

Abstract

Herein, we disclose a series of selective GluN2B negative allosteric modulators containing a 1H-pyrrolo[3,2-b]pyridine core. Lead optimization efforts included increasing brain penetration as well as decreasing cytochrome P450 inhibition and hERG channel binding. The series was also optimized to reduce metabolic turnover in human and rat. Compounds 9, 25, 30, and 34 have good in vitro GluN2B potency and good predicted absorption, but moderate to high projected clearance. They were assessed in vivo to determine their target engagement. All four compounds achieved >75% receptor occupancy after an oral dose of 10 mg/kg in rat. Compound 9 receptor occupancy was measured in a dose–response experiment, and its ED50 was found to be 2.0 mg/kg.
Keywords: Azaindole, NR2B, Lead Optimization, Autoradiography
N-Methyl-d-aspartate (NMDA) receptors are a critical component of the functional machinery of the glutamatergic network in the mammalian central nervous system (CNS), operating as the mediators of fast excitatory neurotransmission. Channel activation occurs in a voltage-dependent manner following glutamate release and relief of a physiological Mg2+ block of the channel, which initiates the ion flux. Once the channel is open, it is highly permeable to sodium, potassium, and calcium ions. Overactivation of the receptor results in excessive Ca2+ influx and may lead to excitotoxicity.1,2
Structurally, an NMDA receptor functions as a tetramer comprising two GluN1 and two GluN2 subunits. GluN1 is expressed ubiquitously throughout the CNS, while GluN2 has four subunits A–D that have distinct localizations predicated upon expression patterns of the four genes that encode for them. Each of the GluN2 subunits exhibit unique biochemical properties and have distinct physiological roles.3
Clinical and preclinical research on NMDAR subunit selective modulators has overwhelmingly been focused on GluN2B functional antagonists.4 Selective GluN2B modulation by a small molecule inhibitor is distinct from a channel blocking inhibitor in that selective GluN2B inhibitors have been found to utilize a GluN2B extracellular allosteric site positioned at the interface of GluN1 and GluN2B. The site is not present in subunits GluN2A, GluN2C, and GluN2D,5 and inhibitors that bind at this site can have at least 1000-fold selectivity over GluN2A, GluN2C, and GluN2D subunits.6
Therapeutically, the NMDA receptors have received an increasing amount of attention since the end of the 20th century, in part due to positive outcomes reported in patients diagnosed with major depressive disorder following the administration of NMDAR antagonists.7,8 Proof-of-concept clinical trials have shown GluN2B-selective negative allosteric modulators (NAMs) to be therapeutic in treatment-resistant depression (TRD). Traxoprodil (CP-101,606; Chart 1) was shown to be efficacious in Phase II clinical trials for TRD, achieving a response in 60% of subjects diagnosed with TRD and in a current depressive episode.9 QT interval prolongation was also observed, and trials with Traxoprodil were ceased.10,11
Chart 1. GluN2B-Selective NAMs Assessed in Clinical Proof of Concept Trials for Depression.

Another GluN2B-selective NAM, CERC-301 (Chart 1) has been assessed in a pilot trial12 and two Phase II trials in patients with major depressive disorder (MDD).13,14 Both Phase II trials failed to meet their primary end points; although in the latter of the two Phase II studies it was noted that on Day 2 subjects averaged a 3.47-point improvement over placebo on the Hamilton Depression Rating Scale (HDMS). Based on the data reported for CP-101,606 and prior to the results reported with CERC-301, medicinal chemistry efforts toward the discovery of novel GluN2B-selective NAMs were initiated at Janssen with the goal of identifying an orally available small molecule with putative therapeutic use in patients with TRD.
A high throughput screen (HTS) of the Janssen global screening deck yielded a potent and selective series of GluN2B NAMs containing a 1,3-dihydro-2H-imidazo[4,5-b]pyridin-2-one core. Compound 3, for example, was found to have a human GluN2B IC50 of 22 nM and was selective for the GluN2B-subunit (2A, 2C, 2D IC50 all >10 μM). It also displaced [3H]-Ro 25–6981 with a Ki of 13 nM, suggestive of allosteric binding. Compound 3 has adequate drug-like physicochemical properties with cLogP = 1.9, LE = 0.39, and MPO = 4.6. It was soon determined, however, that poor solubility and poor epithelial cellular uptake of dihydro-2H-imidazo[4,5-b]pyridin-2-one core compounds would be challenging to overcome. We attributed these undesirable properties to the cyclic urea that is contained within the central core. We were pleased to find that adjusting the ring system to a pyrrole-fused pyridine (1H-pyrrolo[3,2-b]pyridine) resulted in a more soluble, potentially more brain penetrant, GluN2B-active compound (4).
Initial optimization efforts focused on replacing the cyclopropyl amide. The hydrogen bond donor on the secondary amide was thought to be contributing to efflux mechanisms reported in a MDCK-MDR1 assay. After a slight tuning of the phenyl substituents and replacement of the secondary cyclopropyl amide with a tertiary amide (azetidine), compound 5 emerged (cLogP = 3.2, LE = 0.45) (Figure 1). It had significantly improved rat GluN2B binding affinity compared to 4 and was selective over the 2A, 2C, and 2D isoforms (IC50s all >10 μM). Compound 5 also had good solubility and good cellular uptake but exhibited hERG channel binding, as measured by dofetilide binding. Even so, its overall favorable profile merited further structure–activity relationship (SAR) investigations. 4-Fluoro-3-methylphenyl was set as the aryl group left-hand side when conducting modifications to the amide piece of the molecule. This choice was made due to excellent GluN2B binding values obtained for preliminary compounds containing this moiety (compound 5 is one such example [Ki = 3.3 nM]).
Figure 1.

HTS hits and initial core optimization of fused pyridines.
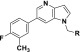
A representative set of 6-(4-fluoro-3-methylphenyl)-1H-pyrrolo[3,2-b]pyridines containing varied substituents tethered from the 1-position of the core is shown (i.e., N-pyrrolo-substituted, Table 1). The examples containing an amide were all found to have good or very good binding affinities, with rat Ki values <100 nM (5–9, Table 1). The azetidines (5 and 8) and the pyrrolidine (7) had better human IC50 and rat Ki values than the piperidine amide (6). All of the examples have high or moderately high turnover in human and rat liver microsomes (LM). In general, as the number of saturated carbons on the amide increased, a decrease in the compounds’ stability in LM was noted (5–9). As it follows, compound 9, which contains a dimethyl amide, was found to be the most stable LM. However, ER is still relatively high (0.7, human) and suggestive of a moderately high clearance of 9 in human.
Table 1. SAR of 1-Position Nitrogen Substituents of 1H-Pyrrolo[3,2-b]pyridines.

IC50 values are reported in nM and were determined by a calcium mobilization assay in inducible CHO T-Rex cells heterologously expressing the hGluN1a/GluN2B receptor. Values reported are the mean of three experiments unless otherwise stated; the SEM for their pIC50 values are all ≤ ±0.2.
Ki values are reported in nM and were determined using a radioligand competitive binding assay in rat cortex membranes using [3H]-5. Values reported are the mean of three experiments, unless otherwise stated.
CYP IC50 values were obtained from human liver microsomes for six isoforms: 1A2, 2C19, 2C8, 2C9, 2D6, 3A4.
Stability in human and rat liver microsomes. Data reported as hepatic extraction ratio.
Ki values are reported in μM and were determined using a radioligand ([3H]dofetilide) competitive binding assay to the hERG protein expressed in HEK-293 cells.
Efflux ratio was measured using MDCK cells transfected with the P-glycoprotein (MDR-1). The efflux ratio is reported as the ratio of velocities (B–A/A–B).
The mean of four experiments.
The mean of nine experiments.
The mean of five experiments.
The mean of ten experiments
Many examples were found to have cytochrome P450 (CYP) inhibition, particularly of 2C9 (6–8, 10, 12–15), but also significantly of 2C19 (7, 10, 12–15) and 1A2 (10, 12–15). In general, the examples containing a tethered amide were the weakest inhibitors; however, the examples containing larger amides did inhibit CYPs 2C9 and 2C19. Interestingly, the extent of 2C9 inhibition was found to be proportional to the size of the amide group.
Compounds 5, 8–10, and 13–15 displayed hERG binding. Further cardiovascular safety derisking of the compounds was not conducted since the compounds demonstrated additional disadvantages that we later found could be overcome (vide infra). For example, either a high efflux ratio (8) and/or CYP inhibition (8, 10, 13–15) were measured for the majority of the hERG binders.
Aromatic and aliphatic heterocycles were evaluated as amide replacements (10–16). Methyl 2-pyridine 10 had moderate GluN2B potency, but was unstable in LM, inhibited CYPs, and had hERG binding. Oxadiazoles 11–12 and an oxazole (13) had moderate GluN2B potency and low stability in LM. Compound 11 was unique to the set of methyl-2-heterocycles because it did not inhibit CYPs (up to 10 μM). Compound 11 also had no hERG binding, while 8–10 and 13–15 had moderate to high hERG binding with Ki values < 6 μM. Compounds 14–16 are small cyclic ethers or aliphatic rings with moderate hGluN2B potency. The two examples analyzed in LM were found to be unstable (14–15).
The dimethylamide was the R-group used in the next round of SAR optimization (Table 2). It was chosen as a follow-up to 9, which had good GluN2B potency, low potential for drug–drug interaction (DDI), and among the lowest extraction ratios measured (Table 1). Several substituted 1H-pyrrolo[3,2-b]pyridines were made using the azetidine amide as well, but generally they were less stable and/or less potent than their corresponding dimethylamides (19 and 28 are the two azetidine examples included here).
Table 2. SAR of the 6-Aryl Position 1H-Pyrrolo[3,2-b]pyridines.


One potential weak point identified in the preliminary in vitro characterization of dimethylamide 9 was its moderate level of hERG binding (IC50 = 5.9 μM). It was therefore just serendipity that most examples in Table 2 had hERG IC50 > 10 μM, despite them all containing the dimethylamide R-group (data not shown). The single exception is 19 (hERG IC50 = 5.3), which is an azetidine amide.
Compounds 17–23 demonstrate the basic SAR of a single trifluoromethyl substituent on the phenyl (17–20) or pyridine (21–23) R-group. Substitution meta to the ring attachment was preferred for good potency (18–19), although the presence of a pyridine ring reduced potency dramatically (18 vs 21–23). Three-position substitution on phenyl tended to generate compounds with good GluN2B potencies (18–19, 24–27, 29). The thiophenes (30–34) had moderate to excellent human IC50 and rat Ki values.
Compounds 18, 21, 24-27 and 29–35 were moderately stable in LM and generally had low DDI potential. Compound 27 additionally demonstrated excellent rat Ki (5.1 nM), but because it also had a high efflux ratio (B–A/A–B = 16) it was deprioritized. Compounds 9, 18, 25, 30, and 34 were selected for in vivo studies, based on their robust rat Ki values and good all-around in vitro ADME.
Rat pharmacokinetic (PK) experiments were conducted with 9, 18, 25, 30 and 34 dosed at 1.0 mg/kg i.v. and 5.0 mg/kg p.o. Calculated bioavailability values ranged from 23 to >100%. The five compounds had moderate to high clearance (CL). It is of note that CL values for 18 and 34 were 167 and 90 mL/min/kg, respectively, higher than rat hepatic clearance (∼70 mL/min/kg), suggesting the potential for extra-hepatic clearance mechanisms. Despite moderate to high CL values, compounds 9, 25, 30, and 34 were selected for target engagement studies and brain level assessment in rats after oral dosing. They had excellent rat Ki values, making them good candidates for rat receptor occupancy studies.
Target engagement was measured using ex vivo receptor autoradiography. Time dependency was evaluated after oral administration of a 10 mg/kg solution dose. The animals were sacrificed at different time points (0.25, 0.5, and 2 h) after drug administration. Brain sections were prepared and briefly incubated with the radiolabeled compound 3-[3H] 1-(azetidin-1-yl)-2-[6-(4-fluoro-3-methyl-phenyl)pyrrolo[3,2-b]pyridin-1-yl]ethanone (i.e., a tritiated version of 5).16 Thiophene 34 had the highest level of GluN2B occupancy (96%) despite its very high CL value. Brain concentrations of 34 at 30 min were high (802 ng/mL). Interestingly, free brain fraction measured in vitro for 24 in rat was only 0.75%, or 6 ng/mL (20 nM) at the 0.5 h time point. For comparison, rat brain protein binding for 9 and 30 were 96.06% and 98.27% bound, respectively, but they did not reach quite as robust occupancies as 34 (Table 3). Compound 9 GluN2B occupancy was steady over the 2 h time course, which is consistent with its moderately low rat CL (Table 3).
Table 3. Rat PK and GluN2B Occupancy Data for 9, 18, 25, 30, and 34.
| |
|
GluN2B
occupancy (10 mg/kg p.o.) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| i.v.
PK (1 mg/kg) |
p.o.
PK (5 mg/kg) |
% occupancy
timecourse (h)a |
|||||||||
| Cl (mL/min/kg) | Vss (L/kg) | Cmax (ng/mL) | AUCinf (h·ng/mL) | tmax (h) | F (%) | 0.25 | 0.5 | 2.0 | Cmax-brain (ng/mg) | Tmax (h) | |
| 9 | 32 | 0.7 | 1380 | 1587 | 0.42 | 60 | 79 | 83 | 82 | 1426 | 0.5 |
| 18 | 167 | 1.6 | 227 | 372 | 0.50 | 64 | |||||
| 25 | 48 | 0.5 | 546 | 571 | 0.25 | 32 | 77 | 69 | 32 | 818 | 0.25 |
| 30 | 25 | 0.5 | 2330 | 4728 | 0.67 | 140 | 77 | 84 | 76 | 1972 | 0.5 |
| 34 | 90 | 1.4 | 184 | 226 | 0.30 | 23 | 80 | 96 | 41 | 802 | 0.5 |
Ex vivo GluN2B labeling was expressed as the percentage of GluN2B labeling in corresponding brain areas of vehicle-treated animals.
Compound 9 was selected for assessment in a dose–response ex vivo GluN2B occupancy study. It was prioritized in part due to the absence of a thiophene structural alert that is present in 30.17 We set 70% GluN2B occupancy as the level of target engagement desired for compound advancement. Dose–response analysis ex vivo GluN2B occupancy was measured at 30 min after oral dosing (from 0.01 to 30 mg/kg, (Figure 2). Level of GluN2B occupancy at each dose was measured and ED50/70 values were calculated. The measured ED50 and EC70 values for compound 9 were 2.0 and 3.4 mg/kg, respectively, in rat. The plasma concentration associated with 70% GluN2B occupancy was 798 ng/mL total, or 31 ng/mL free for 9.
Figure 2.

Dose versus receptor occupancy at 0.5 h in rat in a dose–response experiment of 9.
Finally, compounds 9, 18, 25, 30 , and 34 were assessed qualitatively for their propensity to produce reactive metabolites in human liver microsomes supplemented with glutathione (GSH) and NADPH. All compounds were identified by LC–MS/MS to form at least three GSH adducts with net additions to parent mass of +GSH – 2H + O, +GSH – 2H + O – CH2, and +GSH – 2H + 2O. Further studies would be necessary to better understand the nature and potential of these metabolites.
To summarize, compounds 9 and 30 have the highest plasma and brain exposures of the 1H-pyrrolo[3,2-b]pyridines assessed in vivo (Table 3). Compound 34, although a very high clearing compound with relatively low plasma exposure in PK, has robust brain exposure. It is the only compound that displays full receptor occupancy, even though it is very highly protein bound (99.25% in the brain). Its advantage may have resulted from a superior rat Ki value. Unfortunately, GSH adducts have been identified for all of the promising 1H-pyrrolo[3,2-b]pyridines, warranting additional SAR investigations and in-depth metabolite characterization.
In conclusion, 1H-pyrrolo[3,2-b]pyridine GluN2B NAMs, which are chemically distinct from literature compounds, were identified from an HTS. Optimization efforts included improving solubility and permeability of the compounds, first by changing the core and then modifying the secondary amide, which eliminated two hydrogen bond donors. The series was also optimized for a lower metabolic turnover and CYP inhibition potential. Compounds 9, 25, 30, and 34 had favorable rat metabolic stability and were taken on to rat PK and receptor occupancy studies. The four achieved >75% receptor occupancy after oral dosing of 10 mg/kg. In addition, a dose–response target engagement study was conducted to determine the ED70 and EC70 of 9 (3.4 mg/kg and 798 ng/mL, respectively). GSH adducts were identified for the compounds of the series, which would be prudent to further characterize prior to compound advancement.
Acknowledgments
The authors would like to thank Heather McAllister for HRMS data and Moravek for the synthesis and characterization of [3H]-5.
Glossary
ABBREVIATIONS
- NMDA
N-methyl-d-aspartate
- NAM
negative allosteric modulator
- TRD
treatment-resistant depression
- MDD
major depressive disorder
- HDMS
Hamilton Depression Rating Scale
- LE
ligand efficiency
- MPO
multiparameter optimization
- LM
liver microsomes
- CYP
cytochrome P450
- MDCK
Madin–Darby canine kidney epithelial cells
- DDI
drug–drug interaction
- PK
pharmacokinetic
- CL
clearance
- RO
receptor occupancy
- GSH
glutathione
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00542.
General experimental protocols and analytical data for compounds 5–35, protocols for pharmacological assays, PPB data, time vs concentration curves for GluN2B occupancy of 9, 30, and 34, GSH adduct metabolic scheme, and characterization data for [3H]-5 (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Pinheiro P. S.; Mulle C. Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nat. Rev. Neurosci. 2008, 9, 423–36. 10.1038/nrn2379. [DOI] [PubMed] [Google Scholar]
- Paoletti P.; Neyton J. NMDA receptor subunits: function and pharmacology. Curr. Opin. Pharmacol. 2007, 7, 39–47. 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Dravid S. M.; Erreger K.; Yuan H.; Nicholson K.; Le P.; Lyuboslavsky P.; Almonte A.; Murray E.; Mosely C.; Barber J.; French A.; Balster R.; Murray T.; Traynelis S. F. Subunit-specific mechanisms and proton sensitivity of NMDA receptor channel block. J. Physiol. 2007, 581, 107–128. 10.1113/jphysiol.2006.124958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mony L.; Kew J. N. C.; Gunthorpe M. J.; Paoletti P. Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br. J. Pharmacol. 2009, 157, 1301–17. 10.1111/j.1476-5381.2009.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perin-Dureau F.; Rachline J.; Neyton J.; Paoletti P. Mapping the binding site of the neuroprotectant ifenprodil on NMDA receptors. J. Neurosci. 2002, 22 (14), 5955–65. 10.1523/JNEUROSCI.22-14-05955.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G.; Mutel V.; Trube G.; Malherbe P.; Kew J. N.; Mohacsi E.; Heitz M. P.; Kemp J. A. Ro 25–6981, a Highly Potent and Selective Blocker of N-Methyl-D-aspartate Receptors Containing the NR2B Subunit. Characterization in Vitro. J. Pharmacol Exp Ther. 1997, 283, 1285–92. [PubMed] [Google Scholar]
- Ates-Alagoz Z.; Adejare A. NMDA Receptor Antagonists for Treatment of Depression. Pharmaceuticals 2013, 6, 480–99. 10.3390/ph6040480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lener M. S.; Kadriu B.; Zarate C. A. Jr. Ketamine and Beyond: Investigations into the Potential of Glutamatergic Agents to Treat Depression. Drugs 2017, 77, 381–401. 10.1007/s40265-017-0702-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preskorn S. H.; Baker B.; Kolluri S.; Menniti F. S.; Krams M.; Landen J. W. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J. Clin. Psychopharmacol. 2008, 28, 631–37. 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- Nutt J. G.; Gunzler S. A.; Kirchhoff T.; Hogarth P.; Weaver J. L.; Krams M.; Jamerson B.; Menniti F. S.; Landen JW Effects of a NR2B Selective NMDA Glutamate Antagonist, CP-101,606, on Dyskinesia and Parkinsonism. Mov. Disord. 2008, 23, 1860–66. 10.1002/mds.22169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W.; Rogawski M. A.. Chapter 3: Epilepsy. In Ionotropic Glutamate Receptors as Therapeutic Targets; Lodge D., Danysz W., Parsons C. G., Eds.; FP Graham Publishing Co.: Johnson City, TN, 2002; pp 91–132. [Google Scholar]
- Ibrahim L.; DiazGranados N.; Jolkovsky L.; Brutsche N.; Luckenbaugh D. A.; Herring W. J.; Potter W. Z.; Zarate C. A. Jr. A Randomized, Placebo-Controlled, Crossover Pilot Trial of the Oral Selective NR2B Antagonist MK-0657 in Patients with Treatment-Resistant Major Depressive Disorder. J. Clin. Psychopharmacol. 2012, 32, 551–57. 10.1097/JCP.0b013e31825d70d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson B.; Fraser H.; Wang C.; Marcus R.. A Randomized, double-blind, pacebo-controlled, sequential parallel study of CERC-301 in the adjunctive treatment of subjects with severe depression and recent active suicidal ideation despite antidepressant treatment. Presented at the 2015National Network of Depression Centers Annual Conference, November 15, Ann Arbor, MI, USA.
- https://ir.cerecor.com/all-sec-filings/content/0001558370-16-010303/0001558370-16-010303.pdf.
- Lord B.; Wintmolders C.; Langlois X.; Nguyen L.; Lovenberg T.; Bonaventure P. Comparison of the ex vivo receptor occupancy profile of ketamine to several NMDA receptor antagonists in mouse hippocampus. Eur. J. Pharmacol. 2013, 715, 21–25. 10.1016/j.ejphar.2013.06.028. [DOI] [PubMed] [Google Scholar]
- Gramec D.; Mašič L. P.; Dolenc M. S. Bioactivation Potential of Thiophene-Containing Drugs. Chem. Res. Toxicol. 2014, 27, 1344–58. 10.1021/tx500134g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.