

Abstract

The allosteric targeting of ionotropic glutamate receptors (iGluRs) is a valuable approach for treating various central nervous system (CNS) disorders. In this frame, this Innovations provides a summary of the state-of-the art in the development of allosteric modulators for iGluRs and offers an outlook regarding innovative strategies for treating neurological diseases.
Keywords: Allosteric modulation, ionotropic glutamate receptors (iGluRs), positive allosteric modulator (PAM), negative allosteric modulator (NAM), CNS-related disorders
In the last two decades, the regulation of proteins’ functions and the control of their behavior by allosteric modulators have attracted growing interest in the quest for novel therapeutics. The possibility of regulating proteins’ functions inducing relevant conformational changes by binding accessory sites (allosteric), different from the endogenous activation site (orthosteric), endows benefits as higher selectivity and better modulatory control.
Allosteric modulators can be classified, based on their influence on protein functions and responses, as positive allosteric modulators (PAMs) or negative allosteric modulators (NAMs). PAMs cause an increase and NAMs a decrease of the response elicited by the endogenous ligands acting at the orthosteric sites. This implies that a fine-tuning of the signaling triggered by endogenous ligands can be obtained by a noninvasive control of the proteins by small-molecules acting at the allosteric binding sites. Accordingly, the discovery of allosteric drugs represents an appealing approach toward innovative and safer drugs for a wide-range of disorders, including cardiovascular, neurological/neurodegenerative, and infectious diseases. Moreover, specific observations suggest that all the proteins could be potentially modulated by allosteric ligands.1 Regrettably, the identification of PAMs and NAMs may be hampered by the occurrence of “flat structure–activity relationship (SAR)”, if studied by classical SAR analysis. This may complicate the identification and optimization processes, making pivotal the setup of proper studies for evaluating binding and efficacy cooperativity.2
Worth mentioning that, a lower evolutionary pressure affects allosteric sites with respect to the orthosteric sites, thus offering binding sites characterized by high specificity. This approach is particularly useful within families of homologous proteins in which the development of allosteric ligands increases the chance of achieving satisfactory selectivity.3 Benzodiazepines (Figure 1), an early example of allosteric drugs, discovered as PAMs of ionotropic GABAA receptors, can potentiate the effect of γ-aminobutyric acid, without eliciting the severe side effects of orthosteric GABAA agonists. Furthermore, also GPCRs, allosteric ligands demonstrated relevant advantages, in terms of higher functional selectivity and improved safety profile.4 The dopamine receptors have been targeted for developing allosteric ligands such as DETQ5 (PAM selective for dopamine receptor-1) and SB2696526 (NAM selective for dopamine receptors-2/3), as promising drugs for treating neuropsychiatric disorders (Figure 1).4 Significant advances for the potential treatment of schizophrenia were obtained investigating metabotropic glutamate receptors (mGluRs) PAMs.4 Furthermore, PAMs and NAMs have been proposed for the in vivo modulation of nAChRs to treat nicotinic-related disorders.7 In the practice, after the clinical success of several benzodiazepines as GABAA PAMs, other allosteric modulators were approved for diverse pathologies: the calcium-sensing receptor (CaSR) PAM cinacalcet (Amgen) for hyperparathyroidism, the CC-chemokine receptor type-5 (CCR5) NAM maraviroc (Pfizer) for HIV infection, and the mTOR NAM temsirolimus for cancer (Figure 1).1 Beyond these examples, over 40 clinical trials with allosteric drugs are currently ongoing.
Figure 1.

Chemical structures of DETQ (PAM selective for dopamine receptor-1), SB269652 (NAM selective for dopamine receptors-2/3), and representative approved allosteric modulators.
These success stories robustly prompted the development of allosteric regulators as a promising frontier in drug discovery.1 Within the big class of ligand-gated ion channels (LGICs), ionotropic glutamate receptors (iGluRs) represent attractive targets for developing drugs against neurological/neurodegenerative diseases. The physiology and structures of iGluRs have been thoroughly described in recent review articles.8,9 Briefly, iGluRs facilitate excitatory synaptic transmission in CNS upon binding of l-glutamate (l-Glu), an important neurotransmitter modulating most of the fast excitatory synaptic signaling in the CNS. l-Glu is involved in most aspects of normal brain functioning including cognition, memory, and learning. The role of l-Glu in the CNS has been the object of extensive investigation in the modern neuroscience. In neurons, l-Glu is stored in specialized presynaptic vesicles and released into the synaptic cleft, upon different stimuli. Once in the synaptic cleft, l-Glu activates various proteins including receptors (iGluRs and mGluRs) and electrogenic transport systems (excitatory amino acid transporters, EAATs).
iGluRs share a similar structural organization consisting of an extracellular amino-terminal domain (ATD), a ligand-binding domain (LBD), a transmembrane domain (TMD), which encompasses the ion channel, and a carboxyl-terminal domain (Figure 2). (S)-2-Amino-3-(5-methyl-3-hydroxyisoxazol-4-yl)propanoic acid receptors (AMPARs) can form functional homotetramers activated by l-Glu, while the N-methyl-d-aspartate receptors (NMDARs) are obligate heterotetramers requiring the binding of both glycine and l-Glu for activation. The iGluRs are encoded by a family of 18 genes: four subtypes exist for AMPARs (GluA1–4), five for kainate receptors (KARs, GluK1–5), two for delta receptors (GluD1–2), and seven subtypes for NMDARs (GluN1, GluN2A-D, and GluN3A-B). Assembly of these subunits within the families generates several receptor subtypes.
Figure 2.
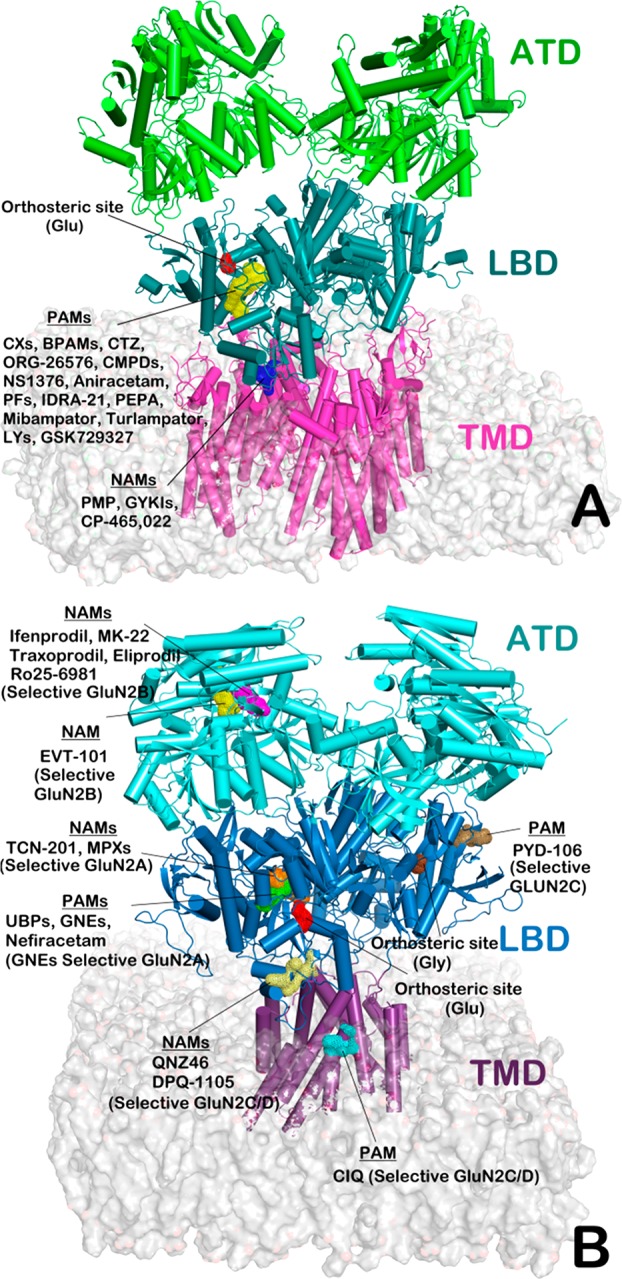
Structural view of AMPA (A) and NMDA (B) receptors highlighting known allosteric binding sites for PAMs and NAMs. (Pictures were generated by PyMOL using the PDBs 5WEO (AMPAR) and 4PE5 (NMDAR) and the information in ref (9).)
Although the complex roles of iGluRs are not completely understood, they are undoubtedly involved in key brain functions including learning and memory formation. Consequently, their malfunctioning is associated with neurological disorders including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease, and amyotrophic lateral sclerosis (ALS), but also with psychiatric disorders such as schizophrenia, depression (NMDARs), and epilepsy (NMDARs and AMPARs).8 Among KARs, the GluK1 receptor was demonstrated to be involved in pain signaling.10 It is important to point out that iGluRs display a series of unique features, which render their modulation a challenging task. First, the iGluRs are not expressed in the brain as well-defined subtype populations, but rather as a mixture of subtypes. To complicate this picture, the changes in their expression pattern during development are substantial, also encompassing alterations in channel subtype composition that finally lead to receptors with different functional properties.
Two main issues are related to the development of clinically useful small-molecules targeting iGluRs: (i) the ability of a compound to bind specific receptor subtypes involved in a given disorder; (ii) the risk of off-target effects, by orthosteric ligands, due to the similarities of l-Glu binding pockets among iGluRs. Therefore, allosteric modulation of iGluRs represents a reliable and promising strategy for achieving subfamily- and subtype-specific binding.
In recent years, the classical development of molecules that act as competitive ligands at the orthosteric sites of iGluRs10 has been growingly flanked by the allosteric modulation of iGluRs in the quest for novel therapies.9 The significant improvements achieved through the key structural studies performed to identify allosteric sites on iGluRs were pivotal for the development of improved allosteric modulators. Accordingly, the AMPARs NAM perampanel (PMP, Eisai) and the NMDARs NAM felbamate (MedPointe) have been marketed for treating epilepsy, while the AMPARs PAM aniracetam (Roche) is administered for treating cognitive disorders.1
As reported in Figure 2, several allosteric binding sites on AMPARs and NMDARs have been discovered in the last years, and different molecules (Table 1) have demonstrated ability to modulate iGluRs by targeting these sites. Some modulators bind the LBD dimer interface of AMPARs GluA2 (Figure 2). The PAMs aniracetam, CXs, and BPAMs bind next to a hinge region of the “clamshell”, thus stabilizing AMPARs’ closed conformation (l-Glu bound). These molecules slow down channel deactivation by increasing the affinity of the receptor for l-Glu. On the contrary, compounds such as cyclothiazide (CTZ) act as PAMs by stabilizing the LBD dimer, thus inhibiting receptor desensitization. BPAMs exert their action by binding the dimer interface at the LBD. NAMs such as PMP and GYKIs target the linker between the LBD and the TMD of AMPARs (Figure 2).9
Table 1. Representative Allosteric Modulators for iGluRs.

Regarding NMDARs (GluN1 and GluN2), the LBD is also responsive to allosteric modulation. TCN-201 and MPXs act as NAMs by targeting a site at the dimer interface. PAMs of the NMDARs targeting the LBD have been identified, and their binding site partially overlaps that of NAMs. In fact, X-ray studies revealed that GNEs (PAMs) share a portion of the TCN-201 and MPXs (NAMs) binding site. Other NAMs (DQP-1105 and QNZ46) target a different region in proximity of the LBD. The ATD can be targeted for obtaining an allosteric modulation of the NMDARs. Small-molecules such as ifenprodil, MK-22, and EVT-101 act as NAMs by binding the ATD subunit interface of GluN1 and GluN2B causing clamshell closure, decreasing the probability of channel opening. It is very impressive how, upon binding of these molecules to the ATD a regulation of the channel region, distant over 100 Å from their allosteric binding site, can be attained. In addition, the TMD binds different PAMs (CIQ, endogenous neurosteroids, and pregnenolone sulfate).1 Recently, BPAMs were characterized as KARs PAMs targeting the homologous site of AMPARs for PAMs. The selective allosteric modulation of KARs could be exploited for developing effective drugs for improving cognitive functions.
Although novel modulators displaying preferential binding for a given subunit have been recently disclosed (GluN2C-selective PAM, PYD-106; GluN2A-selective PAMs, GNEs),9 the selective modulation of NMDAR subtypes, especially for GluN2 is still challenging. The design of subunit-selective allosteric modulators of NMDARs may overcome this drawback.
On the wave of the success gained with the approved AMPARs allosteric modulators, other iGluRs allosteric ligands have been advanced to clinic. However, most of the identified clinical candidates failed to provide efficacy. Clinical trials with AMPARs NAM, PMP, were suspended due to its inefficacy in treating alcohol dependence (NCT02120365), although the same compound displayed efficacy for the treatment of epilepsy.1 AMPARs NAM talampanel (GYKI-53773 also named LY300164, Table 1) underwent phase II clinical trials for ALS (NCT00696332, NCT00982150), epilepsy (NCT00034814), glioblastoma multiforme (NCT00267592), PD (NCT00004576), and advanced PD stage (NCT00036296) and for assessing the improvement in brain functions (NCT00057460). Talampanel did not show any therapeutic effects in ALS, while in the other trials it showed some benefits but was discontinued due to its poor pharmacokinetic profile and adverse effects. A series of AMPARs PAM have been advanced in clinical studies. Although ORG-26576 failed in phase II for major depressive disorder (NCT00610649), it gave encouraging results in phase II for the treatment of attention deficit hyperactivity disorder (ADHD) (NCT00610441). CX 516 failed to demonstrate efficacy against schizophrenia, mild cognitive impairment, autism, or AD.11 CX 717 demonstrated some efficacy in ADHD patients and in subject with respiratory depression.11 CX 691 (farampator) failed to demonstrate efficacy in patients with major depression.11 Lilly, Organon, and GSK have also progressed molecules into the clinic; however, up to now, only negative data have been reported. PF-4958242 failed to show efficacy in treating age-related hearing loss.11 LY451395 (mibampator) did not show any benefits in patients with AD (NCT00843518; NCT00051909). GSK729327, a AMPARs PAM (Table 1), was investigated in phase I in patients with schizophrenia (NCT00448890). This compound was expected to improve cognitive measures in schizophrenic patients with acceptable safety. Due to the limited positive output, the study was discontinued. AMPARs PAM S 47445 (Table 1) is currently under investigation in phase II of clinical studies in AD and in major depressive disorder (NCT02626572; NCT02805439).
Furthermore, also NMDARs allosteric modulators have been progressed into clinical studies. Clinical trials for ifenprodil (NMDAR NAM) were discontinued due to its blockage of G protein-activated inwardly rectifying K+ channels (GIRK) and to its interaction with numerous off-targets, including voltage-gated calcium channels (VGCCs), 5-HT3, and α1 adrenergic.11,12 GluN2B NAM radiprodil (Table 1), a second generation of “prodil” containing molecules, showed good potency with enhanced oral bioavailability compared with earlier agents (i.e., ifenprodil) and has been advanced into clinic by different pharma companies. Clinical trials with radiprodil, investigating the treatment of infantile spasms (IS) (NCT02829827) and diabetic peripheral neuropathic pain (NCT00838799) have been started. Unfortunately, UCB Biopharma has suspended these trials after reviewing the feasibility and estimated completion date for the treatment of IS. Due to the lack of significant reduction in daily pain scores, also the investigation for the neuropathic pain was terminated. Another GluN2B NAM, EVT-101 (Table 1), was engaged in two trials, for enhancing cognitive functions in AD (NCT00526968) and for the potential therapy of treatment-resistant major depression (NCT01128452), but without success. NMDARs PAM nefiracetam was advanced in phase II for the treatment of AD, but the study was suspended due to the limited observed benefits (NCT00001933). Two NMDARs allosteric modulators with undisclosed structures, SAGE-718 (Sage therapeutics, PAM) and AGN-241751 (Allergan), are currently under clinical trials in phase I (NCT03771586, NCT03787758) and phase II (major depressive disorders, NCT03586427), respectively.
It needs to be said that the high level of attrition that has hampered the approval of iGluRs allosteric modulators might have also been hurdled by the particular design of the clinical studies in which they were engaged. In the case of NCT02829827, radiprodil was in fact engaged in a study for the treatment of a particularly resistant form of infantile epilepsy, and since only three patients were recruited, the study was discontinued. The failure of GluN2B NMDA allosteric modulators (e.g., radiprodil) in clinical studies for the treatment of diabetic neuropathy might be due to the lack of evidence for an increase in NMDARs activity in the spinal cord in this particular kind of neuropathic pain.13 More in depth studies would be needed but, based on several evidence,13 the GluN2B subunit may appear as a questionable target for the treatment of the diabetic neuropathy. However, understanding the reasons why a number of iGluRs allosteric ligands failed to provide favorable clinical outcomes represents a significant challenge.
A viable, fascinating, and alternative option to face this issue might be offered by the possibility of regulating AMPARs activity by interfering with regulatory proteins acting as receptor auxiliary subunits. The excitatory synaptic transmission of AMPARs has been recently demonstrated to be modulated by AMPAR auxiliary subunits including auxiliary transmembrane AMPAR regulatory proteins also known as TARPs and cornichon-2/3 (CNIH-2/3), that regulate AMPAR protein levels, pharmacology, trafficking, and gating. TARPs can be clustered in type I (γ-2, γ-3, γ-4, and γ-8) and type II (γ-5 and γ-7) by sequence homology. Based on the differences in modulating AMPARs (i.e., receptor gating, rectification, resensitization, and pharmacological properties), type I TARPs are also subdivided in type Ia (γ-2 and γ-3) and type Ib (γ-4 and γ-8). Notably, type I TARPs modulate all AMPAR subunits including heteromeric AMPARs, while type II TARPs modulate AMPAR functions in an AMPAR subunit-dependent manner. The GluA2-γ-2 complex was experimentally solved by cryo-electron microscopy (cryo-EM) approaches providing further insight about activated and desensitized GluA2-γ-2 complexes and TARPs gating modulation of AMPAR. Although all neuronal types express TARPs, the family members show distinct expression patterns in the brain (type I TARPs: γ-2, cerebellum; γ-3, cerebral cortex; γ-4, low in adult with some enrichment in striatum; γ-8, forebrain with enrichment in hippocampus; type II TARPs: γ-5, olfactory bulb, hippocampal CA2, cerebellum; γ-7, cerebellum). The identification of the TARPs and their different regional expression across the CNS provide an opportunity to identify AMPAR indirect antagonists selectively targeting specific brain regions. Accordingly, TARP γ-2 is highly expressed within the cerebellum, which is involved in motor coordination, while TARP γ-8 is highly expressed within the hippocampus, a primary locus of epileptogenic activity. Of particular interest was the very low expression of TARP γ-8 in the cerebellum. Therefore, the identification of a TARP γ-8-dependent AMPAR antagonist can provide antiepileptic efficacy without motor impairment. Gardinier and co-workers54 discovered a compound, LY3130481/CERC-611 (Figure 3), as a potent antagonist of recombinant γ-8-containing AMPARs, but not AMPARs associated with other TARPs/CNIH-2, or AMPARs without auxiliary subunits. Consistent with the expression profile of γ-8, the γ-8-selective antagonist (LY3130481/CERC-611) potently blocked AMPARs functions in hippocampal and cortical neurons, but not in cerebellar Purkinje or red nucleus neurons in rodents. The compound also blocks hippocampal excitatory synaptic transmission in rodents and it is orally bioavailable. LY3130481/CERC-611 showed anticonvulsant activity in animal model of epilepsy (mesial temporal lobe epilepsy model induced by KA) without motor side effects. JNJ-55511118 and JNJ-56022486 (Figure 3) are structurally distinct compounds with comparable pharmacological properties. Both JNJ-5551118 and LY3130481/CERC-611 showed wake-promoting effects and relatively mild cognitive decline in different rodent models of epilepsy. For these reasons, LY3130481/CERC-611 is currently under clinical development for epilepsy.55 Another key functional role of TARPs is their modulation of various aspects of AMPARs pharmacology. TARPs regulate the efficacy of KA and CNQX (competitive AMPARs/KARs antagonist), GYKI potency, l-Glu affinity, CTZ specificity, and efficacy for KA-evoked currents and polyamine efficacy.
Figure 3.

Chemical structures of γ-8 TARP-dependent-AMPARs antagonists (LY3130481/CERC-611, JNJ-55511118, and JNJ-56022486) and γ-2 TARP AMPARs modulators (VU0612951, VU0627849, and VU0539491).
The different auxiliary subunit families for AMPARs so far identified can represent interesting targets for modulating AMPARs functions. Azumaya and collaborators56 identified PAMs and NAMs typified by VU0627849 and VU0612951 (Figure 3), respectively, which were slightly more potent at TARP γ-2- or CNIH-3-containing GluA2 than at GluA2 alone. They also discovered VU0539491 (Figure 3), a PAM of γ-2 containing, but a NAM for CNIH-3 containing GluA2 AMPARs. Positive or negative AMPARs modulation through interference with auxiliary subunits represents a challenging and innovative approach to drug discovery in this field. Progress in structural biology for the resolution of iGluRs in complex with auxiliary subunits in the next future will pave the way to innovative approaches to treat CNS-related disorders.
This kind of modulation of iGluRs features a series of benefits including the increased diversity of the chemical space, regional specificity of drugs due to the defined localization of specific auxiliary subunits, and an increased possibility of identifying specific modulators for the protein complex with close structural homology. The success in identifying LY3130481/CERC-611, which selectively antagonizes the AMPARs associated with TARP γ-8, and the growing database of auxiliary proteins can open new avenues for the discovery of new drugs improving patient outcomes mainly in chronic disease states like epilepsy.57,58
As introduced in the previous paragraphs, the advent of specific research tools and techniques represents a glimmer for acquiring improved knowledge on allosteric modulation of iGluRs. This will potentially reinvigorate efforts to develop novel ligands with improved efficacy and safety toward several CNS disorders. The impressive breakthroughs in crystallography59 for hunting allosteric binding sites and the growing progress in the resolution of receptor/modulators cocrystal complexes will enable the rational design of allosteric modulators endowed with relevant affinity and selectivity (structure-based techniques). Notably, the improvements in the armamentarium of the available constructs for crystallographic studies for exploring allosteric sites have enabled the in-depth study of the “real” interaction of allosteric modulators at AMPARs and NMDARs by a snapshot of a particular receptors’ conformational states (see ref (60) and references therein). These outcomes hold exceptional potentialities for the future design of novel “state-specific ligands”. Along with the possibility to obtain comprehensive structural data, new tools and methods are appearing in this scenario (Table 2). Databases related to the allosteric modulators can assist researchers working in this field to facilitate the allosteric drug discovery, including:
Allosteric database (http://mdl.shsmu.edu.cn/ASD/) encompassing over 77,000 allosteric modulators;
AlloSigMA (http://allosigma.bii.a-star.edu.sg/home/) for estimating the allosteric ligand free energy;
AlloFinder (http://mdl.shsmu.edu.cn/ALF/) for understanding the role of metabolites in the pathways of allosteric feedback;
GPCRdb (http://gpcrdb.org/) database regarding the GPCR families and their ligands, including allosteric modulators.
Table 2. Potential Methods for Finding Allosteric Modulators.
| strategies | methods |
|---|---|
| crystallography | X-ray, cryo-EM |
| biochemistry/pharmacology | NMR-(fragment) screening, second harmonic generation (SHG) technology, high throughput screening, high-throughput fluorescent membrane potential screen, medium-throughput electrophysiology, fluorescence calcium assays, functional assays (competition binding technologies), affinity mass spectrometry assay |
| in silico | molecular dynamics, ensemble docking, ligand-based (i.e., proteochemometric modeling) and structure-based (i.e., reverse perturbation approach) methods for virtual screening, allosteric databases |
The growing knowledge in the field of structural and chemical biology, as well as pharmacology, along with the progress in computational methods and the increasing availability of bioinformatics resources will speed up the discovery of novel druggable allosteric sites and the development of novel allosteric modulators with higher efficacy. Additionally, innovative and appealing approaches are arising. Among them, the use of specific allosteric antibodies represents an important pitch for future research in this field.1
Furthermore, an interesting and feasible approach toward the identification of allosteric modulators by the discovery of hidden allosteric sites (HASs) is worthy of mention in this Innovations.61 The concept is that HASs of potential targets are not visible in ligand-unbound crystal structures but that they become evident in ligand-bound crystal structures when a specific ligand binds and stabilizes a unique conformation. The identification of these sites is a fascinating challenge; it offers a new opportunity for searching allosteric drugs against human diseases and could be extended to iGluRs to identify new drugs for brain diseases. This approach has been already successfully applied to other targets such as those related to cancer. To identify HASs in proteins, several computational and experimental strategies have been developed as in the case of the HAS of K-Ras.61 The G12C mutation of K-Ras4B is one of the most frequent Ras mutations found in 20% of nonsmall cell lung cancers. The presence of the Cys12 at the P-loop domain of the K-Ras4B G12C mutants led to a screen a library of tethering compounds (disulfide-based compounds, carbon-based electrophiles, vinyl sulfonamides, and acrylamides) to irreversibly bind this residue. The subsequent crystallographic studies showed that these tethering compounds covalently bind to an adjacent pocket comprising mainly the switch-II domain (so-called the switch-II pocket, S-IIP). S-IIP does not overlap with the GDP- or GTP-binding site; therefore, it can be considered an allosteric site. The comparison of the crystal structure of the acrylamide covalently bound to K-Ras4B G12C with the structures of the complexes GDP/K-Ras4B and GTP/K-Ras4B displayed that the S-IIP is not visible in the GDP- and GTP-bound mK-Ras4B. Thus, S-IIP represents a HAS that can be observed upon the conformational changes of K-Ras4B during the GTP-to-GDP hydrolysis.
Another innovative and intriguing emerging approach is the possibility to develop covalent allosteric modulators.62 This strategy is interesting for overcoming resistance to drugs as in the case of the antiapoptotic protein Mcl-1.62 With the aim of finding an alternative mechanism for Mcl-1 inhibition by a rational design approach, a small-molecule compound named Mcl-1 allosteric inhibitor molecule 1 (MAIM1) was identified. This compound was able to bind Cys286, which is a residue belonging to the new allosteric site distant from the active site. MAIM1 covalently modified Cys286 at a helix α6 regulatory site positioned on the opposite side of the protein from the canonical BH3-binding cleft. Remarkably, C286S mutagenesis abolished the effect of MAIM1 on the inhibitory activity of Mcl-1 in the presence of the proapoptotic BID BH3 domain, highlighting the requirement of Cys286 for MAIM1 activity. This approach may offer a novel possibility for the allosteric inhibition of a protein and could be translated to iGluRs. Accordingly, the design of appropriate allosteric ligands targeting a site containing a cysteine residue by forming a covalent adduct could offer several advantages over classical allosteric modulation (high potency, extended duration of action, and low drug resistance).
In conclusion, the allosteric modulation of iGluRs could be a robust and innovative approach toward targeted therapies, with a special focus on CNS-related disorders. A few pivotal steps have already been made in the field of allosteric pharmacology, which allowed relevant advances in the understanding of the “allosteric world”. Further progresses, prompted by the growing availability of novel tools and techniques, are forecast to be reported in the next years. The substantial opportunities offered by the allosteric modulation of iGluRs can provide in the next decade several allosteric clinical drug candidates for treating CNS-related diseases.14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53
Glossary
ABBREVIATIONS
- 5-HT
5-hydroxytryptamine receptor
- AMPARs
(S)-2-amino-3-(5-methyl-3-hydroxyisoxazol-4-yl)propanoic acid receptors
- CaSR
calcium-sensing receptor
- CCR5
CC chemokine receptor 5
- GPCRs
G-protein coupled receptors
- DETQ
2-(2,6-dichlorophenyl)-1-((1S,3R)-3-(hydroxymethyl)-5-(2-hydroxypropan-2-yl)-1-methyl-3,4-dihydroisoquinolin-2(1H)-yl)ethan-1-one
- HAS
hidden allosteric site
- KARs
kainate receptors
- iGluRs
ionotropic glutamate receptors
- mGluR
metabotropic glutamate receptor
- NAMs
negative allosteric modulators
- NMDARs
N-methyl-d-aspartate receptors
- nAChRs
nicotinic acetylcholine receptors
- PAMs
positive allosteric modulators
- TARPs
transmembrane AMPAR regulatory proteins
- VGCCs
voltage-gated calcium channels
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Changeux J. P.; Christopoulos A. Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell 2016, 166, 1084–1102. 10.1016/j.cell.2016.08.015. [DOI] [PubMed] [Google Scholar]
- Johnstone S.; Albert J. S. Pharmacological property optimization for allosteric ligands: A medicinal chemistry perspective. Bioorg. Med. Chem. Lett. 2017, 27, 2239–2258. 10.1016/j.bmcl.2017.03.084. [DOI] [PubMed] [Google Scholar]
- Nussinov R.; Tsai C. J.; Csermely P. Allo-network drugs: harnessing allostery in cellular networks. Trends Pharmacol. Sci. 2011, 32, 686–693. 10.1016/j.tips.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster D. J.; Conn P. J. Allosteric Modulation of GPCRs: New Insights and Potential Utility for Treatment of Schizophrenia and Other CNS Disorders. Neuron 2017, 94, 431–446. 10.1016/j.neuron.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns R. F.; Mitchell S. N.; Wafford K. A.; Harper A. J.; Shanks E. A.; Carter G.; O’Neill M. J.; Murray T. K.; Eastwood B. J.; Schaus J. M.; Beck J. P.; Hao J.; Witkin J. M.; Li X.; Chernet E.; Katner J. S.; Wang H.; Ryder J. W.; Masquelin M. E.; Thompson L. K.; Love P. L.; Maren D. L.; Falcone J. F.; Menezes M. M.; Zhang L.; Yang C. R.; Svensson K. A. Preclinical profile of a dopamine D1 potentiator suggests therapeutic utility in neurological and psychiatric disorders. Neuropharmacology 2018, 128, 351–365. 10.1016/j.neuropharm.2017.10.032. [DOI] [PubMed] [Google Scholar]
- Rossi M.; Fasciani I.; Marampon F.; Maggio R.; Scarselli M. The First Negative Allosteric Modulator for Dopamine D2 and D3 Receptors, SB269652 May Lead to a New Generation of Antipsychotic Drugs. Mol. Pharmacol. 2017, 91, 586–594. 10.1124/mol.116.107607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias H. R. Positive and negative modulation of nicotinic receptors. Adv. Protein Chem. Struct. Biol. 2010, 80, 153–203. 10.1016/B978-0-12-381264-3.00005-9. [DOI] [PubMed] [Google Scholar]
- Traynelis S. F.; Wollmuth L. P.; McBain C. J.; Menniti F. S.; Vance K. M.; Ogden K. K.; Hansen K. B.; Yuan H.; Myers S. J.; Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–96. 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen K. B.; Yi F.; Perszyk R. E.; Furukawa H.; Wollmuth L. P.; Gibb A. J.; Traynelis S. F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogi S.; Brindisi M.; Butini S.; Kshirsagar G. U.; Maramai S.; Chemi G.; Gemma S.; Campiani G.; Novellino E.; Fiorenzani P.; Pinassi J.; Aloisi A. M.; Gynther M.; Venskutonyte R.; Han L.; Frydenvang K.; Kastrup J. S.; Pickering D. S. (S)-2-Amino-3-(5-methyl-3-hydroxyisoxazol-4-yl)propanoic Acid (AMPA) and Kainate Receptor Ligands: Further Exploration of Bioisosteric Replacements and Structural and Biological Investigation. J. Med. Chem. 2018, 61, 2124–2130. 10.1021/acs.jmedchem.8b00099. [DOI] [PubMed] [Google Scholar]
- Reuillon T.; Ward S. E.; Beswick P.. 7.11 - Modulating AMPA Receptors for the Treatment of CNS Disorders. In Comprehensive Medicinal Chemistry III; Chackalamannil S., Rotella D., Ward S. E., Eds.; Elsevier: Oxford, 2017; pp 447–480. [Google Scholar]
- Kobayashi T.; Washiyama K.; Ikeda K. Inhibition of G protein-activated inwardly rectifying K+ channels by ifenprodil. Neuropsychopharmacology 2006, 31, 516–24. 10.1038/sj.npp.1300844. [DOI] [PubMed] [Google Scholar]
- Zhou H. Y.; Chen S. R.; Pan H. L. Targeting N-methyl-D-aspartate receptors for treatment of neuropathic pain. Expert Rev. Clin. Pharmacol. 2011, 4, 379–388. 10.1586/ecp.11.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menniti F. S.; Shah A. K.; Williams S. A.; Wilner K. D.; White W. F.; Chenard B. L. CP-101,606: An NR2B-Selective NMDA Receptor Antagonist. CNS Drug Rev. 1998, 4, 307–322. 10.1111/j.1527-3458.1998.tb00073.x. [DOI] [PubMed] [Google Scholar]
- Bristow L. J.; Gulia J.; Weed M. R.; Srikumar B. N.; Li Y. W.; Graef J. D.; Naidu P. S.; Sanmathi C.; Aher J.; Bastia T.; Paschapur M.; Kalidindi N.; Kumar K. V.; Molski T.; Pieschl R.; Fernandes A.; Brown J. M.; Sivarao D. V.; Newberry K.; Bookbinder M.; Polino J.; Keavy D.; Newton A.; Shields E.; Simmermacher J.; Kempson J.; Li J.; Zhang H.; Mathur A.; Kallem R. R.; Sinha M.; Ramarao M.; Vikramadithyan R. K.; Thangathirupathy S.; Warrier J.; Islam I.; Bronson J. J.; Olson R. E.; Macor J. E.; Albright C. F.; King D.; Thompson L. A.; Marcin L. R.; Sinz M. Preclinical Characterization of (R)-3-((3S,4S)-3-fluoro-4-(4-hydroxyphenyl)piperidin-1-yl)-1-(4-methylbenzyl)pyrr olidin-2-one (BMS-986169), a Novel, Intravenous, Glutamate N-Methyl-d-Aspartate 2B Receptor Negative Allosteric Modulator with Potential in Major Depressive Disorder. J. Pharmacol. Exp. Ther. 2017, 363, 377–393. 10.1124/jpet.117.242784. [DOI] [PubMed] [Google Scholar]
- Stroebel D.; Buhl D. L.; Knafels J. D.; Chanda P. K.; Green M.; Sciabola S.; Mony L.; Paoletti P.; Pandit J. A Novel Binding Mode Reveals Two Distinct Classes of NMDA Receptor GluN2B-selective Antagonists. Mol. Pharmacol. 2016, 89, 541–551. 10.1124/mol.115.103036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G.; Mutel V.; Trube G.; Malherbe P.; Kew J. N.; Mohacsi E.; Heitz M. P.; Kemp J. A. Ro 25–6981, a highly potent and selective blocker of N-methyl-D-aspartate receptors containing the NR2B subunit. Characterization in vitro. J. Pharmacol. Exp. Ther. 1997, 283, 1285–92. [PubMed] [Google Scholar]
- Monaghan D. T.; Irvine M. W.; Costa B. M.; Fang G.; Jane D. E. Pharmacological modulation of NMDA receptor activity and the advent of negative and positive allosteric modulators. Neurochem. Int. 2012, 61, 581–592. 10.1016/j.neuint.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann R. A.; Fanger C. M.; Anderson D. R.; Sirivolu V. R.; Paschetto K.; Gordon E.; Virginio C.; Gleyzes M.; Buisson B.; Steidl E.; Mierau S. B.; Fagiolini M.; Menniti F. S. MPX-004 and MPX-007: New Pharmacological Tools to Study the Physiology of NMDA Receptors Containing the GluN2A Subunit. PLoS One 2016, 11, e0148129. 10.1371/journal.pone.0148129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter C. J.; Lloyd K. G.; Zivkovic B.; Scatton B. Ifenprodil and SL 82.0715 as cerebral antiischemic agents. III. Evidence for antagonistic effects at the polyamine modulatory site within the N-methyl-D-aspartate receptor complex. J. Pharmacol. Exp. Ther. 1990, 253, 475–482. [PubMed] [Google Scholar]
- Mony L.; Kew J. N.; Gunthorpe M. J.; Paoletti P. Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br. J. Pharmacol. 2009, 157, 1301–1317. 10.1111/j.1476-5381.2009.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weed M. R.; Bookbinder M.; Polino J.; Keavy D.; Cardinal R. N.; Simmermacher-Mayer J.; Cometa F. N.; King D.; Thangathirupathy S.; Macor J. E.; Bristow L. J. Negative Allosteric Modulators Selective for The NR2B Subtype of The NMDA Receptor Impair Cognition in Multiple Domains. Neuropsychopharmacology 2016, 41, 568–577. 10.1038/npp.2015.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa B. M.; Irvine M. W.; Fang G.; Eaves R. J.; Mayo-Martin M. B.; Skifter D. A.; Jane D. E.; Monaghan D. T. A novel family of negative and positive allosteric modulators of NMDA receptors. J. Pharmacol. Exp. Ther. 2010, 335, 614–621. 10.1124/jpet.110.174144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackos D. H.; Lupardus P. J.; Grand T.; Chen Y.; Wang T. M.; Reynen P.; Gustafson A.; Wallweber H. J.; Volgraf M.; Sellers B. D.; Schwarz J. B.; Paoletti P.; Sheng M.; Zhou Q.; Hanson J. E. Positive Allosteric Modulators of GluN2A-Containing NMDARs with Distinct Modes of Action and Impacts on Circuit Function. Neuron 2016, 89, 983–999. 10.1016/j.neuron.2016.01.016. [DOI] [PubMed] [Google Scholar]
- Moriguchi S.; Shioda N.; Maejima H.; Zhao X.; Marszalec W.; Yeh J. Z.; Fukunaga K.; Narahashi T. Nefiracetam potentiates N-methyl-D-aspartate (NMDA) receptor function via protein kinase C activation and reduces magnesium block of NMDA receptor. Mol. Pharmacol. 2006, 71, 580–587. 10.1124/mol.106.027607. [DOI] [PubMed] [Google Scholar]
- Volgraf M.; Sellers B. D.; Jiang Y.; Wu G.; Ly C. Q.; Villemure E.; Pastor R. M.; Yuen P. W.; Lu A.; Luo X.; Liu M.; Zhang S.; Sun L.; Fu Y.; Lupardus P. J.; Wallweber H. J.; Liederer B. M.; Deshmukh G.; Plise E.; Tay S.; Reynen P.; Herrington J.; Gustafson A.; Liu Y.; Dirksen A.; Dietz M. G.; Wang T. M.; Hanson J. E.; Hackos D.; Scearce-Levie K.; Schwarz J. B. Discovery of GluN2A-Selective NMDA Receptor Positive Allosteric Modulators (PAMs): Tuning Deactivation Kinetics via Structure-Based Design. J. Med. Chem. 2016, 59, 2760–2779. 10.1021/acs.jmedchem.5b02010. [DOI] [PubMed] [Google Scholar]
- Villemure E.; Volgraf M.; Jiang Y.; Wu G.; Ly C. Q.; Yuen P. W.; Lu A.; Luo X.; Liu M.; Zhang S.; Lupardus P. J.; Wallweber H. J.; Liederer B. M.; Deshmukh G.; Plise E.; Tay S.; Wang T. M.; Hanson J. E.; Hackos D. H.; Scearce-Levie K.; Schwarz J. B.; Sellers B. D. GluN2A-Selective Pyridopyrimidinone Series of NMDAR Positive Allosteric Modulators with an Improved in Vivo Profile. ACS Med. Chem. Lett. 2017, 8, 84–89. 10.1021/acsmedchemlett.6b00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. M.; Brown B. M.; Deng L.; Sellers B. D.; Lupardus P. J.; Wallweber H. J. A.; Gustafson A.; Wong E.; Volgraf M.; Schwarz J. B.; Hackos D. H.; Hanson J. E. A novel NMDA receptor positive allosteric modulator that acts via the transmembrane domain. Neuropharmacology 2017, 121, 204–218. 10.1016/j.neuropharm.2017.04.041. [DOI] [PubMed] [Google Scholar]
- Yang C. R.; Svensson K. A. Allosteric modulation of NMDA receptor via elevation of brain glycine and D-serine: the therapeutic potentials for schizophrenia. Pharmacol. Ther. 2008, 120, 317–332. 10.1016/j.pharmthera.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Rogawski M. A. Revisiting AMPA receptors as an antiepileptic drug target. Epilepsy Curr. 2011, 11, 56–63. 10.5698/1535-7511-11.2.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro J. T.; Paternain A. V.; Lerma J.; Chenard B. L.; Ewing F. E.; Huang J.; Welch W. M.; Ganong A. H.; Menniti F. S. Functional characterization of CP-465,022, a selective, noncompetitive AMPA receptor antagonist. Neuropharmacology 2002, 42, 143–153. 10.1016/S0028-3908(01)00170-8. [DOI] [PubMed] [Google Scholar]
- Donevan S. D.; Rogawski M. A. Allosteric regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionate receptors by thiocyanate and cyclothiazide at a common modulatory site distinct from that of 2,3-benzodiazepines. Neuroscience 1998, 87, 615–629. 10.1016/S0306-4522(98)00109-2. [DOI] [PubMed] [Google Scholar]
- Balannik V.; Menniti F. S.; Paternain A. V.; Lerma J.; Stern-Bach Y. Molecular mechanism of AMPA receptor noncompetitive antagonism. Neuron 2005, 48, 279–288. 10.1016/j.neuron.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Belayev L.; Alonso O. F.; Liu Y.; Chappell A. S.; Zhao W.; Ginsberg M. D.; Busto R. Talampanel, a novel noncompetitive AMPA antagonist, is neuroprotective after traumatic brain injury in rats. J. Neurotrauma 2001, 18, 1031–1038. 10.1089/08977150152693728. [DOI] [PubMed] [Google Scholar]
- Ruel J.; Bobbin R. P.; Vidal D.; Pujol R.; Puel J. L. The selective AMPA receptor antagonist GYKI 53784 blocks action potential generation and excitotoxicity in the guinea pig cochlea. Neuropharmacology 2000, 39, 1959–1973. 10.1016/S0028-3908(00)00069-1. [DOI] [PubMed] [Google Scholar]
- Pirotte B.; Francotte P.; Goffin E.; de Tullio P. AMPA receptor positive allosteric modulators: a patent review. Expert Opin. Ther. Pat. 2013, 23, 615–628. 10.1517/13543776.2013.770840. [DOI] [PubMed] [Google Scholar]
- Krintel C.; Harpsoe K.; Zachariassen L. G.; Peters D.; Frydenvang K.; Pickering D. S.; Gajhede M.; Kastrup J. S. Structural analysis of the positive AMPA receptor modulators CX516 and Me-CX516 in complex with the GluA2 ligand-binding domain. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1645–1652. 10.1107/S0907444913011839. [DOI] [PubMed] [Google Scholar]
- Woolley M. L.; Waters K. A.; Gartlon J. E.; Lacroix L. P.; Jennings C.; Shaughnessy F.; Ong A.; Pemberton D. J.; Harries M. H.; Southam E.; Jones D. N.; Dawson L. A. Evaluation of the pro-cognitive effects of the AMPA receptor positive modulator, 5-(1-piperidinylcarbonyl)-2,1,3-benzoxadiazole (CX691), in the rat. Psychopharmacology (Berl) 2009, 202, 343–354. 10.1007/s00213-008-1325-2. [DOI] [PubMed] [Google Scholar]
- Hampson R. E.; Espana R. A.; Rogers G. A.; Porrino L. J.; Deadwyler S. A. Mechanisms underlying cognitive enhancement and reversal of cognitive deficits in nonhuman primates by the ampakine CX717. Psychopharmacology (Berl) 2009, 202, 355–369. 10.1007/s00213-008-1360-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak C. P.; Ahmed A. H.; Oswald R. E. Probing the allosteric modulator binding site of GluR2 with thiazide derivatives. Biochemistry 2009, 48, 8594–8602. 10.1021/bi901127s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francotte P.; Goffin E.; Fraikin P.; Lestage P.; Van Heugen J. C.; Gillotin F.; Danober L.; Thomas J. Y.; Chiap P.; Caignard D. H.; Pirotte B.; de Tullio P. New fluorinated 1,2,4-benzothiadiazine 1,1-dioxides: discovery of an orally active cognitive enhancer acting through potentiation of the 2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl)propionic acid receptors. J. Med. Chem. 2010, 53, 1700–1711. 10.1021/jm901495t. [DOI] [PubMed] [Google Scholar]
- Larsen A. P.; Fievre S.; Frydenvang K.; Francotte P.; Pirotte B.; Kastrup J. S.; Mulle C. Identification and Structure-Function Study of Positive Allosteric Modulators of Kainate Receptors. Mol. Pharmacol. 2017, 91, 576–585. 10.1124/mol.116.107599. [DOI] [PubMed] [Google Scholar]
- Norholm A. B.; Francotte P.; Olsen L.; Krintel C.; Frydenvang K.; Goffin E.; Challal S.; Danober L.; Botez-Pop I.; Lestage P.; Pirotte B.; Kastrup J. S. Synthesis, pharmacological and structural characterization, and thermodynamic aspects of GluA2-positive allosteric modulators with a 3,4-dihydro-2H-1,2,4-benzothiadiazine 1,1-dioxide scaffold. J. Med. Chem. 2013, 56, 8736–8745. 10.1021/jm4012092. [DOI] [PubMed] [Google Scholar]
- Krintel C.; Francotte P.; Pickering D. S.; Juknaite L.; Pohlsgaard J.; Olsen L.; Frydenvang K.; Goffin E.; Pirotte B.; Kastrup J. S. Enthalpy-Entropy Compensation in the Binding of Modulators at Ionotropic Glutamate Receptor GluA2. Biophys. J. 2016, 110, 2397–2406. 10.1016/j.bpj.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin E.; Drapier T.; Larsen A. P.; Geubelle P.; Ptak C. P.; Laulumaa S.; Rovinskaja K.; Gilissen J.; Tullio P.; Olsen L.; Frydenvang K.; Pirotte B.; Hanson J.; Oswald R. E.; Kastrup J. S.; Francotte P. 7-Phenoxy-Substituted 3,4-Dihydro-2H-1,2,4-benzothiadiazine 1,1-Dioxides as Positive Allosteric Modulators of alpha-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid (AMPA) Receptors with Nanomolar Potency. J. Med. Chem. 2018, 61, 251–264. 10.1021/acs.jmedchem.7b01323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen G. B.; Harbak B.; Hede S. E.; Gouliaev A. H.; Olsen L.; Frydenvang K.; Egebjerg J.; Kastrup J. S.; Holm M. M. A novel dualistic profile of an allosteric AMPA receptor modulator identified through studies on recombinant receptors, mouse hippocampal synapses and crystal structures. Neuroscience 2015, 310, 709–722. 10.1016/j.neuroscience.2015.09.073. [DOI] [PubMed] [Google Scholar]
- Shaffer C. L.; Hurst R. S.; Scialis R. J.; Osgood S. M.; Bryce D. K.; Hoffmann W. E.; Lazzaro J. T.; Hanks A. N.; Lotarski S.; Weber M. L.; Liu J.; Menniti F. S.; Schmidt C. J.; Hajos M. Positive allosteric modulation of AMPA receptors from efficacy to toxicity: the interspecies exposure-response continuum of the novel potentiator PF-4778574. J. Pharmacol. Exp. Ther. 2013, 347, 212–224. 10.1124/jpet.113.204735. [DOI] [PubMed] [Google Scholar]
- Shaffer C. L.; Patel N. C.; Schwarz J.; Scialis R. J.; Wei Y.; Hou X. J.; Xie L.; Karki K.; Bryce D. K.; Osgood S. M.; Hoffmann W. E.; Lazzaro J. T.; Chang C.; McGinnis D. F.; Lotarski S. M.; Liu J.; Obach R. S.; Weber M. L.; Chen L.; Zasadny K. R.; Seymour P. A.; Schmidt C. J.; Hajos M.; Hurst R. S.; Pandit J.; O’Donnell C. J. The discovery and characterization of the alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor potentiator N-{(3S,4S)-4-[4-(5-cyano-2-thienyl)phenoxy]tetrahydrofuran-3-yl}propane-2-sulfona mide (PF-04958242). J. Med. Chem. 2015, 58, 4291–4308. 10.1021/acs.jmedchem.5b00300. [DOI] [PubMed] [Google Scholar]
- Gouliaev A. H.; Senning A. Piracetam and other structurally related nootropics. Brain Res. Rev. 1994, 19, 180–222. 10.1016/0165-0173(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Partin K. M. AMPA receptor potentiators: from drug design to cognitive enhancement. Curr. Opin. Pharmacol. 2015, 20, 46–53. 10.1016/j.coph.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill M. J.; Murray T. K.; Clay M. P.; Lindstrom T.; Yang C. R.; Nisenbaum E. S. LY503430: pharmacology, pharmacokinetics, and effects in rodent models of Parkinson’s disease. CNS Drug Rev. 2005, 11, 77–96. 10.1111/j.1527-3458.2005.tb00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese F.; Savino E.; Mocaer E.; Bretin S.; Racagni G.; Riva M. A. Upregulation of neurotrophins by S 47445, a novel positive allosteric modulator of AMPA receptors in aged rats. Pharmacol. Res. 2017, 121, 59–69. 10.1016/j.phrs.2017.04.019. [DOI] [PubMed] [Google Scholar]
- Ward S. E.; Harries M.; Aldegheri L.; Andreotti D.; Ballantine S.; Bax B. D.; Harris A. J.; Harker A. J.; Lund J.; Melarange R.; Mingardi A.; Mookherjee C.; Mosley J.; Neve M.; Oliosi B.; Profeta R.; Smith K. J.; Smith P. W.; Spada S.; Thewlis K. M.; Yusaf S. P. Discovery of N-[(2S)-5-(6-fluoro-3-pyridinyl)-2,3-dihydro-1H-inden-2-yl]-2-propanesulfonamide, a novel clinical AMPA receptor positive modulator. J. Med. Chem. 2010, 53, 5801–5812. 10.1021/jm1005429. [DOI] [PubMed] [Google Scholar]
- Gardinier K. M.; Gernert D. L.; Porter W. J.; Reel J. K.; Ornstein P. L.; Spinazze P.; Stevens F. C.; Hahn P.; Hollinshead S. P.; Mayhugh D.; Schkeryantz J.; Khilevich A.; De Frutos O.; Gleason S. D.; Kato A. S.; Luffer-Atlas D.; Desai P. V.; Swanson S.; Burris K. D.; Ding C.; Heinz B. A.; Need A. B.; Barth V. N.; Stephenson G. A.; Diseroad B. A.; Woods T. A.; Yu H.; Bredt D.; Witkin J. M. Discovery of the First alpha-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid (AMPA) Receptor Antagonist Dependent upon Transmembrane AMPA Receptor Regulatory Protein (TARP) gamma-8. J. Med. Chem. 2016, 59, 4753–4768. 10.1021/acs.jmedchem.6b00125. [DOI] [PubMed] [Google Scholar]
- Lee M. R.; Gardinier K. M.; Gernert D. L.; Schober D. A.; Wright R. A.; Wang H.; Qian Y.; Witkin J. M.; Nisenbaum E. S.; Kato A. S. Structural Determinants of the gamma-8 TARP Dependent AMPA Receptor Antagonist. ACS Chem. Neurosci. 2017, 8, 2631–2647. 10.1021/acschemneuro.7b00186. [DOI] [PubMed] [Google Scholar]
- Azumaya C. M.; Days E. L.; Vinson P. N.; Stauffer S.; Sulikowski G.; Weaver C. D.; Nakagawa T. Screening for AMPA receptor auxiliary subunit specific modulators. PLoS One 2017, 12, e0174742. 10.1371/journal.pone.0174742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A. S.; Witkin J. M. Auxiliary subunits of AMPA receptors: The discovery of a forebrain-selective antagonist, LY3130481/CERC-611. Biochem. Pharmacol. 2018, 147, 191–200. 10.1016/j.bcp.2017.09.015. [DOI] [PubMed] [Google Scholar]
- Kato A. S.; Burris K. D.; Gardinier K. M.; Gernert D. L.; Porter W. J.; Reel J.; Ding C.; Tu Y.; Schober D. A.; Lee M. R.; Heinz B. A.; Fitch T. E.; Gleason S. D.; Catlow J. T.; Yu H.; Fitzjohn S. M.; Pasqui F.; Wang H.; Qian Y.; Sher E.; Zwart R.; Wafford K. A.; Rasmussen K.; Ornstein P. L.; Isaac J. T.; Nisenbaum E. S.; Bredt D. S.; Witkin J. M. Forebrain-selective AMPA-receptor antagonism guided by TARP gamma-8 as an antiepileptic mechanism. Nat. Med. 2016, 22, 1496–1501. 10.1038/nm.4221. [DOI] [PubMed] [Google Scholar]
- Grimes J. M.; Hall D. R.; Ashton A. W.; Evans G.; Owen R. L.; Wagner A.; McAuley K. E.; von Delft F.; Orville A. M.; Sorensen T.; Walsh M. A.; Ginn H. M.; Stuart D. I. Where is crystallography going?. Acta Crystallogr. D Struct. Biol. 2018, 74, 152–166. 10.1107/S2059798317016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan M. C.; Furukawa H. Deeper Insights into the Allosteric Modulation of Ionotropic Glutamate Receptors. Neuron 2016, 91, 1187–1189. 10.1016/j.neuron.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.; Ji M.; Ni D.; Zhang J. Discovery of hidden allosteric sites as novel targets for allosteric drug design. Drug Discovery Today 2018, 23, 359–365. 10.1016/j.drudis.2017.10.001. [DOI] [PubMed] [Google Scholar]
- Lu S.; Zhang J. Designed covalent allosteric modulators: an emerging paradigm in drug discovery. Drug Discovery Today 2017, 22, 447–453. 10.1016/j.drudis.2016.11.013. [DOI] [PubMed] [Google Scholar]