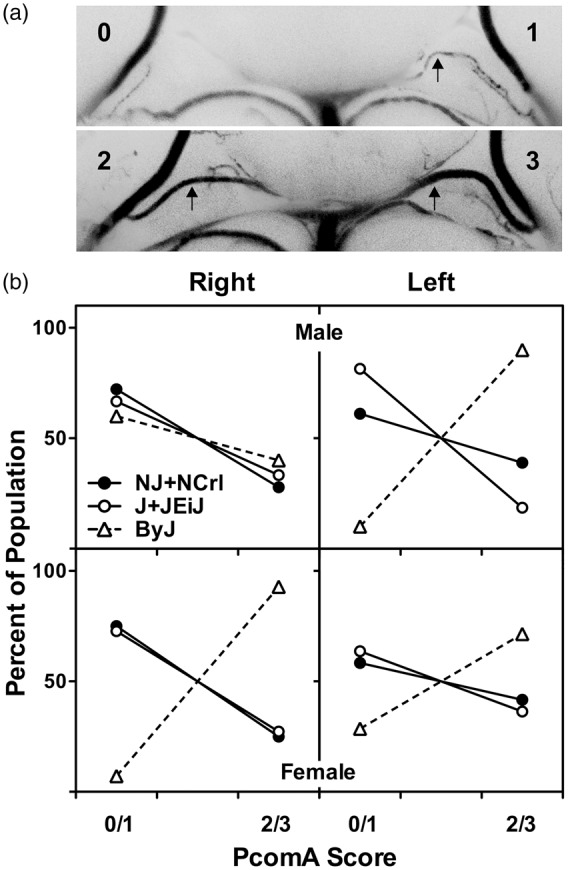
Substrain and sex differences in circle of Willis anatomy. (a) Examples of posterior communicating artery (PcomA) scoring. Arrows indicate PcomA. Vascular caliber was graded from 0 to 3, and communication was subsequently rated as poor (grade 0 or 1) or robust (grade 2 or 3) as described in the text. These examples were perfused with blue silicone. (b) Sex and substrain differences in the distribution of PcomA scores. Results did not differ between NJ and NCrl, or between J and JEiJ, so results are pooled to simplify presentation. The right PComA was comparably developed in males of all substrains. Female NJ/NCrl and J/JEiJ mice exhibited PcomA scores comparable to males of the same substrains, whereas all female ByJ had robust communication. Left PcomA scores showed the same pattern of strain variation as the right with the striking exception of ByJ males, in which the left PcomA exhibited the consistent patency seen bilaterally in ByJ females. Numbers of NJ, NCrl, J, JEiJ and ByJ mice were 9, 9, 12, 15 and 10 males, and 4, 8, 13, 9 and 14 females, respectively.
MCA anatomy was also evaluated in a subset of adequately perfused ByJ and NCrl females, and results for representative examples are illustrated in Figure 5. These substrains showed similarly wide variations in branching patterns among animals (not shown) but a consistent anatomical extent, with the line of dorsal anastomoses located 1.7 ± 0.2 and 1.7 ± 0.1 mm from the midline in ByJ and NCrl mice (n = 12 and 15 hemispheres), respectively. Comparable numbers of anastomoses (4.5 ± 0.9 and 4.4 ± 1.0, mean ± SD) were also identified along this border. These results indicate that variations in either the anatomy of the MCA or in the extent of its collaterals are not likely to account for the differences in stroke vulnerability among substrains.
Figure 5.
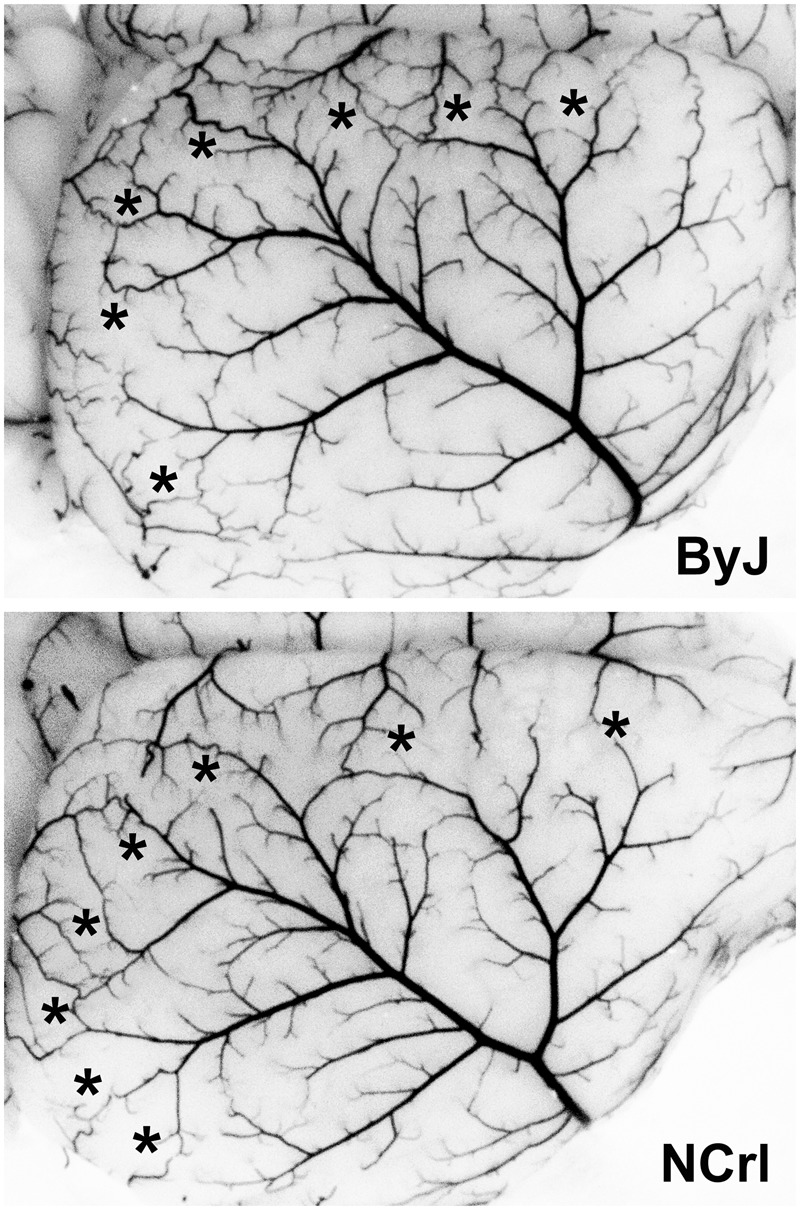
MCA anatomy in C57BL/6 substrains. A subset of the mice perfused for circle of Willis evaluation also achieved adequate filling of the microvasculature. Their comparable extent of MCA territory is evident for representative examples of female ByJ and NCrl mice. A similar abundance of vascular collaterals is evident in the line of anastomoses (asterisks). Quantitative results are presented in the text.
Perfusion imaging identified no significant difference in contralateral perfusion between mice of J and N lineages (Figure 6(a)). Apparent perfusion in ipsilateral penumbra was lower in the less vulnerable J/ByJ/EiJ substrains, but as a percentage of contralateral perfusion, this difference was not statistically significant. Although not highly predictive of outcome for an individual animal, penumbral perfusion was inversely correlated with infarct size within a substrain lineage (Figure 6(b)). J/ByJ/EiJ mice on average exhibited smaller infarcts than NCrl/NJ mice for a given flow deficit, suggesting an intrinsic substrain difference in the CBF threshold for vulnerability. Comparable results were obtained for separate analyses of males and females.
Figure 6.
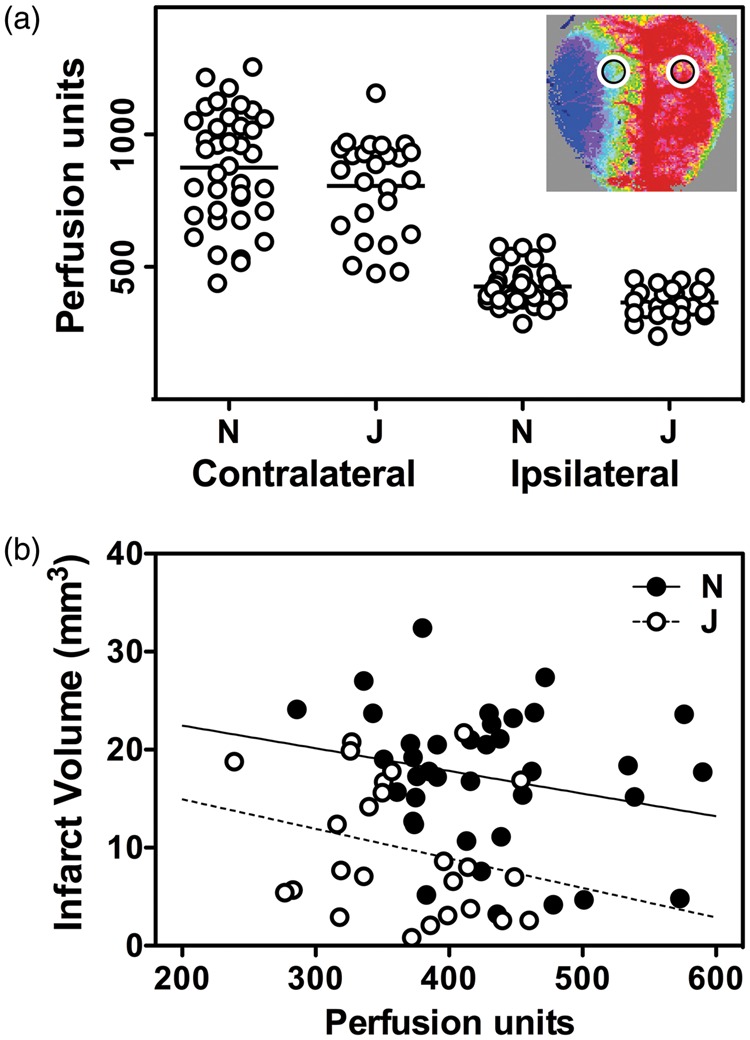
Substrain differences in infarct size are independent of acute perfusion deficits. (a) Speckle contrast imaging. Mean contralateral perfusion did not differ significantly in mice with large (N) and small (J) stroke phenotypes. J/ByJ/JEiJ mice had lower penumbral perfusion than NJ/NCrl mice, but as a percentage of contralateral flow, this did not achieve statistical significance. Inset shows a representative image and identifies regions of interest sampled for the measurements. (b) Penumbral perfusion and stroke outcome. Infarct volume was inversely related to the residual blood flow within a strain pool, but J/ByJ/JEiJ mice tended to exhibit smaller infarcts than NJ/NCrl mice for a given flow value. Lines indicate linear regression for the respective groups. Animal numbers (male, female) were as follows in the J pool: C57BL/6 J (4, 0), ByJ (12, 7), JEiJ (2, 0); and in the N pool: NJ (9, 13), NCrl (10, 6).
Discussion
The main observation of this study is that C57BL/6 mouse substrains differ in their intrinsic vulnerability to experimental stroke with J, ByJ and JEiJ yielding systematically smaller infarcts than NCrl, NJ and NTac mice. Substrain is therefore a critical variable in stroke studies using C57BL/6 mice, potentially confounding any comparison of wild type and genetically modified animals unless background homogeneity has been achieved by appropriate backcrossing strategies. A second finding is that ByJ and J females exhibit a higher proportion of small infarcts, resulting in sex differences that are not seen in NCrl and NJ substrains.
Substrain genetic variation
The N-lineage ByJ substrain that grossly retains the J phenotype with respect to stroke was derived from the NIH colony in 1961, some 10 years after the initial transfer of mice from JAX. This would imply that the responsible mutation(s) shared by subsequently derived N substrains occurred after this time (Figure 3). A potential caveat is that ByJ uniquely exhibits more robust communication between carotid and vertebral circulations (Figure 4), suggesting an additional mutation event, although because of a surprising sex-dependent difference in laterality this appears primarily relevant to females in the model of right MCA occlusion used in the current study. To our knowledge, a single commercially available N line, C57BL/6CrSlc (Japan SLC) originated from a separation that occurred a few years prior to that of NCrl in 1974,31 so additional results for this substrain could further refine the time line. In any case, all subsequently derived N substrains exhibit comparable stroke vulnerability, as confirmed here for NCrl, NJ, and NTac (Figure 3). Since N-substrain mice provide the embryonic stem cells for the IKMC,15 all knockout strains produced via this pipeline and maintained on the same background would be biased toward larger infarcts. Further confounds could be introduced by background mixing in the course of breeding to produce conditional strains.16
C57BL/6 genomes have been characterized in some detail and the extent of their divergence is consistent with the order and timing of substrain derivation.31,32 The pattern of stroke vulnerability can be distinguished from several well-known variations among B6 substrains. For example, the deletion in nicotinamide nucleotide transferase (Nnt) occurred comparatively recently in the J lineage, and is not present in the JEiJ or ByJ substrains,33 although these share the smaller infarct phenotype. This argues against the recent speculation that Nnt deletion contributes to the comparable substrain differences reported in a perinatal hypoxia-ischemia model.23 Conversely, the rd8 retinal degeneration mutation common to N lineage substrains was stated to also be present in ByJ.21 A comprehensive comparison of C57BL/6 J and C57BL/6 N genomes validated more than 100 coding and non-coding variants,11 and a recent analysis of likely inactivating mutations identified 76 impacted genes.34 However, most gene variants between J and N lineage substrains that underlie recognized differences in metabolism and immune function,11 potentially relevant to stroke, have yet to be identified.
Since ByJ is most closely related to the N lineage its comparison with other N substrains should reveal a more limited number of candidate loci. A screen of copy number variations among mouse substrains identified only one that matches the pattern of stroke vulnerability.35 This is a deletion impacting the inner mitochondrial membrane peptidase 2-like (Immp2l) gene in N-lineage substrains other than ByJ, as confirmed in the later comparison of N and J genome sequences.11 This gene was also the target of a random mutagenesis strategy in FVB mice using a screen for impaired fertility,36 and the resulting mice exhibited mitochondrial hyperpolarization and increased superoxide production. Several studies implicate attenuated nitric oxide vasodilatory signaling as an end result of the pathophysiological cascade,36,37 along with other manifestations of increased oxidative stress.38,39 Mice heterozygous for this mutation exhibit worse outcome after experimental stroke,40 and in vitro studies show increased superoxide production and cell death when Immp2l is knocked down prior to transient hypoxia.41 Although Immp2l represents a plausible candidate, it should be noted that the mutant and the spontaneous substrain variant represent different deletions, and different phenotypes result (e.g. N substrains of C57BL/6 have no recognized fertility issues).
One other known mutation with an identical pattern of substrain variation is an inactivating amino acid substitution in Cyfip2 of N-lineage mice, associated with attenuated responses to cocaine and compulsive food consumption.10,14 Cyfip2 is expressed in brain, especially in cortex, hippocampus, and striatum.42 Complete deletion of Cyfip2 function results in perinatal or early postnatal lethality.10,42 Heterozygous mice that express one functional copy of the gene have a ∼50% reduction in protein levels but are viable, with no overt brain structural abnormalities and mild behavioral changes.42 Cyfip2 variation has not previously been linked to stroke phenotypes, although it has been suggested to have a potential involvement in p53-mediated apoptosis.43 One potentially relevant observation in humans may be a two-locus interaction between variants in this gene and phosphodiesterase 2 A, associated with increased susceptibility to automimmune vasculitis (Kawasaki Disease).44 With roles in regulation of both actin polymerization45 and protein synthesis,46,47 CYFIP2 has been proposed to link signal- or activity-dependent remodeling of the cytoskeleton with translation,47,48 with broad potential relevance to brain pathophysiology.
Vascular anatomy and perfusion
PcomA anatomy has received considerable attention as a variable impacting stroke outcome, largely in the context of intraluminal filament occlusions that can obstruct the posterior cerebral artery (PCA). In such models, limited PcomA input from the basilar artery circulation to the PCA is responsible for the extension of flow deficits beyond MCA territory, with associated increases in infarct size.49–52 Smaller filament designs that avoid impacting the PCA have been reported to eliminate PcomA anatomy as a source of variability after intraluminal occlusions,29 as modeled in J-lineage mice from Shanghai Laboratory Animal Center (mouse source identified by Yongting Wang, personal communication). This variable was considered not to impact outcome after direct surgical occlusion of the distal MCA in C57BL/6 mice, whether obtained from JAX,4 or Charles River Japan.53 Reduced PcomA patency was proposed as a factor contributing to greater vulnerability of Balb/c mice in a surgical MCA occlusion model that also included tandem CCA occlusion,1 although much of this strain sensitivity has since been attributed to poor pial collaterals.6
The present results are largely consistent with the low PcomA patency reported for C57BL/6 mice from diverse sources of both the J lineage,4,49,54,55 and N substrains.56 Large vessel variation by itself would appear not to account for the overall difference in stroke outcome since the distributions of patency scores were essentially identical. The striking exception was ByJ, which showed a marked sex difference in laterality, such that the right PcomA was more frequently robust in females (Figure 4). This correlated with the sex-dependence of infarct size distributions in ByJ mice in this model of right MCA/CCA occlusion (Figure 2), which might seem to suggest that the model is sensitive to circle of Willis anatomy. However, if this were the case it should contribute more generally to the heterogeneity in infarct volume within a substrain. The wide distribution of infarct sizes in J males might be consistent with that possibility, but ByJ males had predominantly larger infarcts despite comparable vascular heterogeneity. Conversely, more J females had small infarcts despite a range of PcomA scores identical to those of J males. Another recent study suggested a trend toward greater PcomA patency in J females relative to males based on the incidence of their bilateral presence,57 but also concluded that this did not correlate with infarct size after distal MCA occlusion. Together these results indicate that most of the observed substrain and sex differences in stroke vulnerability are unlikely to be related to vascular anatomy. A limitation of the current study is that stroke outcome and PcomA patency were not assessed in the same animals. Also, a scoring system applied to a single vessel would not be expected to capture all relevant information regarding the efficacy of blood delivery via the circle of Willis. Future studies examining the impact of occlusion side on infarct size may be able to exploit the curious asymmetry in ByJ males to better define the interaction between PcomA anatomy and stroke outcome in this model.
Distal MCA anatomy has also received considerable attention in the context of experimental stroke. Early strain comparisons noted marked variation in the extent of the MCA distribution among mouse strains, with a smaller perfused territory as would be expected associated with smaller infarcts.3 More recent work has established that the collateral vasculature is a major determinant of variations in infarct size among mouse strains.6,58 However, the current results indicate that the position and number of anastomoses between MCA and other vascular territories do not differ between less vulnerable ByJ and more vulnerable NCrl substrains (Figure 5). These should be considered preliminary observations. Vasodilators were not administered so the numbers of anastomoses are likely underestimated. Perhaps more importantly, temporal changes in response to occlusion have yet to be investigated.
Perfusion imaging further indicates that blood flow is not the primary factor determining substrain differences in stroke vulnerability, in that the acute CBF deficit is comparable in J and N substrains (Figure 6). It must be noted that such measurements in anesthetized animals at a single early time point do not provide information about the time course of CBF change and infarct progression over the hours during which final infarct distribution is determined.58 Also, we detected no evidence in any animal for the expected perfusion responses that accompany peri-infarct depolarizations,59 suggesting that these may have been suppressed by anesthesia either directly60,61 or via indirect physiological effects such as hyperglycemia,62,63 which was not assessed. Comprehensive comparisons of the time course of perfusion changes and infarct progression across substrains would provide more insight into the mechanisms underlying substrain differences. Nevertheless, the current results appear to rule out gross differences in either vascular anatomy or CBF.
Substrain as a confound in experimental stroke
Unrecognized substrain variation would be expected to account for some effects previously attributed to targeted genetic manipulations. The literature was searched to identify mice used in stroke studies that also were listed among commercially available knockout strains found to be on an N or mixed N/J background based on the presence of wild-type Nnt alleles,17 but for which C57BL/6 J appear to have been used as controls. The intrinsic difference in substrain vulnerability is sufficient to account for larger infarcts reported in knockout strains targeting interleukin-1064 and the chemokine receptor CXCR5.65 It would also be consistent with one report of larger infarcts in toll-like receptor 2 (Tlr2) knockouts.66 Although a seemingly comparable study using the same strains found the converse,67 subtle modeling differences could account for the discrepancy since a third report noted a marked temporal progression from smaller to larger infarcts in the Tlr2 knockout.68 Conversely, beneficial gene deletions potentially could have been missed with such mismatches, although we identified no published examples of this type. Some effects that might have been impacted were evidently sufficiently robust to be insensitive to strain background. This appears to be the case for knockout of the chemokine receptor CX3CR1.69 Similarly, although not on the above list, α-2-antiplasmin knockout mice produced under the Knockout Mouse Project are resistant to thromboembolic stroke relative to C57BL/6 J.70 Consideration of substrain would therefore only add to the strength of these prior results. We have also recently observed that heterozygote knockouts of phosphatase and tensin homologue (Pten), on a mixed N/J background,17 exhibit the large-infarct N phenotype in the current stroke model (L. Zhao, F. Liao, T. S. Nowak, Jr., unpublished observation).
Excluded from the above assessments were many studies lacking control substrain information, or involving strains that had been maintained by laboratory breeding programs, whether obtained commercially or from other investigators. The backgrounds of such colonies may differ from the original sources. Ideally, this would be by intent, in the course of the backcrossing necessary to produce strain pairs on consistent substrain backgrounds. However, given the large number of genetically engineered lines maintained on the J background, their likely use in crosses with knockout mouse strains produced on N backgrounds,16 and the comparatively recent appreciation of the critical needs to document substrain background, the potential for strain mismatch is considerable. This has been appreciated previously in the context of myocardial infarction.71 Screens for C57BL/6 substrain background are now available through vendors, and such verification should be required reporting for all laboratory-maintained strains going forward.
Finally, the absence of sex-dependent variation in infarct volume for N substrain mice has specific implications for their use in elucidating molecular mechanisms underlying such effects. Although sex differences can be model dependent (e.g. more evident after transient vs. permanent occlusions72), the present results suggest that N-background knockout strains should by default exhibit attenuated differences between males and females, potentially confounding any impact of intended genetic manipulations.
Technical and statistical considerations
Permanent MCA/CCA occlusion produces essentially the maximum ischemic insult that can be selectively delivered to cortical MCA territory. The intrinsic difference in stroke vulnerability between N and J substrain lineages observed here would be generally relevant to other modeling approaches to the extent that they impact the same region. The negligible infarcts often reported in C57BL/6 J mice after distal MCA occlusion alone5 should be detectably larger in N substrain mice, and preliminary results are consistent with this expectation (not shown). This should also be the case after transient occlusions using comparable surgical methods, although the smaller infarcts and greater variability expected might require large groups to discriminate. It is difficult to predict the outcome after short intervals of intraluminal filament occlusion in which a striatal injury component often predominates, and to which selective vulnerability of the abundant medium spiny neurons can sometimes contribute injury mechanisms distinct from infarction.73,74 On the other hand, the cortical component of infarcts produced following the longer intervals of filament occlusion that are typically used, often involving vascular territories beyond that of the MCA,51 should be sensitive to the same intrinsic differences in insult threshold identified in the current study. As noted above, some examples of knockout strain comparisons using filament models are consistent with this interpretation.64,66
Substrain differences in mean infarct size are detectable with comparatively small numbers of animals (Figure 3(a)), but larger samples document the considerable heterogeneity in outcome that can be expected when only limited exclusions are allowed (Figure 2). Few previous studies have involved the combination of a highly reproducible occlusion method with experimental groups large enough to adequately document lesion size distributions. Infarct volumes for J and ByJ substrains clearly are not normally distributed, requiring the use of non-parametric tests in all comparisons (Figure 1). Unrecognized postoperative cooling and other procedural variables could contribute to occasional small infarcts, but their systematically higher incidence in J and ByJ mice establishes that this is a property intrinsic to these populations. PcomA anatomy (Figure 4) could contribute to sex difference in the case of ByJ mice, but does not appear to be a primary variable underlying either between-sex or within-group differences for other substrains. The J lineage emerges as an impractical choice for conventional stroke studies using such surgical MCA occlusion methods. Infarcts are intrinsically small after distal occlusions alone,5 and methods that increase insult severity, such as filament occlusions or the tandem method applied here, show considerable variability. On the other hand, in addition to their required use as controls for many knockout strains, C57BL/6 substrains of the N lineage offer the advantage of both larger and more homogeneous infarcts. For a typical treatment study in NCrl mice using the current model, group sizes required to detect a 30% decrease in infarct volume with α = 0.05 and 1−β = 0.9 would be <20 mice. Corresponding numbers for the J substrain would be >30 males and >80 females.
Conclusions
Previously unrecognized substrain differences in stroke vulnerability have introduced potential confounds in prior studies involving genetically modified C57BL/6 mice. Clearly, not all results are compromised but the frequent lack of information about substrain background, particularly about the mice used as controls in such studies, will require a case-by-case reassessment of many reports. Looking ahead, the larger and more uniform infarcts of N substrain mice, and their established use as the common background for so many genetically modified strains, would seem to make these the preferred choice for many future studies in experimental stroke. This must be weighed against the presence of at least one recognized mutation that could be directly relevant to stroke pathophysiology. The absence of a sex difference, at least after permanent occlusions, may limit their direct utilization in sex comparison studies, but understanding the basis of this substrain-dependent variation should be informative. Whether the observed substrain differences persist in aged mice also remains to be determined. Resolution of these practical questions will provide insights into fundamental mechanisms contributing to stroke vulnerability.
Supplementary Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Ganey Fund, Department of Neurology, University of Tennessee Health Science Center; by the Center for Integrative and Translational Genomics, University of Tennessee Health Science Center; and by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award R21NS066166 (TSN). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article
Authors’ contributions
LZ performed stroke surgery, measured infarct volumes and carried out perfusion imaging studies; MKM made substrain recommendations, provided mouse colonies and contributed to study design and manuscript preparation; TSN performed estrous cycle evaluations, carried out vascular filling studies, prepared final figures and wrote the manuscript.
Supplementary material
Supplementary material for this paper can be found at the journal website: http://journals.sagepub.com/home/jcb
References
- 1.Barone FC, Knudsen DJ, Nelson AH, et al. Mouse strain differences in susceptibility to cerebral ischemia are related to vascular anatomy. J Cereb Blood Flow Metab 1993; 13: 683–692. [DOI] [PubMed] [Google Scholar]
- 2.Fujii M, Hara H, Meng W, et al. Strain-related differences in susceptibility to transient forebrain ischemia in SV-129 and C57Black/6 mice. Stroke 1997; 28: 1805–1811. [DOI] [PubMed] [Google Scholar]
- 3.Maeda K, Hata R, Hossmann K-A. Differences in the cerebrovascular anatomy of C57black/6 and SV129 mice. Neuroreport 1998; 9: 1317–1319. [DOI] [PubMed] [Google Scholar]
- 4.Majid A, He YY, Gidday JM, et al. Differences in vulnerability to permanent focal cerebral ischemia among 3 common mouse strains. Stroke 2000; 31: 2707–2714. [DOI] [PubMed] [Google Scholar]
- 5.Keum S, Marchuk DA. A locus mapping to mouse chromosome 7 determines infarct volume in a mouse model of ischemic stroke. Circ Cardiovasc Genet 2009; 2: 591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang H, Prabhakar P, Sealock R, et al. Wide genetic variation in the native pial collateral circulation is a major determinant of variation in severity of stroke. J Cereb Blood Flow Metab 2010; 30: 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bryant CD, Zhang NN, Sokoloff G, et al. Behavioral differences among C57BL/6 substrains: implications for transgenic and knockout studies. J Neurogenet 2008; 22: 315–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mulligan MK, Ponomarev I, Boehm SL, 2nd, et al. Alcohol trait and transcriptional genomic analysis of C57BL/6 substrains. Genes Brain Behav 2008; 7: 677–689. [DOI] [PubMed] [Google Scholar]
- 9.Matsuo N, Takao K, Nakanishi K, et al. Behavioral profiles of three C57BL/6 substrains. Front Behav Neurosci 2010; 4: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar V, Kim K, Joseph C, et al. C57BL/6N mutation in cytoplasmic FMRP interacting protein 2 regulates cocaine response. Science 2013; 342: 1508–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon MM, Greenaway S, White JK, et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol 2013; 14: R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sturm M, Becker A, Schroeder A, et al. Effect of chronic corticosterone application on depression-like behavior in C57BL/6N and C57BL/6J mice. Genes Brain Behav 2015; 14: 292–300. [DOI] [PubMed] [Google Scholar]
- 13.Ulland TK, Jain N, Hornick EE, et al. Nlrp12 mutation causes C57BL/6J strain-specific defect in neutrophil recruitment. Nature Commun 2016; 7: 13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirkpatrick SL, Goldberg LR, Yazdani N, et al. Cytoplasmic FMR1-interacting protein 2 is a major genetic factor underlying binge eating. Biol Psychiatry 2017; 81: 757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pettitt SJ, Liang Q, Rairdan XY, et al. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nat Meth 2009; 6: 493–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fontaine DA, Davis DB. Attention to background strain is essential for metabolic research: C57BL/6 and the International Knockout Mouse Consortium. Diabetes 2016; 65: 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bourdi M, Davies JS, Pohl LR. Mispairing C57BL/6 substrains of genetically engineered mice and wild-type controls can lead to confounding results as it did in studies of JNK2 in acetaminophen and concanavalin A liver injury. Chem Res Toxicol 2011; 24: 794–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Specht C, Schoepfer R. Deletion of the alpha-synuclein locus in a subpopulation of C57BL/6J inbred mice. BMC Neurosci 2001; 2: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freeman HC, Hugill A, Dear NT, et al. Deletion of nicotinamide nucleotide transhydrogenase. A new quantitative trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes 2006; 55: 2153–2156. [DOI] [PubMed] [Google Scholar]
- 20.Mahajan VS, Demissie E, Mattoo H, et al. Striking immune phenotypes in gene-targeted mice are driven by a copy-number variant originating from a commercially available C57BL/6 strain. Cell Rep 2016; 15: 1901–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mattapallil MJ, Wawrousek EF, Chan C-C, et al. The rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci 2012; 53: 2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keane TM, Goodstadt L, Danacek P, et al. Mouse genome variation and its effect on phenotypes and gene regulation. Nature 2011; 477: 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolf S, Hainz N, Beckmann A, et al. Brain damage resulting from postnatal hypoxic-ischemic brain injury is reduced in C57BL/6J mice as compared to C57BL/6N mice. Brain Res 2016; 1650: 224–231. [DOI] [PubMed] [Google Scholar]
- 24.Zhao L, Mulligan MK, Nowak TS., Jr Substrain and sex differences in infarct size after permanent focal ischemia in C57BL/6 mice. J Cereb Blood Flow Metab 2017; 37: 284–285. [Google Scholar]
- 25.Brint S, Jacewicz M, Kiessling M, et al. Focal brain ischemia in the rat: methods for reproducible neocortical infarction using tandem occlusion of the distal middle cerebral and ipsilateral common carotid arteries. J Cereb Blood Flow Metab 1988; 8: 474–485. [DOI] [PubMed] [Google Scholar]
- 26.Takeda Y, Zhao L, Jacewicz M, et al. Metabolic and perfusion responses to peri-infarct depolarization during focal ischemia in the Spontaneously Hypertensive Rat: dominant contribution of sporadic CBF decrements to infarct expansion. J Cereb Blood Flow Metab 2011; 31: 1863–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jay TM, Lucignani G, Crane AM, et al. Measurement of local cerebral blood flow with [14C]iodoantipyrine in the mouse. J Cereb Blood Flow Metab 1988; 8: 121–129. [DOI] [PubMed] [Google Scholar]
- 28.Murakami K, Kondo T, Kawase M, et al. The development of a new mouse model of global ischemia: focus on the relationships between ischemia duration, anesthesia, cerebral vasculature, and neuronal injury following global ischemia in mice. Brain Res 1998; 780: 304–310. [DOI] [PubMed] [Google Scholar]
- 29.Yuan F, Tang Y, Lin X, et al. Optimizing suture middle cerebral artery occlusion model in C57BL/6 mice circumvents posterior communicating artery dysplasia. J Neurotrauma 2012; 29: 1499–1505. [DOI] [PubMed] [Google Scholar]
- 30.Byers SL, Wiles MV, Dunn SL, et al. Mouse estrous cycle identification tool and images. PloS One 2012; 7: e35538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mekada K, Abe K, Murakami A, et al. Genetic differences among C57BL/6 substrains. Exp Anim 2009; 58: 141–149. [DOI] [PubMed] [Google Scholar]
- 32.Zurita E, Chagoyen M, Cantero M, et al. Genetic polymorphisms among C57BL/6 mouse inbred strains. Transgenic Res 2011; 20: 481–489. [DOI] [PubMed] [Google Scholar]
- 33.Huang TT, Naeemuddin M, Elchuri S, et al. Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Hum Mol Genet 2006; 15: 1187–1194. [DOI] [PubMed] [Google Scholar]
- 34.Timmermans S, Van Montagu M, Libert C. Complete overview of protein-inactivating sequence variations in 36 sequenced mouse inbred strains. Proc Natl Acad Sci U S A 2017; 114: 9158–9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Egan CM, Sridhar S, Wigler M, et al. Recurrent DNA copy number variation in the laboratory mouse. Nat Genet 2007; 39: 1384–1389. [DOI] [PubMed] [Google Scholar]
- 36.Lu B, Poirier C, Gaspar T, et al. A mutation in the inner mitochondrial membrane peptidase 2-like gene (Immp2l) affects mitochondrial function and impairs fertility in mice. Biol Reprod 2008; 78: 601–610. [DOI] [PubMed] [Google Scholar]
- 37.Soler R, Füllhase C, Lu B, et al. Bladder dysfunction in a new mutant mouse model with increased superoxide – lack of nitric oxide? J Urol 2010; 183: 780–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.George SK, Jiao Y, Bishop CE, et al. Mitochondrial peptidase IMMP2L mutation causes early onset of age-associated disorders and impairs adult stem cell self-renewal. Aging Cell 2011; 10: 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu C, Li X, Lu B. The Immp2l mutation causes age-dependent degeneration of cerebellar granule cell neurons prevented by antioxidant treatment. Aging Cell 2016; 15: 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma Y, Mehta SL, Lu B, et al. Deficiency in the inner mitochondrial membrane peptidase 2-like (Immp2l) gene increases ischemic brain damage and impairs mitochondrial function. Neurobiol Dis 2011; 44: 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma Y, Zhang Z, Chen Z, et al. Suppression of inner mitochondrial membrane peptidase 2-like (IMMP2L) gene exacerbates hypoxia-induced neural death under high glucose condition. Neurochem Res 2017; 42: 1504–1514. [DOI] [PubMed] [Google Scholar]
- 42.Han K, Chen H, Gennarino VA, et al. Fragile X-like behaviors and abnormal cortical dendritic spines in Cytoplasmic FMR1-interacting protein 2-mutant mice. Hum Mol Genet 2015; 24: 1813–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saller E, Tom E, Brunori M, et al. Increased apoptosis induction by 121F mutant p53. EMBO J 1999; 18: 4424–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuo H-C, Chang J-C, Guo MM-H, et al. Gene-gene associations with the susceptibility of Kawasaki disease and coronary artery lesions. PloS One 2015; 10: e0143056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eden S, Rohatgi R, Podtelejnikov AV, et al. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 2002; 418: 790–793. [DOI] [PubMed] [Google Scholar]
- 46.Schenck A, Bardoni B, Moro A, et al. A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc Natl Acad Sci U S A 2001; 98: 8844–8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abekhoukh S, Bardoni B. CYFIP family proteins between autism and intellectual disability: links with Fragile X syndrome. Front Cell Neurosci 2014; 8: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schenck A, Bardoni B, Langmann C, et al. CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links Rac1 GTPase pathway to the fragile X protein. Neuron 2003; 38: 887–898. [DOI] [PubMed] [Google Scholar]
- 49.McColl BW, Carswell HV, McCulloch J, et al. Extension of cerebral hypoperfusion and ischaemic pathology beyond MCA territory after intraluminal filament occlusion in C57Bl/6J mice. Brain Res 2004; 997: 14–22. [DOI] [PubMed] [Google Scholar]
- 50.Kitagawa K, Matsumoto M, Yang G, et al. Cerebral ischemia after bilateral carotid artery occlusion and intraluminal suture occlusion in mice: evaluation of the patency of the posterior communicating artery. J Cereb Blood Flow Metab 1998; 18: 570–579. [DOI] [PubMed] [Google Scholar]
- 51.Özdemir YG, Bolay H, Erdem E, et al. Occlusion of the MCA by an intraluminal filament may cause disturbances in the hippocampal blood flow due to anomalies of circle of Willis and filament thickness. Brain Res 1999; 822: 260–264. [DOI] [PubMed] [Google Scholar]
- 52.Leithner C, Füchtemeier M, Jorks D, et al. Infarct volume prediction by early magnetic resonance imaging in a murine stroke model depends on ischemia duration and time of imaging. Stroke 2015; 46: 3249–3259. [DOI] [PubMed] [Google Scholar]
- 53.Furuya K, Kawahara N, Kawai K, et al. Proximal occlusion of the middle cerebral artery in C57Black6 mice: relationship of patency of the posterior communicating artery, infarct evolution, and animal survival. J Neurosurg 2004; 100: 97–105. [DOI] [PubMed] [Google Scholar]
- 54.Beckmann N. High resolution magnetic resonance angiography non-invasively reveals mouse strain differences in the cerebrovascular anatomy in vivo. Magn Reson Med 2000; 44: 252–258. [DOI] [PubMed] [Google Scholar]
- 55.Tamaki M, Kidoguchi K, Mizobe T, et al. Carotid artery occlusion and collateral circulation in C57Black/6J mice detected by synchrotron radiation microangiography. Kobe J Med Sci 2006; 52: 111–118. [PubMed] [Google Scholar]
- 56.Yang G, Kitagawa K, Matsushita K, et al. C57BL/6 strain is most susceptible to cerebral ischemia following bilateral common carotid artery occlusion among seven mouse strains: selective neuronal death in the murine transient forebrain ischemia. Brain Res 1997; 752: 209–218. [DOI] [PubMed] [Google Scholar]
- 57.Faber JE, Moore SM, Lucitti JL, et al. Sex differences in the collateral cerebral circulation. Transl Stroke Res 2017; 8: 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kao Y-CJ, Oyarzabal EA, Zhang H, et al. Role of genetic variation in collateral circulation in the evolution of acute stroke. A multimodal magnetic resonance imaging study. Stroke 2017; 48: 754–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shin HK, Dunn AK, Jones PB, et al. Vasoconstrictive neurovascular coupling during focal ischemic depolarizations. J Cereb Blood Flow Metab 2006; 26: 1018–1030. [DOI] [PubMed] [Google Scholar]
- 60.Saito R, Graf R, Hübel K, et al. Reduction of infarct volume by halothane: effect on cerebral blood flow or perifocal spreading depression-like depolarizations. J Cereb Blood Flow Metab 1997; 17: 857–864. [DOI] [PubMed] [Google Scholar]
- 61.Kudo K, Zhao L, Nowak TS., Jr Peri-infarct depolarizations during focal ischemia in the awake Spontaneously Hyertensive Rat. Minimizing anesthesaia confounds in experimental stroke. Neuroscience 2016; 325: 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoffmann U, Sukhotinsky I, Eikermann-Haerter K, et al. Glucose modulation of spreading depression susceptibility. J Cereb Blood Flow Metab 2013; 33: 191–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao L, Nowak TS., Jr Preconditioning cortical lesions reduce the incidence of peri-infarct depolarizations during focal ischemia in the Spontaneously Hypertensive Rat: interaction with prior anesthesia and the impact of hyperglycemia. J Cereb Blood Flow Metab 2015; 35: 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grilli M, Barbieri I, Basudev H, et al. Interleukin-10 modulates neuronal threshold of vulnerability to ischaemic damage. Eur J Neurosci 2000; 12: 2265–2272. [DOI] [PubMed] [Google Scholar]
- 65.Chapman KZ, Ge R, Monni E, et al. Inflammation without neuronal death triggers striatal neurogenesis comparable to stroke. Neurobiol Dis 2015; 83: 1–15. [DOI] [PubMed] [Google Scholar]
- 66.Hua F, Ma J, Ha T, et al. Differential roles of TLR2 and TLR4 in acute focal cerebral ischemia/reperfusion injury in mice. Brain Res 2009; 1262: 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tang S-J, Arumugam TV, Xu X, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A 2007; 104: 13798–13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bohacek I, Cordeau P, Lalancette-Hébert M, et al. Toll-like receptor 2 deficiency leads to delayed exacerbation of ischemic injury. J Neuroinflammation 2012; 9: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jolivel V, Bicker F, Binamé F, et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol 2015; 129: 279–295. [DOI] [PubMed] [Google Scholar]
- 70.Reed GL, Houng AK, Wang D. Microvascular thrombosis, fibrinolysis, ischemic injury, and death after cerebral thromboembolism are affected by levels of circulating α2-antiplasmin. Arterioscler Thromb Vasc Biol 2014; 34: 2586–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gorog DA, Tanno M, Kabir AMN, et al. Varying susceptibility to myocardial infarction among C57BL/6 mice of different genetic background. J Mol Cell Cardiol 2003; 35: 705–708. [DOI] [PubMed] [Google Scholar]
- 72.Brait V, Jackman KA, Walduck AK, et al. Mechanisms contributing to cerebral infarct size after stroke: gender, reperfusion, T lymphocytes, and Nox2-derived superoxide. J Cereb Blood Flow Metab 2010; 30: 1306–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Korematsu K, Goto S, Nagahiro S, et al. Changes of immunoreactivity for synaptophysin (‘protein p38') following a transient cerebral ischemia in the rat striatum. Brain Res 1993; 616: 320–324. [DOI] [PubMed] [Google Scholar]
- 74.Katchanov J, Waeber C, Gertz K, et al. Selective neuronal vulnerability following mild focal brain ischemia in the mouse. Brain Pathol 2003; 13: 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.