

Abstract
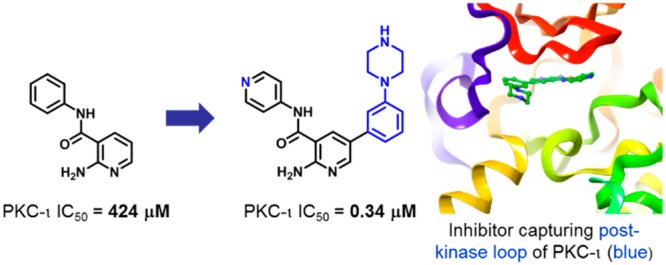
The atypical protein kinase C-iota (PKC-ι) enzyme is implicated in various cancers and has been put forward as an attractive target for developing anticancer therapy. A high concentration biochemical screen identified pyridine fragment weakly inhibiting PKC-ι with IC50 = 424 μM. Driven by structure–activity relationships and guided by docking hypothesis, the weakly bound fragment was eventually optimized into a potent inhibitor of PKC-ι (IC50= 270 nM). Through the course of the optimization, an intermediate compound was crystallized with the protein, and careful analysis of the X-ray crystal structure revealed a unique binding mode involving the post-kinase domain (C-terminal tail) of PKC-ι.
Keywords: Protein kinase C iota, fragment-based drug discovery, kinase inhibitor, C-terminal tail, hepatocellular carcinoma, nonsmall cell lung cancer
Protein kinase C (PKC) family consists of 11 isoenzymes that can be grouped into three subfamily types: classical, novel, and atypical PKCs. The interest in PKC enzymes as targets for therapeutic intervention stems from the fact that PKCs are important for critical cellular processes implicated in the promotion of tumor growth.1 The levels of most of the PKC isoenzymes are altered in cancer cells, and several PKCs are involved in regulation of apoptosis.2 However, the complex network of processes dependent on PKCs is the cause of controversy whether PKC enzymes are viable drug targets, and consequently, out of the 11 isoforms, only PKC-epsilon and PKC-iota (PKC-ι) are described unambiguously as bona fide oncogenes.3−9 Accordingly, isoform-selective, high quality PKC inhibitors are needed to elucidate the complex biology and explore the therapeutic potential of PKC modulation.
We were particularly interested in targeting atypical PKC-ι, due to its central role in the formation of an oncogenic complex with epithelial cell transforming sequence 2 (ECT2) and partitioning defective 6 homologue (Par6). The assembly of the three enzymes triggers the oncogenic Rac-Pak-Mek-Erk signaling cascade, which ultimately leads to tumor growth and invasion, as demonstrated in nonsmall cell lung cancer (NSCLC) and hepatocellular carcinoma (HCC).10,11 Correspondingly, high levels of ECT2 were correlated with poor prognosis for patients suffering from NSCLC and HCC. More importantly, upregulation of PKC-ι, has been directly linked to poor cancer-specific survival in NSCLC. In total, inhibition of PKC-ι would be an attractive approach for therapeutic intervention in HCC, through modulation of ECT2 phosphorylation. To date, several atypical PKC inhibitors have been reported;12−15 herein, we describe the optimization of a fragment hit against PKC-ι.
Results
A fragment screen15 identified the pyridine-amide fragment (compound 1, Table 1, IC50 = 424 μM, LE = 0.29)16 as a hit against PKC-ι. In the absence of an X-ray structure, we hypothesized the aminopyridine moiety in 1 to serve as a hinge binder and begun a systematic structure–activity relationship (SAR)-driven exploration of the binding site by examining substitutions of the phenylamine (synthesis described in Supporting Information). Interestingly, replacement of the phenyl with pyridine resulted in a 20-fold increase in potency (compound 2, IC50 = 22 μM), leading to a rapid improvement in the overall efficiency of the molecule (LE = 0.40, LLE = 4.2). This simple substitution indicated the possibility of a hydrogen bond (H-bond) interaction between the pyridine nitrogen and PKC-ι. Next, a close analogue of compound 2 containing 2-aminopyrazine hinge binding motif in place of the 2-aminopyridine was found to be equally active (compound 3). Based on the aminopyrazine scaffold, it was found that repositioning of the pyridine nitrogen (4) or replacing pyridine with phenol (5) did not result in a significant change in inhibitory activity as compared to compound 3. However, compound 6 containing the more flexible pyridine-methylene amine was inactive. Interestingly, replacement of the pyridine with a large bicyclic indole was found to be well tolerated in this position, while 7-azaindole showed no inhibitory activity (compounds 7 and 8, respectively). Taken together, the equipotency of phenol, indole (both H-bond donors), and pyridine (H-bond acceptor) could suggest water-mediated interaction, where water could interact with either donor or acceptor.
Table 1. Fragment Hit 1 and Modifications of the Amine Moiety of the Amidea.

The two close analogues 2 and 3 showed improved potency and ligand efficiency. Subsequent modifications of compound 3 indicated little potential for improvement in IC50 by modifications of the peripheral pyridine.
Driven by the ability of the pocket to accommodate larger indole substituents, we next sought to explore potential hydrophobic interactions through substitutions at the 2-position of the peripheral pyridine/phenyl. As shown in Table 1, none of the larger compounds containing ethyl (9), tert-butyl (10), or cyclopropyl (11) substituents brought any significant improvement in potency. however, compounds bearing 2,6-dimethylpyridineamine (12) or bromopyridine amines (13 and 14) were inactive. Overall, the initial SAR suggested the peripheral pyridine/phenyl engaged in favorable interactions with protein; however, due to apparent limited space and possible presence of water molecule, elaboration of the pyridine/phenyl did not transpire as an effective optimization strategy at this point.
Seeking further improvement, we next turned to molecular modeling to guide subsequent evolution of compound 2. Specifically, 2 was docked into published PKC-ι X-ray structure with the assumption that the 2-aminopyridine is engaged in hydrogen-bonding interactions with PKC-ι hinge residues Glu333 and Val335. Furthermore, as guided by the SAR in Table 1, the peripheral pyridine was oriented with the nitrogen pointing toward the wall of the binding site consisting of Ile332 and Lys283 (Figure 1).17 This docking conformation supported the formation of hydrogen-bonding interactions between the peripheral pyridine nitrogen and a flexible lysine 283 as well as between the amide carbonyl and Thr395. As shown in Figure 1b, expansion from the hinge-binding 2-amino-pyridine core could increase binding energy through gaining hydrophobic interactions with isoleucine (Ile260) and a hydrogen-bond with aspartic acid (Asp339). Notably, the model also suggested phenylalanine Phe552 as a possible partner for π–π interactions with expanded ligands. It is noteworthy that since Phe552 is located in a flexible post-kinase loop (C-terminal tail), fulfilling this interaction might not be straightforward. While Phe552 was shown to reside in the ATP site in both the apo and ATP-bound structures of PKC-ι (PDB: 3A8X and 3A8W), the residue is displaced by small molecule inhibitors, as shown in X-ray structures of PKC-ι-inhibitor complexes (e.g., PDB: 3ZH8 and 1ZRZ). Additionally, a varying degree of structural rearrangement of the post-kinase domain dependent on the geometry of a ligand would likely affect the clarity of SAR.
Figure 1.

Compound 2 (in green) modeled into X-ray structure of PKC-ι. (a) Hydrogen bonds between 2 and protein residues are shown as yellow dashed lines; (b) a grid is superimposed on the binding site showing areas favorable for binding ligand atoms with the following properties: hydrophobic (yellow), acceptor or negatively charged (red), and donor or positively charged (blue).
To enable the envisioned interactions in the inhibitor series, substitutions on the hinge-binding pyridine were next attempted. First, a bromine atom was introduced at the 5-position of the 2-aminopyridine moiety of compound 2 to probe for space and as a handle for subsequent modifications (Table 2). Somewhat unexpectedly, the resulting brominated compound 15 was inactive. However, a phenyl ring installed at the same position was well tolerated (16). However, compared to compound 2, the addition of the phenyl ring did not bring about a significant improvement in activity, which is also reflected in the decrease in ligand efficiency of compound 16 (LE = 0.28). The discrepancy between the actual inhibitory results and the predicted improvement in potency from docking experiments could be caused by the energy penalty paid for reorganization of the post-kinase domain of PKC-ι, offset by interactions of the phenyl moiety in 16 with protein residues.
Table 2. Expansion from the Hinge Binding Corea.

The benzylamine improved potency by 45-fold, indicating a strong interaction with protein, hypothetically the aspartic acid (Asp339). This interaction seemed translatable phenylpiperazine-containing compounds, as well.
To further validate our docking model and capture additional enthalpic interactions through possible engagement of aspartic acid (Asp-339), a 3-methylamino group was introduced into the phenyl ring of compound 16. This strategy was successful, and a 45-fold increase in potency was observed (17, IC50 = 0.71 μM) as compared to compound 16. The critical role of the base was further confirmed by converting the benzylamine to an amide (18). As expected, compound 18 was inactive. Next, seeking a less basic alternative of benzylamine, a piperazine group was tested as a replacement of the methylamino-substituent. Gratifyingly, the resulting compound 19 was 2-fold more potent than its benzylamine-containing counterpart, with IC50 reaching 0.34 μM. The analogous compound containing 2-aminopyrazine as a hinge binder was equally potent (20, IC50 = 0.27 μM). At this stage, the three most potent compounds 17, 19, and 20 were also tested against three other PKC isoenzymes representative of PKC subfamilies: classical (PKC-α), novel (PKC-ε), and atypical (PKC-ζ SI Table S1). In general, compounds showed little selectivity within PKC family. However, SAR across the four isoenzymes indicated opportunity to enhance selectivity toward atypical or classical PKCs (see SI for details).
Next, the pharmacokinetic profile of compound 19 was assessed through a series of in vitro assays (Table 3). The results confirmed the high solubility of the inhibitor at pH 7.4. As expected, the permeability as measured in a Caco-2 assay was found to be low. Additionally, 19 was stable in a metabolic assay with human liver microsomes but underwent fast degradation mediated by mouse liver microsomes. To evaluate the translation of biochemical activity into antiproliferative effect against hepatocellular carcinoma cells, HUH-7 cells were treated with 19 and weak growth inhibition was observed (GI50 = 11.3 μM).
Table 3. Pharmacokinetic Properties and Cellular Activity of Compound 19a.
| IC50 [μM] | PKC-ι | 0.34 |
| GI50 [μM] | Huh-7 | 11.3 |
| in vitro PK | Sol (pH 7.4, μg/mL) | 164 |
| Caco-2 (A–B, 10–6 cm/s) | 0.28 | |
| MLM/HLM (CL, μL/min/mg) | 71/5.1 |
PKC-ι inhibitor CRT006685412 was used as a positive control; GI50 against HUH-7 cells was 3.1 μM.
X-ray Crystal Structure and Binding Mode of PKC-ι Inhibitors
Concurrently to improving the potency of the scaffold, we attempted to elucidate the binding mode of the inhibitors through X-ray crystallography. Our efforts were fruitful, and compound 19 was successfully cocrystallized in complex with PKC-ι (Figure 2, PDB: 6ILZ; SI). The analysis of the complex confirmed the key interactions between 19 and PKC-ι: hydrogen-bond between 2-aminopyridine and the hinge residues of the protein-Val335 and Glu333, and between the carbonyl oxygen of the amide and Thr395. The pyridine moiety of compound 19 was shown to engage in lipophilic interactions with Val268 and Thr395 and to form a large number of van der Waals contacts with Asp396. While not unambiguously shown in the X-ray, the arrangement of the protein residues around the peripheral pyridine of 19 suggested the existence of a water-mediated hydrogen bond between the pyridine nitrogen, Lys283, and Asp396. Molecular dynamics (MD) performed on the complex showed direct or water-mediated hydrogen bonds to Lys283 during 80% of the simulation time (see SI for details). Interestingly, analysis of the crystal structure revealed the phenyl-piperazine substituent of compound 19 positioned between Ile260 and the side chain of the post-kinase residue Phe552. Furthermore, a clear π–π interaction of the phenyl group of 19 and Phe552, as well as lipophilic attraction to Ile260 were detected. The observed unusual alignment of the phenyl moiety of compound 19 on top of Phe552 enabled additional van der Waals contacts with Tyr334. Furthermore, while not unequivocally shown in the X-ray structure of the complex, the piperazine is likely to form hydrogen bond with Asp553—another post-kinase domain residue immediately following Phe552—and not with the expected Asp339 (see SI for additional comments). In contrast, many kinase inhibitors have an aromatic ring in this region, which is sandwiched between the homologous residue of Ile260 and the protein backbone immediately following the kinase hinge region. Previously published PKC X-ray structures with inhibitors have the post-kinase domain residue Phe552 displaced from the ATP-site, and the inhibitors complexed in these structures bind in the canonical binding mode (e.g., PDB: 3ZH8 and 1ZRZ). This binding conformation could likely apply to compound 16, displacing the post-kinase loop and attracting Ile260, and explain no overall potency improvement. However, the addition of a second strong interaction (H-bonding to Asp553, compounds 17, 19, and 20) could have resulted in the capture and rigidification of the post-kinase loop instead of displacing it.
Figure 2.

X-ray crystal structure of compound 19 in complex with PKC-ι. (a) Hydrogen bonds are shown as yellow dashed lines. π–π stacking interaction of the phenyl group of the inhibitor with a post-kinase domain residue Phe552 is shown as a blue dashed line. (b) View of the binding site of PKC-ι in complex with 19 with surface applied. (c) Comparison of binding mode of compound 19 (I) with two small molecule inhibitors RBT205 (II) and CRT0066854 (III). Post-kinase loop (colored blue and indicated by an arrow) is shown to interact with compound 19 while being displaced in the other two instances (II, III; flexible loop only partially resolved).
In general, the binding mode derived from the X-ray crystal structure is in a good agreement with the computational model developed in the early fragment-to-hit-to-lead stage of the project. While the empirical SAR remained our ultimate guide in fragment optimization, the model aided the successful design of potent inhibitors with substituents targeting specific protein residues. Importantly, the iterative process of monitoring the SAR and refinement of the computational model was key to the acceleration of fragment optimization in the absence of an X-ray crystal structure of a starting fragment hit.15,18,19
Summary
Optimization of fragment hit (IC50 = 424 μM) toward potent inhibition of PKC-ι resulted in the discovery of submicromolar inhibitors of the target protein (IC50 = 340–270 nM). It is noteworthy that the advancement of the fragment hit was driven exclusively by SAR and molecular modeling. The binding mode of a more potent and advanced compound 19 was then elucidated through X-ray crystallographic analysis and revealed a unique binding conformation involving the post-kinase residues Phe552 and Asp553 of PKC-ι. The PKC kinase family is distinct in having the post-kinase domain that inserts side chains into the ATP-site, and our X-ray crystal structure shows that this can be exploited for inhibitor design.
Acknowledgments
This work was in part supported by the Agency for Science, Technology and Research (A*STAR) Joint Council grant 1231B105. The analysis of PKC-ι–compound 19 complex was undertaken on the MX1 beamline at the Australian Synchrotron, part of ANSTO.
Glossary
ABBREVIATIONS
- ECT2
epithelial cell transforming sequence 2
- Par6
bind partitioning defective 6 homologue
- HCC
hepatocellular carcinoma
- NSCLC
nonsmall cell lung cancer
- HLM
human liver microsomes
- MLM
mouse liver microsomes
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00546.
Author Present Address
(A.E.J.) GE Healthcare, Uppsala, Sweden.
Author Present Address
(J.K.) Queensland Emory Drug Discovery Initiative, UniQuest; Brisbane, Australia.
The authors declare no competing financial interest.
Supplementary Material
References
- Parker P. J.; Justilien V.; Riou P.; Linch M.; Fields A. P. Atypical Protein Kinase Cι as a Human Oncogene and Therapeutic Target. Biochem. Pharmacol. 2014, 88, 1–11. 10.1016/j.bcp.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka Y. Intracellular Signaling by Hydrolysis of Phospholipids and Activation of Protein Kinase C. Science 1992, 258, 607–614. 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- Fields A. P.; Regala R. P. Protein Kinase Cι: Human Oncogene, Prognostic Marker and Therapeutic Target. Pharmacol. Res. 2007, 55, 487–497. 10.1016/j.phrs.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regala R. P.; Weems C.; Jamieson L.; Khoor A.; Edell E. S.; Lohse C. M.; Fields A. P. Atypical Protein Kinase Cι Is an Oncogene in Human Non–Small Cell Lung Cancer. Cancer Res. 2005, 65, 8905–8911. 10.1158/0008-5472.CAN-05-2372. [DOI] [PubMed] [Google Scholar]
- Gorin M. A.; Pan Q. Protein Kinase Cε: An Oncogene and Emerging Tumor Biomarker. Mol. Cancer 2009, 8, 9. 10.1186/1476-4598-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray N. R.; Fields A. P. Atypical Protein Kinase C Iota Protects Human Leukemia Cells Against Drug-induced Apoptosis. J. Biol. Chem. 1997, 272, 27521–27524. 10.1074/jbc.272.44.27521. [DOI] [PubMed] [Google Scholar]
- Jamieson L.; Carpenter L.; Biden T. J.; Fields A. P. Protein Kinase C Iota Activity is Necessary for Bcr-Abl-mediated Resistance to Drug-induced Apoptosis. J. Biol. Chem. 1999, 274, 3927–3930. 10.1074/jbc.274.7.3927. [DOI] [PubMed] [Google Scholar]
- Nanos-Webb A.; Bui T.; Karakas C.; Zhang D.; Carey J. P. W.; Mills G. B.; Hunt K. K.; Keyomarsi K. PKCiota Promotes Ovarian Tumor Progression through Deregulation of Cyclin E. Oncogene 2016, 35, 2428–2440. 10.1038/onc.2015.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roffey J.; Rosse C.; Linch M.; Hibbert A.; McDonald N. Q.; Parker P. J. Protein Kinase C Intervention-the State of Play. Curr. Opin. Cell Biol. 2009, 21, 268–279. 10.1016/j.ceb.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Justilien V.; Fields A. P. Ect2 Links the PKCι-Par6α Complex to Rac1 Activation and Cellular Transformation. Oncogene 2009, 28, 3597–3607. 10.1038/onc.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Xia H.; Zhang X.; Karthik S.; Pratap S. V.; Ooi L. L.; Hong W.; Hui K. M. ECT2 Regulates the Rho/ERK Signalling Axis to Promote Early Recurrence in Human Hepatocellular Carcinoma. J. Hepatol. 2015, 62, 1287–1295. 10.1016/j.jhep.2015.01.014. [DOI] [PubMed] [Google Scholar]
- Kjær S.; Linch M.; Purkiss A.; Kostelecky B.; Knowles P. P.; Rosse C.; Riou P.; Soudy C.; Kaye S.; Patel B.; Soriano E.; Murray-Rust J.; Barton C.; Dillon C.; Roffey J.; Parker P. J.; McDonald N. Q. Adenosine-binding Motif Mimicry and Cellular Effects of a Thieno[2,3-d]pyrimidine-based Chemical Inhibitor of Atypical Protein Kinase C Isoenzymes. Biochem. J. 2013, 451, 329–342. 10.1042/BJ20121871. [DOI] [PubMed] [Google Scholar]
- Pillai P.; Desai S.; Patel R.; Sajan M.; Farese R.; Ostrov D.; Acevedo-Duncan M. A Novel PKC-ι Inhibitor Abrogates Cell Proliferation and Induces Apoptosis in Neuroblastoma. Int. J. Biochem. Cell Biol. 2011, 43, 784–794. 10.1016/j.biocel.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Fröhner W.; Lopez-Garcia L. A.; Neimanis S.; Weber N.; Navratil J.; Maurer F.; Stroba A.; Zhang H.; Biondi R. M.; Engel M. 4-Benzimidazolyl-3-Phenylbutanoic Acids as Novel Pif-Pocket-Targeting Allosteric Inhibitors of Protein Kinase PKCζ. J. Med. Chem. 2011, 54, 6714–6723. 10.1021/jm2005892. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski J.; Liu B.; Tee D. H.-Y.; Chen G.-Y.; Ahmad N. H. B.; Wang Y. X.; Poh Z. Y.; Ang S. H.; Tan E. S. W.; Ong E.; Rahad N. D. B.; Poulsen A.; Pendharkar V.; Sangthongpitag K.; Lee M. A.; Supramaniam S.; Ho S. Y.; Cherian J.; Hill J.; Hung A. W.; Keller T. H. Fragment-based Drug Discovery of Potent Atypical PKC-iota Inhibitors. J. Med. Chem. 2018, 61, 4386–4396. 10.1021/acs.jmedchem.8b00060. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The Role of Ligand Efficiency Metrics in Drug Discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Lys283 is a crucial catalytic residue, conserved in all PKC isoenzymes.
- Jhoti H.; Williams G.; Rees D. C.; Murray C. W. The ‘Rule of Three’ for Fragment-based Drug Discovery: Where Are We Now?. Nat. Rev. Drug Discovery 2013, 12, 644–645. 10.1038/nrd3926-c1. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A.; Davis B. J.; Jahnke W. Fragment-Based Drug Discovery: Advancing Fragments in the Absence of Crystal Structures. Cell Chem. Biol. 2019, 26, 9–15. 10.1016/j.chembiol.2018.10.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.