

Abstract

8-Nitrobenzothiazinones (BTZs) typified by the second-generation analogue PBTZ169 are a new class of antitubercular agents. The activity of BTZs and lipophilicity are tightly coupled since the molecular target DprE1 is located in the mycobacterial cell envelope. A series of analogues was designed to address the notorious insolubility of the BTZs while preserving the required lipophilicity. This was accomplished by decreasing the molecular planarity and symmetry through bioisosteric replacement of the piperazine moiety of PBTZ169 with spirocyclic and bicyclic diamines. Several promising compounds with improved aqueous solubilities were identified with potent antitubercular activity. Compound 5 was identified as the most promising candidate based on its excellent antitubercular activity (MIC of 32 nM), more than 1000-fold improvement in solubility, 2-fold lower clearance in mouse and human microsomes relative to PBTZ169, and promising pharmacokinetic parameters.
Keywords: benzothiazinone, tuberculosis, antitubercular, spirocyclic
Tuberculosis (TB) is an infectious disease caused by members of Mycobacterium tuberculosis (Mtb) complex.1 In 2017, more than 10 million people were actively infected by TB and approximately 1.6 succumbed to the disease. The current treatment regimen for drug-susceptible TB involves 6–9 months of the first-line TB drugs isoniazid, rifampicin, ethambutol, and pyrazinamide. The lengthy treatment course is necessary to eliminate the various bacterial subpopulations that exhibit differential drug sensitivity including dormant bacilli, which are phenotypically drug resistant. Consequently, the emergence of drug resistant TB (DR-TB) caused by either multidrug or extensively drug resistant Mtb strains represents a global health crisis.
The 8-nitrobenzothiazinones (BTZs) new class of compounds with a fascinating history and mechanism of action first identified by Ute Möllmann as biotransformation products of dithiocarbamates, which in turn had been initially synthesized by Vadim Makarov.2,3 The benzothiazinones are mechanism-based inhibitors of the flavoenzyme DprE1 and are bioactivated by the dihydroflavin cofactor FADH2 through reduction of the C-8 nitro group to a nitroso intermediate that covalently reacts with Cys387 in the enzyme active site to form a semimercaptal enzyme–inhibitor adduct (Figure 1).4 With support from the NM4TB consortium led by Stewart Cole, Makarov developed a concise synthesis of the BTZs and carried out an extensive structure–activity relationship (SAR) campaign culminating in the synthesis of the second-generation candidate PBTZ169.5 This promising compound possesses extraordinary whole-cell activity with a minimum inhibitory concentration (MIC) of ∼1 nM against drug-sensitive (DS) and drug-resistant (DR) Mtb strains, displays strong synergism with other TB drugs, is potently bactericidal, and significantly shortens therapy in a TB mouse relapse model.5 While impressive, PBTZ169 does have liabilities emanating from its extremely poor solubility (<0.01 μg/mL at pH 7.4 in 1× PBS buffer at 37 °C) that portend poor membrane penetration. This may affect oral bioavailability (F) and volume of distribution (Vd), neither of which have been reported.
Figure 1.
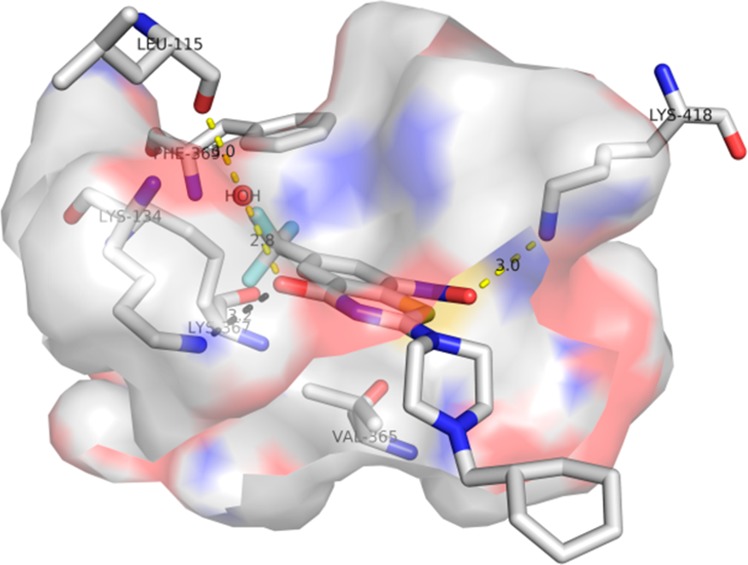
Binding mode of PBTZ169 to the target DprE1 (PDB code: 4NCR).
The SARs of the benzothiazinones reveal a strong positive correlation between lipophilicity and activity, consistent with location of the target DprE1, which is found in the periplasmic space of the mycobacterial cell wall.6 Consequently, introduction of polar functional groups to modulate solubility is unlikely to be successful without adversely impacting potency. Disruption of molecular symmetry and planarity is an alternate strategy to improve aqueous solubility.7,8 The 6-trifluormethyl-8-nitrobenzothiazinone heterocycle must be strictly maintained, and even subtle alterations obliterate activity, providing limited opportunities to disrupt planarity in this portion of the molecule. However, the piperazine substituent appears substantially more tolerant to modification.5 Indeed, many structural modifications at position 2 of BTZ core have been reported.4,5,9−18 Based on these SAR requirements, we initially elected to replace the central piperazine moiety with a wide variety of bioisosteric spirocyclic and bicyclic diamines that preserve the overall lipophilicity but disrupt the molecular symmetry and/or planarity of PBTZ169 (Figure 2).
Figure 2.
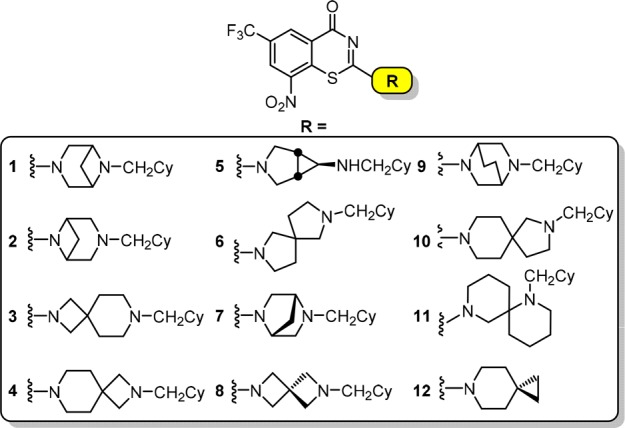
Proposed analogues in this study containing bioisosteric replacements of the piperazine moiety.
We initially conceived a series of closely related bicyclic and spirocyclic analogues to reduce planarity. A representative synthesis is illustrated in Scheme 1 for compound 1. Reductive amination19 of Boc-3,6-diazabicyclo[3.1.1]heptane 13 and cyclohexane carboxaldehyde with NaBH(OAc)3 afforded 14, which was subsequently deprotected by trifluoroacetic acid to furnish 15. Reaction with carbon disulfide in aqueous NaOH yielded 16.20 Nucleophilic aromatic substitution of 16 with 2-chlorobenzamide derivative 17 followed by intramolecular cyclization provided 1 in 80% yield, which was purified by silica gel chromatography.13 Consistent with a decrease in planarity, the melting point of 1 (mp 126–128 °C) was significantly decreased compared to PBTZ169 (mp 176–178 °C).5 Compounds 3–4 and 6–12 were prepared in analogous fashion as described in the Supporting Information.
Scheme 1. Representative Synthesis of Analog 1.

For compounds 2 and 5, we employed an alternate synthetic route due to low yields in the final cyclization step with 17. Boc-3,6-diazabicyclo[3.1.1]heptane 13 was elaborated to 18 via reaction with carbon disulfide in aqueous NaOH.2 Nucleophilic aromatic substitution of 17 with 18 followed by cyclization smoothly provided 19. TFA-mediated deprotection of the Boc group furnished 20, and reduction amination with cyclohexane carboxaldehyde yielded 2 in 81% yield (Scheme 2). Spirocyclic analogue 2 was recrystallized (mp 122–124 °C) from EtOH–EtOAc (1:1, v/v). Analogue 5 was prepared analogously as described in the Supporting Information.
Scheme 2. Representative Synthesis of Piperazine Analog 2.

All compounds were evaluated for whole-cell activity against M. tuberculosis strains H37Rv, CDC1551, and Erdman in 7H9 medium to determine minimum inhibitory concentrations (MICs) that resulted in complete inhibition of observable growth. The MICs were nearly identical for all M. tuberculosis strains and MIC data for strain H37Rv are shown in Table 1 (MICs for CDC1551 and Erdman were equal to or 4-fold lower than H37Rv, Table S2). The MICs ranged by nearly two-orders of magnitude from 16–1024 nM. To track physicochemical properties, we calculated the lipophilic ligand efficiency (LLE) and logP of each analogue. Spirocyclic 12 is the most potent analogue with an MIC of 16 nM followed by compounds 5 and 9 with MICs of 32 nM and compound 7 whose MIC is 64 nM. Among these analogues, only 12 has an improved LLE relative to PBTZ169 due to an overall decrease in clogP, whereas 5, 7, and 9 all have lower LLEs primarily attributed to their decreased activity. Examination of the SAR reveals analogues containing conservative modifications to the piperazine nucleus are generally better tolerated, whereas more extreme modifications resulted in substantial decrease in activity. Thus, the 4,4-, 4,6-, 5,5-, 5,6-, and 6,6-spirocycles in 3, 4, 6, 8, 10, and 11, respectively, were poorly tolerated resulting in nearly 64–512-fold losses in potencies compared to PBTZ169. Based on the outstanding whole-cell activities, we selected compounds 5, 7, 9, and 12 for further evaluation.
Table 1. MIC90 and clogP of 1–12.

Calculated log10P (clogP) was determined by ChemDraw Professional version 16.0.
Lipophilic ligand efficiency (LLE) was calculated from the equation: LLE = log10MIC −clog10P.
The objectives of our study were to improve aqueous solubility; thus, selected compounds 5, 7, 9, and 12 were examined for their kinetic solubility in phosphate-buffered saline pH 7.4 by LC–MS/MS, and the results are shown in Table 2 along with the experimentally determined melting points (mp) and total polar surface areas (tPSA). Compound 7 displayed the highest solubility (14.6 μg/mL) that was 1600-fold greater than PBTZ169, while 5, 9, and 12 also showed marked improvements in solubility. The solubility did not show obvious correlation with the molecular descriptor tPSA or melting points.
Table 2. Solubility, Melting Points, and tPSA of 5, 7, 9, and 12.
| Compounds | Solubility (μg/mL) | mp (°C) | tPSAa |
|---|---|---|---|
| PBTZ169 | <1 × 10–2 | 183–185 | 87.7 |
| 5 | 4.8 ± 0 | 110–112 | 96.5 |
| 7 | 14.6 ± 0.1 | >250 | 87.7 |
| 9 | <2.9 × 10–2 | 209–211 | 87.7 |
| 12 | 0.30 ± 0.02 | 192–194 | 84.5 |
tPSA was calculated by Chembiodraw Ultra version 15.0.
We performed in vitro metabolic stability studies for compounds 5, 7, 9, and 12 in parallel with PBTZ169, using both mouse and human liver microsomes (MLM and HLM). The results are shown in Table 3. Compound 5 has the lowest intrinsic clearance in both MLM and HLM that is nearly 2-fold lower than PBTZ169, while 9 behaves similarly to PBTZ169. However, compounds 7 and 12 were rapidly metabolized in MLMs but displayed improved stability in HLMs relative to PBTZ169. We also measured plasma protein binding (PPB) and showed all compounds exhibited nearly quantitative PPB with compound 5 having a slightly improved free fraction of 0.5% versus 0.1% for PBTZ169. Given the high PPB, it was not surprising that all compounds exhibited high plasma stability. Based on these results, we selected compound 5 for evaluation of in vivo pharmacokinetic parameters in ICR mice.
Table 3. Microsomal Stability, Plasma Protein Binding, and Plasma Stability of Compounds 5, 7, 9, 12, and PBTZ169.
|
t1/2 (min) |
Clint (μL·min–1·mg–1) |
|||||
|---|---|---|---|---|---|---|
| MLM | HLM | MLM | HLM | PPB (%) | Plasma Stability (%) | |
| PBTZ169 | 34.4 | 43.0 | 20.1 | 16.5 | 99.9 | 88.7 |
| 5 | 65.2 | 76.6 | 10.6 | 9.05 | 99.5 | 91.4 |
| 7 | 7.05 | 26.6 | 98.4 | 26.1 | 99.6 | 81.9 |
| 9 | 27.3 | 30.6 | 25.4 | 22.7 | 99.9 | 102 |
| 12 | 8.33 | 43.0 | 83.3 | 16.1 | 99.4 | 93.3 |
The in vivo PK profile of compound 5 was evaluated in ICR mice after intravenous (i.v.) (2 mg/kg) and oral (p.o.) (10 mg/kg) administration (Table 4). Compound 5 displays a useful PK profile with a 27% oral bioavailability and a high volume of distribution (Vd) that is offset by moderate-to-high clearance resulting in a terminal elimination half-life of 2.5 h. Makarov and co-workers only dosed PBTZ169 orally at 25 mg/kg and observed similar exposure as measured by the area-under-the curve (AUC) assuming linear PK and a slightly shorter terminal elimination half-life.5
Table 4. Pharmacokinetic Parameters for Compound 5 in ICR Mice Following Intravenous (2 mg/kg) and Oral (10 mg/kg) Administration.
| Compound 5 |
||
|---|---|---|
| PK Parametersa | p.o. | i.v. |
| Cmax (ng·mL–1) | 195 ± 61 | 483 ± 143 |
| t1/2 (h) | 2.19 ± 0.29 | 2.53 ± 0.25 |
| MRT (h) | 3.09 ± 0.09 | 2.24 ± 0.23 |
| AUC (ng·h·mL–1) | 791 ± 338 | 576 ± 54 |
| Clint (mL·min–1·kg–1) | 58.2 ± 5.7 | |
| Vd (L·kg–1) | 12.8 ± 2.5 | |
| F (%) | 27.4 ± 11.7 | |
Cmax, maximum concentration of drug in blood plasma; t1/2, the elimination half-life of drug; MRT, mean residence time; AUC, area under the curve; Clint, hepatic clearance; Vd, apparent volume of distribution; F, oral bioavailability.
In conclusion, we designed and synthesized a series of benzothiazinones with spirocyclic and bicyclic isosteres of the piperazine to improve physicochemical properties by disruption of molecular planarity. Compound 5 containing the azabicyclo[3.1.0]hexan-3-amine emerged as the most promising analogue based on its improved solubility, enhanced microsomal stability, and lower PPB compared to PBTZ169. In addition, compound 5 had a respectable pharmacokinetic profile with an oral bioavailability of 27% and high volume of distribution.
Acknowledgments
This work was financially supported by a grant from the CAMS Innovation Fund for Medical Sciences (CAMS-2017-I2M-1-011).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00634.
Experimental procedure, biochemical methods, LC–MS/MS method, and 1H and 13C NMR spectra (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO. Global Tuberculosis Report 2018, 23ed; 2018. [Google Scholar]
- Makarov V.; Riabova O.; Granik V.; Yuschenko A.; Urlyapova N.; Daudova A.; Zipfel P.; Möllmann U. Synthesis and antileprosy activity of some dialkyldithiocarbamates. J. Antimicrob. Chemother. 2006, 57, 1134–1138. 10.1093/jac/dkl095. [DOI] [PubMed] [Google Scholar]
- Makarov V.; Manina G.; Mikusova K.; Mollmann U.; Ryabova O.; Saint-Joanis B.; Dhar N.; Pasca M. R.; Buroni S.; Lucarelli A. P.; Milano A.; De Rossi E.; Belanova M.; Bobovska A.; Dianiskova P.; Kordulakova J.; Sala C.; Fullam E.; Schneider P.; McKinney J. D.; Brodin P.; Christophe T.; Waddell S.; Butcher P.; Albrethsen J.; Rosenkrands I.; Brosch R.; Nandi V.; Bharath S.; Gaonkar S.; Shandil R. K.; Balasubramanian V.; Balganesh T.; Tyagi S.; Grosset J.; Riccardi G.; Cole S. T. Benzothiazinones Kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324, 801–804. 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Trefzer C.; Skovierová H.; Buroni S.; Bobovská A.; Nenci S.; Molteni E.; Pojer F.; Pasca M. R.; Makarov V.; Cole S. T.; Riccardi G.; Mikusová K.; Johnsson K. Benzothiazinones are suicide inhibitors of mycobacterial decaprenylphosphoryl-β-D-ribose 2′-oxidase DprE1. J. Am. Chem. Soc. 2012, 134, 912–915. 10.1021/ja211042r. [DOI] [PubMed] [Google Scholar]; b Neres J.; Pojer F.; Molteni E.; Chiarelli L. R.; Dhar N.; Boy-Röttger S.; Buroni S.; Fullam E.; Degiacomi G.; Lucarelli A. P.; Read R. J.; Zanoni G.; Edmondson D. E.; De Rossi E.; Pasca M. R.; McKinney J. D.; Dyson P. J.; Riccardi G.; Mattevi A.; Cole S. T.; Binda C. Structural basis for benzothiazinone-mediated killing of Mycobacterium tuberculosis. Sci. Transl. Med. 2012, 4, 150ra121. 10.1126/scitranslmed.3004395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V.; Lechartier B.; Zhang M.; Neres J.; van der Sar A. M.; Raadsen S. A. S. A.; Hartkoorn R. C.; Ryabova O. B.; Vocat A.; Decosterd L. A.; Widmer N.; Buclin T.; Bitter W.; Andries K.; Pojer F.; Dyson P. J.; Cole S. T. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecik M.; Centárová I.; Mukherjee R.; Kolly G. S.; Huszár S.; Bobovská A.; Kilacsková E.; Mokošová V.; Svetlíková Z.; Šarkan M.; Neres J.; Korduláková J.; Cole S. T.; Mikušová K. DprE1 is a vulnerable tuberculosis drug target due to its cell wall localization. ACS Chem. Biol. 2015, 10, 1631–1636. 10.1021/acschembio.5b00237. [DOI] [PubMed] [Google Scholar]
- Ishikawa M.; Hashimoto Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem. 2011, 54, 1539–1554. 10.1021/jm101356p. [DOI] [PubMed] [Google Scholar]
- Degorce S. L.; Bodnarchuk M. S.; Cumming I. A.; Scott J. S. Lowering lipophilicity by Adding Carbon: One-Carbon Bridges of Morpholines and Piperazines. J. Med. Chem. 2018, 61, 8934–8943. 10.1021/acs.jmedchem.8b01148. [DOI] [PubMed] [Google Scholar]
- Lv K.; You X.; Wang B.; Wei Z.; Chai Y.; Wang B.; Wang A.; Huang G.; Liu M.; Lu Y. Identification of better pharmacokinetic benzothiazinone derivatives as new antitubercular agents. ACS Med. Chem. Lett. 2017, 8, 636–641. 10.1021/acsmedchemlett.7b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piton J.; Vocat A.; Lupien A.; Foo C.; Riabova O.; Makarov V.; Cole S. T. Structure-based drug design and characterization of sulfonyl-piperazine benzothiazinone inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2018, 62, e00681. 10.1128/AAC.00681-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C.; Ye T.-H.; Wang N.-Y.; Zeng X.-X.; Zhang L.-D.; Xiong Y.; You X.-Y.; Xia Y.; Xu Y.; Peng C.-T.; Zuo W.-Q.; Wei Y.; Yu L.-T. Synthesis and structure-activity relationships evaluation of benzothiazinone derivatives as potential anti-tubercular agents. Bioorg. Med. Chem. Lett. 2013, 23, 4919–4922. 10.1016/j.bmcl.2013.06.069. [DOI] [PubMed] [Google Scholar]
- Peng C. T.; Gao C.; Wang N. Y.; You X. Y.; Zhang L. D.; Zhu Y. X.; Xv Y.; Zuo W. Q.; Ran K.; Deng H. X.; Lei Q.; Xiao K. J.; Yu L. T. Synthesis and antitubercular evaluation of 4-carbonyl piperazine substituted 1,3-benzothiazin-4-one derivatives. Bioorg. Med. Chem. Lett. 2015, 25, 1373–1376. 10.1016/j.bmcl.2015.02.061. [DOI] [PubMed] [Google Scholar]
- Li P.; Wang B.; Zhang X.; Batt S. M.; Besra G. S.; Zhang T.; Ma C.; Zhang D.; Lin Z.; Li G.; Huang H.; Lu Y. Identification of novel benzothiopyranone compounds against Mycobacterium tuberculosis through scaffold morphing from benzothiazinones. Eur. J. Med. Chem. 2018, 160, 157–170. 10.1016/j.ejmech.2018.09.042. [DOI] [PubMed] [Google Scholar]
- Lv K.; Tao Z.; Liu Q.; Yang L.; Wang B.; Wu S.; Wang A.; Huang M.; Liu M.; Lu Y. Design, synthesis and antitubercular evaluation of benzothiazinones containing a piperidine moiety. Eur. J. Med. Chem. 2018, 151, 1–8. 10.1016/j.ejmech.2018.03.060. [DOI] [PubMed] [Google Scholar]
- Karoli T.; Becker B.; Zuegg J.; Möllmann U.; Ramu S.; Huang J. X.; Cooper M. A. Identification of antitubercular benzothiazinone compounds by ligand-based design. J. Med. Chem. 2012, 55, 7940–7944. 10.1021/jm3008882. [DOI] [PubMed] [Google Scholar]
- Xiong L.; Gao C.; Shi Y.-J.; Tao X.; Rong J.; Liu K.-L.; Peng C.-T.; Wang N.-Y.; Lei Q.; Zhang Y.-W.; Yu L.-T.; Wei Y.-Q. Identification of a new series of benzothiazinone derivatives with excellent antitubercular activity and improved pharmacokinetic profiles. RSC Adv. 2018, 8, 11163–11176. 10.1039/C8RA00720A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Lv K.; Wang B.; Li L.; Wang B.; Liu M.; Guo H.; Wang A.; Lu Y. Design, synthesis and antitubercular evaluation of benzothiazinones containing an oximido or amino nitrogen heterocycle moiety. RSC Adv. 2017, 7, 1480–1483. 10.1039/C6RA25712G. [DOI] [Google Scholar]
- Richter A.; Rudolph I.; Mollmann U.; Voigt K.; Chung C. W.; Singh O. M. P.; Rees M.; Mendoza-Losana A.; Bates R.; Ballell L.; Batt S.; Veerapen N.; Futterer K.; Besra G.; Imming P.; Argyrou A. Novel insight into the reaction of nitro, nitroso and hydroxylamino benzothiazinones and of benzoxacinones with Mycobacterium tuberculosis DprE1. Sci. Rep. 2018, 8, 13743. 10.1038/s41598-018-31316-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Magid A. F.; Carson K. G.; Harris B. D.; Maryanoff C. A.; Shah R. D. Reductive amination of aldehydes and ketones with sodium triacetoxyborohydride. Studies on direct and indirect reductive amination procedures. J. Org. Chem. 1996, 61, 3849–3862. 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- Rudorf W. D. Reaction of carbon disulfide with N-nucleophiles. J. Sulfur Chem. 2007, 28, 295–339. 10.1080/17415990701245107. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.