

Abstract

In order to study the role of S1PRs in inflammatory skin disease, S1PR modulators are dosed orally and topically in animal models of disease. The topical application of S1PR modulators in these models may, however, lead to systemic drug concentrations, which can complicate interpretation of the observed effects. We set out to design soft drug S1PR modulators as topical tool compounds to overcome this limitation. A fast follower approach starting from the drug ponesimod allowed the rapid development of an active phenolic series of soft drugs. The phenols were, however, chemically unstable. Protecting the phenol as an ester removed the instability and provided a compound that is converted by enzymatic hydrolysis in the skin to the phenolic soft drug species. In simple formulations, topical dosing of these S1PR modulators to mice led to micromolar skin concentrations but no detectable blood concentrations. These topical tools will allow researchers to investigate the role of S1PR in skin, without involvement of systemic S1PR biology.
Keywords: S1PR, soft-drug, plasma stability, topical tool compound
Sphingosine-1-phosphate receptor (S1PR) agonists, such as fingolimod and ponesimod (Figure 1), initially activate S1P receptors but subsequently trigger receptor internalization and downregulation of signaling; shutting down the sphingosine-1-phosphate signaling pathway. Fingolimod was approved in 2010 for the treatment of relapsing/remitting multiple sclerosis and is the only S1PR agonist approved to date.1 It is efficacious at low doses (0.5 mg/day) and at low steady state systemic concentrations (Cmax 3.1 ng/mL). Recently the potential for the S1PR pathway to be of therapeutic use in the treatment of a range of diverse inflammatory skin diseases has emerged.2−6 Some studies have explored the skin biology of S1PR agonists by topical application of these compounds in various animal models of diseases such as atopic dermatitis,4 allergic dermatitis,5 and psoriasis.6 Topical application can, however, also lead to systemic effects. Following penetration through the stratum corneum, drugs will eventually distribute into the vasculature. If the rate of absorption exceeds the rate of elimination, topical dosing will lead to systemic drug exposure. Topical dosing of potent drugs, such as fingolimod, may lead to sufficient systemic drug concentrations to elicit measurable biological effects, complicating the interpretation of such studies.
Figure 1.
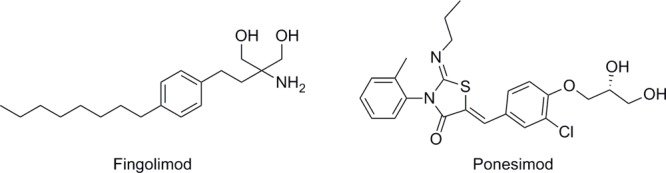
Selected S1PR modulators.
In order to remove the potential for systemic exposure, we decided to develop a soft drug S1PR modulator.7,8 Soft drugs are locally active, in this case in the skin, but are designed to undergo rapid systemic metabolism to metabolites, which are either inactive or rapidly cleared from systemic circulation.9 Due to ease of access of the diseased organ, many dermatological diseases are ideally suited to treatment with topical soft drugs, which can safely engage biological targets, previously shown to lead to adverse side effects, following oral dosing.
In their paper describing ponesimod’s discovery, Bolli et al. disclosed that phenols, such as compound 4a, although active were unsuitable for progression, as an oral drug, due to high clearance in both in vitro and in vivo experiments.10 The authors speculated that the high clearance may be due to the fact that phenols are well-known substrates for phase 2 metabolism conjugating enzymes.10 Glucuronidation is a common phase II metabolism pathway that covalently conjugates glucuronic acid, in a base-catalyzed process from UDPGA (uridine-50-diphosphoglucuronic acid) to lipophilic substrates via UGT enzymes (uridine-50-diphosphoglucuronosyl transferases).11 Sulfation, another common phase II metabolism pathway, covalently links a substrate to a sulfo group (SO3), usually derived from 3′-phosphoadenosine-5′-phosphosulfate (PAPS), via sulfotransferase enzymes.12 As the glucuronide and sulfate metabolites are highly polar, and therefore water-soluble, they subsequently undergo renal or biliary elimination. Due to their affinity for phase II metabolism, phenols are commonly used motifs when designing soft drugs.13,14 There is little evidence of clinically relevant drug-related inhibition of glucuronidation or sulfation, so the risk of drug–drug interactions is considered to be low.15 Accordingly we set out to utilize phase II metabolism pathways as the major routes of clearance for our S1PR agonist soft drugs.
Although 4a had been shown to be rapidly cleared, which was confirmed in our hands (Table 1), the compound displayed poor aqueous solubility. Aqueous solubility is an important parameter for topically applied drugs as it can support use in higher water content formulations, such as a creams, which may be preferred by patients over oily formulations like ointments. We therefore set out to improve the aqueous solubility of 4a.
Table 1. Optimization of the Thiazolidinone Core.
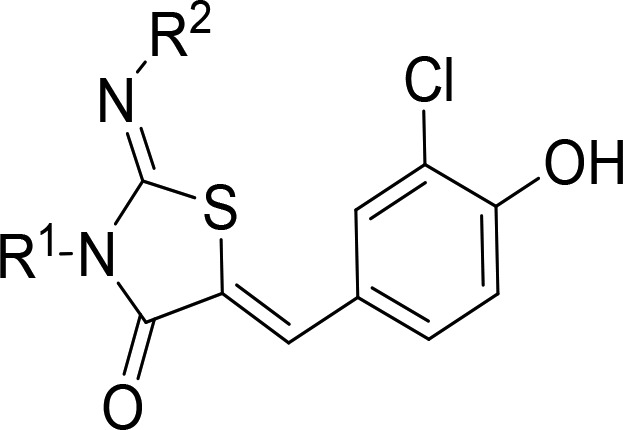

Racemic mixture.
Reverse-phase HPLC method to determine the chromatographic hydrophobicity index (CHI): n of 1.
The aqueous kinetic solubility of the test compounds was measured using laser nephelometry: n of 1.
Human S1PR1 activity was measured using a human PathHunter β-Arrestin recruitment assay. All pIC50s reported in this table correspond to n ≥ 2, reported as their geometric mean.
Keeping the 3-chloro-4-hydroxybenzylidene motif from 4a constant, we synthesized a series of phenols with different substituents to replace the 2-tolyl 4a motif with aromatic or aliphatic groups (Scheme 1). Using Method A, the appropriate aniline was reacted with 2-chloroacetyl chloride to give the corresponding 2-chloro-N-phenylacetamide, which was condensed with 1-isothiocyanatopropane to give the required thiazolidinone core 2a,b. Subsequent condensation with 3-chloro-4-hydroxybenzaldehyde 3 generated compounds 4a,b. Compounds 4c–g with aliphatic R1 groups used Method B, where amines 1c–g were reacted with 1-isothiocyanatopropane, then with 2-bromoacetyl bromide in the same reaction vessel. The resulting thiazolidinone cores 2c–g were condensed with 3-chloro-4-hydroxybenzaldehyde 3, and the products 4c–g were obtained using preparatory HPLC. Compound 4h was prepared by BBr3 demethylation of the anisole 4b to give the corresponding phenol.
Scheme 1.
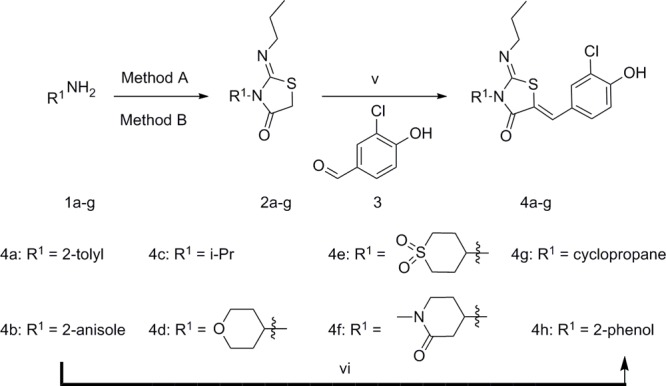
Method A (i) 2-chloroacetyl chloride, TEA, THF, −78 °C to RT, 2 h; (ii) 1-isothiocyanatopropane, NaH, DMF, RT, 16 h. Method B (iii) 1-isothiocyanatopropane, CH2Cl2, RT, 2 h; (iv) 2-bromoacetyl bromide, pyridine, CH2Cl2, 0 °C to RT, 1 h; (v) NaOAc, AcOH, 65 °C, 16 h; (vi) BBr3, CH2Cl2, −70 °C to 0 °C, 3 h.
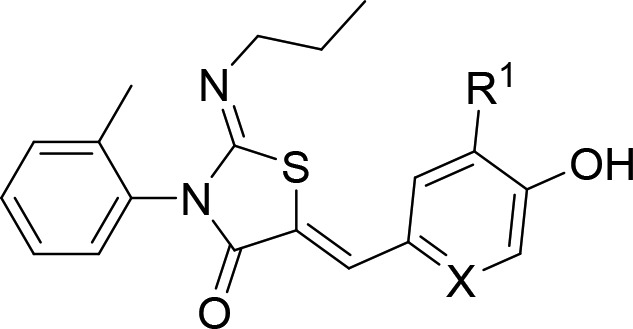
Compounds 9a–9e replaced the n-propyl group of 4a with several small N-linked aliphatic substituents, while compounds 9f–l looked at effects of substituents on the 4-hydroxybenzylidene group (Scheme 2). The appropriately substituted thiazolidin-4-one core 7a–d was synthesized utilizing a one-pot, two-step reaction. Alkyl amines were reacted with 1-isothiocyanato-2-methylbenzene 5 to give the resulting thioureas 6a–d, which condensed with 2-bromoacetyl bromide, followed by addition of pyridine to furnish the desired thiazolidin-4-one. The thiazolidin-4-one cores 6a–d were condensed with the 4-hydroxybenzaldehyde 3 to give 9a–d. Compound 9e was synthesized by treatment of 9c with BBr3. Compound 2a was reacted with 8f,g,i–l to furnish products 9f,g,i–l. Compound 9h was synthesized using a Negishi coupling with dicyanozinc and palladium tetrakis from 9f.
Scheme 2.
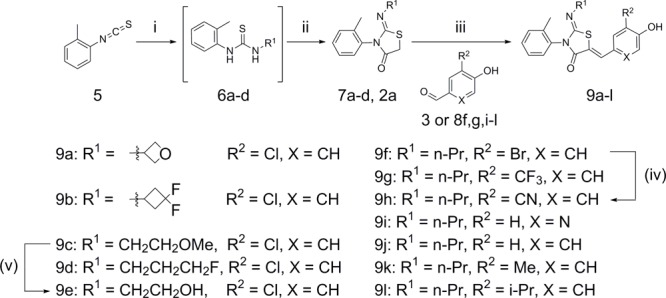
(i) R2NH2, CH2Cl2, RT, 1 h; (ii) 2-bromoacetyl bromide, pyridine, CH2Cl2, 0 °C to RT, 2 h; (iii) NaOAc, AcOH, 65 °C, 16 h; (iv) dicyanozinc, Pd(PPh3)4, DMA 100 °C, 1.5 h; (v) BBr3, DCM, −78 °C, 3 h then 0 °C, 3 h.
The configuration of the double bonds in ponisimod and 4a were determined by X-ray crystallography.10 The HMBC and NOESY data of ponesimod and 4a were compared with 9k and 10a (see Supporting Information). The HMBC data for the alkene proton to the carbonyl carbon (H9–C3 or H9′–C3′) in all cases was consistent and suggested a Z double bond arrangement of the alkene bond (the size of the 1H–13C coupling constant was estimated to be 6–7 Hz). The only cross peaks observed in the NOESY experiments were between the 2-tolyl and imine groups. These weak signals between the respective methyl groups (see Supporting Information) were also observed for ponesimod, 4a, 9k, and 10a. It may be expected that if the imine was in the E configuration that there would have been cross peaks observed between the methyl of the 2-tolyl group and the NCH2 protons of the imine group; however, this was not observed. Taken together, the data was consistent with the Z configuration observed using X-ray crystallography but did not confirm it. Based on the analysis of analogous compounds 4a–h, 9a–l, and 10a–i were assigned to the Z,Z-isomer, unless stated otherwise.
Compounds 4c–h and 9a–e were designed to improve solubility by reducing logD or aromatic ring count.16 Although the CHIlogD values were lower or equivalent for 4d, 4e, 4g, and 9a–e, the compounds did not show an improvement in aqueous solubility (Table 1). Reducing the aromatic ring count in 4c–e and 4g also failed to improve aqueous solubility, while 4f gave an improvement in aqueous solubility 250 μM possibly due to a 3-log unit reduction in CHIlogD but had a pIC50 of <6.0. The addition of a 2-phenol group into the R1 position, compound 4h, lowered the CHIlogD by 1.1 units and improved aqueous solubility to 220 μM. Two of the changes to the 2-propylimino group showed an improvement in aqueous solubility. Compound 9a with a 2-oxetan-3-ylimino group moderately increased solubility to 150 μM, compared to 79 μM for 4a. Compound 9e gave an improvement in aqueous solubility (>250 μM) presumably due to the addition of the polar hydroxyl-group and the commensurate reduction in CHIlogD, but unfortunately, the compound had a pIC50 of <6.0. The fact that 9a improves aqueous solubility and was equipotent identifies the 2-oxetan-3-ylimino group as a potentially useful change to incorporate in the design of future compounds.
Having examined two of the vectors off the thiazolidin-4-one core, we turned our attention to the benzylidene substituent to optimize activity, aqueous solubility, and hepatic metabolism. For reason of synthetic expediency, we kept the 2-tolyl and n-propyl groups in place with the intention of combining the optimum substituents in subsequent design rounds. We therefore synthesized a series of phenols (9f–9l) using the method shown in Scheme 2. Compounds 9f–9l contained a range of electron withdrawing and donating groups ortho to the 4-phenol of the benzylidene substituent. Compounds 9f, 9g, and 9i–9k were largely equipotent to 4a, while 9h and 9l had a pIC50 of <6.0, presumably in the case of 9l due to increased steric bulk (Table 2). The trifluoromethyl group of 9g had low aqueous solubility, while 9f and 9h–9l had acceptable solubility.
Table 2. Effect of Substituents on the Phenol.
| compound | R1 | X | H S1PR1 pIC50a | Kinetic Solubility (μM)b | pKac | HLM Cld | H Heps Cle | stabilityf | CHI logDg |
|---|---|---|---|---|---|---|---|---|---|
| 4a | Cl | CH | 7.4 | 79 | 6.8 | 1.6 | 8.4 | 6 | 3.8 |
| 9f | Br | CH | 7.7 | 79 | 6.4 | 1.6 | 6.0 | 16 | 3.9 |
| 9g | CF3 | CH | 7.5 | 14 | 6.6 | 1.0 | 4.2 | 10 | 3.8 |
| 9h | CN | CH | <6.0 | 220 | <0.5 | 20 | |||
| 9i | H | N | 7.0 | 110 | 7.1 | 2.8 | 7.9 | 4 | 3.0 |
| 9j | H | CH | 7.1 | 79 | 8.3 | 2.4 | 1.7 | 0 | 3.5 |
| 9k | Me | CH | 7.6 | 78 | 8.5 | 2.8 | 31 | 3 | 3.8 |
| 9l | i-Pr | CH | <6.0 | 79 | 8.6 | 12 | 3 | 4.3 |
Human S1PR1 activity was measured using a human PathHunter β-Arrestin recruitment assay. All pIC50s reported in this table correspond to n ≥ 2, reported as their geometric mean.
The aqueous kinetic solubility of the test compounds was measured using laser nephelometry: n = 1.
pKa was determined using a potentiometric fast UV-metric titration method: n = 1.
Intrinsic clearance in human liver microsomes (mL/min/g): n = 1.
Intrinsic clearance in human liver hepatocytes (mL/min/g): n = 1.
% decrease in purity when stored in DMSO solution for 28 days: n = 1.
Reverse-phase HPLC method to determine the chromatographic hydrophobicity index (CHI): n = 1.
As soft drugs must be rapidly cleared systemically and phenols commonly undergo phase 2 metabolism, we used human hepatocytes (H Heps) to study this potential route of metabolism. We sought to obtain clearance rates of greater than 85% human liver blood flow (>4.8 mL/min/g); data shown in Table 2. We then measured intrinsic clearance in human liver microsomes (HLM) to determine if phase 1 metabolism was contributing to the observed intrinsic clearance in hepatocytes. As glucuronidation is a base-catalyzed process, where conserved carboxylate and histidine residues facilitate the deprotonation of the phenol, we expected to see an effect of the pKa of the phenolic hydrogen on the rate of hepatic clearance.14 We explored the effect of the phenol pKa on hepatic clearance with a set of ortho-substituents and a meta-pyridine (Table 2). Electron-withdrawing groups did reduce the pKa of the phenol 4a, and 9f–i have increased hepatic clearance rates vs unsubstituted 9j. However, weakly electron-donating groups demonstrated an even greater increase in hepatic clearance rates (9k and 9l) despite the expected increase in pKa. For this phenolic scaffold, ortho-substituents led to an increase in glucuronidation rate in all cases and was independent of phenolic pKa.
Although several compounds shown in Table 2 satisfy the rapid clearance requirements of a soft drug and retain primary activity, none were suitable for progression into in vivo studies due to chemical stability liabilities. Analysis of the originally pure compounds shown in Table 1, after being in DMSO solution for 28 days, showed a range of purities. Compounds with electron-withdrawing groups ortho to the phenol (4a, 9f–h) were the least stable with a 6–20% impurity formed over 28 days. Compounds with neutral or donating groups in the ortho position (9j–l) were more stable, in some cases giving compounds that were stable over a 28-day period (9j). The instability in solution represented a major development hurdle as topical drugs are usually stored in solution or suspensions (cream, ointment, paste, lotion, or gels), rather than in solid form, as is the case for oral drugs. We turned our attention to identifying the impurity and preventing its formation.
We conducted NMR studies of compound 9h after incubation in DMSO-d6 for 6 months (see Supporting Information for HMBC and NOESY spectra). In that time the impurity had increased from 20% to 32% of the mixture based on the integration of H18 vs H18′ in the 1H NMR spectrum. The NOESY spectrum of the mixture indicated no changes in the arrangement of the imine (no correlation was observed between H11–H19 or H11′–H19′). HMBC experiments measuring the three bond coupling constant between H9–C3 and H9′–C3′ were analyzed and confirmed that the double bond in the major component (68%) had a coupling constant of 6.4 Hz indicating a Z arrangement, while in the minor component (32%) the coupling was measured at 11.9 Hz indicating an E arrangement.
As we expected them to be significantly less active due to the orientation of the phenol group, no examples of (Z,E) compounds were isolated.
The association between electron withdrawing groups and the rate of the isomerization could be explained by the requirement for a base-catalyzed isomerization mechanism (see Supporting Information for proposed mechanism). We hypothesized that protecting the phenol via alkylation (as in ponesimod) or acylation would remove the ability of the conjugated pi-system to isomerize the double bond from Z to E. To test this theory we synthesized compounds 10a–i via esterification of the parent phenol (Scheme 3). Reaction of phenol 9k,f,g with the corresponding acid chloride gave compounds 10a–h. Reaction of phenol 9k with dimethylpropanoic acid and DCC gave compound 10i.
Scheme 3.
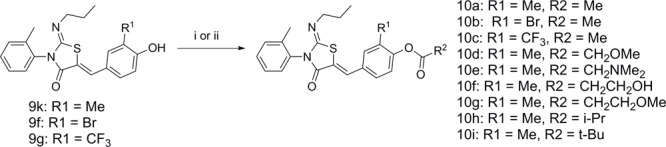
(i) Acid chloride (1 equiv), DMAP (0.05 equiv), TEA (1.2 equiv) in CH2Cl2 at rt, 16 h; (ii) 2,2-dimethylpropanoic acid (1 equiv), DCC (1.2 equiv), DMAP (0.2 equiv), DMF, 40 °C, 16 h.
We were delighted to discover that acylation blocked isomerization and 10a,f shown in Table 3 did not isomerize after being in a DMSO solution for 28 days. Electron-withdrawing groups at R1 (10b,c) did lead to a slight decrease in purity (6.3 and 1.4%, respectively) over the 28-day duration of this experiment, but the degradation product was due to hydrolysis of the ester to the phenol rather than isomerization of the double bond. Decomposition studies used 1H NMR to monitor the increase in the acetic acid methyl group peak over 28 days (see Supporting Information). Compound 10a showed no hydrolysis or isomerization. However, the chemical stability was poor when heteroatoms were alpha to the carbonyl of the acetate group 10d,e, and these compound degraded on standing within 24 h, preventing full characterization. Chemically, instability was not an issue when the heteroatoms were in the beta position 10f,g.
Table 3. Effect of Substitution at R1 and R2 on Skin S9 and Chemical Stability.
| compound | R1 | R2 | H Skin S9 (half-life min)b | stability (% decrease)c |
|---|---|---|---|---|
| ponesimod | Cl | >180 | 0 | |
| 10a | Me | Me | 7.3 | 0 |
| 10b | Br | Me | 8.8 | 6.3 |
| 10c | CF3 | Me | 8.3 | 1.4 |
| 10d | Me | CH2OMe | a | |
| 10e | Me | CH2NMe2 | a | |
| 10f | Me | CH2CH2OH | 8.2 | 0 |
| 10g | Me | CH2CH2OMe | 4.8 | |
| 10h | Me | i-Pr | 21 | |
| 10i | Me | t-Bu | >180 |
Unstable after 24 h in DMSO solution: n = 1.
Stability measured in skin S9 over 180 min in the presence of enzymatic cofactors: n = 1.
% loss in purity when stored in DMSO solution for 28 days: n = 1.
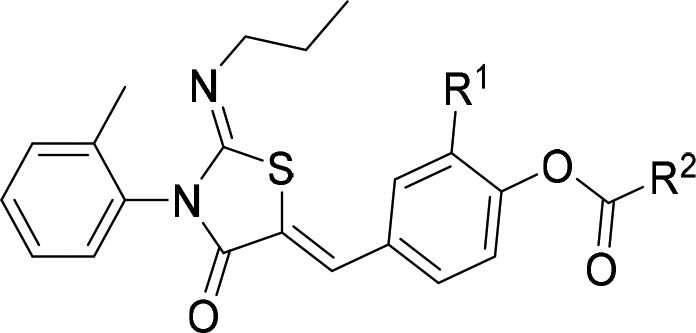
We expected the ester to be unstable in skin, which would liberate the phenol to engage the receptor in the target tissue. Determining skin stability using human skin S9 fraction (Table 3) demonstrated compounds 10a–c,f,g underwent rapid metabolism. Bulking out the ester with i-Pr 10h or t-Bu 10i gave longer half-lives as expected.
Unfortunately, acylation of 9k led to a decrease in aqueous solubility, for example, 10a (39 μM) and 9k (79 μM). This could be improved by adding polar groups as in 10f (79 μM); however, solubility only improved to the level of phenol 4a and is still lower than what is desirable in a topical drug. Our identification of the propensity for the phenol series to isomerize meant they were unsuitable for development as tool compounds or potential drugs. The acylated series does not have the desired solubility of a potential drug but is suitable for use as a topical tool.
Compound 10a was selected for further study, due its ease of synthesis, stability to degradation in solution, and instability in skin.
The selectivity of (Z,Z)-10a and (Z,Z)-9k across S1PR1–4 was determined (Table 4). As with ponesimod,10 both (Z,Z)-10a and (Z,Z)-9k were most active against S1PR1, with >40- and >80-fold selectivity, respectively, over the other S1PR isoforms measured. Compounds (Z,Z)-10a and (Z,Z)-9k were equipotent. The reactivity of the exocyclic double bond of (Z,Z)-10a was examined using a glutathione trapping experiment in human liver microsomes; no evidence of glutathione adducts or derivatives was observed (see Supporting Information).
Table 4. Selectivity against S1PR1-4a.
| compound | S1PR1 pIC50 | S1PR2 pIC50 | S1PR3 pIC50 | S1PR4 pIC50 |
|---|---|---|---|---|
| ponesimodb | 8.2 | <5.0 | 7.0 | 6.0 |
| 10a | 7.6 | <5.0 | 6.0 | <5.0 |
| 9k | 8.0c | <5.0 | 6.1 | <5.0 |
S1PR1–4 activity was measured using a human PathHunter β-Arrestin recruitment assay (n = 2).
S1PR1–4 activity reported in the literature using a GTPγS assay.9
To demonstrate (Z,Z)-10a is a suitable tool for in vivo experiments, a topical pharmacokinetic experiment in mice (see Supporting Information) using 22.5 μL of 1% propylene glycol/ethanol 7/3 formulation was carried out. At 2 and 8 h time points, (Z,Z)-10a concentrations in blood were below the lower limit of quantification (LLoQ). Compound (Z,Z)-10a’s concentrations in blood were 8.8 μM (2 h) and 4.9 μM (8 h). Compound (Z,Z)-9k’s concentrations were below the LLoQ in blood at both time points and 134 μM (2 h) and 101 μM (8 h) in the skin. Compound (Z,Z)-9k is present in the skin of mice at >10,000-fold above the IC50 demonstrating that the modulator is likely to be present at sufficient concentration to inhibit local S1PR1. Compound (Z,Z)-10a is also present in the skin of mice at >350-fold above the IC50 and will also be able to locally inhibit S1PR1.
Metabolite identification of (Z,Z)-10a using incubation with human skin S9 fraction confirmed that the expected phenol (Z,Z)-9k was obtained after hydrolysis of the ester group: no other metabolites were observed (Figure 2a). Based on the stability of (Z,Z)-9k in DMSO over 28 days (Table 2), it is likely this hydrolysis is enzymatically driven. We then performed metabolite identification studies using (Z,Z)-9k in human hepatocytes to confirm the routes of clearance of our S1PR1 modulators. As before, (Z,Z)-9k isomerizes into (Z,E)-9k in solution; Figure 2b shows the disappearance of parent phenol (both (Z,Z)-9k orange and (Z,E)-9k green isomeric forms) and identifies the glucuronide conjugation product, hydroxylation products, and hydroxylation with sulfation metabolites.
Figure 2.

(a) Metabolite identification of (Z,Z)-10a in human skin S9 fraction (n = 1). (b) Metabolite identification of (Z,Z)-9k and (Z,E)-9k in human hepatocytes (n = 1). (c) Depiction of the enzymatic hydrolysis of (Z,Z)-10a and the hepatic metabolism of (Z,Z)-9k and (Z,E)-9k.
In conclusion, we have used a fast follower approach to identify several highly cleared and active phenolic S1PR1 modulators. Many of the phenol soft drugs were unstable in solution due to isomerization. We were able to prevent this isomerization by acylation of the phenol, to deliver chemically stable chemical tools. The strategy underpinning our S1PR1 soft drug modulators is illustrated in Figure 2c. When (Z,Z)-10a is applied to the skin of mice, it should be enzymatically hydrolyzed to give (Z,Z)-9k. At this point, 9k can bind to S1PR1 in the epidermis causing receptor internalization and degradation. Compound (Z,Z)-9k will also start to slowly isomerize to (Z,E)-9k. The mixture of isomers of 9k would then enter the bloodstream and be distributed to the liver, where it would be rapidly metabolized and cleared. The hepatic intrinsic clearance rate for phenol (Z,Z)-9k, 31 mL/min/g (Table 2), would correspond to 97% liver blood flow if there is a good in vitro to in vivo correlation, predicting that a single pass through the liver could eliminate the majority of the drug, greatly reducing the risk of systemic on-target toxicities, which to date have limited the use of S1PR modulators.
Compound (Z,Z)-10a provides the community with a valuable new tool that will enable targeted studies of S1PR biology in skin, lung, or other suitable tissues.
Glossary
ABBREVIATIONS
- S1PR
sphingosine-1-phosphate receptor;
- IL
interleukin
- PASI
psoriasis area and severity index
- UDPGA
uridine-50-diphosphoglucuronic acid
- UGT
uridine-50-diphosphoglucuronosyl transferase
- HPLC
high pressure liquid chromatography
- rt
room temperature
- TEA
triethylamine
- THF
tetrahydrofuran
- DMF
dimethylformamide
- CHI
chromatographic hydrophobicity index
- HLM
human liver microsomes
- DMSO
dimethyl sulfoxide
- Hz
hertz
- DMAP
dimethylaminopyridine
- DCC
N,N′-dicyclohexylcarbodiimide
- DMA
dimethylacetamide
- HMBC
Heteronuclear Multiple Bond Correlation
- HSQC
Heteronuclear Single Quantum Correlation
- NOESY
Nuclear Overhauser Effect Spectroscopy
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00616.
Experimental and characterization data for all new compounds and all biological and DMPK methods (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the Wellcome Trust [098439/Z/12/Z].
The authors declare no competing financial interest.
Supplementary Material
References
- Kappos L.; Radue E. W.; O’Connor P.; Polman C.; Hohlfeld R.; Calabresi P.; Selmaj K.; Agoropoulou C.; Leyk M.; Zhang-Auberson L.; Burtin P. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 387–401. 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- Kunkel G. T.; Maceyka M.; Milstien S.; Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discovery 2013, 12, 688–702. 10.1038/nrd4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W. C.; Nagahashi M.; Terracina K. P.; Takabe K. Emerging Role of Sphingosine-1-phosphate in Inflammation. Biomolecules 2013, 3, 408–434. 10.3390/biom3030408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Japtok L.; Baumer W.; Kleuser B. Sphingosine-1-phosphate as signaling molecule in the skin: Relevance in atopic dermatitis. Allergo. J. Int. 2014, 23, 54–59. 10.1007/s40629-014-0008-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reines I.; Kietzmann M.; Mischke R.; Tschernig T.; Luth A.; Kleuser B.; Baumer W. Topical application of sphingosine-1-phosphate and FTY720 attenuate allergic contact dermatitis reaction through inhibition of dendritic cell migration. J. Invest. Dermatol. 2009, 129, 1954–1962. 10.1038/jid.2008.454. [DOI] [PubMed] [Google Scholar]
- Schaper K.; Dickhaut J.; Japtok L.; Kietzmann M.; Mischke R.; Kleuser B.; Baumer W. Sphingosine-1-phosphate exhibits anti-proliferative and anti-inflammatory effects in mouse models of psoriasis. J. Dermatol. Sci. 2013, 71, 29–36. 10.1016/j.jdermsci.2013.03.006. [DOI] [PubMed] [Google Scholar]
- Bodor N.; Buchwald P. Retrometabolic drug design: Principles and recent developments. Pure Appl. Chem. 2008, 80, 1669–1682. 10.1351/pac200880081669. [DOI] [Google Scholar]
- Bell M.; Foley D.; Naylor C.; Robinson C.; Riley J.; Epemolu O.; Scullion P.; Shishikura Y.; Katz E.; McLean W. H. I.; Wyatt P.; Read K. D.; Woodland A. Discovery of super soft-drug modulators of sphingosine-1-phosphate receptor 1. Bioorg. Med. Chem. Lett. 2018, 28, 3255–3259. 10.1016/j.bmcl.2018.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felding J.; Sorensen M. D.; Poulsen T. D.; Larsen J.; Andersson C.; Refer P.; Engell K.; Ladefoged L. G.; Thormann T.; Vinggaard A. M.; Hegardt P.; Sohoel A.; Nielsen S. F. Discovery and Early Clinical Development of 2-{6-[2-(3,5-Dichloro-4-pyridyl)acetyl]-2,3-dimethoxyphenoxy}-N-propylacetamide (LEO 29102), a Soft-Drug Inhibitor of Phosphodiesterase 4 for Topical Treatment of Atopic Dermatitis. J. Med. Chem. 2014, 57, 5893–5903. 10.1021/jm500378a. [DOI] [PubMed] [Google Scholar]
- Bolli M. H.; Abele S.; Binkert C.; Bravo R.; Buchmann S.; Bur D.; Gatfield J.; Hess P.; Kohl C.; Mangold C.; Mathys B.; Menyhart K.; Muller C.; Nayler O.; Scherz M.; Schmidt G.; Sippel V.; Steiner B.; Strasser D.; Treiber A.; Weller T. 2-Imino-thiazolidin-4-one Derivatives as Potent, Orally Active S1P(1) Receptor Agonists. J. Med. Chem. 2010, 53, 4198. 10.1021/jm100181s. [DOI] [PubMed] [Google Scholar]
- Fisher M. B.; Paine M. F.; Strelevitz T. J.; Wrighton S. A. The role of hepatic and extrahepatic UDP-glucuronosyltransferases in human drug metabolism. Drug Metab. Rev. 2001, 33, 273–297. 10.1081/DMR-120000653. [DOI] [PubMed] [Google Scholar]
- Falany C. N.Human Cytosolic Sulfotransferases. In Drug Metabolism and Transport: Molecular Methods and Mechanisms, 1st ed.; Lash L. H., Ed.; Humana Press: Totowa, NJ, 2005; pp 341–378. [Google Scholar]
- Jones P.; Storer R. I.; Sabnis Y. A.; Wakenhut F. M.; Whitlock G. A.; England K. S.; Mukaiyama T.; Dehnhardt C. M.; Coe J. W.; Kortum S. W.; Chrencik J. E.; Brown D. G.; Jones R. M.; Murphy J. R.; Yeoh T.; Morgan P.; Kilty I. Design and Synthesis of a Pan-Janus Kinase Inhibitor Clinical Candidate (PF-06263276) Suitable for Inhaled and Topical Delivery for the Treatment of Inflammatory Diseases of the Lungs and Skin. J. Med. Chem. 2017, 60, 767–786. 10.1021/acs.jmedchem.6b01634. [DOI] [PubMed] [Google Scholar]
- Glossop P. A.; Watson C. A.; Price D. A.; Bunnage M. E.; Middleton D. S.; Wood A.; James K.; Roberts D.; Strang R. S.; Yeadon M.; Perros-Huguet C.; Clarke N. P.; Trevethick M. A.; Machin I.; Stuart E. F.; Evans S. M.; Harrison A. C.; Fairman D. A.; Agoram B.; Burrows J. L.; Feeder N.; Fulton C. K.; Dillon B. R.; Entwistle D. A.; Spence F. J. Inhalation by design: novel tertiary amine muscarinic M(3) receptor antagonists with slow off-rate binding kinetics for inhaled once-daily treatment of chronic obstructive pulmonary disease. J. Med. Chem. 2011, 54, 6888–6904. 10.1021/jm200884j. [DOI] [PubMed] [Google Scholar]
- Williams J. A.; Hyland R.; Jones B. C.; Smith D. A.; Hurst S.; Goosen T. C.; Peterkin V.; Koup J. R.; Ball S. E. Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab. Dispos. 2004, 32, 1201–1208. 10.1124/dmd.104.000794. [DOI] [PubMed] [Google Scholar]
- Ritchie T. J.; Macdonald S. J. The impact of aromatic ring count on compound developability--are too many aromatic rings a liability in drug design?. Drug Discovery Today 2009, 14, 1011–1020. 10.1016/j.drudis.2009.07.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.